


Abstract
In bacteria, the assembly factors tightly orchestrate the maturation of ribosomes whose competency for protein synthesis is validated by translation machinery at various stages of translation cycle. However, what transpires to the quality control measures when the ribosomes are produced with assembly defects remains enigmatic. In Escherichia coli, we show that 30S ribosomes that harbour assembly defects due to the lack of assembly factors such as RbfA and KsgA display suboptimal initiation codon recognition and bypass the critical codon–anticodon proofreading steps during translation initiation. These premature ribosomes on entering the translation cycle compromise the fidelity of decoding that gives rise to errors during initiation and elongation. We show that the assembly defects compromise the binding of initiation factor 3 (IF3), which in turn appears to license the rapid transition of 30S (pre) initiation complex to 70S initiation complex by tempering the validation of codon–anticodon interaction during translation initiation. This suggests that the premature ribosomes harbouring the assembly defects subvert the IF3 mediated proofreading of cognate initiation codon to enter the translation cycle.
INTRODUCTION
The bacterial ribosome is a large ribonucleoprotein complex consisting of two asymmetrical subunits 30S and 50S, respectively. The subunits are composed of three ribosomal RNAs (5S, 16S and 23S) and >50 ribosomal proteins (r-Proteins). Ribosomes are synthesized by a highly regulated process, which is collectively referred to as ribosome biogenesis and involves the synthesis and the concomitant assembly of ribosomal components. The orchestrated association of r-Proteins with rRNA—termed as ribosome assembly—is catalysed by several non-ribosomal proteins called as ribosome assembly factors (RAF) (1,2). RAFs are involved in end processing of the premature rRNA (3–6) and rRNA modifications like pseudouridylation (Ψ) and methylation (7–10). Additionally, RAFs assist in rRNA folding and positioning of r-Proteins thus overcoming energy barriers and kinetic traps during assembly (11–13). Finally, RAFs also mark important maturation events during assembly (14) thus driving the creation of functional ribosomal subunits.
The subunits that endure full maturation become competent to enter the translation cycle where, the 30S subunit decodes the genetic message and the 50S subunit catalyses the peptide bond formation. The 30S subunit interacts with mRNA, tRNA and initiation factors (IFs) to form the pre-initiation complex (30S-PIC). During this process, the IFs perform critical quality control tests on codon–anticodon (CO–AC) triplets on the mRNA and the tRNA, which if validated, leads to the formation of the 30S-IC (15–17). The 30S-IC associates with 50S subunit to form the 70S initiation complex (70S-IC), which eventually enters the elongation phase of translation and drives protein synthesis. Disruptions in translation are known to hinder the biogenesis of ribosomes by skewing the ration of r-Proteins to r-RNA (18,19), but the functional consequences of assembly defects on translation are yet to be fully understood. Additionally, the suboptimal maturation of ribosomes in humans is implicated in several human disorders collectively referred to as ribosomopathies (20–22). In bacteria, assembly defects arising due to deletion of RAFs are manifested as compromised growth phenotype, sensitivity to cold temperatures as well as accumulation of ribosome assembly intermediates harbouring premature rRNA as well as a suboptimal r-Protein complement (1,2,8,11,12,23–26). Efforts to understand the nature of structural defects in assembly intermediates in bacteria are undermined by pleiotropy that is associated with the deletion of RAFs and the presence of multiple pathways for the maturation of ribosomes (27–30). Further, little is known about the metabolic cost of harbouring premature subunits and their ultimate fate in bacterial cells. Although studies have shed light on how cells regulate the number of active ribosomes and maintain fitness in nutrient-limiting conditions (31–35), such knowledge is virtually unknown for cells with compromised ribosome structure and composition.
It is believed that subunit association may be impaired in premature ribosomes, which lack fully formed bridging sites, and this, in turn, may arrest their entry into translation (8,36,37). In contrast, it has also been observed that mutations in rRNA, lack of r-Proteins or the absence of assembly factors like KsgA and Hfq induce defects during translation (38–41). Studies have also hypothesized that perturbed ribosome maturation may create a bottleneck that decreases new translation initiation events (42). This would result in the suppression of translation or alternate scenarios wherein premature subunits may compete with mature subunits for the mRNA, thus averting translation once again. The spectrum of studies suggests that participation of premature ribosomes in translation may have energetic implications that affect cellular fitness. However, mechanistic details of such relationships are yet to be uncovered.
Here, we set out to address some of the primary outstanding questions: Do premature ribosomes enter translation? If so, how such ribosomes bypass the quality control mechanisms in bacteria? To address these questions, we employed the 30S subunit from Escherichia coli as a model system and assessed how the defects in the 30S assembly engender errors in translation initiation and elongation. Using a series of biochemical and next-generation sequencing experiments, we establish that premature ribosomes indeed enter the translation cycle but with compromised fidelity in decoding. Our investigation led us to establish that the assembly defects weaken the recognition by the IFs, especially IF3, and thereby permits this bypass of quality control mechanisms.
MATERIALS AND METHODS
Creation of strains and plasmids
The Keio collection parent strain BW25113 referred to as Wt was used as the parental strain for all genetic manipulations and reference measurements. Null mutant for LepA was procured from the Coli genetic stock centre (CGSC). Additionally, null mutants for RbfA, RsgA and KsgA were created using the λ Red recombineering method (Supplementary Table S1 and S2) (43).
Gene encoding GFP was amplified from the vector 1GFP (Addgene #29663) with four different start codons i.e. AUG, CUG, AUA and AGG. Additionally, frameshift mutations (+1 base and −1 base) were introduced at codon 7 of the GFP construct using oligonucleotides. Similarly, codons 7 and 8 were replaced with UAG and UAA codons. The modified GFP constructs were individually cloned into vector 8R (Addgene # 37506) using Xba1/Nhe1 and BamH1 specific restriction sites. Similarly, the constructs used for studying translation kinetics were created by amplifying the gene encoding β-galactosidase (bgal) from E. coli B cells. Modifications were also introduced into this gene by incorporating different start codons (39,44). The amplified cassettes were cloned into vector pQE2 using XhoI/HindIII sites.
In order to complement assembly factors (RAFs) or initiation factors (IFs), respective genes were amplified from Wt cells and cloned into a p15A vector backbone under an Anhydrotetracycline (Atc) inducible promoter.
Genomic DNA from Wt was used to amplify the gene encoding IF3. The amplified fragment was cloned into the vector 1R using the Ssp1 site to generate the vector p1R-IF3. This placed the gene under a T7 inducible promoter and the gene encoded an N-terminally Strep tagged IF3.
The plasmid employed for tRNAifmet overproduction was created using a construct carrying the E. coli lpp gene promoter region fused to the E. coli fmt gene. The construct was cloned into the vector pQE2 using Xho1/BamH1 sites thus placing the fmt gene under a promoter that would confer constitutive expression. All constructs were verified by Sanger sequencing. A list of constructs, strains and primers used in this study is presented in the Supplementary section (Supplementary Tables S1 and S2).
Growth analysis
Primary inoculums were always prepared by growing freshly transformed colonies in LB medium with respective antibiotics at 37°C with shaking at 180 rpm. The next day, OD600 of primary cultures was normalized. These cultures were then diluted into 1 ml fresh LB medium with appropriate antibiotics in a 24-well plate followed by incubation at 37°C with regular shaking. OD600 measurements were taken in Tecan infinite M200 multimode plate reader at every 30-minute interval until cultures reached saturation. Additionally, in order to study growth in the presence of elevated levels of IFs or RAFs, LB medium was supplemented with 50 ng/ml Atc at the time of inoculation to trigger protein production. The experiment was repeated at least three times to derive the average growth curves.
GFP fluorescence measurements and calculation of indices
Wt or null mutant was freshly transformed using the desirable plasmid carrying the gene encoding GFP before initiating the experiments. For each measurement, three colonies were picked in fresh LB medium supplemented with 100 μg/ml ampicillin (Amp) and allowed to grow at 37°C and 180 rpm till OD600 reached 0.6. At this point, gfp expression was induced by adding 2 mM arabinose and cells were allowed to grow for another 3 h. Upon completion, cells were harvested and lysed in Buffer G (20 mM Tris–HCl (pH 7.5 at 25°C), 500 mM NaCl, 1 mM EDTA, 1 mM PMSF, 6 mM β-ME) supplemented with 1X CelLytic B solution (Sigma Aldrich). The lysates were normalized for total protein content and were taken for fluorescence measurements. All fluorescence spectrums were generated by exciting at 488 nm and scanned for emission from 500 to 600 nm, with averaging over three scans after baseline correction in a FluoroMax-4 spectrofluorometer (Horiba Scientific, Edison, NJ, USA). The slit width used for excitation and emission was 2 and 7 nm, respectively. All fluorescence experiments were performed with five independent trials. For measurement of Initiation Error Index, GFP emission from null mutants was compared with that of Wt cells. Similarly, Initiation Rescue Index was calculated as the ratio of GFP fluorescence after complementing the null mutant with respect to Wt. Briefly, cells were co-transformed using plasmids carrying genes coding for GFP and assembly or initiation factor, respectively. The cells were grown on a dual selection marker at 37°C and induced with Atc to trigger the production of the RAF or IF when OD600 reached 0.3. Subsequently, GFP production was induced with 2 mM arabinose when OD600 of the Atc induced cells reached 0.6. Further, the cells were processed for fluorescence measurements as described above. The indices were defined as follows:
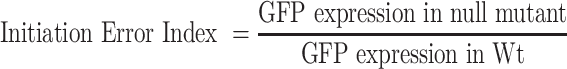 |
 |
Mean indices and standard deviations were calculated from three biological replicates and were subsequently plotted.
Determination of initiation and elongation rates from β-galactosidase (bgal) production kinetics
In order to calculate the translation initiation and elongation rates, the bgal production assay (45–47) was performed with minor modifications. Briefly, Wt or null mutant was transformed using respective constructs that encoded bgal (variants containing different start codons: pAUG-bgal or pAGG-bgal). Three independent biological replicates were prepared by diluting overnight grown cultures into fresh 5 ml LB medium supplemented with 100 μg/ml Amp and allowed to grow at 37°C and 180 rpm till OD600 reached 0.6. These cultures were then shifted to a water bath at 37°C or 25°C (RT). Bgal production was induced with 1 mM IPTG followed by instant mixing for 5 s. Following this, aliquots of 200 μl each were taken every 30 s for the assays done at 37°C and every 120 s for assays done at RT. The aliquots were added to tubes containing 300 μl of Z-buffer (60 mM Na2HPO4·H2O, 40 mM NaH2PO4·H2O (pH 7.0 at 25°C), 10 mM KCl, 1 mM MgCl2, 5 mM β-ME), 20 μl of chloroform, and 10 μl of 0.1% SDS. The tubes were then instantly vortexed for 20 s and transferred to ice. In the end, all the tubes were incubated for 5 min on ice and then for 5 min at RT after which, 500 μl of 1 mg/ml ONPG (o-nitro-phenyl galactopyranoside) was added to the reaction. ONPG hydrolysis for cells with pAUG-bgal plasmid was allowed to proceed for 15 min at RT. Similarly, for cells with pAGG-bgal, the reaction was allowed to proceed for 1 h at RT. After completion, the reaction was stopped by adding 500 μl of 1 M Na2CO3. Colour was allowed to develop for 15 min and then 300 μl of the reaction mix was transferred to 96-well plates. Absorbance was recorded at 420 and 550 nm using a Tecan infinite M200 multimode plate reader. The averaged readings from three biological replicates were used to derive the plot representing residual enzymatic activity for calculating elongation rates and rates of initiation (48). Length of the protein (1024 amino acids) divided by the lag time for the appearance of enzymatic activity after induction was used to calculate the elongation rates. The square root of the residual enzyme activity √(Et – E0), where Et signifies the miller units at time t and E0 signifies the miller units recorded at t = 0 was plotted against time (t) to derive a linear plot for calculating the rate of translation initiation.
Polysome profiling
Wt, ΔrbfA or ΔksgA cells were grown overnight in LB medium. For complementation studies, these cells were transformed with a plasmid carrying the respective rbfA, ksgA, infA, infB or infC and were grown overnight in LB medium supplemented with 25 μg/ml Chloramphenicol (Cmp). The next day, cultures were diluted in 100 ml fresh LB medium supplemented with Cmp and grown at 37°C with vigorous shaking. After OD600 reached 0.3, the protein production was induced with Atc (50 ng/ml) and allowed to grow until the OD600 of the cultures was 0.6. At this point, the culture was chilled rapidly on ice with the addition of ice cubes and 200 μg/ml Cmp followed by incubation for 15 min. The cells were harvested by centrifugation at 4°C, 8000 × g for 10 min. Cells were then washed once with Buffer CL (20 mM Tris–HCl (pH 7.6 at 25°C), 150 mM NH4Cl, 10.5 mM Mg(OAc)2, 0.5 mM EDTA and 6 mM β-ME) and later re-suspended in 1 ml buffer CL supplemented with 1 mg/ml Lysozyme, 1 mM PMSF, 0.5× CellLyticB reagent followed by incubation on ice for 1 h. The cells were then ruptured by 5 cycles of freeze-thaw using liquid nitrogen. The lysates were clarified by centrifugation at 30 000 × g followed by A260 (absorbance at 260 nm) quantification in Implen Nanospectrophotometer. For ultracentrifugation, 300 μl of 10 A260 was loaded on a 10–50% sucrose gradient prepared in buffer CL and spun at 102 000 × g in rotor TH-641 (Thermo-Sorvall WX+ ultracentrifuge) for 16 h. Subsequently, fractionation was performed using a syringe pump connected to the Akta Pure system (GE Healthcare).
Purification of IF3
Escherichia coli BL21(DE3) cells harbouring p1R-IF3 were grown in 2 l of LB medium supplemented with 100 μg/ml Kanamycin at 37°C till OD600 reached 0.6. At this point, protein production was induced with 0.3 mM IPTG and growth was continued at 18°C for another 16 h. Following this, cells were harvested using centrifugation at 8000 × g/10 min and were washed once using buffer PP (20 mM Tris–Cl (pH 7.6), 500 mM NaCl, 6 Mm β-ME). Cells were later resuspended into 20 ml of buffer PP supplemented with 1 mM PMSF and lysed using sonication. Cell lysate was clarified by centrifugation at 25 000 × g for 30 min following which the clarified lysate was further subjected to ultracentrifugation at 102 000 × g in the rotor TH-641 (Thermo-Sorvall WX+ ultracentrifuge) for 16 h. The resulting supernatant was diluted 10-fold using buffer PD (20 mM Tris–Cl (pH 7.6), 6 mM β-ME) in order to adjust the final salt concentration to 50 mM NaCl followed by loading onto a 5 ml HiTrap SP HP column (GE Healthcare). The column was washed with at least 5 CVs (column volumes) of buffer PD supplemented with 50 mM NaCl followed by elution on an Akta Pure system (GE Healthcare) using a 0–2 M NaCl gradient. Eluted fractions were analysed by SDS-PAGE and peak fractions were loaded onto a HiLoad Superdex 75 pg gel filtration column (GE healthcare). Peak fractions from gel filtration chromatography were analysed for homogeneity by SDS-PAGE and then subjected to concentration using ultrafiltration. The purified protein was divided into small aliquots, flash frozen and stored at −80°C, until required.
Purification of 30S subunits
Wt, ΔrbfA or ΔksgA cells were grown overnight in LB medium. The next day, cultures were diluted in 800 ml fresh LB medium and grown at 37°C with vigorous shaking until the OD600 reached 0.6. At this point, the culture was chilled rapidly on ice with the addition of ice cubes and 200 μg/ml Cmp followed by incubation for 15 min. The cells were harvested by centrifugation at 4°C, 8000 × g for 10 min. Cells were then washed once with Buffer CL (20 mM Tris–HCl (pH 7.6 at 25°C), 150 mM NH4Cl, 10.5 mM Mg(OAc)2, 0.5 mM EDTA and 6 mM β-ME) and later re-suspended in 1 ml buffer CL supplemented with 1 mg/ml Lysozyme, 1 mM PMSF, 0.5× CellLyticB reagent followed by incubation on ice for 1 h. The cells were then ruptured by 5 cycles of freeze-thaw using liquid nitrogen. The lysates were clarified by centrifugation at 30 000 × g and loaded onto a 10–50% sucrose cushion prepared in buffer CL. The sample was centrifuged at 102 000 × g for 16 h at 4 °C in the rotor TH-641 followed by fractionation to isolate the 70S particles. The purified 70S particles were then dissociated by diluting in buffer CLD (20 mM Tris–HCl (pH 7.6 at 25°C), 150 mM NH4Cl, 2.5 mM Mg(OAc)2, 0.5 mM EDTA and 6 mM β-ME). The diluted fractions were then loaded on a 10–50% sucrose gradient prepared in buffer CLD and centrifuged at 102 000 g for 16 h at 4°C in the rotor TH-641 followed by fractionation to isolate the 30S particles. The purified 30S fractions were again centrifuged at 10 200 × g for 12 h in the rotor TH-641. The 30S pellets were then resuspended in buffer CLD followed by concentration estimation and divided into small aliquots. The aliquots were flash-frozen and stored at −80°C until required.
In vitro ribosome binding assays
Purified 30S subunits from Wt, ΔrbfA or ΔksgA were tested for binding efficiency towards purified IF3 using a ribosome pelleting assay and a co-sedimentation assay. Briefly, 40 pmol of ribosomes were mixed with 400 pmol of IF3 in a 50 μl reaction in Buffer RB (20 mM Tris–Cl (pH 7.6), 60 mM NH4Cl, 2.5 mM Mg(OAc)2, 0.5 mM EDTA, 6 mM β-ME). The reaction was incubated at 37°C for 10 min, followed by placement on ice. For the pelleting assay, the reaction was layered on a 15% sucrose cushion prepared in buffer RB and centrifuged for 4 h at 160 000 × g in a TLS-55 rotor in an Optima TLX 120 ultracentrifuge (Beckman-Coulter). The pellet fraction of each reaction was resuspended in buffer RB followed by A260 measurements. The pellet fractions were loaded on a 15% acrylamide SDS-PAGE and probed for the presence of IF3 using a mouse anti-strep tag primary antibody (Strep-MAB classic, Cat. No: 2-1507-001, IBA life sciences) and anti-mouse secondary antibody (HRP-linked Antibody, Product #7076, Cell signalling Technology). For the co-sedimentation assay, the binding reaction was layered onto a 10–50% sucrose gradient prepared in buffer RB. The sample was centrifuged in the TH-641 rotor at 10 200 × g for 16 h and followed by fractionation. The fractions were TCA precipitated and resolved using a 15% acrylamide SDS-PAGE and probed using a mouse anti-strep tag primary antibody (Strep-MAB classic, Cat. No.: 2-1507-001, IBA life sciences) and anti-mouse secondary antibody (HRP-linked Antibody, Product #7076, Cell signalling Technology).
Growth, cross-linking and cell lysis conditions for TCP-seq
Preparation of ribosome and initiation complex footprints were done essentially according to the original protocol (49) with minor modification as required for bacterial cells. One colony each, of Wt, ΔrbfA or ΔksgA cells carrying the plasmid pAGG-GFP-8R was grown in 2 l of fresh LB medium supplemented with 100 μg/ml Amp and incubated at 25°C with shaking at 180 rpm. Minimal gfp expression was induced using 50 μM arabinose and growth was allowed to proceed till OD600 reached 0.4. At this point, cells were rapidly chilled by adding ice cubes accompanied by the addition of 150 ml of freshly prepared 30% formaldehyde. Cultures were mixed vigorously and placed on ice for 15 min, followed by the addition of 200 ml of 2.5 M glycine. Following this, cells were harvested by centrifugation at 8000 × g for 10 min. Pellets were washed with 40 ml of Buffer TW (20 mM HEPES–KOH (pH 7.4 at 25°C), 100 mM KCl and 2 mM MgCl2) followed by resuspension in 0.4 ml of buffer TL (20 mM HEPES–KOH (pH 7.4 at 25°C), 100 mM KCl, 2.5 mM MgCl2 and 0.5 mM EDTA) supplemented with 2 mg/ml lysozyme, 6 mM β-ME, 1 unit/μl RNaseOUT inhibitor (ThermoFisher Scientific), 40 units DNase and 1 mM PMSF. Cells were placed on ice for 1 h followed by rupturing using 5 cycles of freeze-thaw in liquid nitrogen. Cell lysates were clarified by centrifugation at 30 000 × g for 30 min at 4°C.
Isolation of mRNA bound fractions and MNase treatment
Clarified cell lysate was layered onto a 10–20% sucrose gradient prepared in Buffer TP (50 mM Tris–HCl (pH 7.0 at 25°C), 50 mM NH4Cl, 4.5 mM MgCl2, 0.5 mM EDTA, 6 mM β-ME) and centrifuged at 52 000 rpm for 80 min at 4°C using a TLS-55 rotor in an Optima TLX 120 ultracentrifuge (Beckman-Coulter). Ribosome pellets derived after centrifugation were resuspended in 1 ml of buffer TL supplemented with 1 unit/μl RNaseOUT and 1 mM PMSF. These ribosomes were then treated with 30 units of MNase per 1 A260 of ribosomes for 1 h at RT. The reaction was stopped by addition of 2 mM EGTA and the nuclease-treated complexes were immediately loaded onto a 10–40% sucrose gradient prepared with buffer TP. Samples were spun at 40 000 rpm for 4 h at 4°C using a TLS-55 rotor. Upon completion, fractions of 100 μl each were collected manually and later processed for footprint isolation.
De-crosslinking and footprint isolation
Fractionated 30S or 70S samples were supplemented with 1% (w/v) SDS, 10 mM EDTA, 10 mM Tris–HCl (pH 7.4 at 25°C), 10 mM glycine and 1 mg/ml Proteinase K (Sigma-Aldrich). The samples were incubated at 50°C for 30 min with regular mixing. Following this, an equal volume of acid phenol:chloroform (5:1) was added to this mix and incubated at 65°C for another 45 min with regular vortexing. The sample was then centrifuged at 16 000 ×g for 30 min at 4°C followed by separation of the aqueous phase. An equal volume of chloroform was then added to the aqueous phase and centrifuged at 16 000 × g for 30 min at 4°C. The aqueous phase was again separated in a fresh tube and supplemented with 0.1 volume of 3 M sodium acetate (pH 5.2 at 25°C), 1 mg/ml glycol blue (Ambion) and 2.5 volumes of chilled ethanol. Precipitation was done overnight at −20°C following which the sample was centrifuged at 16 000 × g for 45 mins at 4°C. RNA pellets were washed twice with chilled 70% ethanol, followed by air drying. Finally, the pellets were resuspended in nuclease-free water and checked for integrity. Footprints corresponding to 30S or 70S fractions were size selected from 10 to 80 bases by gel elution using denaturing polyacrylamide gel electrophoresis (PAGE) in Protean ii XL setup (Bio-Rad). The extracted RNA was end-repaired using T4 Polynucleotide Kinase (PNK) and purified by ethanol precipitation.
Library preparation and sequencing
Library preparation and sequencing were outsourced to Genotypic Technology Pvt Ltd, Bengaluru, India. Libraries were prepared strictly according to the manufacturer's recommendations using the TruSeq Small RNA Sample Preparation kit (Illumina, USA). Briefly, 50 ng of RNA was used as starting material for ligation of 3′ and 5′ adapters. Specific index sequence was added to each sample for identification during sequencing. The Illumina Universal Adapter used in the study was: AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA and the Index Adapter was: CAAGCAGAAGACGGCATACGAGAT[INDEX]GTGACTGGAGTTCCTTGGCACCCGAGAATTCC.
Adapter-ligated fragments were reverse transcribed with Superscript III Reverse transcriptase (Invitrogen). The cDNA was enriched and barcoded by 15 cycles of PCR amplification and the amplified library was size-selected using denaturing PAGE. The library was size selected in the range of 140–210 bp followed by overnight gel elution and finally resuspended in nuclease-free water. Illumina compatible sequencing library was initially quantified by Qubit fluorimeter (Thermo Fisher Scientific) and its fragment size distribution was analysed on Agilent TapeStation. Sequencing was performed on the Illumina NextSeq 500 platform. The sequencing depth for each sample ranged from 10 to 15 million reads.
Analysis of sequencing data
The reads were subjected to several pre-processing steps described as follows. Firstly, reads with a Phred score <20 were removed by utilizing fastq_quality_trimmer from the FASTX-toolkit-version-0.0.13. The remaining reads were trimmed for 3′-end adapter sequences [5′-TGGAATTCTCGGGTGCCAAGGAACTC-3′] using Cutadapt-1.5 (50). Following these, in order to filter and remove the reads derived from rRNA, tRNA, sRNA or any other non-coding RNA, reads were aligned using STAR-2.5.3 (51) against non-coding RNAs from E. coli K-12 MG1655 genome (ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/Escherichia_coli/reference/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_rna_from_genomic.fna). The unmatched reads were aligned against the E. coli K-12 MG1655 reference genome (NCBI: NC_000913.3;ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/Escherichia_coli/reference/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_genomic.fna) using STAR-2.5.3 with the following parameters: spliced alignment turned off (–alignIntronMax 1), forced end to end alignment (–alignEndsType EndToEnd) and allowing eight mismatches per 100 nt (–outFilterMismatchNoverLmax 0.08). Sam to Bam conversion was accomplished using Samtools-1.4.1 (52). Only uniquely matching reads were considered for further analyses. Metagene analysis for mapping the reads to known START codon of 4257 CDS in E. coli was performed by employing custom written shell scripts utilizing Samtools-1.4.1. Plots were generated using ggplot2 implemented in R.
RESULTS
Defects in late 30S assembly perturb codon recognition during translation
We wondered whether the premature 30S subunits bypass the quality control checkpoints to enter the translation cycle. To address this, using E. coli we created null mutants for genes encoding late-stage 30S specific RAFs such as RsgA, RbfA, KsgA and LepA (Figure 1A). Premature ribosomes from these strains were tested for their ability to recognize AUG (cognate) and non-AUG (near-cognate and non-cognate) initiation codons for translating the gene encoding GFP in vivo (Figure 1B). In order to quantify the extent of error in decoding the start codon due to an assembly defect in the 30S subunit, we formulated an index: Initiation Error Index (IEI). IEI was defined as the ratio of GFP fluorescence from null mutants of the respective RAF to Wild type (Wt). We have chosen four different initiation signals of variable strength: AUG > CUG > AUA > AGG to assess the potency of premature 30S subunits in discriminating cognate from non-cognate codons (Supplementary Figure S1) (53,54).
Figure 1.

Assembly defects in 30S subunit compromise decoding fidelity. (A) Structure of the E. coli 30S subunit represented in sky blue surface [PDB ID: 5LMV] showing the binding sites of late-stage acting RAFs as observed by Cryo-EM studies (8,36,37,94). Colour coding for each RAF binding site is indicated along with the 30S decoding centre. (B) A representation of the GFP constructs used for measuring translation fidelity. Codon variations at codon 1, 7 and 8 are indicated. (C–F) Errors and rescue from misrecognition of the start codon, measured as IEI and IRI are shown for ΔrsgA (C), ΔlepA (D), ΔrbfA (E) and ΔksgA (F), respectively. Dashed lines marking IEI = 1, signifies similar expression in null mutants and Wt, indicating optimal recognition of the start codon. Dashed lines marking IRI = 1, signifies rescue from erroneous translation initiation at near or non-canonical start codons. (G) Evaluation of frame maintenance and fidelity of stop codon recognition measured as EEI is shown for ΔksgA and ΔrbfA. The dashed line indicating EEI = 1 indicates a similar level of GFP expression between null mutants and Wt cells.
Deletion of RsgA did not display any increase in the IEI, indicating no difference in the codon recognition between ΔrsgA and Wt (Figure 1C). Deletion of the back-translocase EF-4/LepA, led to elevated misrecognition of CUG and AUA as start codons (Figure 1D). However, expression from AUG and AGG start codons remained unchanged in ΔlepA strain (Figure 1D). In order to further investigate if the decoding errors were specifically due to deletion of the respective assembly factor, we complemented the loss of RsgA and LepA from a plasmid-borne copy. The effect of RAF complementation was gauged using another index, the Initiation Rescue Index (IRI), which was calculated as the ratio of GFP fluorescence in complemented null mutant to that of Wt. If the translation errors were indeed caused due to the absence of the respective RAF, a shift from IEI >1 to IRI ∼1 would hint at rescue from misinitiation at the non-canonical start codon. Upon complementation, IRI ∼1 indicated that translation defects were ameliorated in ΔlepA cells (Figure 1D), on the other hand, the IRI remained unaffected for ΔrsgA cells (Figure 1C). In contrast, loss of RbfA and KsgA displayed an elevated IEI for near-cognate as well as non-cognate start codons indicating significant misinitiation (Figure 1E and F). To ascertain this, we complemented the loss of RbfA and KsgA with a plasmid-borne copy to measure the IRI values. The rescue was indicated by IRI ∼1 in RbfA and KsgA complemented strains (Figure 1E and F). These findings highlight the fact that the premature ribosomes accumulating due to loss of LepA, KsgA and RbfA have the potency to enter translation even with a significantly compromised native structure.
Having observed the mis-recognition of start codons by the premature ribosomes, we further investigated if the assembly defects in ΔrbfA and ΔksgA also manifest during translation elongation. Towards this, we employed variants of gfp that harboured frameshift mutations or premature stop codons and measured the Elongation Error Index (EEI) (Figure 1G). EEI >1 would indicate decoding errors leading to compromised frame maintenance or bypass of stop codons with respect to Wt. Both ΔrbfA and ΔksgA showed slightly elevated EEI values, indicating that premature ribosomes in both strains have compromised fidelity during elongation and termination. However, the observations also suggest that these bypasses are less frequent in comparison to the erroneous decoding of the initiation signals.
Defects in assembly upset the kinetics of translation initiation and elongation
In order to further dissect the link between assembly and translation, we studied translation by tracing the pre-steady-state kinetics of β-galactosidase (bgal) synthesis in vivo. For this, we employed an IPTG inducible construct, pAUG-bgal (AUG as the start codon). The trace signifying accumulation of bgal was used to calculate the peptide chain elongation rate (ER) as described previously (45,48). For the purpose of uniformity, Wt, ΔrbfA and ΔksgA cells were grown till mid-log phase at 37°C and bgal production was triggered for the ER measurements (Figure 2A; Table 1). The ER for Wt was in close agreement with the previous report (34). The elongation was marginally faster for ΔksgA, whereas it plummeted approximately three-fold for ΔrbfA relative to Wt (Figure 2A and C; Table 1), suggesting that the perturbed assembly indeed upsets translation. This phenomenon aggravated further at 25°C thus echoing the exacerbation of assembly defects at a colder temperature (Figure 2B and D, Table 1).
Figure 2.

Translation kinetics of premature ribosomes. (A and B) Translation kinetics of bgal production with AUG start codon in Wt, ΔrbfA and ΔksgA. Protein production kinetics was performed at 37°C (A) and 25°C (B), respectively. The plots were drawn as the averaged measurement from three independent time course experiments. The error bars represent the standard deviation of the three independent trials. (C and D) Schleif plot for bgal translation kinetics derived by plotting the square root of residual bgal enzyme activity against time from data in (A) and (B) (vide Materials and Methods). The plotted data were fitted to a linear regression model. Equations representing the trend line for each plot have been shown. The slope of the line indicates the rate of translation initiation and the X-intercept marks the first appearance of the enzyme activity that can be further used to calculate the rate of peptide chain elongation. (E) Translation kinetics of bgal production with AGG start codon in Wt, ΔrbfA and ΔksgA. The kinetics was performed at 37°C. (F) Schleif plot for bgal translation kinetics with AGG start codon derived from the data in (E).
Table 1.
Peptide chain elongation rate (ER) measured from Wt, ΔksgA and ΔrbfA cells using bgal translation kinetics. All measurements are in amino acids/second (aa/sec)
| Start codon | Growth temperature | Wt | ΔksgA | ΔrbfA |
|---|---|---|---|---|
| AUG | 37°C | 16.1 ± 2.5 | 17.9 ± 2.9 | 4.9 ± 0.5 |
| AUG | 25°C | 7.4 ± 1.9 | 3.5 ± 0.5 | 1.7 ± 0.1 |
| AGG | 37°C | 16.1 ± 3 | 9.94 ± 0.7 | 4.3 ± 0.2 |
| AGG | 25°C | −ND- | −ND- | −ND- |
In order to further understand the effect of assembly defects on translation initiation, we measured the translation initiation rates (IR), as indicated by the slope of the bgal accumulation trace (Figure 2C and D) (45,48). IRs measured at 37°C, from the AUG start codon recalled the patterns of elongation, as they were similar for Wt and ΔksgA and significantly slower for ΔrbfA (Figure 2C; Table 2). Furthermore, in line with ER, IR for Wt, ΔrbfA and ΔksgA was significantly reduced at 25°C (Figure 2D; Table 2). Notably, IR for ΔrbfA was consistently lower relative to WT, reinforcing that premature ribosomes entering translation in ΔrbfA have impaired translation capacity. Additionally, the marked differences between ΔrbfA and ΔksgA also indicated that assembly defects in the two strains elicit distinct effects on protein synthesis.
Table 2.
Translation initiation rates (IR) measured from Wt, ΔksgA, and ΔrbfA cells using bgal translation kinetics
| Start codon | Growth temperature | Wt | ΔksgA | ΔrbfA |
|---|---|---|---|---|
| AUG | 37°C | 0.21 ± 0.008 | 0.20 ± 0.007 | 0.16 ± 0.008 |
| AUG | 25°C | 0.05 ± 0.002 | 0.06 ± 0.003 | 0.04 ± 0.001 |
| AGG | 37°C | 0.05 ± 0.001 | 0.08 ± 0.002 | 0.12 ± 0.004 |
| AGG | 25°C | −ND- | −ND- | −ND- |
The IR values are derived from the slope of the curve marking accumulation of bgal over time (units: Miller units1/2/s).
It was also important to address a possibility whether the aberrant ER and IR represented errors in decoding or just a general slowdown of the translational machinery that might arise due to a deficit in the number of mature ribosomes entering the translation cycle. In order to address this, we hypothesized that, if the aberrant ER and IR were caused by a general slowdown of the translational machinery, the respective rates should remain unaffected if measured from non-canonical start codons. However, if the decrease was indeed caused by erroneous decoding due to assembly defects, the non-canonical start codons must be misread as canonical ones preferentially in null mutants in comparison to Wt. To test this conjecture, we performed bgal translation kinetics using constructs with an AGG start codon (pAGG-bgal) at 37°C. Remarkably, the IR from AGG start codon displayed an inverse pattern in comparison to AUG start codon for the three strains. Null mutants of RbfA exhibited elevated translation initiation over time in contrast to Wt (Figure 2E and F, Table 2). Similar to the IR and ER defects, AGG start codon recognition was milder in ΔksgA in comparison to ΔrbfA (Figure 2E and F, Table 2). Unexpectedly, the ER from AGG start codon for Wt and ΔrbfA remained unaffected in comparison to AUG, whereas the elongation was drastically slowed down for ΔksgA (Figure 2F, Table 1). These observations strongly support the conjecture that the errors in assembly lead to erroneous translation initiation and elongation events. Collectively, these findings hint at a scenario where evasion of quality control checkpoints permits entry of premature ribosomes into translation.
Profiling of translation initiation complexes formed by premature subunits reveals genome-wide alteration of ribosome occupancy during translation initiation
In order to complement our observations from the above kinetic experiments, we sought to probe how the assembly defects impact the formation of initiation complexes at the genomic level. For this, we turned to TCP-seq (49,55), a modified Ribo-seq strategy that has been devised to probe translation initiation, elongation and termination complexes in Saccharomyces cerevisiae (Figure 3A). For bacterial Ribo-seq, widely used bacterial translation elongation inhibitors like Chloramphenicol do not trap ribosomes in the initiation stage and also introduces a codon-specific bias when used to arrest ribosomes during elongation (56). In contrast, TCP-seq uses formaldehyde cross-linking to trap ribosomes during various stages of translation in vivo and we have adopted this technique for probing the translational complexes in E. coli. In order to arrest the ribosome from Wt, ΔksgA and ΔrbfA (Figure 3B) during various stages of translation in an unbiased manner, we snap chilled the exponentially growing cells and cross-linked mRNA bound translating complexes using formaldehyde (55,57). After cross-linking, free 30S particles were depleted using sedimentation analysis and mRNA cross-linked ribosomes were released by treating with Micrococcal nuclease (MNase). The released particles were further partitioned using density gradient sedimentation and then used to extract the mRNA footprints (FP) bound to 30S and 70S particles. For library preparation, a wide range of FPs of roughly 10–80 nucleotides (nt) was size selected to accommodate FPs arising from translating complexes as well as the tRNA molecules (Supplementary Figure S2).
Figure 3.

Translation complex profiling for studying initiation and elongation. (A) An outline of the strategy used to capture the ribosomes at various stages of translation using TCP-seq. Purified ribosome footprints are shown in Supplementary Figure S2. (B) Representative polysome profiles for Wt, ΔrbfA, and ΔksgA are derived by performing sucrose density gradient sedimentation. Samples for TCP-seq were derived from the 30S and 70S fractions, respectively. (C and D) The normalised frequency distribution (density) of the length of the 30S (C) and 70S (D) protected fragments isolated from Wt, ΔrbfA and ΔksgA is shown.
The length distribution of FPs that were mapped to the coding region was congruent with the size selection criteria used during library preparation, harbouring reads ranging from as low as 12 nt through 75 nt in length (Figure 3C and D). The 30S subunit derived FPs (30S_FP) contained a distinct population of ∼14 nt in length (Figure 3C). Whereas, we spotted three distinct population of FPs from 70S particles (70S_FP) centred on ∼15, 30 and 40 nt, respectively (Figure 3D). Deletion of both KsgA and RbfA led to a significant drop in the number of FPs, possibly due to the reduced polysome population in the respective null mutants (Figure 3B). The reduction in total footprint counts and the missing polysome fractions in the null mutants are suggestive of attenuated translation in these strains (Figure 3C and D). It has been shown that the size of the FPs mirrors the conformational rearrangements the ribosomes undergo in solo as well as when they interact with translation factors during different stages of translation (55,58). To track these, we mapped the 5′ and 3′ end of the FPs of given sizes to the known START of the 4257 coding sequences (CDS) in E. coli (Figure 4). In line with the characteristic size distribution of FPs for the 30S and 70S, the majority of the mapped FPs for 30S fell in the smaller size regime (12–20 nt) whereas they corresponded to larger size regime (30 nt and above) for 70S (Figure 4). Each FP of a given length showed a range of characteristic offset distances from the start codons. 30S_FPs were densely populated near the start codon as opposed to 70S_FPs that recapitulated the fact that 30S is subjected to extensive regulatory events during initiation. At the 5′ UTR, the trailing edge of 30S_FP (5′ end of FPs in Figure 4A) showed a graded increase in the offset distance with an increase in the length of FPs than the leading edge (3′ end of FPs in Figure 4C). In fact, the leading edge was arrested around the start codon, suggesting that there is a queuing of 30S ribosomes at the 5′ UTR because the residence time of the 30S at the start codon is protracted owing to the extensive regulatory controls. However, these signatures were significantly altered in case of ΔksgA and ΔrbfA, especially for the 30S. The altered ribosome occupancy at the initiation site suggests two possibilities: Either the premature ribosomes are actively rejected by the translational machinery to enter the translation cycle or these ribosomes are rapidly surpassing the elaborate checkpoints during the initiation stage (Figure 4). Both of these cases are expected to reduce the residence time of ribosomes around the start codon and thereby the ribosome occupancy. However, in line with the translational kinetics studied using the bgal assay (for AUG codon) that showed minor reduction in translation initiation for the premature ribosomes (Figure 2; Tables 1 and 2), we reasoned that the altered ribosome occupancy for ΔksgA and ΔrbfA could be attributed to rapid surpassing of the kinetic checkpoints during the initiation.
Figure 4.
Metagene analysis for translation initiation complex. (A–D) 5′ ends of the FPs derived from 30S (A) and 70S (B) bound mRNA as well as the 3′ ends of the FPs derived from the 30S (C) and 70S (D) from Wt, ΔrbfA and, ΔksgA, respectively, are mapped against the known start codon of the coding sequences in E. coli. The first base of the start codon is aligned to the ribosome P site. The size and colour of the points correspond to the frequency of occurrence of FPs. Only those FPs with at least 100 reads are represented here.
Premature 30S can form preinitiation complexes
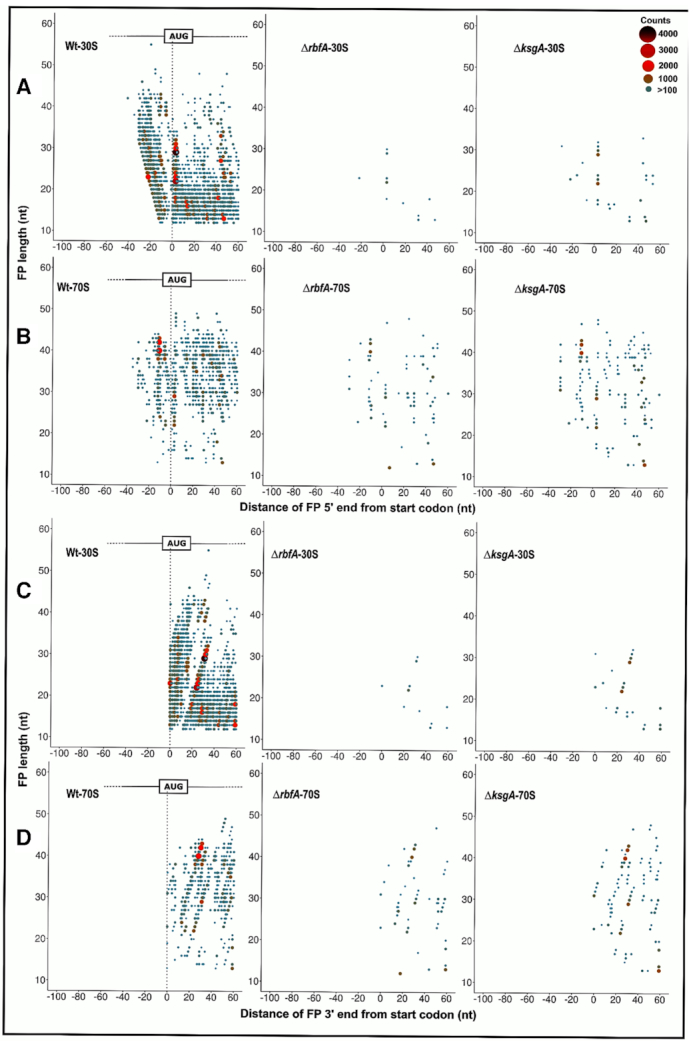
In order to further understand the nature of initiation and elongation complexes formed by the premature subunits, we studied the distribution of N-formylmethionine initiator–tRNA (tRNAifmet) and the elongator tRNA (tRNAe) that cross-linked and co-purified with the 30S and 70S fractions (Figure 3A). We reasoned that the tRNA distribution (tRNAifmet and tRNAe) would act as an indicator to pinpoint the ribosomes that are at the initiation and elongation stages of the translation (55). The initiation stage being the rate-limiting step of translation is expected to be dominated by tRNAifmet during the transition of 30S-PIC to 30S-IC (55,59) as opposed to other stages associated with the elongating 70S particles. Similarly, an inverse distribution of tRNAe is expected for elongating 70S than the 30S-PIC/IC. In line with our conjecture, Wt 30S ribosomes were found to harbour more tRNAifmet than the 70S (Figure 5A). Surprisingly, the distribution of tRNAifmet was reversed in both ΔksgA and ΔrbfA such that its level in the 70S was significantly higher in comparison to that in 30S. We posit that the reduced distribution of tRNAifmet in the 30S fraction of ΔksgA and ΔrbfA may arise from the diminished binding affinity of pre-30S towards the tRNAifmet or due to a short-lived pre-30S-PIC/IC formed in ΔksgA and ΔrbfA, which rapidly associates with the 50S subunit to form 70S-IC. Since the transition from 30S-PIC/IC to 70S IC occurs through a well-orchestrated sequence of events, the association between tRNAifmet and 70S should have occurred during the formation of 70S-IC. Therefore, this alludes to the possibility that there is a rapid transition of pre-30S-PIC/IC to 70S-IC in ΔksgA and ΔrbfA and the prospect of low affinity between pre-30S and tRNAifmet can thus be ruled out. This is further bolstered by the altered ribosome occupancy observed for ΔksgA and ΔrbfA during metagene analysis (Figure 4). Further, tRNAe was distributed approximately in equal measure between Wt 30S and 70S particles, suggesting the dynamic interaction between tRNAe and ribosome subunits. However, its distribution was skewed for ΔksgA and ΔrbfA and showed more preference for the 70S than the 30S (Figure 5A).
Figure 5.
Premature ribosomes form distinct initiation complexes. (A) A stacked bar plot representing the distribution of initiator (tRNAifmet) and elongator (tRNAe) tRNAs in the 30S and 70S derived fractions as observed in the TCP-seq data. Read counts were converted to Reads per million (RPM). (B) An outline of the methodology used to capture translation initiation and elongation complexes on an mRNA harbouring AGG start codon. (C and D) Distribution of the AGG-GFP mRNA derived FPs from the 30S (C) and 70S fractions (D) of Wt, ΔrbfA and ΔksgA. The FP counts are shown along the entire length of the gene encoding GFP.
Intrigued by the altered ribosome occupancy of the translation initiation complex formed by premature ribosomes bound to mRNA containing cognate start (AUG) codon (Figure 4), we asked whether the identity of the cognate start codon has any role in limiting the progression of premature ribosomes towards elongation stage. To address this, we studied the distribution of the 30S and 70S specific FPs from AGG-gfp (Figure 5B). We reasoned that the distribution of FPs should mirror the observations from GFP and bgal expression studies (Figures 1 and 2). In line with our conjecture, 30S_FPs derived from Wt were densely populated around the start codon region (Figure 5C), whereas 30S_FPs from ΔrbfA and ΔksgA showed the reduced distribution of FPs around the start codon. This hints at the possibility that 30S from Wt stalls at the initiation site longer than the pre-30S from ΔrbfA and ΔksgA. This further suggests that unlike the pre-30S-PIC/IC from ΔrbfA and ΔksgA, 30S-PIC/IC from Wt does not efficiently progress towards the formation of 70S-IC if the start codon is AGG. Next, in order to understand how this 30S-PIC/IC transitioned into 70S-IC or an elongation complex, we analysed the distribution of FPs from the 70S fraction. Interestingly, we noted that despite the significant distribution of FPs around the initiation site from the Wt 30S and 70S fraction, their occupancy throughout the length of GFP is abysmal, suggesting that the AGG-gfp is poorly translated (Figure 5C and D). Strikingly, 70S_FPs from ΔrbfA were evenly distributed in higher proportion than those from ΔksgA and Wt, suggesting that premature ribosomes from ΔrbfA undergo conversion of pre-30S-PIC/IC to 70S-IC, leading to efficient translation of AGG-gfp. This also agrees with the observations from GFP and bgal expression studies (Figures 1 and 2).
Assembly defects subvert Initiation factors mediated scrutiny of translation initiation
Having observed the entry of premature ribosomes into the translation cycle, we wondered how such 30S-PIC engaging the non-cognate initiation codon bypassed the scrutiny of initiation factors during the translation initiation. To understand this, we analysed the structures of the ribosome in complex with Initiation factors (IFs) or RAFs (8,17,37) (Figure 1A and Supplementary Figure S3). Taking cues from our analyses and previous observations, we found a significant overlap in the binding site of IFs and late-stage assembly factors on the 30S subunit (Figure Supplementary figure S3B). We hypothesized that the structural distortions in the premature ribosomes might have tempered affinities for the IFs. Such premature ribosomes, when entering translation with either cognate or non-cognate initiation signal could bypass the fidelity checkpoints posed by IFs, thus leading to an inaccurate translation event. To test this conjecture in vivo, we simply elevated the cellular concentration of all three IFs individually by their ectopic expression in Wt as well as the RAF null mutants, expecting that an increased cellular concentration of IFs may restore the skewed mass balance ratio between IFs and premature 30S subunits. IRI values were calculated as a ratio between GFP fluorescence from cells with elevated IF concentration to that of cells with a basal concentration of IF. An IRI value ∼1 would mean that the expression levels of GFP were unaffected upon overexpressing IFs, whereas IRI <1 would indicate repression of translation upon overproduction of the respective IF. The IRI values were measured for constructs with either AUG or AGG start codon individually. In order to achieve this, IF production was triggered when cells just entered the log phase (OD600 ∼ 0.2–0.3) followed by induction of GFP production at mid-log phase (OD600 ∼ 0.6), thus allowing for cellular levels of the respective IFs to be elevated before gfp expression could initiate (vide Materials and Methods).
Elevated levels of IF1 had no significant effects on the expression of GFP from AGG or AUG start codons for Wt or ΔksgA (Figure 6A). In ΔrbfA, IF1 elevation specifically suppressed expression from AGG start codon (Figure 6A). Unusually, indiscriminate repression of GFP production from both AGG and AUG start codons was observed when the cellular levels of IF2 were elevated (Figure 6B). It would seem plausible that overproduction of IF2 somehow inhibited translation. Strikingly, rising IF3 level specifically inhibited expression from AGG start codon (Figure 6C). However, unlike IF1, IF3 mediated inhibition of translation from AGG start codon is equally strong even in case of Wt or assembly deficient strains, highlighting the role of IF3 in discriminating the CO-AC interactions on the 30S-PIC/IC. Collectively, these observations reaffirm our hypothesis that the restoration of the pre-30S-IF mass balance may avert the participation of premature ribosomal particles in translation.
Figure 6.

Elevated levels of initiation factors avert premature ribosomes from engaging in translation. (A–C) IRI measurements for Wt, ΔrbfA and ΔksgA using gfp that harbours AUG or AGG start codon in presence of elevated cellular concentrations of IF1 (A), IF2 (B) and IF3 (C) are presented. The statistical significance was tested using a paired t-test analysis with a 95% confidence interval. (D–F) Representative polysome profiles for Wt, ΔrbfA or ΔksgA cells carrying an empty p15A vector (only Wt, ΔrbfA or ΔksgA) or p15A vector carrying genes encoding IF1, IF2 or IF3 are shown. The inset shows the profile for the respective null mutants carrying p15A vector carrying a gene encoding RbfA or KsgA. The profiles were drawn from mid-log phase cells overexpressing the respective initiation factors.
In parallel, we also checked if ectopic expression of IFs improved cellular fitness, which could, in turn, lead to a decrease in error-prone translation. To test this, Wt, ΔrbfA and ΔksgA cells transformed with plasmids carrying genes encoding IFs were tested for their growth characteristics after inducing the expression of the respective gene encoding IF. For uniformity, OD600 of all cultures was normalized at the time of starting the experiment and IF expression was also triggered at the same time (time = 0) (vide Materials and Methods). The absence of a growth advantage (Supplementary Figure S4) indicated that the rescue is only a result of the translation quality control and not due to an increase in the overall pool of premature ribosomes. As observed, overexpression of IF1 or IF3 did not improve cellular fitness in any manner but a radical effect of IF2 overexpression was observed for all the three strains as they failed to grow even after prolonged incubation (Supplementary Figure S4). These surprising observations suggest that overproduction of IF2 is somehow toxic to cells and thus prompted us to probe the IF mediated moderation of the ribosome. Towards this, we studied the polysome profiles from Wt, ΔksgA and ΔrbfA containing elevated concentrations of the respective IFs (Figure 6D–F). For these experiments, IF, KsgA and RbfA overproduction was initiated using 50 μg/ml Atc when cells reached OD600 0.3. Here, the delayed production of IF2 did not seem to be as deleterious to cells, as seen with the growth-related experiments (Supplementary Figure S4) and thus allowed us to collect a viable cell mass for the profiling analysis.
In line with our expectations, complementation of RbfA and KsgA restored the distorted polysome profiles in the respective null mutants (inset in Figure 6E and F), suggesting efficient rescue from the assembly defects. Further, except in ΔrbfA, an elevated level of IF1 did not seem to alter the distribution of ribosomes in Wt and ΔksgA (Figure 6D–F). However, overproduction of IF2 exhibited an associative effect on free 30S and 50S subunits (60–62), leading to a higher proportion of 70S particles (Figure 6D–F, compare ΔrbfA with ΔrbfA + IF2). On the contrary, elevated levels of IF3 displayed a pronounced subunit dissociation activity, where a decrease in the 70S population was accompanied by a concomitant increase in the 50S and 30S populations (Figure 6D-F). Similar to IF2, the effect of IF3 overproduction was more prominent in ΔrbfA as opposed to Wt and ΔksgA (Figure 6D–F, compare ΔrbfA with ΔrbfA + IF3).
Repression of translation from mRNA with non-cognate start codon is at the initiation stage
Observing the distinctive adjustment of ribosome populations and the tuning of translation by the three IFs, we were enthused to investigate further the stage at which these IFs pre-empt the entry of premature ribosomes into translation. Towards this, we studied translation initiation in Wt, ΔrbfA and ΔksgA from AUG and AGG start codons with elevated levels of IFs. Similar to the GFP expression studies, synthesis of IFs was triggered before the measurement of bgal production kinetics. We then measured the IRs during elevated (IRelevated) and basal IF (IRbasal) concentrations and these were subsequently used to calculate a ratio defined as IRelevated/IRbasal. A ratio <1 for a start codon would indicate attenuation of translation initiation upon elevating the cellular levels of the respective IF.
We observed that this ratio remained unchanged for Wt when IF1 levels were elevated. In contrast, IF1 overproduction appeared to appreciably disrupt translation initiation exclusively from AGG start codon in ΔrbfA (Figure 7A and B, Supplementary Figure S6A and D). These observations echoed the IF1 mediated repression of GFP production that was restricted to the AGG start codon in ΔrbfA (Figure 6A). Intriguingly enough, the elevation of IF3 levels displayed a striking rescue from mis-initiation from the AGG start codon in all three strains, (Figure 7A and B, Supplementary Figure S6C and F). This IF3 mediated progressive inhibition of translation from non-canonical initiation signals again reinforces the positive effects of restoration of the mass balance between IF3 and premature subunits. On the contrary, the initiation events dampened for AUG start codon and plummeted further for AGG start codon upon elevation of IF2 levels (Figure 7A and B, Supplementary Figure S6B and E). These observations in conjunction with the severe repression in growth (Supplementary Figure S3) and GFP synthesis (Figure 6B) called for further investigations into the centrality of IF2 levels in translation initiation where it is known to recruit the incoming tRNAifmet (63). One plausible scenario for this could be that the elevated IF2 levels are not appropriately accounted for by the concomitant rise in the levels of tRNAifmet, leading to suboptimal charging of IF2 with tRNAifmet (Figure 7C). This may give rise to vacant IF2 binding to 30S and stalling of initiation. To test this, we co-elevated cellular levels of tRNAifmet along with IF2 in Wt cells (vide Materials and Methods). To our surprise, these strains, in spite of an initial lag in growth, displayed a rescued growth phenotype (Figure 7C) as opposed to those overproducing IF2 alone (Figure 7C, Supplementary figure S4). To confirm if this rescue occurred as per our hypothesis or due to suppressor mutations in IF2+ tRNAifmet overproducing cells, we extracted the plasmids carrying the genes encoding IF2 and tRNAifmet from the rescued cells (Figure 7C). These plasmids were used to transform fresh Wt cells, and subsequently the cell growth was monitored upon IF2 overproduction. To our surprise, these cells did not display any decrease in growth upon induction of IF2 overproduction, indicating the rise of suppressor mutations that may nullify the toxic effect of high cellular levels of IF2 (Supplementary figure S5A). To understand the nature of the suppressors, we attempted to sequence the genes encoding IF2 as well as tRNAifmet. The sequencing highlighted five mutations in the tRNAifmet, spread in the D-loop and the TΨC stem region (Supplementary Figure S5B). However, despite repeated attempts to recover and sequence the gene encoding IF2, we could not succeed. Hence, we suspect that the suppressor mutants arise from the collective effects of loss of the gene overproducing IF2 and the mutations in the tRNAifmet. These observations suggest a critical role of IF2 in translation regulation (64) that may be very sensitive to cellular concentrations of IF2.
Figure 7.

Premature ribosomes evade translation quality control by initiation factors. (A and B) Bar plots representing the ratio of translation initiation rates (IRs) in cells harbouring elevated levels of IFs (IRelevated) to translation initiation rates in cells with basal levels of IFs (IRbasal) from AUG start codon (A) and AGG start codon (B). A ratio of the two translation initiation rates, derived from three independent time-course experiments are presented here. The curve fittings used to derive the respective IRs are presented in the Supplementary Figure S6. (C) Growth comparison for Wt transformed with vectors carrying genes encoding IF2 and tRNAifmet. IF2 production was induced at time = 0 by adding Atc (vide Methods) and this is indicated as IF2(+), whereas cells grown without the addition of Atc are represented as IF2(-). Overproduction of tRNAifmet was constitutive. (D) An outline of the strategy used to study the binding of IF3 to 30S subunits from Wt, ΔrbfA and ΔksgA. 30S subunits engaged in translation were purified by cycles of ultracentrifugation and IF3 binding to 30S subunits was tested using co-migration or pelleting assays. (E) Immunoblot for the pelleting assay using strep-tagged IF3 and 30S particles from Wt, ΔrbfA and ΔksgA is shown. Reaction mixtures containing 30S and IF3 either separately or in combination were ultracentrifuged on sucrose cushions. Pellet fractions from the respective reactions were resolved using SDS-PAGE and the indicated area was probed using an anti-strep antibody. The reaction components are indicated on top of the gel with the respective source of the ribosomes. Two picomoles of IF3 was directly loaded in the lane marked as ‘Unbound IF3’ as a control. Protein markers are loaded in the lane ‘M’ and the respective molecular weights are shown on the right. (F) Immunoblot for the co-migration assay using strep-tagged IF3 and 30S particles from Wt, ΔrbfA and ΔksgA is shown. A reaction mixture comprising 30S and IF3 was loaded on a 10–50% sucrose gradient, which was followed by ultracentrifugation and fractionation. Four ribosomal fractions were TCA precipitated and resolved using SDS-PAGE and the indicated area was developed using immunoblotting. The reaction components and the respective fractions are indicated above the blot and the respective ribosomal profiles and the fractions are indicated below the gel. Two picomoles of IF3 was loaded in the lanes indicated as ‘Unbound IF3’. A protein size standard containing strep tagged proteins is loaded in the lane ‘M’ and the corresponding molecular weights are indicated on the right.
Our work strengthens the previously known ancillary role of IF1 in codon selection (17) that augments the dominant checkpoint role played by IF3 during initiation (17,53). Here it is plausible that it is this IF3 mediated quality control checkpoint that the premature subunits subvert to enter the translation cycle. In order to test this, we studied the stability of binding between IF3 and 30S subunits that were actively engaged in protein synthesis by associating with 50S particles. In order to perform this, we decoupled actively translating 70S particles and selectively purified the 30S subunits by rigorous cycles of ultracentrifugation (vide Materials and Methods). We incubated the 30S particles derived from Wt, ΔrbfA and ΔksgA with a 10-fold molar excess of purified IF3 (Supplementary figure S7). These reaction mixtures were further analysed both using a pelleting assay and a co-sedimentation assay to test for the co-migration of IF3 with the 30S particles (Figure 7D). The presence of IF3 was probed using immunoblotting against its N-terminal strep tag. For Wt ribosomes, the presence of significant levels of IF3 in both 30S pellets (Figure 7E) as well as sucrose density gradient fractions (Figure 7F) indicates a stable association between 30S and IF3. However, to our surprise, IF3 failed to co-migrate with pre-30S derived from ΔrbfA and ΔksgA (Figure 7E and F). This suggests that these 30S premature particles indeed display negligible IF3 binding, which would render them fallible during the start codon selection.
DISCUSSION
Genetic disruptions of ribosome assembly (65–67) and production of misfolded proteins by compromised translational machinery are associated with cell death and neurodegeneration (68). To avert this, eukaryotic systems deploy proactive mechanisms to prevent the entry of premature ribosomes into the translation cycle (69). However, similar mechanisms in prokaryotic organisms still elude discovery.
Our data suggest that ribosome quality control checkpoints are also present in bacteria and premature ribosomes evade this scrutiny to enter the translation cycle. Consistent with the previous report of mistranslation events in ΔksgA (38), our observations for ΔlepA, ΔrbfA and ΔksgA echo a direct correlation between assembly defects and impaired translational machinery (Figure 1). The premature ribosomes work with suboptimal fidelity and a compromised ability to recognise the correct start codon. It is also important to reflect upon, whether the mistranslation events observed here arise from CO–AC interactions between non-cognate mRNA and tRNAifmet or incorporation of tRNAe into the pre-30S-ICs. Our observations of significant tRNAifmet localisation in the 70S fractions from ΔrbfA and ΔksgA (Figure 5A) and previous reports of N-terminal sequencing of mistranslated products (70) suggest that these misinitiation events arise due to interactions between the tRNAifmet and non-AUG start codons. Here, the recognition of the AGG start codon is particularly noteworthy given the marked conservation of U among cognate and near-cognate start codons (i.e. AUG, GUG, UUG, AUU, AUC and AUA) and indicates severely compromised codon recognition by the pre-30S decoding centre.
What does the pre-30S lack, that it loses its ability to discriminate non-canonical CO-AC interactions during initiation? The recently implicated RAF, LepA, mediates conformational changes in initiating 70S particles and its deletion results in a reduced representation of uS3, uS10 and uS14 (42,71). On the other hand, RsgA mediates optimal head region formation by recruiting tertiary r-proteins and monitoring the 30S A-site in a fashion similar to IF1 (72). Similarly, RbfA binds near the decoding centre and directs the formation of h1, h2 and the 3′ domain of the 16S rRNA. During this 3′ domain remodelling, it also reorients h44, h45 and the platform region (36,73,74). Later, KsgA binds to the helices h24, h27 and h45 in nearly matured 30S particles (8) and methylates two universally conserved residues, A1518 and A1519 on the h45 (8,75,76). These modifications seem to be important for the release of RbfA from 30S particles (8,38), and also stabilise packing interactions between h44 and h45, thus ensuring accurate head domain closure during CO-AC validation (77–80). While both ΔksgA and ΔrbfA show deformed platform and head regions, premature ribosomes assembling in the absence of the RbfA checkpoint harbour severe defects in the head domain. Cryo-EM structure of the ΔrbfA pre-30S particles display the head to be rotated in an orthogonal conformation in comparison to mature 30S particles (81). Collectively, these reports establish that the aforementioned RAFs mediate maturation of the 3′ end of 16S rRNA and the decoding centre of the 30S subunit to varying extent.
Notably, mutations in 3′ region of the 16S rRNA disrupt initiation factor-induced neck and head rotation thus promoting translation initiation from non-AUG start codons and premature 50S association (82). Our work shows that similar defects are also seen in the absence of late-stage RAFs leading to translation initiation with erroneous start codon recognition (Figures 1 and 2). In addition, milder defects in recognising frameshifts and stop codons (Figure 1G) indicate a limited effect of assembly defects on decoding at the A-site too. Further, our experiments to gauge the mechanistic implications of impaired P-site decoding also reveal widespread effects on protein production due to a decrease in the rate of translation initiation and peptide bond synthesis (Figure 2C, D and F). However, it is not possible to infer from the current data, if this decrease in the peptide chain elongation rate is solely due to defects in the P-site or other structural deformities that decrease the speed of the translocation step (83), thus slowing down the overall process of translation. It is also plausible to extend that, as defects in assembly exacerbate, deficiencies in codon recognition and translation impairment also aggravate (Figures 3C, D and 4). However, these defects are not an outcome of translation cessation in RAF null mutants but rather a bona fide compromise in the fidelity during the gatekeeping stages of translation initiation (Figure 2F).
Intrigued by these observations, we asked what aspect of initiation falters in RAF null mutants? During initiation, the 30S binds the Initiation factors (IF1, IF2 and IF3), the mRNA and the tRNAifmet in a poorly defined order to form the 30S-PIC, wherein the formation of cognate CO-AC interaction is not yet complete. Large-Scale conformational rearrangements within the 30S-PIC guided by the Initiation factors ensure the proofreading of cognate CO-AC interaction and the transition towards the 30S-IC. These rearranged complexes subsequently dock with the 50S to form the 70S-IC that is competent to enter the elongation cycle (17,59). However, in case of ΔksgA and ΔrbfA, the altered ribosome occupancy, a preferential enrichment of tRNAifmet with 70S particles (Figures 4 and 5A) and the robust expression of AGG-gfp using pre-30S (Figures 1, 2 and 5) support the notion of the transition of 30S-PIC/IC to 70S-IC by evading the translation quality control checkpoint. Here, the IF governed transition of 30S-PIC to 30S-IC, the first checkpoint to ensure translational fidelity, seems to go awry when the 30S harbours assembly defects.
In Wt cells, two events are critical for bona fide transition of 30S-PIC to 30S-IC. These are (i) the formation of cognate CO-AC interaction and (ii) the validation of cognate CO-AC interaction by Initiation factors (especially IF3). Upon 30S binding, IF3 anchors on the 30S platform region using its N-terminal domain followed by adjustment of the mRNA and tRNAifmet towards the decoding centre (17,84,85). IF3 scans at least three conformational spaces and induces rotation of the 30S head while monitoring the CO–AC interactions with its C-terminal domain (17,86–88). In line with these observations, defects in the head and platform of the pre-30S weaken IF3 binding (Figure 7E and F) and compromise the validation of CO–AC interactions (Figure 6–8 and Supplementary Figure S6). Supporting this conjecture, we noted that the severity of translational defects in ΔlepA, ΔksgA and ΔrbfA correlates with where the respective defects lie. In line with this, ΔlepA and ΔrsgA show minimal disruption to start codon recognition (Figure 1C and D). On the other hand, exacerbated translation-related defects in ΔrbfA (Figure 1E, G and 2) could be attributed to its early and vital role in catalysing the formation of the 30S head and platform region. From this, it is evident that head and platform defects impact the formation of CO-AC interactions and the subsequent proofreading by the IFs. This idea is further bolstered by translational rescue observed upon the elevation of cellular concentrations of IFs (Figure 6A–C, 7A–B). Additionally, this also reiterates the recent reports of the close coupling between 30S maturation, translation initiation (89,90) and stress tolerance (91,92) in bacteria.
Figure 8.

A schematic representation of the plausible events leading to the participation of premature subunits in translation. (A) Initiation factors and tRNAifmet are recruited to mature 30S subunits complexed with mRNA harbouring canonical start signal to form accurate initiation intermediates. These intermediates undergo meticulous kinetic proofreading checkpoints posed by initiation factors and only then are allowed to enter the translation cycle. On the contrary, premature subunits with structural deformities possess weak binding affinities for initiation factors thus readily bind to 50S particles to initiate translation from canonical start signals. (B) Mature 30S particles can form pre-initiation complexes on non-canonical signals, but proofreading of the impaired codon–anticodon interaction by initiation factors pre-empts their transition into initiation complexes and further translation. However, due to the weak binding of Initiation factors, premature 30S particles elude such proofreading checkpoints to form 70S-IC, which initiates protein synthesis from non-canonical initiation signals thus displaying compromised fidelity of translation initiation.
Our findings also lay to rest, the previous uncertainties about the involvement of premature ribosomes in protein synthesis (38,71) and clarify that IF1 and to a greater extent, IF3 rescue translation initiation and restore the specificity of codon recognition (Figure 7A, B and Supplementary Figure S4). While IF3 is already known to monitor the conversion of 30S-PIC to 30S-IC (17,59,88), this work sheds light on the mechanism by which assembly defects subvert the role of IF3 in translation quality control. Additionally, the fitful binding of IF3 (Figure 7E and F) to premature subunits is likely to compromise its anti-association activity (Figure 6D–F) which is critical in ensuring fidelity of initiation by the rapid ejection of non-canonical mRNA-tRNA interaction from the P-site (93). However, the mechanism of IF1 mediated rescue is subtler (Figure 6D–F) as it is thought to enhance the function of IF3 (17). Thus, it is conceivable that the conversion of initiating pre-30S particles to elongating 70S particles takes place by skipping the IF3 mediated quality control of protein synthesis (Figure 8). Further structural and biochemical evidence would be needed to fully understand the factors that contribute to these bypasses in ribosome quality control.
CONCLUDING REMARKS
In bacteria, it appears that the translation initiation factors act as a single-window quality control checkpoint to proofread not only the fidelity of the genetic message during the translational cycle but also to test drive and validate the competence of nascent ribosomes. In this context, it is tempting to speculate that the universality of AUG as an initiation codon perhaps stems from the fact that the initiation factors, especially IF3, is evolved and attuned to recognise AUG than the non-AUG. This inter-dependency is also a cost-cutting measure to optimise the energy reserves while at the same time tightening the quality control.
DATA AVAILABILITY
Sequencing data are deposited in the Gene Expression Omnibus under accession number GEO: GSE122870.
Supplementary Material
ACKNOWLEDGEMENTS
The pBAD Strep TEV LIC (8R) cloning vector (Addgene plasmid # 37506), the pET His6 GFP TEV (1GFP) LIC cloning vector (Addgene plasmid # 29663) and pET StrepII TEV LIC cloning vector (1R) (Addgene plasmid # 29664) were kind gifts from Scott Gradia. The vector pdCas9-bacteria was a gift from Stanley Qi (Addgene plasmid # 44249). The E. coli TKC strain was a kind gift from Donald L. Court. We acknowledge the geniality of the aforementioned scientists for sharing their plasmids. We also acknowledge the NGS services of Genotypic Technologies Pvt Ltd, Bengaluru, India. The authors also thank Sumit Kinger and Kiran Dhobale for the technical assistance and are also grateful to all members of the MAB lab for their support and critical comments on the manuscript.
Author contributions: H.S. and B.A. conceived the problem; H.S. designed assays and performed all experiments; B.A. processed and analysed the deep sequencing data; H.S. and B.A. analysed the remaining data and wrote the manuscript; B.A. arranged for funds and oversaw the project.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Department of Biotechnology (DBT) [BT/PR15925/NER/95/141/2015, BT/08/IYBA/2014/05 and BT/406/NE/UEXCEL/2013]; Science and Engineering Research Board (SERB) [YSS/2014/000286]. The open access publication charge for this paper has been waived by Oxford University Press – NAR.
Conflict of interest statement. None declared.
REFERENCES
- 1. Davis J.H., Williamson J.R.. Structure and dynamics of bacterial ribosome biogenesis. Philos. Trans. R. Soc. B Biol. Sci. 2017; 372:20160181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shajani Z., Sykes M.T., Williamson J.R.. Assembly of bacterial ribosomes. Annu. Rev. Biochem. 2011; 80:501–526. [DOI] [PubMed] [Google Scholar]
- 3. Sulthana S., Deutscher M.P.. Multiple exoribonucleases catalyze maturation of the 3′ terminus of 16S ribosomal RNA (rRNA). J. Biol. Chem. 2013; 288:12574–12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deutscher M.P. Twenty years of bacterial RNases and RNA processing: how we’ve matured. RNA. 2015; 21:597–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith B.A., Gupta N., Denny K., Culver G.M.. Characterization of 16S rRNA processing with Pre-30s subunit assembly intermediates from E. coli. J. Mol. Biol. 2018; 430:1745–1759. [DOI] [PubMed] [Google Scholar]
- 6. Baumgardt K., Gilet L., Figaro S., Condon C.. The essential nature of YqfG, a YbeY homologue required for 3′ maturation of Bacillus subtilis 16S ribosomal RNA is suppressed by deletion of RNase R. Nucleic Acids Res. 2018; 46:8605–8615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jack K., Bellodi C., Landry D.M., Niederer R.O., Meskauskas A., Musalgaonkar S., Kopmar N., Krasnykh O., Dean A.M., Thompson S.R. et al.. rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol. Cell. 2011; 44:660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boehringer D., O’Farrell H.C., Rife J.P., Ban N.. Structural insights into methyltransferase KsgA function in 30S ribosomal subunit biogenesis. J. Biol. Chem. 2012; 287:10453–10459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Spenkuch F., Motorin Y., Helm M.. Pseudouridine: still mysterious, but never a fake (uridine). RNA biology. 2014; 11:1540–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Polikanov Y.S., Melnikov S.V., Söll D., Steitz T.A.. Structural insights into the role of rRNA modifications in protein synthesis and ribosome assembly. Nat. Struct. Mol. Biol. 2015; 22:342–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Charollais J., Pflieger D., Vinh J., Dreyfus M., Iost I.. The DEAD-box RNA helicase SrmB is involved in the assembly of 50S ribosomal subunits in Escherichia coli: E. coli ribosome assembly involves a RNA helicase. Mol. Microbiol. 2003; 48:1253–1265. [DOI] [PubMed] [Google Scholar]
- 12. Charollais J., Dreyfus M., Iost I.. CsdA, a cold-shock RNA helicase from Escherichia coli, is involved in the biogenesis of 50S ribosomal subunit. Nucleic Acids Res. 2004; 32:2751–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gulati M., Jain N., Davis J.H., Williamson J.R., Britton R.A.. Functional interaction between ribosomal protein L6 and RbgA during ribosome assembly. PLos Genet. 2014; 10:e1004694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goto S., Kato S., Kimura T., Muto A., Himeno H.. RsgA releases RbfA from 30S ribosome during a late stage of ribosome biosynthesis. EMBO J. 2011; 30:104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Simonetti A., Marzi S., Myasnikov A.G., Fabbretti A., Yusupov M., Gualerzi C.O., Klaholz B.P.. Structure of the 30S translation initiation complex. Nature. 2008; 455:416–420. [DOI] [PubMed] [Google Scholar]
- 16. Gualerzi C.O., Pon C.L.. Initiation of mRNA translation in bacteria: structural and dynamic aspects. Cell Mol. Life Sci. 2015; 72:4341–4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hussain T., Llácer J.L., Wimberly B.T., Kieft J.S., Ramakrishnan V.. Large-scale movements of IF3 and tRNA during bacterial translation initiation. Cell. 2016; 167:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Siibak T., Peil L., Xiong L., Mankin A., Remme J., Tenson T.. Erythromycin- and chloramphenicol-induced ribosomal assembly defects are secondary effects of protein synthesis inhibition. Antimicrob. Agents Chemother. 2009; 53:563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nikolay R., Schloemer R., Schmidt S., Mueller S., Heubach A., Deuerling E.. Validation of a fluorescence-based screening concept to identify ribosome assembly defects in Escherichia coli. Nucleic Acids Res. 2014; 42:e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Freed E.F., Bleichert F., Dutca L.M., Baserga S.J.. When ribosomes go bad: diseases of ribosome biogenesis. Mol. Biosyst. 2010; 6:481–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Danilova N., Gazda H.T.. Ribosomopathies: how a common root can cause a tree of pathologies. Dis. Models Mech. 2015; 8:1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mills E.W., Green R.. Ribosomopathies: there's strength in numbers. Science. 2017; 358:eaan2755. [DOI] [PubMed] [Google Scholar]
- 23. Guthrie C., Nashimoto H., Nomura M.. Structure and function of E. coli ribosomes, VIII. Cold-sensitive mutants defective in ribosome assembly. Proc. Natl. Acad. Sci. U.S.A. 1969; 63:384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alix J.H., Guerin M.F.. Mutant DnaK chaperones cause ribosome assembly defects in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:9725–9729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bylund G.O., Wipemo L.C., Lundberg L.A., Wikström P.M.. RimM and RbfA are essential for efficient processing of 16S rRNA in Escherichia coli. J. Bacteriol. 1998; 180:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Britton R.A. Role of GTPases in bacterial ribosome assembly. Annu. Rev. Microbiol. 2009; 63:155–176. [DOI] [PubMed] [Google Scholar]
- 27. Lerner C.G., Inouye M.. Pleiotropic changes resulting from depletion of Era, an essential GTP-binding protein in Escherichia coli. Mol. Microbiol. 1991; 5:951–957. [DOI] [PubMed] [Google Scholar]
- 28. Talkington M.W.T., Siuzdak G., Williamson J.R.. An assembly landscape for the 30S ribosomal subunit. Nature. 2005; 438:628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lai J., Chen K., Luthey-Schulten Z.. Structural intermediates and folding events in the early assembly of the ribosomal small subunit. J. Phys. Chem. B. 2013; 117:13335–13345. [DOI] [PubMed] [Google Scholar]
- 30. Earnest T.M., Lai J., Chen K., Hallock M.J., Williamson J. R., Luthey-Schulten Z.. Toward a whole-cell model of ribosome biogenesis: kinetic modeling of SSU assembly. Biophys. J. 2015; 109:1117–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Neidhardt F.C., Magasanik B.. Studies on the role of ribonucleic acid in the growth of bacteria. Biochim. Biophys. Acta. 1960; 42:99–116. [DOI] [PubMed] [Google Scholar]
- 32. Koch A.L., Deppe C.S.. In vivo assay of protein synthesizing capacity of Escherichia coli from slowly growing chemostat cultures. J. Mol. Biol. 1971; 55:549–562. [DOI] [PubMed] [Google Scholar]
- 33. Towbin B.D., Korem Y., Bren A., Doron S., Sorek R., Alon U.. Optimality and sub-optimality in a bacterial growth law. Nat. Commun. 2017; 8:14123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dai X., Zhu M., Warren M., Balakrishnan R., Patsalo V., Okano H., Williamson J.R., Fredrick K., Wang Y.-P., Hwa T.. Reduction of translating ribosomes enables Escherichia coli to maintain elongation rates during slow growth. Nat. Microbiol. 2017; 2:16231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dai X., Zhu M., Warren M., Balakrishnan R., Okano H., Williamson J.R., Fredrick K., Hwa T., Lee S.Y.. Slowdown of Translational Elongation in Escherichia coli under Hyperosmotic Stress. mBio. 2018; 9:e02375-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Datta P.P., Wilson D.N., Kawazoe M., Swami N.K., Kaminishi T., Sharma M.R., Booth T.M., Takemoto C., Fucini P., Yokoyama S. et al.. Structural aspects of RbfA action during small ribosomal subunit assembly. Mol. Cell. 2007; 28:434–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Razi A., Guarné A., Ortega J.. The cryo-EM structure of YjeQ bound to the 30S subunit suggests a fidelity checkpoint function for this protein in ribosome assembly. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E3396–E3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Connolly K., Culver G.. Overexpression of RbfA in the absence of the KsgA checkpoint results in impaired translation initiation. Mol. Microbiol. 2013; 87:968–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. O’Connor M., Gregory S.T., Dahlberg A.E.. Multiple defects in translation associated with altered ribosomal protein L4. Nucleic Acids Res. 2004; 32:5750–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Qin D., Liu Q., Devaraj A., Fredrick K.. Role of helix 44 of 16S rRNA in the fidelity of translation initiation. RNA. 2012; 18:485–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Andrade J.M., Dos Santos R.F., Chelysheva I., Ignatova Z., Arraiano C.M.. The RNA-binding protein Hfq is important for ribosome biogenesis and affects translation fidelity. EMBO J. 2018; 37:e97631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gibbs M.R., Moon K.-M., Chen M., Balakrishnan R., Foster L.J., Fredrick K.. Conserved GTPase LepA (Elongation Factor 4) functions in biogenesis of the 30S subunit of the 70S ribosome. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:980–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Datsenko K.A., Wanner B.L.. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. O’Connor M. Decoding fidelity at the ribosomal A and P sites: influence of mutations in three different regions of the decoding domain in 16S rRNA. Nucleic Acids Res. 1997; 25:1185–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dalbow D.G., Young R.. Synthesis time of beta-galactosidase in Escherichia coli B/r as a function of growth rate. Biochem. J. 1975; 150:13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dalbow D.G., Bremer H.. Metabolic regulation of beta-galactosidase synthesis in Escherichia coli. A test for constitutive ribosome synthesis. Biochem. J. 1975; 150:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miller J.H. Experiments in Molecular Genetics. 1992; NY: Cold Spring Harbor Laboratory. [Google Scholar]
- 48. Schleif R., Hess W., Finkelstein S., Ellis D.. Induction kinetics of the L-arabinose operon of Escherichia coli. J. Bacteriol. 1973; 115:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shirokikh N.E., Archer S.K., Beilharz T.H., Powell D., Preiss T.. Translation complex profile sequencing to study the in vivo dynamics of mRNA–ribosome interactions during translation initiation, elongation and termination. Nat. Protoc. 2017; 12:697–731. [DOI] [PubMed] [Google Scholar]
- 50. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal. 2011; 17:10–12. [Google Scholar]
- 51. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. Genome Project Data Processing, S. . The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sussman J.K., Simons E.L., Simons R.W.. Escherichia coli translation initiation factor 3 discriminates the initiation codon in vivo. Mol. Microbiol. 1996; 21:347–360. [DOI] [PubMed] [Google Scholar]
- 54. Hecht A., Glasgow J., Jaschke P.R., Bawazer L.A., Munson M.S., Cochran J.R., Endy D., Salit M.. Measurements of translation initiation from all 64 codons in E. coli. Nucleic Acids Res. 2017; 45:3615–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Archer S.K., Shirokikh N.E., Beilharz T.H., Preiss T.. Dynamics of ribosome scanning and recycling revealed by translation complex profiling. Nature. 2016; 535:570–574. [DOI] [PubMed] [Google Scholar]
- 56. Marks J., Kannan K., Roncase E.J., Klepacki D., Kefi A., Orelle C., Vázquez-Laslop N., Mankin A.S.. Context-specific inhibition of translation by ribosomal antibiotics targeting the peptidyl transferase center. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:12150–12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Valášek L., Szamecz B., Hinnebusch A.G., Nielsen K.H.. Lorsch J. Methods in Enzymology. 2007; 429:Academic Press; 163–183. [DOI] [PubMed] [Google Scholar]
- 58. Lareau L.F., Hite D.H., Hogan G.J., Brown P.O.. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. eLife. 2014; 3:e01257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Milon P., Maracci C., Filonava L., Gualerzi C.O., Rodnina M.V.. Real-time assembly landscape of bacterial 30S translation initiation complex. Nat. Struct. Mol. Biol. 2012; 19:609–615. [DOI] [PubMed] [Google Scholar]
- 60. Ge X., Mandava C.S., Lind C., Åqvist J., Sanyal S.. Complementary charge-based interaction between the ribosomal-stalk protein L7/12 and IF2 is the key to rapid subunit association. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:4649–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Simonetti A., Marzi S., Billas I.M.L., Tsai A., Fabbretti A., Myasnikov A.G., Roblin P., Vaiana A.C., Hazemann I., Eiler D. et al.. Involvement of protein IF2 N domain in ribosomal subunit joining revealed from architecture and function of the full-length initiation factor. Proc. Natl. Acad. Sci. 2013; 110:15656–15661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Moreno J.M.P., Kildsgaard J., Siwanowicz I., Mortensen K.K., Sperling-Petersen H.U.. Binding of escherichia coli initiation factor IF2 to 30S ribosomal subunits: a functional role for the N-terminus of the factor. Biochem. Biophys. Res. Commun. 1998; 252:465–471. [DOI] [PubMed] [Google Scholar]
- 63. Milon P., Carotti M., Konevega A.L., Wintermeyer W., Rodnina M.V., Gualerzi C.O.. The ribosome‐bound initiation factor 2 recruits initiator tRNA to the 30S initiation complex. EMBO Rep. 2010; 11:312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Milon P., Tischenko E., Tomsic J., Caserta E., Folkers G., La Teana A., Rodnina M.V., Pon C.L., Boelens R., Gualerzi C.O.. The nucleotide-binding site of bacterial translation initiation factor 2 (IF2) as a metabolic sensor. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:13962–13967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dragon F., Gallagher J.E., Compagnone-Post P.A., Mitchell B.M., Porwancher K.A., Wehner K.A., Wormsley S., Settlage R.E., Shabanowitz J., Osheim Y. et al.. A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature. 2002; 417:967–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ganapathi K.A., Austin K.M., Lee C.S., Dias A., Malsch M.M., Reed R., Shimamura A.. The human Shwachman-Diamond syndrome protein, SBDS, associates with ribosomal RNA. Blood. 2007; 110:1458–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sun C., Woolford J.L. Jr.. The yeast NOP4 gene product is an essential nucleolar protein required for pre-rRNA processing and accumulation of 60S ribosomal subunits. EMBO J. 1994; 13:3127–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lee J.W., Beebe K., Nangle L.A., Jang J., Longo-Guess C.M., Cook S.A., Davisson M.T., Sundberg J.P., Schimmel P., Ackerman S.L.. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006; 443:50–55. [DOI] [PubMed] [Google Scholar]
- 69. Strunk B.S., Novak M.N., Young C.L., Karbstein K.. A Translation-like cycle is a quality control checkpoint for maturing 40S ribosome subunits. Cell. 2012; 150:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. O’Connor M., Thomas C.L., Zimmermann R.A., Dahlberg A.E.. Decoding fidelity at the ribosomal A and P sites: influence of mutations in three different regions of the decoding domain in 16S rRNA. Nucleic Acids Res. 1997; 25:1185–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Balakrishnan R., Oman K., Shoji S., Bundschuh R., Fredrick K.. The conserved GTPase LepA contributes mainly to translation initiation in Escherichia coli. Nucleic Acids Res. 2014; 42:13370–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. López-Alonso J.P., Kaminishi T., Kikuchi T., Hirata Y., Iturrioz I., Dhimole N., Schedlbauer A., Hase Y., Goto S., Kurita D. et al.. RsgA couples the maturation state of the 30S ribosomal decoding center to activation of its GTPase pocket. Nucleic Acids Res. 2017; 45:6945–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dammel C.S., Noller H.F.. Suppression of a cold-sensitive mutation in 16S rRNA by overexpression of a novel ribosome-binding factor, RbfA. Genes Dev. 1995; 9:626–637. [DOI] [PubMed] [Google Scholar]
- 74. Clatterbuck Soper S.F., Dator R.P., Limbach P.A., Woodson S.A.. In vivo X-ray footprinting of Pre-30S ribosomes reveals chaperone-dependent remodeling of late assembly intermediates. Mol. Cell. 2013; 52:506–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Thammana P., Held W.A.. Methylation of 16S RNA during ribosome assembly in vitro. Nature. 1974; 251:682–686. [DOI] [PubMed] [Google Scholar]
- 76. Connolly K., Rife J.P., Culver G.. Mechanistic insight into the ribosome biogenesis functions of the ancient protein KsgA. Mol. Microbiol. 2008; 70:1062–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Xu Z., O’Farrell H.C., Rife J.P., Culver G.M.. A conserved rRNA methyltransferase regulates ribosome biogenesis. Nat. Struct. Mol. Biol. 2008; 15:534–536. [DOI] [PubMed] [Google Scholar]
- 78. Helser T.L., Davies J.E., Dahlberg J.E.. Mechanism of kasugamycin resistance in escherichia coli. Nat. New Biol. 1972; 235:6–9. [DOI] [PubMed] [Google Scholar]
- 79. Ogle J.M., Brodersen D.E., Clemons W.M., Tarry M.J., Carter A.P., Ramakrishnan V.. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science. 2001; 292:897–902. [DOI] [PubMed] [Google Scholar]
- 80. Demirci H., Murphy F.T., Belardinelli R., Kelley A.C., Ramakrishnan V., Gregory S.T., Dahlberg A.E., Jogl G.. Modification of 16S ribosomal RNA by the KsgA methyltransferase restructures the 30S subunit to optimize ribosome function. RNA. 2010; 16:2319–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yang Z., Guo Q., Goto S., Chen Y., Li N., Yan K., Zhang Y., Muto A., Deng H., Himeno H. et al.. Structural insights into the assembly of the 30S ribosomal subunit in vivo: functional role of S5 and location of the 17S rRNA precursor sequence. Protein Cell. 2014; 5:394–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Qin D., Fredrick K.. Control of translation initiation involves a factor-induced rearrangement of helix 44 of 16S ribosomal RNA. Mol. Microbiol. 2009; 71:1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shi X., Chiu K., Ghosh S., Joseph S.. Bases in 16S rRNA important for subunit association, tRNA binding, and translocation. Biochemistry. 2009; 48:6772–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Dallas A., Noller H.F.. Interaction of translation initiation factor 3 with the 30S ribosomal subunit. Mol. Cell. 2001; 8:855–864. [DOI] [PubMed] [Google Scholar]
- 85. Muralikrishna P., Wickstrom E.. Escherichia coli initiation factor 3 protein binding to 30S ribosomal subunits alters the accessibility of nucleotides within the conserved central region of 16S rRNA. Biochemistry. 1989; 28:7505–7510. [DOI] [PubMed] [Google Scholar]
- 86. Elvekrog M.M., Gonzalez R.L. Jr.. Conformational selection of translation initiation factor 3 signals proper substrate selection. Nat. Struct. Mol. Biol. 2013; 20:628–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Julián P., Milon P., Agirrezabala X., Lasso G., Gil D., Rodnina M.V., Valle M.. The Cryo-EM structure of a complete 30S translation initiation complex from escherichia coli. PLoS Biol. 2011; 9:e1001095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Milon P., Konevega A.L., Gualerzi C.O., Rodnina M.V.. Kinetic checkpoint at a late step in translation initiation. Mol. Cell. 2008; 30:712–720. [DOI] [PubMed] [Google Scholar]
- 89. Shetty S., Varshney U.. An evolutionarily conserved element in initiator tRNAs prompts ultimate steps in ribosome maturation. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E6126–E6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Brandi A., Piersimoni L., Feto N.A., Spurio R., Alix J.-H., Schmidt F., Gualerzi C.O.. Translation initiation factor IF2 contributes to ribosome assembly and maturation during cold adaptation. Nucleic Acids Res. 2019; 47:4652–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kyuma T., Kizaki H., Ryuno H., Sekimizu K., Kaito C.. 16S rRNA methyltransferase KsgA contributes to oxidative stress resistance and virulence in Staphylococcus aureus. Biochimie. 2015; 119:166–174. [DOI] [PubMed] [Google Scholar]
- 92. Campbell T.L., Henderson J., Heinrichs D.E., Brown E.D.. The yjeQ gene is required for virulence of staphylococcus aureus. Infect. Immun. 2006; 74:4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Petrelli D., La Teana A., Garofalo C., Spurio R., Pon C.L., Gualerzi C.O.. Translation initiation factor IF3: two domains, five functions, one mechanism. EMBO J. 2001; 20:4560–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kumar V., Ero R., Ahmed T., Goh K.J., Zhan Y., Bhushan S., Gao Y.-G.. Structure of the GTP form of elongation factor 4 (EF4) bound to the ribosome. J. Biol. Chem. 2016; 291:12943–12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data are deposited in the Gene Expression Omnibus under accession number GEO: GSE122870.