


Abstract
The binding of p53-binding protein 1 (53BP1) to damaged chromatin is a critical event in non-homologous DNA end joining (NHEJ)-mediated DNA damage repair. Although several molecular pathways explaining how 53BP1 binds damaged chromatin have been described, the precise underlying mechanisms are still unclear. Here we report that a newly identified H4K16 monomethylation (H4K16me1) mark is involved in 53BP1 binding activity in the DNA damage response (DDR). During the DDR, H4K16me1 rapidly increases as a result of catalyzation by the histone methyltransferase G9a-like protein (GLP). H4K16me1 shows an increased interaction level with 53BP1, which is important for the timely recruitment of 53BP1 to DNA double-strand breaks. Differing from H4K16 acetylation, H4K16me1 enhances the 53BP1–H4K20me2 interaction at damaged chromatin. Consistently, GLP knockdown markedly attenuates 53BP1 foci formation, leading to impaired NHEJ-mediated repair and decreased cell survival. Together, these data support a novel axis of the DNA damage repair pathway based on H4K16me1 catalysis by GLP, which promotes 53BP1 recruitment to permit NHEJ-mediated DNA damage repair.
INTRODUCTION
Environmental stressors and endogenous metabolites pose a constant threat to DNA integrity; as such, all organisms have evolved efficient systems to repair damaged DNA and maintain genome stability (1,2). Several distinct pathways to repair DNA double-strand breaks (DSBs) have been proposed. Among them, non-homologous DNA end joining (NHEJ) and homologous recombination (HR) have been widely studied and fairly well characterized (3). Determining how histone modifiers participate in these two processes is of crucial importance to improve our understanding of DSB repair and guide the development of novel cancer treatments (4,5).
p53-binding protein (53BP1) binds damaged chromatin and recruits other responsive proteins to DSBs—a critical mechanism for proper NHEJ repair and appropriate repair pathway selection (6). 53BP1 accumulation at DSBs is affected by early responsive DNA repair factors, such as ataxia-telangiectasia mutated (ATM) and MDC1 (7–13), and its binding to chromatin is considered to be mainly regulated by several histone modifications. For example, dimethylation of H4K20 (H4K20me2), a residue known for 53BP1 tandem Tudor domain binding, is fundamental for 53BP1 ionizing radiation-induced foci formation at DSBs (14). In addition, 53BP1 binding to damaged chromatin is strengthened by H2AK15 ubiquitination, which is catalyzed by the E3 ligases RNF8 and RNF168 and recognized by the ubiquitination-dependent 53BP1 recruitment motif (15–20). Moreover, 53BP1 may also be recruited by γH2AX and deacetylated H3K18 (21–23).
Under normal conditions, the H4K20me2 mark is masked by various bound proteins, including L3MBTL1 (24) and KDM4A/JMJD2A (25). RNF8/RNF168-dependent dissociation and/or degradation of these proteins in response to DNA damage exposes the H4K20me2 mark to permit 53BP1 binding (25,26). The Tudor interacting repair regulator (TIRR) directly binds the 53BP1 tandem Tudor domain and also masks the H4K20me2 binding motif of 53BP1 under normal situations. Upon DNA damage, ATM and RAP1-interacting factor 1 (RIF1) promote 53BP1–TIRR complex dissociation and subsequent 53BP1 recruitment to DSBs (27,28). In addition, 53BP1 sequestration by NuMA in the absence of DNA damage has also been reported (29).
The regulation of 53BP1 binding to damaged chromatin is a more complicated process, owing to other indirect but also important regulatory mechanisms that influence the 53BP1–H4K20me2 interaction. One such example is H4K16ac: H4K16ac is catalyzed by the TIP60 acetyltransferase complex, which diminishes 53BP1 binding to H4K20me2, at least in part, by disrupting a salt bridge between H4K16 and the 53BP1 Tudor domain (30). Consistently, H4K16 deacetylation robustly augments 53BP1 binding to H4K0me2 and ionizing radiation-induced foci formation (31). In addition, the TIP60 complex component MBTD1 competes with 53BP1 to bind methylated H4K20, and the TIP60 complex can acetylate H2AK15 in response to DNA damage (32). Interestingly, RNF168-dependent H2AK15 ubiquitylation directly suppresses the ability of TIP60 to acetylate the H4 tail (32). This ubiquitylation/acetylation switch on H2AK15 is a powerful mechanism to regulate 53BP1 binding and TIP60-dependent histone H4 acetylation in the DNA damage response (DDR). Previously, it was reported that H4K16ac levels change in response to DNA damage: H4K16ac undergoes rapid deacetylation and a lagged increase in acetylation at DNA lesions post-irradiation (30). Other modifications to H4K16 might also occur during DNA damage repair that could potentially affect 53BP1 binding to damaged chromatin.
Enzymes involved in histone methylation participate in DNA damage repair by affecting the methylation status of specific histone lysine residues. For example, after irradiation, ATM-dependent dissociation of the histone demethylase KDM2A from chromatin (33,34) and recruitment of the histone methyltransferase Metnase (35) contribute to increased H3K36me2 levels at DSBs and the consequent recruitment of NHEJ-associated repair factors to repair the damaged DNA. In addition, the histone methyltransferase PRDM2 catalyzes H3K9me2 at DSBs in a macroH2A1-dependent manner, and is critical for BRCA1 retention and DNA repair via HR (36). Among dozens of histone methyltransferases, data suggest that the histone methyltransferase G9a-like protein (GLP) might be directly involved in DNA damage repair. First, GLP was reported to be a potential substrate of ATM/ATR in the DDR (37). Secondly, the chromatin level of GLP also increases after irradiation (38). A more recent report identified that a specific G9a/GLP inhibitor (UNC0638) can impair NHEJ repair efficiency (39), but the detailed mechanism underlying this effect remains largely unknown.
Here, we used a combination of in vitro biochemical and cytological assays to show that H4K16 monomethylation (H4K16me1)—a novel histone modification—is involved in DNA damage repair. We found that histone methyltransferase GLP-catalyzed H4K16me1 levels markedly increase during the early stages of DDR and H4K16me1 has an increased interaction with 53BP1 after DNA damage treatment. This interaction is associated with increased 53BP1 binding to DSBs and H4K16me1 can cooperate with H4K20me2 to facilitate 53BP1 recruitment and NHEJ-mediated DNA repair. Our study deepens the knowledge of DNA damage repair and may have potential clinical applications in cancer treatment, with the development of novel therapeutics based on H4K16me1 functions in the DDR.
MATERIALS AND METHODS
Cell culture and transfection
HCT116 cells were grown in McCoy's 5a medium supplemented with 10% fetal bovine serum and the appropriate amount of penicillin/streptomycin, in a humidified atmosphere of 5% CO2 maintained at 37°C. HeLa, LoVo, HEK293, A-T, EJ5-GFP U2OS and DR-GFP U2OS cells were grown in Dulbecco's Modified Eagles Medium under the same conditions. The cells were sub-cultured by trypsinization every 2 days, and seeded at the appropriate confluency. HeLa, LoVo and HCT116 cells were obtained from the American Type Culture Collection. A-T, EJ5-GFP U2OS and DR-GFP U2OS cells were obtained from Professor Xingzhi Xu (Shenzhen University). Transient and stable transfections were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's protocol.
Antibodies
The antibodies used in this study were: anti-His-tag (D291-3, MBL, Aichi, Japan), anti-Myc-tag (M047-3, MBL), anti-GFP-tag (M048-3, MBL), anti-GLP (D220-3, MBL), anti-Flag-tag (F1804, Sigma-Aldrich, St Louis, MO, USA), anti-G9a (G6919, Sigma-Aldrich), anti-GST-tag (C1303, APPLYGEN, Beijing, China), anti-mCherry-tag (C1329, APPLYGEN, Beijing, China), anti-H3 (ab1791, Abcam, Cambridge, UK), anti-H4 (ab10158, Abcam), anti-H3K9me2 (ab1220, Abcam), anti-H3K27me3 (ab6002, Abcam), anti-H3K9me3 (ab8898, Abcam), anti-H3K79me1 (ab2886, Abcam), anti-H4K20me1 (ab9051, Abcam), anti-H4K20me2 (ab9052, Abcam), anti-MRE11 (ab12159, Abcam), anti-RPA32 (ab2175, Abcam), anti-phospho-Histone H2AX (Ser139) (05–636, EMD Millipore, Billerica, MA, USA), anti-53BP1 (MAB3802, EMD Millipore), anti-GLP (09–078, EMD Millipore), anti-FK2 (04–263, EMD Millipore), anti-53BP1 (NB100–304, Novus Biologicals, Abingdon, UK), anti-GLP (B0422, Novus Biologicals), anti-DNA-PKcs (sc-1552, Santa Cruz Biotechnology), anti-ATR (sc-1887, Santa Cruz Biotechnology), anti-Actin (sc-58673, Santa Cruz Biotechnology), anti-SET8 (C18B7, Cell Signaling Technology, Danvers, MA, USA), anti-ATM (GTX70103, GeneTex), anti-RNF8 (14112-1-AP, Proteintech, Wuhan, Hubei, China) and anti-RNF168 (21393-1-AP, Proteintech).
Inhibitors
The inhibitors used in this study were: KU-55933 (S1092), KU-57788 (S2638), VE-821 (S8007), TBB (S5265), Bix-01294 (S8006), A-196 (S7983); all from Selleck Chemicals, Houston, TX, USA.
H4K16me1 antibody generation
The H4K16me1 antibody was raised in rabbits against the H4K16me1 peptide CKGGA-(methyl)K-RHRK coupled to keyhole limpet hemocyanin. Rabbits were injected with the immunogen three times under a typical boost time schedule. One week after the third injection, the rabbits were bled for the first time to perform the ELISA and dot blot assays. The rabbits with high specificity of antisera were boosted again for the last time 1 week later and exsanguinated 10 days following the final boost. Antisera were captured with Protein A resin and purified with antigen peptide conjugated resin. Furthermore, crossing peptide conjugated resin was used to deplete the cross-reactivity. The purified antibody was tested by ELISA, dot blot assay and western blotting.
Plasmids
The mouse GST-GLP plasmid (containing the Ankyrin and SET domains) was provided by Dr. Bing Zhu (Institute of Biophysics, Chinese Academy of Sciences). The human GFP-53BP1 plasmid was provided by Professor Xingzhi Xu (Shenzhen University). The cDNA of the 53BP1 Tudor domain was amplified and subcloned into pGEX-6P-1 and pET-28a vectors. Full-length and SET domain-deleted human GLP were amplified and subcloned into pcDNA3.1 and mCherry-N1 vectors. The full-length human SUV39H1 cDNA and the SETD2 catalytic fragment were amplified and cloned into pGEX-6P-1. The full-length human SET8 cDNA was amplified and cloned into pET-28a. EZH2 and DOT1L cDNAs were amplified and cloned into p3xFLAG-CMV-10. The I-SceI construct was provided by Professor Xingzhi Xu (Shenzhen University). All mutation constructs were generated with a Mut Express II Fast Mutagenesis Kit (Vazyme Biotech Co., Nanjing, China).
DNA damage treatment
For X-ray irradiation, cultured cells at 80% confluency were subjected to the indicated dose of radiation and then re-cultured in fresh medium for the indicated time. X-ray irradiation was delivered using an RS2000pro Ras Source biological X-ray irradiator (Rad Source Techologies, GA, USA) with a radiation output of 160 KV, 25 mA at a dose rate of 4.125 Gy/min.
For etoposide treatment, cells were treated with 40 μM etoposide (E1383, Sigma-Aldrich) for the indicated time, washed with phosphate-buffered saline (PBS) four times, and re-cultured in fresh medium for the indicated time before being harvested.
For micro-irradiation, cells were grown on a thin glass-bottom dish (Corning Incorporated, New York, NY, USA) and then sensitized by BrdU and locally irradiated with a 365 nm pulsed nitrogen UV laser (16 Hz pulse, 41% laser output) generated from a MicroPoint system (Andor Technology, Belfast, Ireland). This system was directly coupled to the epifluorescence path of a Nikon A1 confocal imaging system (Nikon, Tokyo, Japan). The relative intensity represents the gray-scale changes (It-I0) calculated from at least 10 cells per treatment. The data represent the means ± standard deviation (SD). All data were analyzed in Image J.
In vitro HMTase assay
For the in vitro HMTase assay, 2 μg substrates were incubated with different enzymes in a methylation reaction buffer [50 mM Tris–HCl (pH 9.0 for GLP/G9a, pH 8.0 for others), 5 mM MgCl2, 4 mM dithiothreitol (DTT), 0.5 mM SAM] at 37°C for the indicated time before analysis by western blotting. Histones H3 and H4 were obtained from New England Biolabs, MA, USA. GST, GST-GLP (containing the Ankyrin and SET domains), GST-SUV39H1, GST-SETD2 and His-SET8 were purified from Escherichia coli. Flag-EZH2 and Flag-DOT1L complexes were purified from HCT116 cells. G9a (containing the Ankyrin and SET domains) was purchased from Cayman Chemical Company, Ann Arbor, MI, USA.
Histone acid extraction
Cells were lysed in 1 ml hypotonic lysis buffer (10 mM Tris–HCl pH 8.0, 1 mM KCl, 1.5 mM MgCl2 and 1 mM DTT, protease inhibitors), and the intact nuclei were pelleted by centrifugation at 12 000 rpm (13 523 × g) at 4°C for 10 min. The supernatant was discarded and the nuclei were re-suspended in 400 μl 0.2 M sulfuric acid and incubated for at least 30 min at 4°C. The samples were again collected by centrifugation at 12 000 rpm (13 523 × g) at 4°C for 10 min, and the supernatant containing the histones was collected. Trichloroacetic acid was added to the histones to a final concentration of 33% and the samples were incubated on ice for 30 min. The histone pellet was collected by centrifugation at 12 000 rpm (13 523 × g) at 4°C for 10 min, washed with acetone and then dissolved in ddH2O.
Protein extraction and western blotting
For whole-cell lysate extraction, equal numbers of harvested cells were washed with PBS by centrifugation at 10 000 rpm (9391 × g) at 4°C for 30 s and the cell pellet was re-suspended in 30 μl (per 106 cells) 2× protease inhibitor buffer containing one cocktail protease inhibitor pellet (Roche Holding AG, Basel, Switzerland) in 3.5 ml PBS. An equal volume of 2× sodium dodecyl sulphate (SDS) loading buffer (950 μl Laemmle buffer + 50 μl 2-mercaptoethanol) was added to the re-suspended cells. The samples were boiled for 10 min with a pulse vortex every 5 min, and then pelleted by centrifugation at 12,000 rpm (13,523 x g) at 4°C for 15 min.
For soluble and chromatin protein extraction, the cells were re-suspended in buffer I (50 mM Hepes pH 7.5, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 0.1% Triton, protease inhibitors) on ice for 3 min. After centrifugation at 13,000 rpm (15,871 x g) at 4°C for 3 min, the supernatant was collected as the soluble fraction (Dt). The pellet was washed with buffer I and re-suspended in Buffer II (Buffer I containing 2% RNase A, protease inhibitors) at 25°C for 30 min. After centrifugation at 13,000 rpm (15,871 x g) at 4°C for 3 min, the supernatant (containing RNA binding proteins) was discarded, and the pellet was collected as the chromatin fraction (Chr).
Western blotting was used to evaluate protein levels, as previously described (40), with minor modifications. Equal amounts of proteins were size-fractionated on a 6–15% SDS-polyacrylamide gel electrophoresis gel.
Protein purification and pull-down assays
GST-fusion proteins were expressed in E. coli BL21 cells and purified using glutathione-sepharose 4B beads (GE Healthcare, Kings Park, NY, USA), according to the manufacturer's protocol. His-tagged proteins were expressed in E. coli BL21 (DE3) cells and purified using a Ni (ii)-Sepharose Affinity kit (GE Healthcare). Isolated proteins were further purified using a molecular sieve. The amino acid sequence of the biotinylated peptides corresponded to the histone H4 tail (aa 10–25). The following peptides were used:
H4K16me0: Biotin-LGKGGAKRHRKVLRDN;
H4K16me1: Biotin-LGKGGAK(me1)RHRKVLRDN;
H4K16ac: Biotin-LGKGGAK(ac)RHRKVLRDN;
H4K20me2: Biotin-LGKGGAKRHRK(me2)VLRDN;
H4K16acK20me2: Biotin-LGKGGAK(ac)RHRK(me2)VLRDN;
H4K16me1K20me2: Biotin-LGKGGAK(me1)RHRK(me2)VLRDN;
H3K9me1 (R-1025-100, EpiGentek, Farmingdale, NY, USA);
H3K9me2 (R-1027-100, EpiGentek).
For peptide pull-down assays, peptides were incubated with streptavidin agarose slurry (New England Biolabs, Ipswitch, MA, USA) overnight at 4°C in binding buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 10% Glycerol, 1 mM EDTA, 2 mM MgCl2, 0.5% NP-40). After removing the unbound peptides, 2 μg purified His-Tudor was added for 1 h and incubated at 4°C. After three washes with washing buffer (50 mM Tris–HCl pH 7.4, 300 mM NaCl, 10% Glycerol, 1 mM EDTA, 2 mM MgCl 2, 0.5% NP-40), beads were boiled and analyzed by western blotting. For GST pull-down, GST and GST-Tudor were incubated with the indicated peptides in binding buffer at 4°C for 3 h. After three washes in washing buffer, the beads were boiled and subjected to western blotting.
RNA interference (RNAi)
The RNAi oligonucleotide sequences were as follows:
non-specific small interfering RNA (siRNA) sense strand: 5′-UUCUCCGAACGUGUCACGU-3′;
G9a siRNA sense strand: 5′-CCAUGCUGUCAACUACCAUGG-3′;
GLP siRNA sense strands: GLPi#1 5′-GATGCCAGCAGTCATGCAA-3′, GLPi#2 5′- GGATTCAGATGTCACCTTA-3′;
SET8 siRNA sense strand: 5′-CAAAUGCUCUGGAAUGCGU-3′;
53BP1 siRNA sense strand: 5′- AGAACGAGGAGACGGUAAUAGUGGG-3′;
BRCA1 siRNA sense strand: 5′-UUCUAACACAGCUUCUAGUUCAGCC3′;
ATR siRNA sense strand: 5′-CCUCCGUGAUGUUGCUUGA-3′.
All RNAi oligonucleotides were purchased from Shanghai GenePharma Company. RNAi oligonucleotides were transfected using a Lipofectamine 2000 transfection kit (Invitrogen), according to the manufacturer's instructions. After transfection, the cells continued to grow in fresh medium for 72 h before DNA damage treatment. The GLP siRNAs #1 and #2 were cloned into a pGPU6/Hygro vector and a pool of shGLP#1 and shGLP#2 was transfected into HeLa cells to generate a stable GLP knockdown cell line. Briefly, 36 h after transfection, cells were selected with Hygromycin (200 μg/ml) for 14 days, and the survived colonies were selected. The efficiency of GLP knockdown was verified by western blotting.
Immunofluorescent analysis
Cells were cultured on slides to ∼80% confluence. After DNA damage treatment, the cells were washed twice with PBS and fixed with 4% paraformaldehyde at room temperature for 10 min. After washing twice with PBS, the cells were permeabilized with 100% methanol at −20°C for 15 min. The slides were then washed once and incubated with blocking buffer (0.8% bovine serum albumin in PBS) at room temperature for 1 h, and incubated overnight with the indicated primary antibody (1:50–1:500 dilution) at 4°C. After three washes with blocking buffer, the slides were exposed to a secondary antibody conjugated to Alexa Fluor® 488 or 594 dye. After incubation for 1 h with secondary antibody and three washes with blocking buffer, the samples were embedded in DAPI. Immunofluorescent images were captured under an Olympus confocal microscope.
Co-immunoprecipitation (Co-IP) assay
For whole-cell lysate Co-immunoprecipitation (Co-IP) assay, cells were collected and washed twice with PBS and then lysed in NP-40 lysis buffer (20 mM Tris–HCl pH 8.0, 137 mM NaCl, 10% glycerol, 1% Nonidet P-40, protease inhibitors) for 30 min on ice. The samples were then sonicated 15 times on ice, each for 1 s at 30% intensity. After sonication, the samples were treated with benzonase (EMD Millipore) at a final concentration of 50 U/ml supplemented with 2 mM MgCl2 to digest DNA at 4°C for 3 h. After centrifugation at 13 000 rpm (15 871 × g) at 4°C for 30 min, the supernatant was collected for immunoprecipitation. For the chromatin-unbound (Dt) fraction Co-IP, samples were collected as described above. For anti-Flag immunoprecipitation, 50 μl pre-cleared anti-Flag M2 beads (Sigma) was added and incubated at 4°C for 3 h. For anti-Myc immunoprecipitation, 2 μg anti-IgG or anti-Myc antibody was added to each sample and then incubated overnight at 4°C. Then, protein G sepharose beads were added for a further 3 h incubation. Samples were centrifugated at 1000 rpm (94 × g) at 4°C to remove the supernatant fraction. After washing with lysis buffer and centrifugation at 1000 rpm (94 × g) at 4°C for 1 min three times, the immunoprecipitated proteins were analyzed by western blotting.
Real-time RT-PCR assay
Total RNA was extracted in TRIzol (Invitrogen, USA), precipitated in ethanol and dissolved in RNAase/DNAase-free water. cDNA was then synthesized with 2 μg template RNA using a HiScript cDNA Synthesis Kit (Vazyme, China) containing genomic DNA wiper to exclude potential DNA contamination. The relative gene expression values were measured by real-time polymerase chain reaction (PCR) with SYBR-green dye (Vazyme, China) using the following program:Step 1, pre-denaturation at 95°C for 5 min; Step 2, 35 circles of sequential denaturation at 95°C for 10 s, then annealing and extension at 60°C for 30 s; Step 3, melt curve running from 60°C to 95°C. Three biological replicates were performed for each sample. The relative expression of mRNAs was determined by the 2−ΔΔCt method against the reference gene Actin. Target gene expression values are presented as the means ± SD. The primers used are detailed in Supplementary Table S1.
DR-GFP chromatin immunoprecipitation (ChIP)
For chromatin immunoprecipitation (ChIP) assay, DR-GFP U2OS cells were crosslinked with formaldehyde and then lysed in lysis buffer (50 mM Tris–HCl pH 8.0, 5 mM EDTA, 1% sodium dodecyl sulfate) on ice for 30 min. After sonication three times on ice, each for 10 s at 30% intensity, the supernatant was collected by centrifugation at 12 000 rpm (13 523 × g) at 4°C for 10 min and pre-cleared in dilution buffer (20 mM Tris–HCl pH 8.0, 2 mM EDTA, 150 mM NaCl, 1% Triton X-100) containing protein G or A sepharose beads (GE Healthcare, Kings Park, NY, USA) and salmon sperm DNA (Sigma-Aldrich, St Louis, MO, USA) by rotation at 4°C for 2 h. A total of 5% of the pre-cleared samples was used as the input, and then each remaining sample was divided into two parts and incubated with IgG or the indicated antibody at 4°C overnight. Protein G or A sepharose was then added to the sample and incubated at 4°C for 3 h. The beads were washed sequentially in TSE I, TSE II and Buffer III once and TE twice (detailed below). The samples were then eluted from the beads in elution buffer (1%SDS, 0.1M NaHCO3) at 37°C for 30 min and heated at 65°C overnight to reverse the cross-links. The DNA was purified using a NucleoSpin® Gel and PCR Clean-up kit (Macherey Nagel, Düren, Germany) and real-time PCR was performed as described above, on an ABI7500 Real-Time PCR System with the following primers (∼2 kb away from the I-SceI cutting site): 5′-GCCCATATATGGAGTTCCGC-3′ (sense) and 5′-CGTAAGGTCATGTACTGGGC-3′ (antisense). For each sample, three PCR replicates were taken and the average Ct was used to calculate the IP/input (2−ΔCt). Three independent assays for each experiment were performed and the means ± SD of IP/input% (100/2ΔCt) from parallel experiments are presented to show the enrichment. TSE I: 0.1% SDS, 1% TritonX-100, 2 mM EDTA pH 8.0, 20 mM Tris–HCl pH 8.0, 150 mM NaCl; TSE II: 0.1% SDS, 1% TritonX-100, 2 mM EDTA pH 8.0, 20 mM Tris–HCl pH 8.0, 500 mM NaCl; Buffer III: 250 mM LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA pH 8.0, 10 mM Tris–HCl pH 8.0; TE: 10 mM Tris–HCl pH 8.0, 1 mM EDTA pH 8.0.
EJ5-GFP NHEJ assay and DR-GFP HR assay
EJ5-GFP or DR-GFP U2OS cells were treated with the indicated siRNAs in 6-well plates and then transfected with 1 μg of I-SceI per well after 24 h. After 48 h of I-SceI transfection, the cells were trypsinized and the percentage of GFP positive cells was determined by flow cytometry using a BD flow cytometer.
Colony formation assay
Cells were treated with or without 40 μM etoposide for 2 h, washed four times with PBS and then plated into 6 cm plates in equal number. After 2 weeks, the cells were fixed with 4% paraformaldehyde and stained with methylene blue to identify colonies. Three independent experiments were performed.
Comet assay
Comet assay was performed as previously described (41). Briefly, the cells were gently mixed with pre-melted low-temperature-melting agarose at a volume ratio of 1:1 (v/v) and spread on glass slides. The slides were then submerged in pre-cooled lysis buffer at 4°C for 90 min. After rinsing, the slides were electrophoresed at 1.0 V/cm for 45 min, and then stained with propidium iodide. Fluorescent images for at least 100 nuclei were captured using an Olympus FV1000-IX81 Confocal Microscope (Olympus, Tokyo, Japan). The images were analyzed using Cometscore Version 1.5 software (TriTek Corp., Sumerduck, VA, USA) for tail moment. Three independent experiments were performed.
Statistical analysis
The data are expressed as the means ± SD. Significant differences between means were analyzed by two-tailed, unpaired Student's t-test and differences were considered statistically significant at *P < 0.05, **P < 0.01, ***P < 0.001 or ****P < 0.0001. Microsoft Excel 2016 was used to analyze all data.
RESULTS
H4K16me1 levels increase in response to DNA damage
In view of the dynamic H4K16ac changes that occur in the DDR, we first decided to investigate whether H4K16 can be modified by other modifications. Although H4K16me1 was suggested to exist in a mass spectrometry study (42), no validation and no biological study regarding this modification has since been reported. Therefore, we intended to see whether H4K16 methylation is indeed involved in DNA damage repair. Among all the three methylation states (mono-, di- and trimethylation), H4K16me1 was the only modification efficiently detected by our generated antibodies; H4K16 dimethylation and trimethylation did not produce clear signals. Our generated H4K16me1 antibody showed high specificity toward the H4K16me1 epitope, as verified by slot blot assay (Figure 1A and Supplementary Figure S1A) and peptide competition assay (Supplementary Figure S1B). This antibody could successfully detect histones extracted from HEK293 cells but not recombinant H4 (Figure 1B and Supplementary Figure S1C). In addition, it showed good specificity for H4K16me1 as presented with the entire immunoblot membrane probed in parallel with H4 to show corresponding molecular weights (Supplementary Figure S1C). The H4K16me1 signals were completely abolished when lysine was mutated to arginine at H4K16 (Figure 1C). Using this qualified antibody, we were able to detect the existence of H4K16me1 in various mammalian cell lines (Supplementary Figure S1D).
Figure 1.

H4K16me1 levels increase in response to DNA damage. (A) Slot blot assay showing high specificity of the H4K16me1 antibody. Cross reactivity with peptides carrying other modifications was not detected. (B) H4K16me1 expression in HEK293 cells. Recombinant Histone H4 (rH4) was used as a negative control. (C) HCT116 cells were transfected with a Myc-tagged H4 WT or K16R mutant vector for 3 days before anti-Myc tag immunoprecipitation and western blotting. (D) Western blot analysis of histones extracted from HCT116 cells 1 h after exposure to increasing doses of X-ray irradiation (IR). Ctr, no IR. (E) Western blot analysis of histones extracted from HCT116 cells after X-ray irradiation at 10 Gy and re-cultured for the indicated time. (F) HeLa cells were subjected to micro-irradiation, then fixed 10 min later and immunostained with the indicated antibodies. Scale bars: 10 μm. (G) DR-GFP U2OS cells were transiently transfected with I-SceI and subjected to ChIP assay 24 h later with the indicated antibodies. The IP/input% from three independent experiments is shown as the means ± SD. n.s., not significant, *P < 0.05 (Student's t-test).
To determine whether H4K16me1 is involved in the DDR, we subjected HCT116 cells to X-ray irradiation and monitored subsequent H4K16me1 levels. Here, we observed a dose-dependent increase in H4K16me1 (Figure 1D); H4K16me1 levels increased within minutes and recovered to basal levels hours after irradiation (Figure 1E). Consistent with previous reports (14,43), total H4K20me2 levels did not markedly change after irradiation (Figure 1E). We confirmed this increase in H4K16me1 levels after irradiation in HeLa (Supplementary Figure S1E) and LoVo (Supplementary Figure S1F) cells. In addition, H4K16me1 levels also increased when etoposide was used to induce DNA damage (Supplementary Figure S1G). These data indicate that an increase in H4K16me1 is a general phenomenon that occurs in response to DNA damage.
We next micro-irradiated HeLa cells and then performed immunostaining with the indicated antibodies to check whether H4K16me1 levels increase at DSBs. We observed remarkable overlap between H4K16me1 and γH2AX, and between H4K16me1 and 53BP1 at the micro-irradiated sites (Figure 1F). In addition, most of the micro-irradiated cells showed no obvious increase in H4K20me2 at the micro-irradiated sites, which may due to the high basal levels of H4K20me2 (Supplementary Figure S1H). We also performed a ChIP assay in DR-GFP U2OS cells, in which transfection of the I-SceI construct induces DSBs at I-SceI sites (44). Here, we found that H4K16me1 levels significantly increased at I-SceI cut sites (Figure 1G), supporting that H4K16me1 is directly involved in DNA damage repair.
GLP catalyzes H4K16me1 both in vitro and in vivo
To investigate which methyltransferase can catalyze H4K16me1, we performed a series of in vitro methylation assays using different methyltransferases. We found that the GLP catalytic construct efficiently methylated the H4K16 residue in vitro (Figure 2A), whereas the other investigated histone methyltransferases (SUV39H1, EZH2, SETD2, DOT1L and SET8) showed no catalytic activity toward this site (Supplementary Figure S2A). In addition, we detected a very weak signal of H4K16me1 after catalyzation by G9a when compared to that by GLP (Figure 2A). Interestingly, GLP activity toward H4K16 was largely inhibited when G9a was added into the catalytic system (Figure 2B and Supplementary Figure S2B). In comparison to wild-type (WT) GLP, a catalytic dead GLP mutant (C1201A) exhibited no activity in catalyzing H4K16me1 (Figure 2B and Supplementary Figure S2B). We next purified H4 (WT) and H4 (K16R) proteins from E. coli and found that the K16R mutation completely abolished the H4K16me1 signals after GLP catalyzation (Figure 2C). Moreover, when we added the GLP methyltransferase inhibitor Bix-01294 into the in vitro methylation system, GLP activity in catalyzing H4K16me1 was largely inhibited (Supplementary Figure S2C).
Figure 2.

GLP catalyzes H4K16me1 both in vitro and in vivo. (A) In vitro methylation assays were performed using different methyltransferases. Recombinant histone H3 and H4 were used as substrates. (B) In vitro methylation assays were performed with different methyltransferases. Core histones extracted from HCT116 cells were used as substrates. (C) Purified H4 (WT) or H4 mutant (K16R) proteins were incubated with GST or GST-GLP for 20 or 40 min in methylation reaction buffer, respectively. (D) HCT116 cells were treated with 2.5 μM Bix-01294 for 24 h before histone (left) and whole cell lysates (right) extraction and western blotting. (E) GLP was knocked down in HCT116 cells with GLP siRNA#1 or #2. Cells were collected 72 h after transfection, and whole cell lysates and histones were extracted for western blotting. (F) Flag-GLP was over-expressed in HCT116 cells for 72 h before histone extraction and western blotting.
To check whether GLP catalyzes H4K16me1 in vivo, we treated HCT116 cells with Bix-01294 for 2 days and observed a notable decrease in H4K16me1; GLP and G9a expression, however, was unchanged (Figure 2D). In addition, we performed an RNA-interference assay in HCT116 cells: here, H4K16me1 levels were remarkably decreased by GLP knockdown (Figure 2E). Consistently, H4K16me1 levels increased when GLP was over-expressed (Figure 2F). Converse to GLP, we found G9a may be dispensable for H4K16me1 in vivo because H4K16me1 levels were not impaired in G9a knockout (KO) HCT116 cells (Supplementary Figure S2D). Collectively, these data support that GLP is a methyltransferase responsible for catalyzing H4K16me1 both in vitro and in vivo.
GLP catalyzes H4K16me1 in response to DNA damage in an ATM-dependent manner
Because GLP can methylate H4K16 both in vitro and in vivo, and H4K16 is methylated in response to DNA damage, we hypothesized that GLP can induce an increase in H4K16me1 in response to DNA damage. To test this hypothesis, we performed a micro-irradiation assay in HeLa cells (Figure 3A) and a ChIP assay in DR-U2OS cells (Figure 3B) to check whether GLP can be recruited to DSBs. We detected obvious GLP enrichment at the micro-irradiated (Figure 3A) or I-SceI cut sites (Figure 3B). Notably, GLP accumulated at micro-irradiated sites within 20 sec, independently of its methyltransferase activity and its interaction with G9a (Supplementary Figure S3A and B). Although GLP usually forms a heterodimer with G9a, G9a recruitment to the micro-irradiated sites was slower and reached lower levels than GLP (Supplementary Figure S3A and B). In addition, GLP recruitment to micro-irradiated sites was not impaired in G9a KO HCT116 cells (Supplementary Figure S3C–E). We also detected a partial dissociation of the G9a/GLP complex in the soluble, but not the chromatin-bound fraction after etoposide treatment (Supplementary Figure S3F). Endogenous co-immunoprecipitation (Co-IP) experiments confirmed this partial dissociation between G9a and GLP in the soluble fraction (Supplementary Figure S3G and H). In our previous study, we found that G9a KO impaired RPA loading on to ssDNA and HR repair (45). We thus checked RPA foci formation following GLP knockdown: differing from G9a, RPA foci formation was not impaired by GLP knockdown (Supplementary Figure S3I), suggesting that GLP may have distinct roles from G9a in DNA damage repair.
Figure 3.

GLP catalyzes H4K16me1 in response to DNA damage in an ATM-dependent manner. (A) HeLa cells were subjected to micro-irradiation, then fixed 10 min later and immunostained with the indicated antibodies. Scale bars: 10 μm. (B) DR-GFP U2OS cells were transfected with Flag-GLP for 48 h and then transfected with I-SceI expression construct and cultured for a further 24 h. Cells were collected and subjected to ChIP with the indicated antibodies. Three independent experiments were performed. The data represent the means ± SD. n.s., not significant, **P < 0.01 (Student's t-test). (C) HCT116 cells were transfected with GLP siRNA#1 for 72 h before X-ray irradiation. Cells were further cultured for 30 min before whole cell lysates and histone extraction for western blotting. (D) The same experiment as described in (C) was performed with 40 μM etoposide (instead of X-ray irradiation) treatment for 30 min to induce DNA damage. (E) GLP was knocked down in DR-GFP U2OS cells by GLP siRNA#1. After I-SceI transfection for 24 h, four independent ChIP assays were performed with the indicated antibodies. The data represent the means ± SD. n.s., not significant, *P < 0.05, **P < 0.01 (Student's t-test). (F) HeLa cells were transfected with mCherry-GLP for 2 days and treated with 10 μM KU-55933 (ATM inhibitor), 10 μM KU-57788 (DNA-PKcs inhibitor), 10 μM VE-821 (ATR inhibitor) or 50 μM TBB (CK2 inhibitor) for 2 h before micro-irradiation. (G) Relative intensity of mCherry-GLP at micro-irradiated sites in the experiments described in (F). The data represent the means ± SD. (H) Western blot analysis of histones extracted from A-T cells 1 h after exposure to 10 Gy X-ray irradiation.
To verify whether the increase in H4K16me1 levels during the DDR is dependent on GLP, we knocked down GLP in HCT116 cells and subjected the cells to X-ray irradiation. Here, the increase in H4K16me1 was largely inhibited as a result of GLP knockdown (Figure 3C and Supplementary Figure S4A). We obtained consistent results when treating the cells with etoposide (Figure 3D). In addition, the increase in H4K16me1 at I-SceI cut sites was also largely inhibited by GLP knockdown (Figure 3E). H3K9me3 enrichment at I-SceI cut sites was not obviously affected by GLP knockdown and the H3K9me2 levels were still significantly induced, which may be as a result of catalyzation by other H3K9 methyltransferases (Figure 3E) (36). At last, G9a knockout or SET8 knockdown did not impair the increase in H4K16me1 levels in the DDR (Supplementary Figure S4B and C). From these data, we conclude that the increase in H4K16me1 levels at DSBs is catalyzed by GLP.
A previous study proposed GLP as a potential ATM substrate (37). Consistently, we found that GLP recruitment to DNA stripes was impaired upon exposure to the ATM inhibitor KU-55933, but not the DNA-PKcs inhibitor KU-57788, the ATR inhibitor VE-821 or the CK2 inhibitor TBB (Figure 3F and G). These data suggest that GLP recruitment may be directly regulated by ATM. Such direct regulation is supported by an increased interaction between GLP and ATM (Supplementary Figure S4D) and an increase in p-S/T-Q GLP signal after etoposide treatment (Supplementary Figure S4E). In addition, in vitro phosphorylation assay showed that GLP can be phosphorylated by ATM (Supplementary Figure S4F). Interestingly, we found that G9a had a decreased interaction with GLP when GLP was phosphorylated by ATM (Supplementary Figure S4G), suggesting that their partial dissociation in the DDR might be ATM dependent. Consistent with GLP regulation by ATM, H4K16me1 levels failed to increase in response to X-ray irradiation in ATM-mutated A-T cells (Figure 3H). In contrast to ATM, when we knocked down ATR by siRNA (Supplementary Figure S4H) or treated cells with the ATR inhibitor VE-821 (Supplementary Figure S4I), H4K16me1 levels still efficiently increased after X-ray irradiation. These findings suggest that the increased H4K16me1 in response to DNA damage is GLP and ATM dependent.
GLP-catalyzed H4K16me1 is associated with 53BP1 binding activity in response to DNA damage
Contact between the histone H4K16 residue and the 53BP1 Tudor domain has been previously described (14,30); therefore, we hypothesized that H4K16me1 directly interacts with the 53BP1 tandem Tudor domain. To verify this hypothesis, we performed GST (Figure 4A) and peptide pull-down assays (Figure 4B). Here, we successfully detected an interaction between the GST-tagged 53BP1 tandem Tudor domain and biotin-labeled H4K16me1 peptide (Figure 4A and B). In contrast to H4K16me1, we didn’t find an interaction between the H3K9me1 or H3K9me2 peptide with the 53BP1 tandem Tudor domain (Figure 4B). We also performed an endogenous Co-IP to confirm the interaction between 53BP1 and H4K16me1. Interestingly, this interaction notably increased after DNA damage treatment (Figure 4C). These data suggest that H4K16me1 may have a positive role in 53BP1 binding to damaged chromatin through a direct interaction.
Figure 4.

GLP-catalyzed H4K16me1 is associated with 53BP1 binding activity in response to DNA damage. (A) A GST pull-down assay was performed to detect the interaction between the GST-tagged Tudor domain of 53BP1 and the indicated H4 peptides. (B) A peptide pull-down assay was performed to check the interaction between the 53BP1 tandem Tudor domain and the indicated peptides. (C) HCT116 cells were treated with 40 μM etoposide for 30 min. Whole cell lysates were extracted and H4K16me1 levels were normalized to each sample before immunoprecipitation (IP) with an anti-53BP1 antibody. The interacting proteins were analyzed by western blotting. (D) Chromatin and total levels of DNA repair factors were detected in shRNA control (shCtr) or shGLP HeLa cells with or without 40 μM etoposide treatment for 30 min. A pool of shGLP#1 and shGLP#2 was used to generate the stable GLP knockdown cell line. (E) shCtr and shGLP HeLa cells were treated with 40 μM etoposide for 30 min before confocal detection of 53BP1 foci formation. 53BP1 foci numbers were counted from a minimum of 150 cells. The data represent the means ± SD. ***P < 0.001 (Student's t-test). (F) HeLa cells were pre-treated with or without 2.5 μM Bix-01294 for 24 h before X-ray irradiation. The cells were fixed at 20 min post-irradiation to detect 53BP1 foci formation by confocal microscopy. 53BP1 foci numbers were counted from a minimum of 150 cells. The data represent the means ± SD. ***P < 0.001 (Student's t-test). (G) Dynamics of GFP-53BP1 accumulation at micro-irradiated sites with or without 2.5 μM Bix-01294 treatment for 24 h. (H) Relative intensity of GFP-53BP1 at micro-irradiated sites in the experiments described in (G). The data represent the means ± SD. (I–K) shGLP cells were transfected with reconstituted Flag-GLP (WT) or Flag-GLP (ΔSET) for 72 h and then treated with 40 μM etoposide for 30 min. Cells were then fixed and immunostained with anti-Flag, anti-H4K16me1 and anti-53BP1 antibodies. The arrows indicate the cells with successful transfection or elevated H4K16me1 levels (I and J). Ctr, no etoposide treatment. Scale bars: 10 μm. 53BP1 foci numbers in H4K16me1 elevated or not elevated cells were counted (K). The data represent the means ± SD. ***P < 0.001 (Student's t-test).
To validate the positive role of GLP-catalyzed H4K16me1 in 53BP1 recruitment, we checked the chromatin levels of 53BP1 in shCtr and shGLP (GLP stable knockdown) cells following etoposide-induced DNA damage. 53BP1 recruitment onto damaged chromatin was largely inhibited in shGLP cells, whereas other factors, including ATM, ATR or DNA-PKcs were unaffected (Figure 4D). Of note, total 53BP1, ATM, ATR and DNA-PKcs protein levels were unchanged by GLP knockdown (Figure 4D). We also monitored 53BP1 foci formation in shCtr and shGLP cells after etoposide treatment. Here, 53BP1 foci formation was markedly impaired by GLP knockdown (Figure 4E and Supplementary Figure S5A). In addition, 53BP1 foci formation was also impaired upon Bix-01294 treatment (Figure 4F, and Supplementary Figure S5B). Consistently, GFP-53BP1 recruitment to the micro-irradiated sites was markedly delayed by Bix-01294 treatment (Figure 4G and H). Consistent with a positive role for GLP in 53BP1 foci formation, we found that GLP was important for NHEJ repair. We measured NHEJ efficiency following transfection of the I-SceI construct into EJ5-GFP U2OS cells, in which I-SceI excises redundant DNA and GFP expression is restored upon successful NHEJ repair (46). GLP or SET8 knockdown seriously impaired NHEJ, whereas G9a knockdown had only a mild effect (Supplementary Figure S5C). NHEJ efficiency was also significantly impaired by Bix-01294 treatment (Supplementary Figure S5D). Conversely, G9a knockdown but not GLP knockdown caused a remarkable defect in HR-mediated repair in DR-GFP U2OS cells (Supplementary Figure S5E). Together, these data suggest that GLP and its methyltransferase activity are important for 53BP1 foci formation in response to DNA damage.
Because H4K20me2 is important for 53BP1 foci formation, we checked whether GLP knockdown impaired 53BP1 foci formation by affecting H4K20me2: H4K20me2 levels were not impaired in shGLP cells (Supplementary Figure S5F). We also analyzed the expression of DNA repair factors in shCtr and shGLP cells at the mRNA level, and found that none of these factors showed impaired expression by GLP knockdown (Supplementary Figure S5G). In addition, we didn’t see obvious changes in foci formation of RNF8, RNF168 and FK2 (chromatin ubiquitin) by GLP knockdown (Supplementary Figure S6A–C). Although GLP is a methyltransferase for H3K9me2, H3K9me2 has a reported role in recruiting the 53BP1-antagonistic BRCA1 complex and promoting HR-mediated DNA damage repair (36,47). Therefore, we hypothesized that impaired 53BP1 foci formation and NHEJ repair following GLP knockdown may be H4K16me1-dependent. To delineate the role of H4K16me1 in 53BP1 regulation, we transfected shGLP cells with reconstituted WT or SET-domain deleted (ΔSET) Flag-tagged GLP. Flag-GLP (ΔSET) methyltransferase activity was completely abolished by the catalytic SET domain deletion. Consistent with our previous results, H4K16me1 in shGLP cells was markedly enhanced upon rescue with Flag-GLP (WT), but not Flag-GLP (ΔSET) (Figure 4I). Similarly, shGLP cells transfected with Flag-GLP (WT) but not Flag-GLP (ΔSET) exhibited an obvious improvement in 53BP1 foci formation (Supplementary Figure S5H). Immunofluorescent analysis also confirmed a correlation between improved 53BP1 foci formation and elevated H4K16me1 levels (Figure 4J and K). Overall, we conclude that GLP-catalyzed H4K16me1 is important for 53BP1 binding to chromatin in response to DNA damage.
H4K16me1 differs from H4K16ac in mediating the DNA damage response and in regulating the H4K20me2–53BP1 interaction
A previous study suggested a dynamic change in H4K16ac during the DDR (48). As such, we decided to investigate the connection between H4K16me1 and H4K16ac in the DDR. Differing from the trend of H4K16me1 in DDR, H4K16ac decreased in the early stages of DNA repair, and increased a few hours after etoposide withdrawal (Figure 5A and Supplementary Figure S7A). We verified these opposing trends of H4K16me1 and H4K16ac at DSBs by performing a time course ChIP assay near the I-SceI cut sites (Figure 5B). More convincingly, both decreased H4K16ac and increased H4K16me1 levels were detected at micro-irradiated sites after micro-irradiation (Figure 5C).
Figure 5.

H4K16me1 differs from H4K16ac in mediating the DDR and in regulating the H4K20me2–53BP1 interaction. (A) HeLa cells were treated with 40 μM etoposide for 30 min. The cells were then washed four times with PBS and re-cultured in fresh medium. Histones were extracted at each release time (indicated) and analyzed by western blotting. (B) DR-U2OS cells were transfected with I-SceI and then collected at each indicated time point. H4K16me1 and H4K16ac enrichment at I-SceI sites was detected by three independent ChIP assays. (C) HeLa cells were micro-irradiated, fixed and stained with the indicated antibodies. Images show an enrichment in H4K16me1 and a decrease in H4K16ac at the irradiated strips. Scale bars: 10 μm. (D and E) Mononucleosomes were extracted from HCT116 cells that had been exposed to 40 μM etoposide treatment for 30 min. H4K16me1 or H4K16ac levels were normalized to each sample. The mononucleosomes were immunoprecipitated with an anti-H4K20me2 antibody. The H4K16me1 (D) or H4K16ac (E) levels on H4K20me2-enriched mononucleosomes were analyzed by western blotting. (F) 53BP1 was immunoprecipitated from etoposide-treated or untreated shCtr or shGLP cells, and the interactive components were analyzed by western blotting. (G) A peptide pull-down assay was performed to detect the interactions between the 53BP1 Tudor domain and several H4 peptides. (H) Histones were extracted from HCT116 cells and incubated with GLP in methylation reaction buffer with or without SAM for 1 h. Subsequently, the differently modified histones were incubated with GST-Tudor for 8 h before anti-H4K20me2 immunoprecipitation and western blotting. (I) Histones were extracted from HCT116 cells and incubated with GLP in methylation reaction buffer with or without SAM for 1 h. Subsequently, the differently modified histones were incubated with beads-bound GST or GST-Tudor for 2 h at 4°C. The beads were washed three times before western blotting.
By performing a mononucleosome Co-IP assay, we detected increased levels of H4K16me1 on the H4K20me2-enriched mononucleosomes after DNA damage treatment (Figure 5D and Supplementary Figure S7B). In addition, we detected decreased H4K16ac on the H4K20me2-enriched mononucleosomes (Figure 5E). Because H4K16ac impairs the binding of 53BP1 to H4K20me2 (30), we investigated whether H4K16me1 can influence the binding affinity of 53BP1 to H4K20me2. By assaying anti-53BP1 precipitation in shCtr and shGLP cells (where basal levels of H4K16me1 are deceased by GLP knockdown), we found that 53BP1 had a weaker interaction with H4K20me2 in shGLP cells than in shCtr cells following etoposide-induced DNA damage (Figure 5F). Of note, even though 53BP1 had an increased interaction with both H4K20me2 and H4K16me1 after DNA damage, the increased degree of H4K16me1 was higher than H4K20me2 in the 53BP1 interacting fraction (Figure 5F and Supplementary Figure S7C).
We also treated HCT116 cells with A-196, a specific Suv4–20h1/Suv4–20h2 inhibitor (49). A-196 treatment caused a notable decrease in H4K20me2/H4K20me3 levels and a marked increase in H4K20me1 levels, whereas the H4K16me1 levels were not obviously changed (Supplementary Figure S7D). Moreover, the increase in H4K16me1 levels in response to DNA damage was also unaffected by A-196 treatment (Supplementary Figure S7E). Similar to the influence of H4K16me1 loss on the H4K20me2–53BP1 interaction in the DDR (Figure 5F), H4K16me1 showed increased 53BP1 binding after X-ray irradiation, but this interaction was partially suppressed under A-196 treatment (Supplementary Figure S7F). Consistently, combined A-196 and Bix-01294 treatment inhibited 53BP1 foci formation more effectively than A-196 or Bix-01294 single treatment during the DDR (Supplementary Figure S7G–H).
We then performed a peptide pull-down assay with the 53BP1 His-Tudor domain and several modified H4 peptides to assess the binding affinity of the Tudor domain with each peptide. The peptide possessing both H4K16me1 and H4K20me2 had a stronger interaction with the 53BP1 Tudor domain than the H4K20me2 peptide alone (Figure 5G). Consistent with previous reports (30,31), we observed a much weaker interaction between the 53BP1 Tudor domain and the H4K16acK20me2 peptide (Figure 5G). In addition, the 53BP1 tandem Tudor D1521A that fails to bind H4K20me2 maintained the ability to bind the H4K16me1 and H4K16me1K20me2 peptides (Supplementary Figure S7I), indicating different binding sites in the 53BP1 tandem Tudor domain for H4K16me1 and H4K20me2. Moreover, anti-H4K20me2 immunoprecipitation (Figure 5H) and GST pull-down (Figure 5I) assays showed that the GST-Tudor domain had an increased interaction with H4K20me2 when the histones were pre-methylated by GLP to obtain high levels of H4K16me1. Taken together, these data suggest that H4K16me1 can cooperate with H4K20me2 to recruit 53BP1 in response to DNA damage.
GLP promotes DNA damage repair and cell survival in a 53BP1-dependent manner
To further validate the roles of GLP and H4K16me1 in DNA damage repair, we performed comet assays in shCtr and shGLP cells and compared the DNA repair efficiency by monitoring the length and area of the DNA comet tails (41). We observed extensive DNA damage after etoposide treatment in both cell lines, as represented by long comet tails (Figure 6A and B). At 18 h post-etoposide withdrawal, however, the comet tails in shCtrl cells had almost recovered to basal levels, whereas the comet tails in shGLP cells exhibited significant and sustained levels, indicative of delayed DNA damage repair. To confirm the role of GLP methyltransferase activity in DNA damage repair, we transfected shGLP cells with Flag-GLP (WT) or Flag-GLP (ΔSET) vector and again treated the cells with etoposide before analyzing the comet tails (Figure 6A and B; Supplementary Figure S8A). At 18 h after etoposide withdrawal, shGLP cells transfected with Flag-GLP (WT) exhibited a significant improvement in DNA damage repair, whereas cells transfected with Flag-GLP (ΔSET) did not show any improvement (Figure 6A and B).
Figure 6.

GLP promotes DNA damage repair and cell survival in a 53BP1-dependent manner. (A) Comet assays were performed to detect the repair efficiency of shCtr and shGLP cells transfected with pcDNA, Flag-GLP (WT) or Flag-GLP (ΔSET) after 40 μM etoposide treatment. Representative images of the cells under each condition are shown. Consistent results were observed from at least three independent experiments. Scale bars: 150 μm. (B) Quantification of tail moments from the comet assays described in (A). Three independent experiments were performed. The data represent the means ± SD. n.s., not significant, *P < 0.05, **P < 0.01 (Student's t-test). (C) shCtr and shGLP cells were transfected with pcDNA or Flag-GLP (WT) or Flag-GLP (ΔSET) for 72 h before 40 μM etoposide treatment for 2 h. The cells were then analyzed by colony formation assay. (D) Quantification of the three independent colony formation assays described in (C). The data represent the means ± SD. n.s., not significant, ***P < 0.001 (Student's t-test). (E) GLP was knocked down in G9a knockout (KO) cells, and the expression levels of H3K9me2, H4K16me1, GLP and G9a were analyzed by western blotting. (F) GLP was knocked down in G9a KO cells followed by 40 μM etoposide treatment for 2 h. Cell viability under each condition was examined by colony formation assay. Three independent experiments were performed. The data represent the means ± SD. **P < 0.01 (Student's t-test). (G) 53BP1 was knocked down in HCT116 cells before Flag-GLP over-expression for 36 h. After 40 μM etoposide treatment for 1 h, the cells were washed, counted and seeded for colony formation assay. Three independent experiments were performed. The data represent the means ± SD. n.s., not significant, **P < 0.01 (Student's t-test).
We next performed several colony formation assays in shCtr and shGLP cells to investigate the role of GLP-catalyzed H4K16me1 in cell survival following DNA damage. After etoposide treatment, the shGLP cells exhibited significantly impaired cell viability than control cells (Figure 6C and D). This deficiency was rescued by transfection with Flag-GLP (WT) but not Flag-GLP (ΔSET). Because GLP is also a methyltransferase for H3K9, and H3K9 methylation is important for DNA damage repair (36,47,50), we knocked down GLP in G9a KO cells to determine the role of H4K16me1 in DNA damage repair. In agreement with a previous report (51), GLP knockdown did not induce a further decrease in H3K9 methylation in G9a KO cells, whereas it caused a significant decrease in H4K16me1 (Figure 6E). Consistently, the viability of G9a KO cells was also impaired following GLP knockdown (Figure 6F; Supplementary Figure S8B and C). To check whether 53BP1 is involved in GLP-regulated cell viability in response to DNA damage, we over-expressed GLP in HCT116 control cells or HCT116 cells with 53BP1 knockdown and then treated the cells with etoposide. Here, GLP over-expression increased cell viability in HCT116 control cells, whereas this effect was blocked by 53BP1 knockdown (Figure 6G; Supplementary Figure S8D and E). These results indicate that GLP-catalyzed H4K16me1 is important for 53BP1 function in cell survival in response to DNA damage, suggesting GLP or H4K16me1 may be a valuable target for cancer therapy.
DISCUSSION
Our study shows that GLP-catalyzed H4K16me1 facilitates 53BP1 binding to chromatin in the DDR to promote efficient NHEJ-mediated DNA repair. H4K16me1 is an early induced histone modification in the DDR that has an increased interaction with 53BP1 upon DNA damage. During DNA damage repair by NHEJ, H4K16me1, H4K20me2 and H2AK15ub cooperate precisely to mediate efficient 53BP1 recruitment to damaged chromatin (Figure 7).
Figure 7.
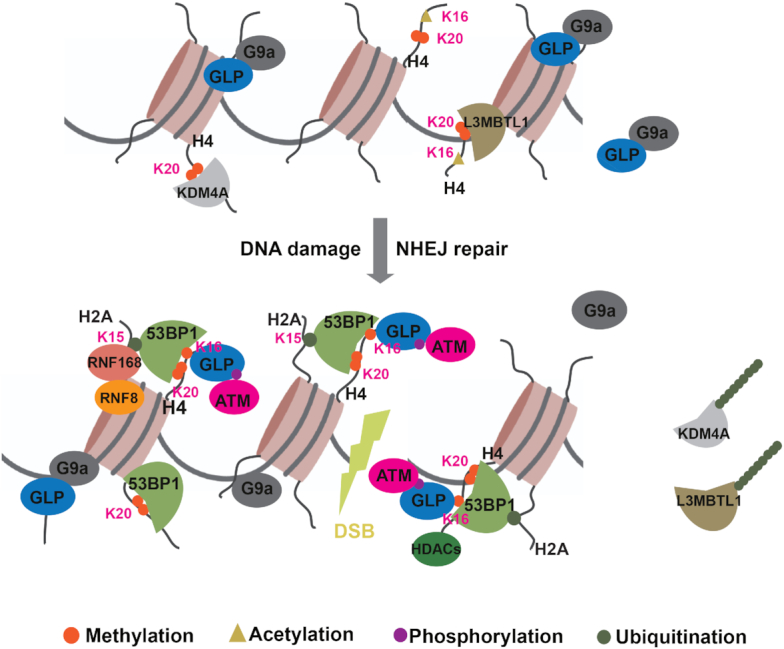
GLP-mediated H4K16me1 promotes 53BP1 recruitment in response to DNA damage. Under normal conditions, H4K20me2 is abundant but masked by a few proteins, including L3MBTL1 and KDM4A. When cells suffer from DNA double-strand breaks, GLP quickly accumulates at DNA lesions to catalyze H4K16me1 in an ATM-dependent manner. Upon H4K20me2 unmasking due to RNF8/RNF168-mediated dissociation of L3MBTL1 and KDM4A, or locally increased H4K20me2 under certain circumstances, H4K16me1, H4K20me2 and RNF8/RNF168-induced H2AK15ub cooperate precisely to mediate efficient 53BP1 recruitment to damaged chromatin for NHEJ repair.
GLP and G9a have different characteristics in vitro and in vivo
Previous reports showed that GLP and G9a possess similar substrate specificities (51–53). Here, we identified GLP as a methyltransferase for H4K16me1 both in vitro and in vivo. However, we detected a very weak signal of H4K16me1 after catalyzation by G9a when compared to that by GLP (Figure 2A). Evidences already suggest that GLP and G9a have different biological characteristics. For example, G9a preferentially binds H3K9me2, whereas GLP preferentially binds H3K9me1 (54). In addition, homozygous knock-in mice bearing GLP (not G9a) 3A mutations—such that GLP fails to bind methylated H3K9—are mostly postnatal-lethal and exhibit a severe growth retardation (55). In humans, microdeletion or mutation in the EHMT1 gene (coding GLP) is associated with a rare Kleefstra syndrome (56); of note, the EHMT2 gene (coding G9a) is not reported to be involved in this syndrome. Although no detailed pathogenic mechanism about Kleefstra syndrome has been reported, it shares several similar phenotypes with some other DNA repair-associated syndromes, such as Seckel syndrome (57), Fanconi anemia (58) and Nijimegen breakage syndrome (58). We thus consider it worthwhile to investigate whether GLP-related NHEJ deficiency is one of the causes of Kleefstra syndrome in the future.
Different characteristics of GLP and G9a in DDR
Regarding DNA damage repair, it was recently reported that GLP but not G9a has an increased interaction with MDC1 during the DDR (59); however, G9a but not GLP depletion reduces MDC1 methylation levels (59). In response to DNA damage, some GLP molecules are considered released from G9a in an ATM-dependent manner and then recruited to damaged chromatin to efficiently methylate H4K16me1. In the in vitro methylation study (Figure 2B), the addition of G9a into the methylation system might have resulted in competition with H4 to bind un-phosphorylated GLP, thus attenuating GLP activity in catalyzing H4K16me1. Because both GLP and G9a are recruited to DSBs, the dissociated GLP and G9a proteins may form dimers under certain circumstances or during certain repair stages to promote DNA damage repair. Previously, Ginjala et al. showed that GLP expression was impaired in G9a-deficient cells: this might be the reason as to why they detected impaired GLP recruitment to micro-irradiated sites after G9a knockdown (60). It is possible that GLP stability might be altered in certain G9a knockdown cell lines because they usually exist as a dimer in cells. However, in current study, we didn’t see changes in GLP protein level in G9a KO HCT116 cells, which is also consistent with a previous report conducted in mouse embryonic fibroblasts (51). In addition, we found GLP accumulated normally at the micro-irradiated sites in HCT116 G9a KO cells, suggesting a G9a-independent recruitment of GLP to DSBs in DDR.
GLP-catalyzed H4K16me1 promotes 53BP1 recruitment and NHEJ repair
Previously, the G9a/GLP inhibitor UNC0638 reportedly impairs BRCA1/BARD1 recruitment to inhibit HR repair (47,61). Others, however, have reported that UNC0638 attenuates NHEJ repair with no marked effect on HR (39). Their differences in HR repair might be explained by the different experimental procedures used, but regardless, the question as to how GLP/G9a inhibition impacts on NHEJ repair has not been well answered. Here, we observed impaired 53BP1 foci formation in the early DDR and delayed 53BP1 recruitment post micro-irradiation by GLP knockdown, which might be the reason for impaired NHEJ repair. Although it was reported that G9a and GLP influence the RNF8/RNF168-ubiquitnation pathway in the DDR in U2OS cells (59,60), we did not see obvious changes in RNF8, RNF168 or FK2 foci formation in shGLP HeLa cells. We consider that GLP-regulated 53BP1 recruitment might predominantly derive from the increased interaction between H4K16me1 and 53BP1. It was previously suggested that 53BP1 is not regulated by H3K9me2 (20,25). In addition, H3K9me2 was also considered to direct the choice between the antagonistic DSB repair mediators BRCA1 and 53BP1 (36), by promoting BRCA1/BARD1 recruitment for HR repair (47). Therefore, GLP/G9a-catalyzed H3K9me2 may be more important for HR repair, whereas GLP-catalyzed H4K16me1 may be an important factor for NHEJ by facilitating 53BP1 recruitment.
H4K16me1 cooperates with H4K20me2 in 53BP1 recruitment in DDR
In response to DNA damage, H4K16me1 shows opposing changes to H4K16ac. As methylation competes with acetylation for lysine modification, it is also likely that the increased H4K16me1 levels prohibit H4K16 from being acetylated, thus ensuring 53BP1 binding at DSBs. In contrast to the increase in H4K16me1 levels, H4K20me2 is highly abundant and globally unchanged in response to DSBs (14,62–65). Although H4K20me2 at DNA lesions is reportedly elevated by its relative methyltransferases (65,66), this elevation might be considerably limited due to high H4K20me2 basal levels. For example, Kovarikova et al. showed that H4K20me2 is not significantly increased locally at micro-irradiated chromatin (67). We also observed that most cells showed no obvious H4K20me2 enrichment at the micro-irradiated sites. We do not currently know the structural basis for the H4K16me1–53BP1 interaction; future crystallography-based studies will be performed to decipher this interaction. In a previous mass spectrometry study (42), only monomethylation of H4K16 was identified. H4K16me1 may be the most abundant methylation state of H4K16 among mono-, di- and trimethylation. Because the existence of both H4K16me1 and H4K20me2 leads to a higher affinity of the 53BP1 tandem Tudor domain to histone H4, and combined treatment with A-196 and Bix-01294 is more effective at inhibiting 53BP1 foci formation in the DDR than either single treatment, we suggest that a combined H4K16me1K20me2 histone mark might be preferential for 53BP1 recruitment. It is also possible that, under certain circumstances, these two modifications can function independently of each other to recruit 53BP1 and that there may be a compensatory effect between these two modifications. As DNA repair networks are precisely regulated both in time and space, different histone modifications can regulate 53BP1 recruitment, retention and dissociation from the damaged chromatin at the appropriate time and place. In this regard, histone methylation and ubiquitination (H4K20me2, H4K16me1, H2AK15ub, et al.) may have an opposing function to histone acetylation (H4K16ac, H3K18ac, H2AK15ac et al.) to trigger the 53BP1 recruitment-dissociation switch in the DNA repair process. Because ATM is a master phosphokinase in DDR, and any factors influencing its activation (68,69) or deactivation (70) can significantly affect DNA damage repair, it will be intriguing to investigate whether other factors influencing ATM activity are associated with GLP recruitment to DSBs in the future.
In conclusion, this is the first study to report the biological function of H4K16me1 and specifically, its role in the DDR. This discovery of a role for GLP-catalyzed H4K16me1 in 53BP1 regulation improves our understanding of the mechanisms underlying NHEJ, and may have potential clinical applications for the design of new cancer treatments in the future.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Dr. Bing Zhu at the Institute of Biophysics, Chinese Academy of Sciences for kindly providing the GST-GLP (containing the Ankyrin and SET domains) plasmid. The manuscript was edited for English Language prior to submission by Dr Jessica Tamanini of ETediting, UK.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key R&D Program of China [2017YFA0503900]; National Natural Science Foundation of China [81720108027, 81530074, 81621063, 31570812, 81802811]; Science and Technology Program of Guangdong Province in China [2017B030301016]; Shenzhen Municipal Commission of Science and Technology Innovation [JCYJ20160427104855100, JCYJ20170818092450901]; Discipline Construction Funding of Shenzhen [(2016)1452]; China Postdoctoral Science Foundation [2019M653054]. Funding for open access charge: National Key R&D Program of China [2017YFA0503900].
Conflict of interest statement. None declared.
REFERENCES
- 1. Ciccia A., Elledge S.J.. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010; 40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jackson S.P., Bartek J.. The DNA-damage response in human biology and disease. Nature. 2009; 461:1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ceccaldi R., Rondinelli B., D’Andrea A.D.. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016; 26:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lukas J., Lukas C., Bartek J.. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011; 13:1161–1169. [DOI] [PubMed] [Google Scholar]
- 5. Dabin J., Fortuny A., Polo S.E.. Epigenome maintenance in response to DNA damage. Mol. Cell. 2016; 62:712–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Panier S., Boulton S.J.. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol Cell Biol. 2014; 15:7–18. [DOI] [PubMed] [Google Scholar]
- 7. Anderson L., Henderson C., Adachi Y.. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol. Cell. Biol. 2001; 21:1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lukas C., Falck J., Bartkova J., Bartek J., Lukas J.. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat. Cell Biol. 2003; 5:255–260. [DOI] [PubMed] [Google Scholar]
- 9. Bekker-Jensen S., Lukas C., Melander F., Bartek J., Lukas J.. Dynamic assembly and sustained retention of 53BP1 at the sites of DNA damage are controlled by Mdc1/NFBD1. J. Cell Biol. 2005; 170:201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rappold I., Iwabuchi K., Date T., Chen J.. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J. Cell Biol. 2001; 153:613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schultz L.B., Chehab N.H., Malikzay A., Halazonetis T.D.. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 2000; 151:1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stewart G.S., Wang B., Bignell C.R., Taylor A.M., Elledge S.J.. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003; 421:961–966. [DOI] [PubMed] [Google Scholar]
- 13. Lou Z., Minter-Dykhouse K., Franco S., Gostissa M., Rivera M.A., Celeste A., Manis J.P., van Deursen J., Nussenzweig A., Paull T.T. et al.. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell. 2006; 21:187–200. [DOI] [PubMed] [Google Scholar]
- 14. Botuyan M.V., Lee J., Ward I.M., Kim J.E., Thompson J.R., Chen J., Mer G.. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006; 127:1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kolas N.K., Chapman J.R., Nakada S., Ylanko J., Chahwan R., Sweeney F.D., Panier S., Mendez M., Wildenhain J., Thomson T.M. et al.. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007; 318:1637–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huen M.S., Grant R., Manke I., Minn K., Yu X., Yaffe M.B., Chen J.. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007; 131:901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stewart G.S., Panier S., Townsend K., Al-Hakim A.K., Kolas N.K., Miller E.S., Nakada S., Ylanko J., Olivarius S., Mendez M. et al.. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009; 136:420–434. [DOI] [PubMed] [Google Scholar]
- 18. Doil C., Mailand N., Bekker-Jensen S., Menard P., Larsen D.H., Pepperkok R., Ellenberg J., Panier S., Durocher D., Bartek J. et al.. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009; 136:435–446. [DOI] [PubMed] [Google Scholar]
- 19. Mattiroli F., Vissers J.H., van Dijk W.J., Ikpa P., Citterio E., Vermeulen W., Marteijn J.A., Sixma T.K.. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell. 2012; 150:1182–1195. [DOI] [PubMed] [Google Scholar]
- 20. Fradet-Turcotte A., Canny M.D., Escribano-Diaz C., Orthwein A., Leung C.C.Y., Huang H., Landry M.-C., Kitevski-LeBlanc J., Noordermeer S.M., Sicheri F. et al.. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013; 499:50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kleiner R.E., Verma P., Molloy K.R., Chait B.T., Kapoor T.M.. Chemical proteomics reveals a gammaH2AX-53BP1 interaction in the DNA damage response. Nat. Chem. Biol. 2015; 11:807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baldock R.A., Day M., Wilkinson O.J., Cloney R., Jeggo P.A., Oliver A.W., Watts F.Z., Pearl L.H.. ATM localization and heterochromatin repair depend on direct interaction of the 53BP1-BRCT2 domain with gammaH2AX. Cell Rep. 2015; 13:2081–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vazquez B.N., Thackray J.K., Simonet N.G., Kane-Goldsmith N., Martinez-Redondo P., Nguyen T., Bunting S., Vaquero A., Tischfield J.A., Serrano L.. SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO J. 2016; 35:1488–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Min J., Allali-Hassani A., Nady N., Qi C., Ouyang H., Liu Y., MacKenzie F., Vedadi M., Arrowsmith C.H.. L3MBTL1 recognition of mono- and dimethylated histones. Nat. Struct. Mol. Biol. 2007; 14:1229–1230. [DOI] [PubMed] [Google Scholar]
- 25. Mallette F.A., Mattiroli F., Cui G., Young L.C., Hendzel M.J., Mer G., Sixma T.K., Richard S.. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012; 31:1865–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Acs K., Luijsterburg M.S., Ackermann L., Salomons F.A., Hoppe T., Dantuma N.P.. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat. Struct. Mol. Biol. 2011; 18:1345–1350. [DOI] [PubMed] [Google Scholar]
- 27. Drane P., Brault M.E., Cui G., Meghani K., Chaubey S., Detappe A., Parnandi N., He Y., Zheng X.F., Botuyan M.V. et al.. TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature. 2017; 543:211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Botuyan M.V., Cui G., Drane P., Oliveira C., Detappe A., Brault M.E., Parnandi N., Chaubey S., Thompson J.R., Bragantini B. et al.. Mechanism of 53BP1 activity regulation by RNA-binding TIRR and a designer protein. Nat. Struct. Mol. Biol. 2018; 25:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Salvador Moreno N., Liu J., Haas K.M., Parker L.L., Chakraborty C., Kron S.J., Hodges K., Miller L.D., Langefeld C., Robinson P.J. et al.. The nuclear structural protein NuMA is a negative regulator of 53BP1 in DNA double-strand break repair. Nucleic Acids Res. 2019; 47:2703–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tang J., Cho N.W., Cui G., Manion E.M., Shanbhag N.M., Botuyan M.V., Mer G., Greenberg R.A.. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 2013; 20:317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hsiao K.Y., Mizzen C.A.. Histone H4 deacetylation facilitates 53BP1 DNA damage signaling and double-strand break repair. J. Mol. Cell Biol. 2013; 5:157–165. [DOI] [PubMed] [Google Scholar]
- 32. Jacquet K., Fradet-Turcotte A., Avvakumov N., Lambert J.P., Roques C., Pandita R.K., Paquet E., Herst P., Gingras A.C., Pandita T.K. et al.. The TIP60 complex regulates bivalent chromatin recognition by 53BP1 through direct H4K20me binding and H2AK15 acetylation. Mol. Cell. 2016; 62:409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cao L.L., Wei F., Du Y., Song B., Wang D., Shen C., Lu X., Cao Z., Yang Q., Gao Y. et al.. ATM-mediated KDM2A phosphorylation is required for the DNA damage repair. Oncogene. 2016; 35:301–313. [DOI] [PubMed] [Google Scholar]
- 34. Li Z., Chen Y., Tang M., Li Y., Zhu W.-G.. Regulation of DNA damage-induced ATM activation by histone modifications. Genome Instab. Dis. 2019; 1:23–36. [Google Scholar]
- 35. Fnu S., Williamson E.A., De Haro L.P., Brenneman M., Wray J., Shaheen M., Radhakrishnan K., Lee S.H., Nickoloff J.A., Hromas R.. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. PNAS. 2011; 108:540–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Khurana S., Kruhlak M.J., Kim J., Tran A.D., Liu J., Nyswaner K., Shi L., Jailwala P., Sung M.H., Hakim O. et al.. A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Rep. 2014; 8:1049–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Matsuoka S., Ballif B.A., Smogorzewska A., McDonald E.R. 3rd, Hurov K.E., Luo J., Bakalarski C.E., Zhao Z., Solimini N., Lerenthal Y. et al.. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007; 316:1160–1166. [DOI] [PubMed] [Google Scholar]
- 38. Mund A., Schubert T., Staege H., Kinkley S., Reumann K., Kriegs M., Fritsch L., Battisti V., Ait-Si-Ali S., Hoffbeck A.S. et al.. SPOC1 modulates DNA repair by regulating key determinants of chromatin compaction and DNA damage response. Nucleic Acids Res. 2012; 40:11363–11379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Agarwal P., Jackson S.P.. G9a inhibition potentiates the anti-tumour activity of DNA double-strand break inducing agents by impairing DNA repair independent of p53 status. Cancer Lett. 2016; 380:467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhu W.G., Hileman T., Ke Y., Wang P., Lu S., Duan W., Dai Z., Tong T., Villalona-Calero M.A., Plass C. et al.. 5-aza-2′-deoxycytidine activates the p53/p21Waf1/Cip1 pathway to inhibit cell proliferation. J. Biol. Chem. 2004; 279:15161–15166. [DOI] [PubMed] [Google Scholar]
- 41. Liao W., McNutt M.A., Zhu W.G.. The comet assay: a sensitive method for detecting DNA damage in individual cells. Methods. 2009; 48:46–53. [DOI] [PubMed] [Google Scholar]
- 42. Tan M., Luo H., Lee S., Jin F., Yang J.S., Montellier E., Buchou T., Cheng Z., Rousseaux S., Rajagopal N. et al.. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011; 146:1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sanders S.L., Portoso M., Mata J., Bahler J., Allshire R.C., Kouzarides T.. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004; 119:603–614. [DOI] [PubMed] [Google Scholar]
- 44. Weinstock D.M., Nakanishi K., Helgadottir H.R., Jasin M.. Assaying double-strand break repair pathway choice in mammalian cells using a targeted endonuclease or the RAG recombinase. Methods Enzymol. 2006; 409:524–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang Q., Zhu Q., Lu X., Du Y., Cao L., Shen C., Hou T., Li M., Li Z., Liu C. et al.. G9a coordinates with the RPA complex to promote DNA damage repair and cell survival. PNAS. 2017; 114:E6054–E6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bennardo N., Cheng A., Huang N., Stark J.M.. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLos Genet. 2008; 4:e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu W., Nishikawa H., Fukuda T., Vittal V., Asano M., Miyoshi Y., Klevit R.E., Ohta T.. Interaction of BARD1 and HP1 Is Required for BRCA1 Retention at Sites of DNA Damage. Cancer Res. 2015; 75:1311–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Miller K.M., Tjeertes J.V., Coates J., Legube G., Polo S.E., Britton S., Jackson S.P.. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 2010; 17:1144–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bromberg K.D., Mitchell T.R., Upadhyay A.K., Jakob C.G., Jhala M.A., Comess K.M., Lasko L.M., Li C., Tuzon C.T., Dai Y. et al.. The SUV4-20 inhibitor A-196 verifies a role for epigenetics in genomic integrity. Nat. Chem. Biol. 2017; 13:317–324. [DOI] [PubMed] [Google Scholar]
- 50. Ayrapetov M.K., Gursoy-Yuzugullu O., Xu C., Xu Y., Price B.D.. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. PNAS. 2014; 111:9169–9174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tachibana M., Ueda J., Fukuda M., Takeda N., Ohta T., Iwanari H., Sakihama T., Kodama T., Hamakubo T., Shinkai Y.. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005; 19:815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ogawa H., Ishiguro K., Gaubatz S., Livingston D.M., Nakatani Y.. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science. 2002; 296:1132–1136. [DOI] [PubMed] [Google Scholar]
- 53. Weiss T., Hergeth S., Zeissler U., Izzo A., Tropberger P., Zee B.M., Dundr M., Garcia B.A., Daujat S., Schneider R.. Histone H1 variant-specific lysine methylation by G9a/KMT1C and Glp1/KMT1D. Epigenet. Chromatin. 2010; 3:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Collins R.E., Northrop J.P., Horton J.R., Lee D.Y., Zhang X., Stallcup M.R., Cheng X.. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat. Struct. Mol. Biol. 2008; 15:245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu N., Zhang Z., Wu H., Jiang Y., Meng L., Xiong J., Zhao Z., Zhou X., Li J., Li H. et al.. Recognition of H3K9 methylation by GLP is required for efficient establishment of H3K9 methylation, rapid target gene repression, and mouse viability. Genes Dev. 2015; 29:379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kleefstra T., Nillesen W.M., Yntema H.G.. Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH et al.. Kleefstra Syndrome. GeneReviews(R). 1993; Seattle: Univeristy of Washington. [Google Scholar]
- 57. O’Driscoll M., Gennery A.R., Seidel J., Concannon P., Jeggo P.A.. An overview of three new disorders associated with genetic instability: LIG4 syndrome, RS-SCID and ATR-Seckel syndrome. DNA Repair (Amst.). 2004; 3:1227–1235. [DOI] [PubMed] [Google Scholar]
- 58. Gennery A.R., Slatter M.A., Bhattacharya A., Barge D., Haigh S., O’Driscoll M., Coleman R., Abinun M., Flood T.J., Cant A.J. et al.. The clinical and biological overlap between Nijmegen Breakage Syndrome and Fanconi anemia. Clin. Immunol. 2004; 113:214–219. [DOI] [PubMed] [Google Scholar]
- 59. Watanabe S., Iimori M., Chan D.V., Hara E., Kitao H., Maehara Y.. MDC1 methylation mediated by lysine methyltransferases EHMT1 and EHMT2 regulates active ATM accumulation flanking DNA damage sites. Sci. Rep. 2018; 8:10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ginjala V., Rodriguez-Colon L., Ganguly B., Gangidi P., Gallina P., Al-Hraishawi H., Kulkarni A., Tang J., Gheeya J., Simhadri S. et al.. Protein-lysine methyltransferases G9a and GLP1 promote responses to DNA damage. Sci. Rep. 2017; 7:16613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fukuda T., Wu W., Okada M., Maeda I., Kojima Y., Hayami R., Miyoshi Y., Tsugawa K., Ohta T.. Class I histone deacetylase inhibitors inhibit the retention of BRCA1 and 53BP1 at the site of DNA damage. Cancer Sci. 2015; 106:1050–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yang H., Pesavento J.J., Starnes T.W., Cryderman D.E., Wallrath L.L., Kelleher N.L., Mizzen C.A.. Preferential dimethylation of histone H4 lysine 20 by Suv4-20. J. Biol. Chem. 2008; 283:12085–12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pesavento J.J., Yang H., Kelleher N.L., Mizzen C.A.. Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol. Cell. Biol. 2008; 28:468–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schotta G., Sengupta R., Kubicek S., Malin S., Kauer M., Callen E., Celeste A., Pagani M., Opravil S., De La Rosa-Velazquez I.A. et al.. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 2008; 22:2048–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pei H., Zhang L., Luo K., Qin Y., Chesi M., Fei F., Bergsagel P.L., Wang L., You Z., Lou Z.. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011; 470:124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tuzon C.T., Spektor T., Kong X., Congdon L.M., Wu S., Schotta G., Yokomori K., Rice J.C.. Concerted activities of distinct H4K20 methyltransferases at DNA double-strand breaks regulate 53BP1 nucleation and NHEJ-directed repair. Cell Rep. 2014; 8:430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Svobodova Kovarikova A., Legartova S., Krejci J., Bartova E.. H3K9me3 and H4K20me3 represent the epigenetic landscape for 53BP1 binding to DNA lesions. Aging (Albany NY). 2018; 10:2585–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shiloh Y., Ziv Y.. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013; 14:197–210. [PubMed] [Google Scholar]
- 69. Li Z., Li Y., Tang M., Peng B., Lu X., Yang Q., Zhu Q., Hou T., Li M., Liu C. et al.. Destabilization of linker histone H1.2 is essential for ATM activation and DNA damage repair. Cell Res. 2018; 28:756–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tang M., Li Z., Zhang C., Lu X., Tu B., Cao Z., Li Y., Chen Y., Jiang L., Wang H. et al.. SIRT7-mediated ATM deacetylation is essential for its deactivation and DNA damage repair. Sci. Adv. 2019; 5:eaav1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.