

Abstract
Aims
Iron deficiency (ID) is common in heart failure (HF) patients and negatively impacts symptoms and prognosis. The aetiology of ID in HF is largely unknown. We studied determinants and the biomarker profile of ID in a large international HF cohort.
Methods and results
We studied 2357 worsening HF patients from the BIOSTAT-CHF cohort. ID was defined as transferrin saturation <20%. Univariable and multivariable logistic regression models were constructed to identify determinants for ID. We measured 92 cardiovascular markers (Olink Cardiovascular III) to establish a biomarker profile of ID. The primary endpoint was the composite of all-cause mortality and first HF rehospitalization. Mean age (±standard deviation) of all patients was 69 ± 12.0 years, 26.1% were female and median N-terminal pro B-type natriuretic peptide levels (+interquartile range) were 4305 (2360–8329) ng/L. Iron deficiency was present in 1453 patients (61.6%), with highest prevalence in females (71.1% vs. 58.3%; P < 0.001). Independent determinants of ID were female sex, lower estimated protein intake, higher heart rate, presence of peripheral oedema and orthopnoea, chronic kidney disease, lower haemoglobin, higher C-reactive protein levels, lower serum albumin levels, and P2Y12 inhibitor use (all P < 0.05). None of these determinants were sex-specific. The biomarker profile of ID largely consisted of pro-inflammatory markers, including paraoxonase 3 (PON3) and tartrate-resistant acid phosphatase type 5. In multivariable Cox proportional hazard regression analyses, ID was associated to worse outcome, independently of predictors of ID (hazard ratio 1.25, 95% confidence interval 1.06–1.46; P = 0.007).
Conclusion
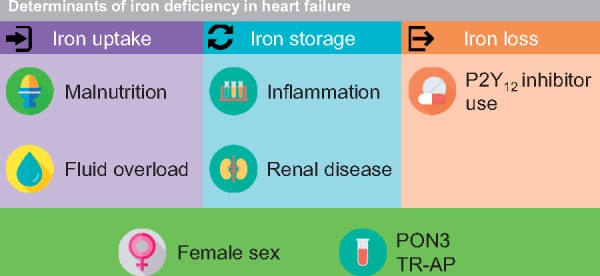
Our data suggest that the aetiology of ID in worsening HF is complex, multifactorial and seems to consist of a combination of reduced iron uptake (malnutrition, fluid overload), impaired iron storage (inflammation, chronic kidney disease), and iron loss (antiplatelets).
Keywords: Heart failure, Iron deficiency, Inflammation, Protein intake, Fluid retention, Antiplatelets
Introduction
Numerous studies showed the adverse clinical and prognostic consequences of iron deficiency (ID) in patients with chronic heart failure (HF).1–4 Despite the significant prevalence of ID in HF, its pathophysiology and aetiology are not well-understood. Suggested mechanisms for ID in HF are poor dietary iron intake, drug interactions, (occult) gastrointestinal blood loss due to antiplatelet drugs and anticoagulants, and hepcidin-induced iron entrapment due to chronic low-grade inflammation.5 While ID is present in approximately half of all HF patients, its prevalence seems highest in females.1,4 It is currently unclear which factors are driving this sex difference. In the present study, we identified determinants of ID in a large international cohort of worsening HF patients and sought to find sex-specific clinical and biochemical predictors of ID. Moreover, we established a cardiovascular biomarker profile of patients with ID.
Methods
Study population
We included HF patients from the BIOSTAT-CHF study (A systems BIOlogy Study to TAilored Treatment in Chronic Heart Failure). This cohort has been described in full detail elsewhere.6–8 In short, the BIOSTAT-CHF study included patients either hospitalized for HF or presenting with worsening HF in the outpatient setting. Patients were eligible to participate with a left ventricular ejection fraction (LVEF) of ≤40% or, alternatively, brain natriuretic peptide or N-terminal pro B-type natriuretic peptide (NT-proBNP) levels of >400 ng/L or >2000 ng/L, respectively. Additionally, patients had to receive suboptimal evidence-based HF treatment (i.e. ≤50% of target dose of angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and/or beta-blockers). After study inclusion, treating physicians were encouraged to up-titrate these drugs during a 3-month treatment optimization phase. The BIOSTAT-CHF study was conducted in accordance with the Declaration of Helsinki. All patients provided written informed consent prior to any study-related activities.
Of all 2516 patients enrolled in the BIOSTAT-CHF cohort, serum for iron status analysis was available in 2357 (93.7%) patients.
Laboratory measurements
Iron parameters were assessed from venous blood. Blood samples were centrifuged at 2500 g for 15 min (4°C) and stored at −80°C afterwards. Samples were never thawed before laboratory analyses. The following blood markers reflecting iron metabolism were assessed on a Roche modular cobas 8000 using standard methods: serum iron, ferritin, and transferrin. Transferrin saturation (TSAT) was calculated as follows: [72.17 * iron (mg/dL)]/transferrin (mg/dL).9
Renal function was expressed as the estimated glomerular filtration rate (mL/min/1.73 m2), calculated using the Chronic Kidney Disease Epidemiology collaboration equation (CKD-EPI). Serum NT-proBNP levels were determined using an immunoassay based on electrochemiluminescence (Elecsys, Roche Diagnostics, Mannheim, Germany). Serum hepcidin levels were measured using a competitive ELISA as described previously.10 Serum soluble transferrin receptor (sTfR) levels were measured using immunonephelometry on a BNII Nephelometer (Siemens AG, Erlangen, Germany).
To establish a biomarker profile for patients with and without ID, 92 cardiovascular-related biomarkers from the Olink Cardiovascular III panel were measured, which were selected based on literature search on bioinformatics (e.g. Uniprot and DisGeNET) and by consulting experts in the cardiovascular field. All biomarkers were measured by Olink Proteomics (Uppsala, Sweden) using the Proximity Extension Assay technology, as previously described.11 Results are reported as Normalized Protein eXpression (NPX) on a log2 scale.
Definitions and study endpoints
Anaemia was defined as a haemoglobin level <12 g/dL in women and <13 g/dL in men as per WHO standards.12 Iron deficiency was defined as a TSAT <20%, as proposed by Grote Beverborg et al.13 This definition has been validated against the gold standard test for ID (bone marrow iron staining) in HF patients and has previously been used.4,13 Daily protein intake of patients was estimated using spot urinary nitrogen and body mass index.14 Median follow-up of the study was 21 months. The primary endpoint of this study was the composite of all-cause mortality and first HF rehospitalization. Secondary endpoints included all-cause mortality and first HF rehospitalization.
Statistical analyses
Data are presented as mean ± standard deviation (SD) when normally distributed, as median and interquartile range (IQR) when non-normally distributed or as percentage when categorical. Baseline characteristics were compared using the Student’s t-test (normally distributed variables), the Mann–Whitney U test (non-normally distributed variables), and the χ2 test (categorical or binary variables). All baseline characteristics were stratified by iron status and sex. After baseline analyses, skewed variables were natural log2-transformed to obtain normal distributions. To identify independent predictors of ID, univariable and multivariable logistic regression models were constructed. Associates of ID with a univariable P-value of ≤0.1 were entered into the multivariable logistic regression models. Final multivariable models were established using backward elimination based on the significance of each variable. Bootstrap analyses with 1000 repeats (using the ‘swboot’ package in Stata) were performed to evaluate the robustness of the final models. Variables selected >700 times were considered robust predictors. As a sensitivity analysis, a regularization approach was performed using multivariable lasso regression. The final model was checked for multicollinearity by calculating the variance inflation factor. Restricted cubic splines (three knots for all variables) were constructed for better visualization of the predictive value of continuous parameters on ID. Multivariable interaction analyses for sex were performed with each predictor of ID.
The differential Olink biomarker expression pattern in iron-deficient patients was visualized using a volcano plot, displaying the magnitude in change of each biomarker (log2-fold change) against the significance of the difference in biomarker expression [negative log10 of the P-value (Mann–Whitney U test)]. Biomarkers in the top left or right of the plot are of interest (large magnitude fold change and high statistical significance). False discovery rate was controlled by correcting the P-values according to the Benjamini–Hochberg procedure (false discovery rate of 0.05). Biomarkers that were significantly up- or down-regulated in ID and had an absolute log2-fold change of >0.25 were entered in uni- and multivariable logistic regression analyses with ID as dependent variable.
Kaplan–Meier curves were constructed to determine the prognostic consequences of ID. Differences in survival rates were tested using the log-rank Mantel–Cox test. The influence of ID on outcome was further assessed with univariable and multivariable Cox proportional hazard regression models. In the multivariable models, adjustment was made for the BIOSTAT prediction models as described elsewhere.8 Additionally, the prognostic consequences of continuous TSAT levels were analysed using univariable and multivariable fractional polynomial analyses. The proportional hazards assumption was evaluated using Schoenfeld residuals and was applicable to all variables included in the outcome models. Missing predictor values were five times imputed as previously described.8 Survival analyses were performed in all five imputation models and the results were averaged in agreement with Rubin’s rules. The degree of missingness of all variables used in the model development is depicted in Supplementary material online, Table S6. A two-sided P-value <0.05 was considered statistically significant, while for interaction testing a P-value <0.1 was used. Data were analysed using Stata version 15.1 (StataCorp LLC, College Station, TX, USA) and R version 3.5.1 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Baseline characteristics
Baseline characteristics of the present study cohort, stratified by iron status, are depicted in Table 1. Mean age (±SD) of all patients was 69 ± 12.0 years, median LVEF (+IQR) was 30% (25–36), and 37.2% of all patients were in New York Heart Association (NYHA) functional class III or IV. The overall prevalence of ID was 61.6% (n = 1453) with highest prevalence in females (71.1% vs. 58.3%, P < 0.001). According to the ‘conventional’ definition of ID in HF patients (i.e. ferritin <100 µg/L or ferritin 100–300 µg/L with a TSAT <20%), the prevalence of ID was even higher (n = 1632; 69.2%). Iron-deficient patients were older, had more comorbidities (including anaemia) and more signs of fluid overload compared to patients without ID (all P < 0.05). Furthermore, patients with ID had lowest estimated protein intake, more inflammation, and highest rate of proton-pump inhibitor and P2Y12 inhibitor use (all P < 0.001). HF aetiology and type of HF event (i.e. new-onset HF or worsening HF) was comparable between patients with and without ID.
Table 1.
Baseline characteristics for the total cohort, stratified by iron status
| Variables | Total cohort | No ID | ID | P-value |
|---|---|---|---|---|
| N | 2357 | 904 | 1453 | |
| Clinical parameters | ||||
| Age (years) | 68.9 ± 12.0 | 68.1 ± 12.1 | 69.3 ± 11.9 | 0.016 |
| Females (%) | 616 (26.1) | 178 (19.7) | 438 (30.1) | <0.001 |
| BMI (kg/m2) | 27.9 ± 5.5 | 27.8 ± 5.3 | 27.9 ± 5.6 | 0.65 |
| Estimated protein intake (g/day) | 55.0 ± 11.2 | 56.8 ± 12.2 | 53.9 ± 10.5 | <0.001 |
| Ischaemic aetiology | 1069 (46.2) | 395 (44.6) | 674 (47.1) | 0.24 |
| LVEF (%) | 30 (25–36) | 30 (25–35) | 30 (25–37) | 0.93 |
| HFrEF | 1707 (80.9) | 687 (82.6) | 1020 (79.8) | 0.034 |
| HFmrEF | 271 (12.8) | 107 (12.9) | 164 (12.8) | |
| HFpEF | 132 (6.3) | 38 (4.6) | 94 (7.4) | |
| Previous hospitalization for HF | 736 (31.2) | 278 (30.8) | 458 (31.5) | 0.70 |
| NYHA functional class III/IV | 1417 (61.8) | 455 (51.6) | 962 (68.3) | <0.001 |
| Systolic blood pressure (mmHg) | 125 ± 22 | 124 ± 20 | 125 ± 23 | 0.36 |
| Heart rate (b.p.m.) | 80 ± 19 | 77 ± 18 | 82 ± 20 | <0.001 |
| Peripheral oedema | 1165 (59.5) | 354 (48.8) | 811 (65.7) | <0.001 |
| Elevated JVP | 518 (33.5) | 156 (26.4) | 362 (37.8) | <0.001 |
| Hepatomegaly | 333 (14.2) | 118 (13.1) | 215 (14.8) | 0.24 |
| Orthopnoea | 818 (34.8) | 231 (25.6) | 587 (40.5) | <0.001 |
| 6MWT (m) | 316 (225–393) | 345 (250–416) | 300 (210–374) | <0.001 |
| KCCQ (overall score) | 49 ± 22 | 56 ± 22 | 45 ± 22 | <0.001 |
| Comorbidities | ||||
| Atrial fibrillation | 1063 (45.1) | 393 (43.5) | 670 (46.1) | 0.21 |
| Diabetes mellitus | 759 (32.2) | 238 (26.3) | 521 (35.9) | <0.001 |
| COPD | 406 (17.2) | 133 (14.7) | 273 (18.8) | 0.011 |
| Renal disease | 649 (27.5) | 177 (19.6) | 472 (32.5) | <0.001 |
| Device therapy | 582 (24.7) | 215 (23.8) | 367 (25.3) | 0.42 |
| Laboratory | ||||
| Haemoglobin (g/dL) | 13.2 ± 1.9 | 13.9 ± 1.8 | 12.8 ± 1.8 | <0.001 |
| Anaemiaa | 778 (36.2) | 181 (23.0) | 597 (43.8) | <0.001 |
| Haematocrit (%) | 40.0 ± 5.4 | 41.6 ± 5.2 | 39.1 ± 5.2 | <0.001 |
| Mean corpuscular volume (fL) | 90 ± 9 | 92 ± 9 | 89 ± 8 | <0.001 |
| Iron (mg/dL) | 45 (28–73) | 78 (62–101) | 34 (22–45) | <0.001 |
| Ferritin (µg/L) | 103 (50–193) | 142 (80–240) | 78 (39–162) | <0.001 |
| Transferrin (mg/dL) | 200 (160–250) | 200 (170–240) | 210 (160–250) | 0.17 |
| Transferrin saturation (%) | 17 (11–25) | 27 (23–33) | 12 (9–16) | NA |
| sTfR (mg/L) | 1.5 (1.2–2.1) | 1.3 (1.0–1.7) | 1.7 (1.3–2.3) | <0.001 |
| Hepcidin (nmol/L) | 6.3 (2.2–16.5) | 8.4 (4.4–20.0) | 4.6 (1.4–13.1) | <0.001 |
| CRP (mg/L) | 13.0 (5.8–26.4) | 8.0 (3.5–17.2) | 16.9 (8.4–32.1) | <0.001 |
| Leucocytes (109/L) | 7.8 (6.4–9.6) | 7.5 (6.3–9.1) | 8.0 (6.6–9.8) | <0.001 |
| AST (U/L) | 25 (19–35) | 26 (20–35) | 25 (19–35) | 0.28 |
| ALT (U/L) | 25 (17–38) | 26 (18–40) | 24 (16–37) | 0.005 |
| γ-GT (U/L) | 55 (28–109) | 53 (29–107) | 56 (27–110) | 0.89 |
| Alkaline phosphatase (µg/L) | 85 (65–118) | 83 (63–117) | 86 (66–120) | 0.26 |
| Total bilirubin (µmol/L) | 14 (10–21) | 14 (10–20) | 14 (10–22) | 0.14 |
| Sodium (mmol/L) | 140 (137–142) | 140 (137–142) | 139 (137–142) | <0.001 |
| Potassium (mmol/L) | 4.2 (3.9–4.6) | 4.3 (4.0–4.6) | 4.2 (3.9–4.6) | 0.012 |
| NT-proBNP (ng/L) | 4305 (2360–8329) | 3300 (1833–6767) | 4812 (2688–8991) | <0.001 |
| Creatinin (µmol/L) | 101 (82–128) | 97 (80–122) | 104 (84–133) | <0.001 |
| eGFR (mL/min/1.73 m2) | 60 (44–79) | 64 (48–83) | 57 (43–76) | <0.001 |
| Albumin (g/L) | 32 ± 9 | 34 ± 8 | 31 ± 9 | <0.001 |
| Urea (mmol/L) | 11.4 (7.6–18.2) | 10.3 (7.1–16.4) | 12.0 (7.8–19.0) | <0.001 |
| Medication | ||||
| Loop diuretics | 2346 (99.5) | 902 (99.8) | 1444 (99.4) | 0.17 |
| Beta-blockers on target dose | 128 (5.4) | 54 (6.0) | 74 (5.1) | 0.36 |
| ACEi/ARB on target dose | 314 (13.3) | 128 (14.2) | 186 (12.8) | 0.35 |
| Aldosterone antagonist | 1259 (53.4) | 514 (56.9) | 745 (51.3) | 0.008 |
| Proton-pump inhibitors | 825 (35.0) | 260 (28.8) | 565 (38.9) | <0.001 |
| Antiplatelets | ||||
| P2Y12 inhibitors | 363 (15.4) | 108 (11.9) | 255 (17.5) | <0.001 |
| Acetylsalicylic acid | 1168 (49.6) | 443 (49.0) | 725 (49.9) | 0.67 |
| Anticoagulants | 915 (38.8) | 349 (38.6) | 566 (39.0) | 0.87 |
| Vitamin K antagonists | 898 (38.1) | 341 (37.7) | 557 (38.3) | 0.77 |
| DOACs | 17 (0.7) | 8 (0.9) | 9 (0.6) | 0.46 |
6MWT, 6-min walk test; ACEi, angiotensin-converting enzyme inhibitor; ALT, alanine aminotransferase; ARB, angiotensin receptor blocker; AST, aspartate aminotransferase; BMI, body mass index; COPD, chronic obstructive pulmonary disease; CRP, C-reactive protein; DOAC, direct-acting oral anticoagulant; eGFR, estimated glomerular filtration rate; γ-GT, gamma-glutamyltransferase; HFmrEF, heart failure with mid-range ejection fraction; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; ID, iron deficiency; JVP, jugular venous pressure; KCCQ, Kansas City Cardiomyopathy Questionnaire; LVEF, left ventricular ejection fraction; NT-proBNP, N-terminal pro B-type natriuretic peptide; sTfR, soluble transferrin receptor.
Anaemia was defined as a haemoglobin level <12 g/dL in women and <13 g/dL in men.
Similar in females and males, patients with ID had more severe HF at baseline (e.g. peripheral oedema, orthopnoea, and highest NT-proBNP levels), showed more signs of inflammation, had highest prevalence of anaemia and lowest estimated protein intake (all P < 0.01, see Supplementary material online, Table S1). Numerically, iron-deficient males had most comorbidities and highest rate of proton-pump inhibitor and P2Y12 inhibitor use, although no significant interaction between these variables and sex was present. A higher prevalence of atrial fibrillation in iron-deficient patients was observed in females, but not in males (P for interaction, 0.028).
Determinants of iron deficiency
Univariable and multivariable logistic regression prediction models for ID are shown in Table 2. Independent determinants of ID were lower estimated protein intake, higher heart rate, presence of peripheral oedema and orthopnoea, history of renal disease, lower haemoglobin, higher C-reactive protein (CRP), lower serum albumin, and use of P2Y12 inhibitors (all P < 0.005). The c-statistic of this model was 0.76. None of the determinants had a significant interaction with sex. All determinants in the final model remained highly selected in additional bootstrap analyses. Additional Lasso regression analysis of the multivariable model confirmed our findings and selected haemoglobin and CRP as best predictors for ID. The variance inflation factors of variables in the multivariable model were not suggestive of multicollinearity (range of factors 1.02–1.27). Restricted cubic splines showing the association between ID and estimated daily protein intake, serum levels of CRP and albumin, and haemoglobin are displayed in Figure 1A–D. Sex-specific restricted cubic splines for these determinants can be found in Supplementary material online, Figure S1A–D.
Table 2.
Univariable and multivariable logistic regression prediction models for iron deficiency
| Variables | Univariable |
Multivariable |
|||||
|---|---|---|---|---|---|---|---|
| Odds ratio (95% CI) | Z-value | P-value | P for interaction with sex | Odds ratio (95% CI) | Z-value | P-value | |
| Clinical parameters | |||||||
| Age (per 5 years) | 1.04 (1.01–1.08) | 2.40 | 0.016 | 0.464 | |||
| Sex (female vs. male) | 1.76 (1.44–2.15) | 5.58 | <0.001 | — | 1.42 (1.13–1.79) | 2.99 | 0.003 |
| BMI (per 5 kg/m2) | 1.02 (0.95–1.10) | 0.60 | 0.551 | 0.368 | |||
| Estimated protein intake (per 10 g/day) | 0.80 (0.74–0.86) | −5.74 | <0.001 | 0.405 | 0.87 (0.79–0.94) | −3.32 | 0.001 |
| Ischaemic aetiology (yes vs. no) | 1.11 (0.93–1.31) | 1.17 | 0.241 | 0.687 | |||
| LVEF (per 5%) | 1.03 (1.00–1.08) | 1.71 | 0.087 | 0.008 | |||
| Male | 0.98 (0.93–1.02) | −0.95 | 0.340 | ||||
| Female | 1.11 (1.02–1.19) | 2.54 | 0.011 | ||||
| Previous HF hospitalization (yes vs. no) | 1.04 (0.87–1.24) | 0.39 | 0.695 | 0.261 | |||
| NYHA functional class III/IV (vs. I/II) | 2.02 (1.70–2.40) | 7.95 | <0.001 | 0.899 | |||
| Systolic blood pressure (per 5 mmHg) | 1.01 (0.99–1.03) | 0.85 | 0.395 | 0.726 | |||
| Heart rate (per 5 b.p.m.) | 1.08 (1.05–1.10) | 6.10 | <0.001 | 0.157 | 1.06 (1.04–1.09) | 3.83 | <0.001 |
| Peripheral oedema (yes vs. no) | 1.97 (1.67–2.34) | 7.90 | <0.001 | 0.649 | 1.36 (1.12–1.66) | 3.03 | 0.002 |
| Elevated JVP (yes vs. no) | 1.69 (1.35–2.12) | 4.59 | <0.001 | 0.735 | |||
| Hepatomegaly (yes vs. no) | 1.16 (0.91–1.48) | 1.23 | 0.220 | 0.116 | |||
| Orthopnoea (yes vs. no) | 1.96 (1.64–2.36) | 7.28 | <0.001 | 0.927 | 1.33 (1.07–1.66) | 2.59 | 0.010 |
| Comorbidities | |||||||
| Atrial fibrillation (yes vs. no) | 1.11 (0.94–1.31) | 1.25 | 0.211 | 0.016 | |||
| Male | 1.02 (0.84–1.23) | 0.19 | 0.850 | ||||
| Female | 1.69 (1.17–2.44) | 2.82 | 0.005 | ||||
| Diabetes mellitus (yes vs. no) | 1.56 (1.30–1.88) | 4.80 | <0.001 | 0.539 | |||
| COPD (yes vs. no) | 1.34 (1.07–1.68) | 2.54 | 0.011 | 0.165 | |||
| Renal disease (yes vs. no) | 1.98 (1.62–2.41) | 6.76 | <0.001 | 0.474 | 1.62 (1.28–2.04) | 4.07 | <0.001 |
| Device therapy (yes vs. no) | 1.08 (0.89–1.31) | 0.81 | 0.419 | 0.133 | |||
| Laboratory | |||||||
| Anaemiaa (yes vs. no) | 2.49 (2.07–3.00) | 9.64 | <0.001 | 0.201 | |||
| Haemoglobin (per g/dL) | 0.75 (0.71–0.78) | −11.77 | <0.001 | 0.216 | 0.79 (0.74–0.83) | −7.87 | <0.001 |
| CRP (per doubling) | 1.48 (1.40–1.57) | 13.66 | <0.001 | 0.805 | 1.39 (1.30–1.48) | 10.14 | <0.001 |
| AST (per doubling) | 0.97 (0.87–1.09) | −0.45 | 0.653 | 0.740 | |||
| ALT (per doubling) | 0.94 (0.86–1.02) | −1.44 | 0.150 | 0.467 | |||
| γ-GT (per doubling) | 0.99 (0.84–1.24) | −0.18 | 0.859 | 0.967 | |||
| Alkaline phosphatase (per doubling) | 1.06 (0.89–1.26) | 0.65 | 0.516 | 0.186 | |||
| Total bilirubin (per doubling) | 1.08 (0.98–1.18) | 1.57 | 0.116 | 0.942 | |||
| Sodium (per mmol/L) | 0.97 (0.94–0.99) | −3.19 | 0.001 | 0.162 | |||
| Potassium (per mmol/L) | 0.81 (0.70–0.94) | −2.73 | 0.006 | 0.182 | |||
| NT-proBNP (per doubling) | 1.23 (1.16–1.30) | 6.95 | <0.001 | 0.028 | |||
| Male | 1.26 (1.18–1.35) | 6.91 | <0.001 | ||||
| Female | 1.08 (0.96–1.22) | 1.25 | 0.211 | ||||
| Creatinin (per doubling) | 1.35 (1.15–1.58) | 3.74 | <0.001 | 0.027 | |||
| Male | 1.69 (1.40–2.05) | 5.40 | <0.001 | ||||
| Female | 1.09 (0.78–1.53) | 0.52 | 0.604 | ||||
| eGFR (per doubling) | 0.79 (0.69–0.91) | −3.39 | 0.001 | 0.162 | |||
| Albumin (per 5 g/L) | 0.81 (0.77–0.86) | −8.09 | <0.001 | 0.776 | 0.93 (0.87–0.98) | −2.46 | 0.014 |
| Urea (per doubling) | 1.26 (1.15–1.38) | 4.90 | <0.001 | 0.387 | |||
| Medication | |||||||
| Loop diuretics (yes vs. no) | NA | ||||||
| Beta-blockers on target dose (yes vs. no) | 0.84 (0.59–1.21) | −0.92 | 0.359 | 0.716 | |||
| ACEi/ARB on target dose (yes vs. no) | 0.89 (0.70–1.13) | −0.94 | 0.346 | 0.472 | |||
| Aldosterone antagonist (yes vs. no) | 0.80 (0.68–0.94) | −2.64 | 0.008 | 0.745 | |||
| Proton-pump inhibitors (yes vs. no) | 1.58 (1.32–1.88) | 4.99 | <0.001 | 0.197 | |||
| P2Y12 inhibitors (yes vs. no) | 1.57 (1.23–2.00) | 3.64 | <0.001 | 0.693 | 1.64 (1.24–2.16) | 3.47 | 0.001 |
| Acetylsalicylic acid (yes vs. no) | 1.04 (0.88–1.22) | 0.42 | 0.674 | 0.225 | |||
| Anticoagulants (yes vs. no) | 1.01 (0.86–1.20) | 0.17 | 0.866 | 0.156 | |||
ACEi, angiotensin-converting enzyme inhibitor; ALT, alanine aminotransferase; ARB, angiotensin receptor blocker; AST, aspartate aminotransferase; BMI, body mass index; CI, confidence interval; COPD, chronic obstructive pulmonary disease; CRP, C-reactive protein; eGFR, estimated glomerular filtration rate; γ-GT, gamma-glutamyltransferase; JVP, jugular venous pressure; LVEF, left ventricular ejection fraction; NT-proBNP, N-terminal pro B-type natriuretic peptide.
Anaemia was defined as a haemoglobin level <12 g/dL in women and <13 g/dL in men.
Figure 1.

(A) Restricted cubic spline of the association between estimated protein intake and the prevalence of iron deficiency. (B) Restricted cubic spline of the association between C-reactive protein and the prevalence of iron deficiency. (C) Restricted cubic spline of the association between serum albumin and the prevalence of iron deficiency. (D) Restricted cubic spline of the association between haemoglobin and the prevalence of iron deficiency. The solid lines indicate estimates of the prevalence of iron deficiency across continuous levels of estimated protein intake, C-reactive protein, serum albumin, and haemoglobin, fitted using logistic regression analysis. The dashed lines indicate 95% confidence intervals.
Biomarker profile of iron deficiency
Median log2 levels of the 92 cardiovascular biomarkers from the Olink Cardiovascular III panel are depicted in Supplementary material online, Table S2. In patients with ID, the following biomarkers were significantly up-regulated with largest magnitude of change: fatty acid binding protein 4 (FABP4), growth differentiation factor 15 (GDF15), NT-proBNP, osteopontin (OPN), ST2 protein (ST2), tumour necrosis factor receptor 1 (TNF-R1), and transferrin receptor protein 1 (TR). Only paraoxonase 3 (PON3) and tartrate-resistant acid phosphatase type 5 (TR-AP) were strongly and significantly down-regulated in ID (Figure 2). After correcting for the determinants for ID originating from Table 2, only PON3, TR-AP, ST2, NT-proBNP, and TR remained significantly associated with ID (all P < 0.05; Supplementary material online, Table S3).
Figure 2.

Biomarker expression profile (volcano plot) in iron-deficient heart failure patients compared to patients without iron deficiency. The volcano plot shows the difference in cardiovascular biomarker expression in patients with and without iron deficiency. Each dot represents one of the 92 biomarkers of the Olink Cardiovascular III panel. On the x-axis, the log2-fold change in biomarker expression is depicted (positive log2-fold change is higher biomarker expression in patients with iron deficiency; negative log2-fold change is lower biomarker expression in patients with iron deficiency), while the y-axis shows the magnitude of the biomarker expression difference as −log10 of the P-value. Red dots are biomarkers with a significant up- or down-regulation in patients with iron deficiency (corrected for a false discovery rate of 5%); green dots indicate biomarkers with an absolute log2-fold change of >0.25. Most biomarkers were significantly up-regulated in patients with ID (n = 39, 42.4%), while 15 biomarkers had significantly lower expression (16.3%). FABP4, fatty acid binding protein 4; GDF15, growth differentiation factor 15; NT-proBNP, N-terminal prohormone brain natriuretic peptide; OPN, osteopontin; PON3, paraoxonase 3; ST2, ST2 protein; TNF-R1, tumour necrosis factor receptor 1; TR, transferrin receptor protein 1; TR-AP, tartrate-resistant acid phosphatase type 5.
Prognostic consequences of iron deficiency
During a median follow-up of 21 months, overall rates of mortality and first HF hospitalization were 26.9% (n = 615) and 24.5% (n = 578), respectively. Event rates were comparable between males and females (see Supplementary material online, Figure S3A and B). Kaplan–Meier estimator curves (for all-cause mortality and the composite endpoint of all-cause mortality and first HF hospitalization) and cumulative incident curves (for first HF hospitalization), stratified by iron status, are shown in Supplementary material online, Figure S2A–C. Iron deficiency was a significant predictor for all endpoints (all P < 0.05). No interaction was observed between iron status and sex on all endpoints. Univariable and multivariable Cox proportional hazard regression analyses for all endpoints are depicted in Supplementary material online, Tables S4 and S7. Iron deficiency remained independently associated with the primary composite endpoint of all-cause mortality and first HF rehospitalization after correcting for the BIOSTAT prediction model [hazard ratio (HR) 1.30, 95% confidence interval (CI) 1.12–1.50; P = 0.0005] and the logistic regression prediction model for ID (HR 1.25, 95% CI 1.06–1.46; P = 0.007). In a multivariable fractional polynomial analysis, lower TSAT levels were associated to an increased risk of all-cause mortality (see Supplementary material online, Figure S4). Finally, in the prognostic models including haemoglobin (all-cause mortality and the composite endpoint), we compared the prognostic power of haemoglobin and ID (defined as TSAT < 20% and using TSAT as a continuous variable). Exchanging haemoglobin for either ID or TSAT did not alter the prognostic power of both models (see Supplementary material online, Table S8).
Discussion
In a large cohort of patients with worsening HF, we identified the following independent determinants of ID: female sex, lower estimated protein intake, higher heart rate, presence of peripheral oedema and orthopnoea, history of renal disease, lower haemoglobin, higher CRP levels, lower serum albumin levels, and antiplatelet use. None of these factors had a significant interaction with sex. The adverse prognostic consequences of ID are independent of these identified predictors. Finally, we provided a biomarker profile of patients with ID, in which predominantly pro-inflammatory markers seem up-regulated.
Determinants of iron deficiency
Our observational data suggest factors which may be involved in the aetiology of ID in HF. These determinants are depicted in Take home figure and are discussed below.
Take home figure.
Determinants of iron deficiency in heart failure. Several graphical elements in this figure are provided by Freepik and DinosoftLabs from www.flaticon.com.
Sex difference
In the final prediction model for ID, we could not identify significant interactions with sex. Instead, female sex was an independent predictor for ID, which has been reported in other studies as well.1,4,15 Several mechanisms might be underlying this association. First, to exclude menstrual blood loss as a confounding factor, we performed a sensitivity analysis in which we excluded premenopausal women (i.e. age <52 years; Supplementary material online, Table S5). This yielded a nearly identical prediction model for ID. Second, female patients in our cohort had a higher prevalence of HFpEF, a HF subtype which has been linked to highest prevalence of ID.16
Reduced estimated protein intake
As depicted in Figure 1A, the prevalence of ID rapidly increases in patients with lower estimated protein intake. Additionally, we identified lower serum albumin levels as an independent predictor of ID (Figure 1C). These findings might suggest a poor nutritional status as an aetiological pathway of ID. Several risk scores for estimating malnutrition have included serum albumin levels as predictor, also in HF patients.17–20 Although we did not study dietary iron intake per se, daily protein intake should provide a fair estimation of daily dietary iron intake, as a significant amount of dietary iron intake is provided by protein-rich food, such as meat (haem iron), nuts and legumes (non-haem iron).21 It should be acknowledged that we only estimated total daily protein intake using surrogate markers; we did not have data on the exact daily protein intake, nor exact daily iron intake.
Venous congestion
Heart failure is associated with right-sided venous congestion which leads to increased gastrointestinal wall thickness and malabsorption.22–25 Intestinal wall oedema due to right-sided congestion might also negatively influence nutrient absorption, including dietary iron. In the multivariable prediction model for ID, we showed that peripheral oedema, as an indicator of right-sided congestion, was an independent predictor of ID. Consequently, malabsorption due to venous congestion may also play a role in the aetiology of ID in HF.
Antiplatelet drugs
Iron-deficient patients had higher prevalence of antiplatelet drug use compared to patients without ID. These patients might be more prone to (sub)clinical gastrointestinal blood loss, for example due to gastrointestinal malignancies or angiodysplasia, which might eventually lead to ID due to iron loss. A recent study by Meijers et al.26 revealed that HF patients may be at risk for incident cancer, including colorectal cancer, possibly due to circulating cardiac and inflammatory markers.
We did not find differences in vitamin K antagonist use in patients with and without ID. Due to the very low use of direct-acting oral anticoagulants (DOACs) in the present cohort (n = 17, 0.7%), it was not possible to study this drug group in relation to ID. Given the conflicting gastrointestinal bleeding risk of DOACs compared to vitamin K antagonists, the prevalence of anticoagulant-related bleeding as a cause of ID might change in the future as DOAC prescriptions become more common in general practice.
Chronic inflammation
Iron-deficient patients in the present study had higher levels of inflammatory markers compared to patients without ID, whereas hepcidin levels were lowest in iron-deficient patients. This is an interesting finding, as hepcidin levels are expected to be elevated in inflammatory states. Despite the pro-inflammatory state in iron-deficient patients, there must be mechanisms lowering hepcidin levels in these patients. It seems conceivable that these patients have ID due to iron unavailability, malabsorption or loss rather than inflammation, which might explain lower hepcidin levels. In this hypothesis, the influence of chronic inflammation on iron status via the inflammation-hepcidin-iron axis seems limited: systemic iron status itself seems to dictate hepcidin release over inflammatory status. This hypothesis has been postulated by Weber et al.27 in their study comprising 60 stable chronic HF patients with anaemia. They showed that iron-deficient patients have lower hepcidin levels despite increased levels of the pro-inflammatory cytokine tumour necrosis factor-α (TNF-α). Our study yields comparable results using CRP and pro-inflammatory biomarkers from the Olink Cardiovascular III panel. Furthermore, some pro-inflammatory cytokines directly influence iron status independently of hepcidin, for example TNF-α.28
Biomarker profile of iron deficiency
As shown in Figure 2, many biomarkers from the Olink Cardiovascular III panel were significantly up- or down-regulated in ID. After correcting for HF severity, renal function and predictors for ID, several biomarkers remained independently associated with iron status. Of these, paraoxonase 3 (PON3) and TR-AP are both down-regulated in ID. PON3 is a liver-derived, HDL-bound protein, which has several antiatherogenic and antioxidative properties. In animal studies, overexpression of PON3 seemed protective against atherosclerosis and cardiac hypertrophy, while PON3-deficient mice show mitochondrial and fatty acid oxidation dysfunction.29–32 Second, TR-AP is predominantly an osteoclast-derived, iron-containing protein reflecting bone turnover rate and is expressed by activated macrophages. In vitro studies show that the expression of TR-AP is regulated by iron status.33,34 TR-AP knockout mice display altered osteoclastic function leading to mild osteopetrosis and an increased proinflammatory response.35,36 Unfortunately, both PON3 and TR-AP are poorly studied in human (patho)physiology. Although our study shows an independent link between PON3, TR-AP, and iron status, the clinical significance of the biomarker expression pattern in ID need to be elucidated.
Clinical implications
Our data confirm the adverse prognostic consequences of ID, which are independent of established predictors of outcome. While ID is frequently observed in HF patients, the aetiology is often unknown. However, it is essential to explore the underlying cause(s), since some of them are treatable and reversible.37 For example, if ID is caused by gastrointestinal blood loss, the underlying cause of this blood loss (e.g. malignancy, angiodysplasia or antiplatelet use) needs to be detected and treated. When ID is caused by the use of antiplatelets or anticoagulants, their use should be reconsidered, especially in patients without a direct treatment indication. Finally, when poor nutritional status is causing ID, this should be treated as well.
Strengths and limitations
To our knowledge, this is the largest cohort with clinical and biochemical parameters available, providing more knowledge on the drivers of ID in HF patients. However, an important limitation of this study is its observational character, making it challenging to directly study aetiological and pathophysiological mechanisms. Nevertheless, we hope our data encourage more studies on the determinants identified in our study. As iron indices were only measured at a single time point, differences in iron status over time could not be studied. Second, there were no data available on the presence or absence of recent blood loss (e.g. blood donations, surgery), which could possibly affect iron status. Third, protein intake was estimated using a formula based on urinary urea and body mass index, and not directly measured. Finally, the majority of patients in BIOSTAT-CHF is male. Therefore, we cannot rule out that the prediction models for ID in males had more statistical power to identify independent determinants of ID compared to the models in female patients.
Funding
The BIOSTAT-CHF study was supported by the European Commission (FP7-242209-BIOSTAT-CHF).
Conflict of interest: The University Medical Center Groningen, which employs several authors, has received research grants and/or fees from AstraZeneca, Abbott, Bristol-Myers Squibb, Novartis, Roche Diagnostics, Trevena, and Thermofisher GmbH. N.G.B. received personal fees from Vifor Pharma. K.D. received honoraria and/or research support from device companies Biotronik and Sorin, Boston Scientific St Jude, and Medtronic, and pharmaceutical companies Abbott, Amgen, Astra Zeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, GSK, Leo, Merck, Novartis, Otsuka, Pfizer, Roche, Sanofi, and Servier. S.D.A. received grants from Abbott Vascular and Vifor Pharma, and consultancy fees from Bayer, Boehringer Ingelheim, Brahms, Cardiorentis, Janssen, Novartis, Relypsa, Servier, Stealth Peptides, Vifor Pharma, and ZS Pharma. C.C.L. received consultancy fees and/or research grants from Amgen, Astra Zeneca, MSD, Novartis, and Servier. D.J.v.V. received board membership fees or travel expenses from BioControl, Cardiorentis, Novartis, Johnson & Johnson, Vifor Pharma, Zoll Medical, CorviaMedical and Arca. A.A.V. received consultancy fees and/or research grants from Alere, Amgen, Bayer, Boehringer Ingelheim, Cardio3Biosciences, Celladon, GSK, Merck/MSD, Novartis, Roche Diagnostics, Servier, Singulex, Sphingotec, Stealth Peptides, Trevana, Vifor Pharma, and ZS Pharma. P.v.d.M. received consultancy fees and/or grants from Novartis, Servier, Vifor Pharma, Astra Zeneca, Pfizer and Ionis. H.H.v.d.W. and L.L.N. have nothing to disclose.
Supplementary Material
References
- 1. Klip IT, Comin-Colet J, Voors AA, Ponikowski P, Enjuanes C, Banasiak W, Lok DJ, Rosentryt P, Torrens A, Polonski L, van Veldhuisen DJ, van der Meer P, Jankowska EA.. Iron deficiency in chronic heart failure: an international pooled analysis. Am Heart J 2013;165:575–582.e3. [DOI] [PubMed] [Google Scholar]
- 2. van Veldhuisen DJ, Anker SD, Ponikowski P, Macdougall IC.. Anemia and iron deficiency in heart failure: mechanisms and therapeutic approaches. Nat Rev Cardiol 2011;8:485–493. [DOI] [PubMed] [Google Scholar]
- 3. Jankowska EA, Rozentryt P, Witkowska A, Nowak J, Hartmann O, Ponikowska B, Borodulin-Nadzieja L, Banasiak W, Polonski L, Filippatos G, McMurray JJ, Anker SD, Ponikowski P.. Iron deficiency: an ominous sign in patients with systolic chronic heart failure. Eur Heart J 2010;31:1872–1880. [DOI] [PubMed] [Google Scholar]
- 4. Okonko DO, Mandal AK, Missouris CG, Poole-Wilson PA.. Disordered iron homeostasis in chronic heart failure: prevalence, predictors, and relation to anemia, exercise capacity, and survival. J Am Coll Cardiol 2011;58:1241–1251. [DOI] [PubMed] [Google Scholar]
- 5. Jankowska EA, von Haehling S, Anker SD, Macdougall IC, Ponikowski P.. Iron deficiency and heart failure: diagnostic dilemmas and therapeutic perspectives. Eur Heart J 2013;34:816–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Voors AA, Anker SD, Cleland JG, Dickstein K, Filippatos G, van der Harst P, Hillege HL, Lang CC, Ter Maaten JM, Ng L, Ponikowski P, Samani NJ, van Veldhuisen DJ, Zannad F, Zwinderman AH, Metra M.. A systems BIOlogy Study to TAilored Treatment in Chronic Heart Failure: rationale, design, and baseline characteristics of BIOSTAT-CHF. Eur J Heart Fail 2016;18:716–726. [DOI] [PubMed] [Google Scholar]
- 7. Ouwerkerk W, Voors AA, Anker SD, Cleland JG, Dickstein K, Filippatos G, van der Harst P, Hillege HL, Lang CC, Ter Maaten JM, Ng LL, Ponikowski P, Samani NJ, van Veldhuisen DJ, Zannad F, Metra M, Zwinderman AH.. Determinants and clinical outcome of uptitration of ACE-inhibitors and beta-blockers in patients with heart failure: a prospective European study. Eur Heart J 2017;38:1883–1890. [DOI] [PubMed] [Google Scholar]
- 8. Voors AA, Ouwerkerk W, Zannad F, van Veldhuisen DJ, Samani NJ, Ponikowski P, Ng LL, Metra M, Ter Maaten JM, Lang CC, Hillege HL, van der Harst P, Filippatos G, Dickstein K, Cleland JG, Anker SD, Zwinderman AH.. Development and validation of multivariable models to predict mortality and hospitalization in patients with heart failure. Eur J Heart Fail 2017;19:627–634. [DOI] [PubMed] [Google Scholar]
- 9. Beilby J, Olynyk J, Ching S, Prins A, Swanson N, Reed W, Harley H, Garcia-Webb P.. Transferrin index: an alternative method for calculating the iron saturation of transferrin. Clin Chem 1992;38:2078–2081. [PubMed] [Google Scholar]
- 10. Kroot JJ, Laarakkers CM, Geurts-Moespot AJ, Grebenchtchikov N, Pickkers P, van Ede AE, Peters HP, van Dongen-Lases E, Wetzels JF, Sweep FC, Tjalsma H, Swinkels DW.. Immunochemical and mass-spectrometry-based serum hepcidin assays for iron metabolism disorders. Clin Chem 2010;56:1570–1579. [DOI] [PubMed] [Google Scholar]
- 11. Assarsson E, Lundberg M, Holmquist G, Bjorkesten J, Thorsen SB, Ekman D, Eriksson A, Rennel Dickens E, Ohlsson S, Edfeldt G, Andersson AC, Lindstedt P, Stenvang J, Gullberg M, Fredriksson S.. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One 2014;9:e95192.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nutritional anaemias. Report of a WHO scientific group. World Health Organ Tech Rep Ser 1968;405:5–37. [PubMed] [Google Scholar]
- 13. Grote Beverborg N, Klip IT, Meijers WC, Voors AA, Vegter EL, van der Wal HH, Swinkels DW, van Pelt J, Mulder AB, Bulstra SK, Vellenga E, Mariani MA, de Boer RA, van Veldhuisen DJ, van der Meer P.. Definition of iron deficiency based on the gold standard of bone marrow iron staining in heart failure patients. Circ Heart Fail 2018;11:e004519.. [DOI] [PubMed] [Google Scholar]
- 14. Kanno H, Kanda E, Sato A, Sakamoto K, Kanno Y.. Estimation of daily protein intake based on spot urine urea nitrogen concentration in chronic kidney disease patients. Clin Exp Nephrol 2016;20:258–264. [DOI] [PubMed] [Google Scholar]
- 15. Yeo TJ, Yeo PS, Ching-Chiew Wong R, Ong HY, Leong KT, Jaufeerally F, Sim D, Santhanakrishnan R, Lim SL, Chan MY, Chai P, Low AF, Ling LH, Ng TP, Richards AM, Lam CS.. Iron deficiency in a multi-ethnic Asian population with and without heart failure: prevalence, clinical correlates, functional significance and prognosis. Eur J Heart Fail 2014;16:1125–1132. [DOI] [PubMed] [Google Scholar]
- 16. Martens P, Nijst P, Verbrugge FH, Smeets K, Dupont M, Mullens W.. Impact of iron deficiency on exercise capacity and outcome in heart failure with reduced, mid-range and preserved ejection fraction. Acta Cardiol 2018;73:115–123. [DOI] [PubMed] [Google Scholar]
- 17. Shirakabe A, Hata N, Kobayashi N, Okazaki H, Matsushita M, Shibata Y, Nishigoori S, Uchiyama S, Asai K, Shimizu W.. The prognostic impact of malnutrition in patients with severely decompensated acute heart failure, as assessed using the Prognostic Nutritional Index (PNI) and Controlling Nutritional Status (CONUT) score. Heart Vessels 2018;33:134–144. [DOI] [PubMed] [Google Scholar]
- 18. Sze S, Zhang J, Pellicori P, Morgan D, Hoye A, Clark AL.. Prognostic value of simple frailty and malnutrition screening tools in patients with acute heart failure due to left ventricular systolic dysfunction. Clin Res Cardiol 2017;106:533–541. [DOI] [PubMed] [Google Scholar]
- 19. Honda Y, Nagai T, Iwakami N, Sugano Y, Honda S, Okada A, Asaumi Y, Aiba T, Noguchi T, Kusano K, Ogawa H, Yasuda S, Anzai T, Investigators N.. Usefulness of geriatric nutritional risk index for assessing nutritional status and its prognostic impact in patients aged ≥65 years with acute heart failure. Am J Cardiol 2016;118:550–555. [DOI] [PubMed] [Google Scholar]
- 20. Iwakami N, Nagai T, Furukawa TA, Sugano Y, Honda S, Okada A, Asaumi Y, Aiba T, Noguchi T, Kusano K, Ogawa H, Yasuda S, Anzai T, Investigators N.. Prognostic value of malnutrition assessed by Controlling Nutritional Status score for long-term mortality in patients with acute heart failure. Int J Cardiol 2017;230:529–536. [DOI] [PubMed] [Google Scholar]
- 21.Institute of Medicine (US) Panel on Micronutrients. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington, DC: The National Academies Press; 2001. [PubMed] [Google Scholar]
- 22. Sandek A, Bauditz J, Swidsinski A, Buhner S, Weber-Eibel J, von Haehling S, Schroedl W, Karhausen T, Doehner W, Rauchhaus M, Poole-Wilson P, Volk HD, Lochs H, Anker SD.. Altered intestinal function in patients with chronic heart failure. J Am Coll Cardiol 2007;50:1561–1569. [DOI] [PubMed] [Google Scholar]
- 23. Sandek A, Bjarnason I, Volk HD, Crane R, Meddings JB, Niebauer J, Kalra PR, Buhner S, Herrmann R, Springer J, Doehner W, von Haehling S, Anker SD, Rauchhaus M.. Studies on bacterial endotoxin and intestinal absorption function in patients with chronic heart failure. Int J Cardiol 2012;157:80–85. [DOI] [PubMed] [Google Scholar]
- 24. Valentova M, von Haehling S, Bauditz J, Doehner W, Ebner N, Bekfani T, Elsner S, Sliziuk V, Scherbakov N, Murin J, Anker SD, Sandek A.. Intestinal congestion and right ventricular dysfunction: a link with appetite loss, inflammation, and cachexia in chronic heart failure. Eur Heart J 2016;37:1684–1691. [DOI] [PubMed] [Google Scholar]
- 25. Valentova M, von Haehling S, Krause C, Ebner N, Steinbeck L, Cramer L, Doehner W, Murin J, Anker SD, Sandek A.. Cardiac cachexia is associated with right ventricular failure and liver dysfunction. Int J Cardiol 2013;169:219–224. [DOI] [PubMed] [Google Scholar]
- 26. Meijers WC, Maglione M, Bakker SJL, Oberhuber R, Kieneker LM, de Jong S, Haubner BJ, Nagengast WB, Lyon AR, van der Vegt B, van Veldhuisen DJ, Westenbrink BD, van der Meer P, Silljé HHW, de Boer RA.. The failing heart stimulates tumor growth by circulating factors. Circulation 2018;138:678–691. [DOI] [PubMed] [Google Scholar]
- 27. Weber CS, Beck-da-Silva L, Goldraich LA, Biolo A, Clausell N.. Anemia in heart failure: association of hepcidin levels to iron deficiency in stable outpatients. Acta Haematol 2013;129:55–61. [DOI] [PubMed] [Google Scholar]
- 28. Sharma N, Laftah AH, Brookes MJ, Cooper B, Iqbal T, Tselepis C.. A role for tumour necrosis factor alpha in human small bowel iron transport. Biochem J 2005;390:437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chistiakov DA, Melnichenko AA, Orekhov AN, Bobryshev YV.. Paraoxonase and atherosclerosis-related cardiovascular diseases. Biochimie 2017;132:19–27. [DOI] [PubMed] [Google Scholar]
- 30. Shih DM, Yu JM, Vergnes L, Dali-Youcef N, Champion MD, Devarajan A, Zhang P, Castellani LW, Brindley DN, Jamey C, Auwerx J, Reddy ST, Ford DA, Reue K, Lusis AJ.. PON3 knockout mice are susceptible to obesity, gallstone formation, and atherosclerosis. FASEB J 2015;29:1185–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Furlong CE, Marsillach J, Jarvik GP, Costa LG.. Paraoxonases-1, -2 and -3: what are their functions? Chem Biol Interact 2016;259:51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pei JF, Yan YF, Tang X, Zhang Y, Cui SS, Zhang ZQ, Chen HZ, Liu DP.. Human paraoxonase gene cluster overexpression alleviates angiotensin II-induced cardiac hypertrophy in mice. Sci China Life Sci 2016;59:1115–1122. [DOI] [PubMed] [Google Scholar]
- 33. Alcantara O, Reddy SV, Roodman GD, Boldt DH.. Transcriptional regulation of the tartrate-resistant acid phosphatase (TRAP) gene by iron. Biochem J 1994;298: 421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fleckenstein E, Dirks W, Dehmel U, Drexler HG.. Cloning and characterization of the human tartrate-resistant acid phosphatase (TRAP) gene. Leukemia 1996;10:637–643. [PubMed] [Google Scholar]
- 35. Hayman AR, Jones SJ, Boyde A, Foster D, Colledge WH, Carlton MB, Evans MJ, Cox TM.. Mice lacking tartrate-resistant acid phosphatase (Acp 5) have disrupted endochondral ossification and mild osteopetrosis. Development 1996;122:3151–3162. [DOI] [PubMed] [Google Scholar]
- 36. Bune AJ, Hayman AR, Evans MJ, Cox TM.. Mice lacking tartrate-resistant acid phosphatase (Acp 5) have disordered macrophage inflammatory responses and reduced clearance of the pathogen, Staphylococcus aureus. Immunology 2001;102:103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, Gonzalez-Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P; Authors/Task Force Members, Document Reviewers . 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016;37:2129–2200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.