


Abstract
Staphylococcus aureus ArlRS is a key two-component regulatory system necessary for adhesion, biofilm formation, and virulence. The response regulator ArlR consists of a C-terminal DNA-binding effector domain and an N-terminal receiver domain that is phosphorylated by ArlS, the cognate transmembrane sensor histidine kinase. We demonstrate that the receiver domain of ArlR adopts the canonical α5β5 response regulator assembly, which dimerizes upon activation, using beryllium trifluoride as an aspartate phosphorylation mimic. Activated ArlR recognizes a 20-bp imperfect inverted repeat sequence in the ica operon, which is involved in intercellular adhesion polysaccharide production. Crystal structures of the inactive and activated forms reveal that activation induces a significant conformational change in the β4-α4 and β5-α5-connecting loops, in which the α4 and α5 helices constitute the homodimerization interface. Crystal structures of the DNA-binding ArlR effector domain indicate that it is able to dimerize via a non-canonical β1–β2 hairpin domain swapping, raising the possibility of a new mechanism for signal transduction from the receiver domain to effector domain. Taken together, the current study provides structural insights into the activation of ArlR and its recognition, adding to the diversity of response regulation mechanisms that may inspire novel antimicrobial strategies specifically targeting Staphylococcus.
INTRODUCTION
Staphylococcus aureus has been recognized as a major human pathogen, the leading cause of skin and soft tissue infections. It can also cause endocarditis and disseminated abscesses anywhere, as well as life-threatening systemic infections including septic shock, and toxic shock syndrome (1). S. aureus is categorized as an ESKAPE pathogen, with its incredible ability to acquire antibiotic resistance by uptake of mobile genetic elements (2,3), and methicillin-resistant S. aureus (MRSA) has emerged as a particularly resistant and widespread healthcare threat. Besides its many antibiotic resistance mechanisms, the success of Staphylococcus as a human pathogen is highly dependent on the production of multiple virulence factors that contribute to evasion of the host immune system. Despite its pathogenic potential, most S. aureus colonizes the skin and mucosal surfaces of approximately 30% of the healthy human population (4). This widespread prevalence and pathogenicity of S. aureus derives from its ability to survive in a variety of conditions and to adapt to many different environments, including almost every organ of the human body (5).
Bacterial two-component regulatory systems constitute the dominant signal transduction mechanism for bacterial adaptation to a variety of environment stimuli using a feedback mechanism (6,7). Two-component regulatory systems typically consist of a membrane sensor histidine kinase, which receives a specific environment stimuli, and its cognate response regulator. After receiving a phosphorylation signal from the histidine kinase, the response regulator modulates target gene expression, hence providing a feedback response to environmental stimuli (7,8).
In Staphylococcus aureus, there are more than 16 two-component regulatory systems that allow adaptation to diverse microenvironments (9). S. aureus two-component regulatory systems have been implicated in virulence factor production (10), resistance to antibacterial agents and nitrosative stress (11,12), cell wall stability (13), biofilm formation, and survival in blood (14). Two-component regulatory systems are thus essential for pathogenesis in Staphylococcus aureus and potential targets for antimicrobial therapy (15,16).
Recently, the Staphylococcus aureus two-component regulatory system ArlRS has been implicated in pathogenesis mechanisms including adhesion, autolysis, biofilm formation, virulence factor regulation, agglutination and capsule regulation (17–20). Gene expression modulation by bacterial response regulators and their activation by phosphorylation have been extensively characterized, with a large number of response regulator structures solved (6,21,22). Despite the accumulated knowledge on two-component regulatory systems (22,23), the mechanism and specificity of the two-component regulatory system family in response to different environmental stimuli is variable, and the mechanism of the ArlRS two-component regulatory system has remained elusive.
The response regulator ArlR consists of an N-terminal receiver domain, which receives a phosphorylation signal from its cognate histidine kinase, ArlS, and a C-terminal DNA-binding effector domain that modulates downstream target gene expression. Here we report the X-ray crystal structures and biophysical characterization of the ArlS receiver domain in the presence and absence of phosphorylation mimic beryllium trifluoride, as well as the DNA-binding effector domain. We have further identified a 20-bp inverted repeat target sequence of activated ArlR located within the intracellular adhesion gene cluster (ica operon), and bioinformatics searching for similar sequences has identified additional potential ArlR-targeting DNA sequences within the S. aureus genome.
MATERIALS AND METHODS
Cloning, expression and purification of ArlR
DNA fragments corresponding to full length ArlR (amino acids 1–219), receiver domain (amino acids 1–121), and effector DNA-binding domain (amino acids 122–219) (numbering according to Uniprot Q9KJN4), were amplified from S. aureus and ligated into a pET28a expression vector with an N-terminal 6× His tag or with a thrombin-cleavable His tag, using primers listed in Supplementary Table S1. For protein expression pET28a-ArlR plasmids were transformed into Escherichia coli BL21 (DE3) cells and grown in LB media supplemented with carbenicillin (100 mg/l) at 28°C. Protein expression was induced with 0.5–1 mM isopropyl β-d-1-thiogalactopyranoside when the optical density at 600 nm reached 0.7–0.8, followed by 6 h post-induction growth. The cells were harvested and centrifuged at 7000 g. The cell pellet was resuspended in 300 mM NaCl, 20 mM Tris, pH 7.5 in the presence of protease inhibitors and further lysed by sonication. The cell debris was centrifuged at 25 000 g for 30 min and the supernatant was filtered with a 0.22 μm syringe filter. The clarified lysate was loaded onto a Ni-NTA column (Qiagen) and eluted with a 0–250 mM imidazole gradient. The sample was concentrated and subjected to size exclusion chromatography on a Superdex 75 column (GE Healthcare), pre-equilibrated with 150 mM NaCl, 20 mM Tris, pH 7.5, and 5% glycerol. For the activated ArlR purification, 5.3 mM BeSO4, 35 mM NaF and 7 mM MgCl2 were supplemented to the size exclusion chromatography buffer. Fractions containing pure protein were pooled and concentrated to 10–20 mg/ml, as determined by absorbance at 280 nm, for further crystallization screening experiments.
DNase I footprinting assay
For preparation of fluorescent FAM-labeled probes, the promoter region of the ica operon was PCR amplified with 2× TOLO HIFI DNA polymerase premix (TOLO Biotech, Shanghai) from the plasmid pclone007 using primers M13F-47 (FAM) and M13R-48. The FAM-labeled probes were purified by the Wizard® SV Gel and PCR Clean-Up System (Promega, USA) and were quantified with a NanoDrop 2000C (Thermo, USA).
DNase I footprinting assays were performed similar to Wang et al. (24). For each assay, 500 ng probes were incubated with different amounts of protein in a total volume of 40 μl. After incubation for 20 min at 30°C, 10 μl solution containing about 0.015 units DNase I (Promega) and 100 nmol freshly prepared CaCl2 was added, and further incubation was performed at 37°C for 1 min. The reaction was quenched by adding 140 μl DNase I stop solution (200 mM sodium acetate, 30mM EDTA and 0.15% SDS). Samples were first extracted with phenol/chloroform, and then precipitated with ethanol. Pellets were dissolved in 30 μl mili-Q water. The preparation of the DNA ladder, electrophoresis and data analysis were the same as described previously (24), except that the GeneScan-LIZ600 size standard (Applied Biosystems) was used.
Crystallization and data collection
Initial crystallization screening experiments were carried out with 384-well plates via sitting drop vapor diffusion method against commercially available sparse matrix screen kits (Molecular Dimensions and Hampton Research) at 20°C. For the crystallization of ArlR receiver domain in complex with BeF3− and magnesium, 5.3 mM BeSO4, 35 mM NaF and 7 mM MgCl2 were supplemented during the purification, and the activated dimer fractions were further used for crystallization (25,26). ArlR receiver domain in complex with BeF3− and magnesium were crystalized in 0.2 M sodium formate, 20% (w/v) polyethylene glycol 3350; crystals of ArlR receiver domain in complex with magnesium were grown with 0.2 M magnesium chloride hexahydrate, 0.1 M HEPES pH 7.5 and 25% (w/v) polyethylene glycol 3350 in the reservoir; the free ArlR receiver domain was grown in 0.2 M calcium chloride, 0.1 M MES and 20% (w/v) polyethylene glycol 3350. Crystals were cryoprotected with the reservoir condition in addition to 15–20% glycerol and flash frozen in a 100 K-nitrogen stream. ArlR DNA-binding domain with His tag was crystalized in a reservoir with 0.1 M Tris pH 8.5 and 2.0 M ammonium sulfate. The ArlR DNA-binding domain with His tag cleaved was observed in the condition of 3.2 M sodium chloride, 0.1 M sodium acetate trihydrate pH 4.6.
Datasets were collected at Beamline 18U1 and 19U1 at Shanghai Synchrotron Radiation Facility. All the X-ray diffraction data were processed and scaled with XDS (27). Data collection details and statistics are summarized in Table 1.
Table 1.
X-ray data collection and refinement statistics
| Crystal | ArlR receiver domain | ArlR receiver domain with Mg2+ | ArlR receiver domain with Mg2+& BeF3− | ArlR DNA-binding domain |
|---|---|---|---|---|
| Data collection | ||||
| Spacegroup | P1 | P1 | P1 | P6422 |
| a, b, c (Å) | 38.4, 41.2, 42.9 | 43.6, 45.8, 61.8 | 61.1, 61.7, 66.1 | 66.9, 66.9, 153.1 |
| α, β, γ (°) | 97.5, 118.9, 96.1 | 99.7, 102.3, 86.5 | 90.4, 105.5, 92.0 | 90, 90, 120 |
| Resolution (Å) | 33.0–1.55 (1.60–1.55) | 31.7–1.59 (1.65–1.59) | 31.77–1.59 (1.65–1.59) | 32.67–1.85 (1.92–1.85) |
| Rmerge | 0.030 (0.480) | 0.053 (0.183) | 0.045 (0.341) | 0.111 (1.211) |
| Rmeas | 0.036 (0.567) | 0.062 (0.123) | 0.054 (0.415) | 0.113 (1.227) |
| Multiplicity | 3.6 (3.4) | 3.5 (2.9) | 3.5 (2.9) | 37.7 (38.9) |
| CC(1/2) | 0.999 (0.808) | 0.996 (0.947) | 0.999 (0.848) | 1 (0.956) |
| CC* | 1 (0.945) | 0.999 (0.986) | 1 (0.958) | 1 (0.989) |
| CCwork | 0.965(0.842) | 0.970 (0.876) | 0.975 (0.876) | 0.948 (0.929) |
| CCfree | 0.962(0.703) | 0.964(0.772) | 0.977 (0.846) | 0.919 (0.891) |
| I/σ(I) | 21.36 (2.69) | 15.00 (2.73) | 15.36 (2.75) | 24.06 (2.94) |
| Completeness (%) | 87.26 (93.27) | 94.33 (74.95) | 93.68 (72.14) | 99.75 (99.20) |
| Wilson B-factor (Å2) | 21.65 | 18.83 | 18.53 | 31.44 |
| Refinement | ||||
| Total Reflections | 101 576 (10405) | 204 421 (13652) | 408 610 (26419) | 678 044 (67964) |
| Unique Reflections | 28 417 (3033) | 58 295 (4635) | 117 259 (9053) | 17 990 (1747) |
| R work/Rfree | 0.1736/0.2058 | 0.1553/0.1883 | 0.1587/0.1889 | 0.1930/0.2165 |
| Number of atoms: | ||||
| Macromolecules | 1967 | 3877 | 7855 | 835 |
| Ligand | 15 | 29 | 55 | 2 |
| Water | 222 | 524 | 1159 | 105 |
| Average B-factor (Å2) | 33.50 | 28.04 | 26.67 | 44.52 |
| Protein (Å2) | 32.44 | 26.67 | 25.32 | 44.42 |
| Ligand (Å2) | 52.09 | 58.81 | 27.14 | 31.69 |
| Water (Å2) | 39.66 | 36.47 | 35.76 | 45.54 |
| Ramachandran plot: | ||||
| Favored/allowed (%) | 98/2 | 99/1 | 98.5/1.5 | 99/1 |
| Root-mean-square-deviation: | ||||
| Bond lengths (Å) | 0.009 | 0.009 | 0.010 | 0.009 |
| Bond angle (°) | 1.03 | 1.02 | 1.07 | 0.92 |
| PDB | 6IS3 | 6IS2 | 6IS1 | 6IS4 |
Statistics for the highest resolution shell are shown in parentheses.
Crystal structure determination and refinement
The structure of ArlR receiver domain was solved by molecular replacement, implemented in Phaser program suite program (28), using BeF3−-activated DrrD receiver domain structure (PDB: 3NNN) as the search model (29). Further manual model building and refinement were carried out with COOT and PHENIX suite (30,31). The crystal structure of ArlR receiver domain, bound with magnesium and in complex with magnesium and BeF3−, were all solved in P1 spacegroup but with different cell constants. The ArlR receiver domain structure without magnesium was solved to 1.55 Å with an Rwork of 0.1736 and Rfree factor of 0.2058 in the unit cell of α = 97.5°, β = 118.9°, γ = 96.1° and a = 38.4 Å, b = 41.2 Å, c = 42.9 Å. The model contains two ArlR receiver domain monomers and includes amino acid residues 2–120, 222 water molecules, 3 calcium ions, and 1 MES molecule. One of the ArlR receiver domain monomer active sites is occupied by 1 calcium ion, but the other is not, as we further describe in the results section.
The ArlR receiver domain structure bound to magnesium was solved to 1.59 Å and refined to a final Rwork of 0.1553 and Rfree factor of 0.1893 with cell constants of α = 99.7°, β = 102.3°, γ = 86.5° and a = 43.6 Å, b = 45.8 Å, c = 61.8 Å. The final structure contains four ArlR receiver domain monomers (amino acid residues 2–120), 524 water molecules, 4 magnesium ions, 1 SO4 ion and 1 HEPES molecule.
The ArlR receiver domain in complex with magnesium and BeF3− was determined to 1.59 Å with a final Rwork of 0.1587 and Rfree factor of 0.1889 with cell parameter of α = 90.4°, β = 105.5°, γ = 92° and a = 61.1 Å, b = 61.7 Å, c = 66.1 Å. The final model contains 8 ArlR receiver domain monomers (amino acid residues 2–120), 1159 water molecules, 8 magnesium ions and 8 BeF3− ions.
The ArlR DNA-binding domain with His tag was solved to 1.85 Å with a final Rwork of 0.1960 and Rfree of 0.2250. The crystal pack in a cell parameter of α = β = 90°, γ = 120° and a = b = 66.9 Å, c = 153.1 Å in P6422 space group. The final model contains a single ArlR DNA-binding domain (amino acid residue 123–219) and 105 water molecules. The ArlR DNA-binding domain with His tag cleaved was solved to 2 Å with Rwork of 0.1995 and Rfree of 0.2425 in the same space group and cell parameter as the ArlR DNA-binding domain with 6 His tag (Supplementary Table S5), the structure factor data used for the final refinement was truncated because of anisotropic diffraction. The assembly between the ArlR DNA-binding domain with and without His tag were the same, which indicated that the His tag has no influence of the ArlR DNA-binding domain dimerization interface.
All structures showed no residues in the Ramachandran plot disallowed regions, and all exhibited favorable stereochemistry. Detailed refinement statistics are summarized in Table 1. All the structure figures were generated using the molecular visualization program PyMOL (32).
Electrophoretic mobility shift assay
The LightShift Chemiluminescent EMSA Kit (Thermo Scientific) was used for EMSA experiments, with DNA labeled with biotin, for the detection of protein-DNA interactions. The sequence used for the EMSA was the ica operon between icaADBC and icaR, the 5′-end biotin-labeled. The EMSA reaction protocol was the same as described previously (33). Briefly, binding reactions were incubated in 10 μl volumes containing 40 fmol DNA and protein in the binding buffer (10 mM Tris, 50 mM KCl, 1 mM DTT, 5% glycerol, pH 7.5) for 30 min. The reaction mixtures were loaded onto a 5% native polyacrylamide gel and separated in 0.5× TBE at 100 V on ice, followed by western blotting for biotin.
Quantitative PCR
The ArlR knockout and site-directed mutagenesis strains were generated using the protocol described previously (34). For the quantitative PCR experiments, 500 ul of ArlR knockout and site-directed mutant strains from overnight pre-culture were grown in 50 ml of TSB (17 g/l tryptone, 3 g/l soya peptone, 2.5 g/l d-glucose, 5 g/l NaCl, 2.5 g/l K2HPO4, pH 7.3) at 37°C for 12 h. Total RNA was isolated from planktonic cells using a Qiagen RNeasy Mini kit, RNA purity was checked by the Abs260/Abs280 ratio of 2.0 to 2.1. First-strand cDNA synthesis from total RNA was performed with the High-Capacity cDNA Reverse Transcription Kit (Bio-Rad) using 300 ng of total RNA as the template following the manufacturer's instructions. Expression of icaA was determined by two-step real-time PCR using the Power SYBR Green PCR Master Mix (Bio-Rad) and Real-Time PCR System (Bio-Rad). Primers were annealed at 60°C, and the 16S-RNA was used as reference to normalize all data. The specificity of all qRT-PCR primers (Supplementary Table S1) was verified using normal PCR. Fold changes in various icaA transcripts in ArlR knock out and mutant strains in relative to pLi50-ArlR were calculated using the 2−ΔΔCt formula (35).
Small angle X-ray scattering
SAXS data collection was performed at the Shanghai Synchrotron Radiation Facility BL19U2 Beamline, carried out within a moment transfer range from 0.04 to 0.45 Å−1. ArlR DNA-binding domain, receiver domain, and full length with or without BeF3− and magnesium freshly purified from size exclusion chromatography were measured under batch mode at 293 K. For each sample, twenty frames were collected without measurable radiation damage detected. The molecular weight determination was carried out with the MoW2 server and Porod volume calculation (36). The ATSAS package was used for data interpretation, PRIMUS was used for the buffer subtraction and extrapolation, Guinier region was determined by Guinier approximation, the program GNOM was used to evaluate the radius of gyration Rg, forward scattering I0, maximum particle dimension Dmax, and the distance distribution function P(r) (37). The overall comparison of the crystal structure to SAXS experimental data was generated by CRYSOL (37,38). The small angle X-ray scattering data collection, analysis, and statistics information are summarized in Supplementary Table S2.
RESULTS
ArlR dimerizes in the presence of aspartate phosphorylation analogue BeF3− and Mg2+
To gain insight into the regulatory mechanism of the S. aureus response regulator ArlR, we successfully expressed and purified both full-length recombinant ArlR and its N-terminal receiver domain. Both constructs behaved as monomers in solution, as judged from their size exclusion chromatography profiles (Figure 1A). Within the response regulators, phosphorylation and dimerization are required for recognition of target DNA and downstream gene activation (22,23). Since BeF3− has been extensively used to mimic the phosphorylated aspartate group of the active state (25), full-length ArlR and its receiver domain were incubated with BeF3− and Mg2+. The addition of BeF3− and Mg2+ induced dimerization in both constructs, as indicated by size exclusion chromatography (Figure 1A, see Supplementary Table S2 for molecular weight estimation based on calibration curve).
Figure 1.

ArlR dimerizes in the presence of aspartate phosphorylation analogue BeF3−. (A) Size exclusion chromatography profile indicates that ArlR existed as a monomer, but elutes as a dimer in the presence of BeF3− and Mg2+. (B) Gunier region analysis and (C) Distance distribution function analysis of SAXS of ArlR full-length with and without BeF3− and Mg2+ (solid line and dotted line, respectively), indicating that full-length ArlR also dimerizes in the presence of BeF3− and Mg2+ as judged from the molecular weight calculation, the Rg of ArlR full length in the absence and presence of BeF3− are 30.76 and 48.35 Å with the Dmax of 88 and 155 Å respectively. (Supplementary Table S1).
We carried out small angle X-ray scattering (SAXS) experiments to further confirm ArlR dimerization induced by BeF3− (Supplementary Table S2). The Guinier region analysis and distance distribution function demonstrate that full-length BeF3−-ArlR exists in a larger assembly, as indicated by Dmax (Figure 1B and C). Further molecular weight calculation, generated from Porod volume using MoW2 software, is consistent with ArlR dimer formation in the presence of BeF3−. The SAXS data are consistent with the size exclusion chromatography results, strongly supporting that the aspartate phosphorylation analogue BeF3− induces dimerization of ArlR.
BeF3−-activated ArlR recognizes an inverted repeat in the ica operon
Staphylococcal biofilm formation depends on the production of sugar polymers by enzymes encoded by the icaADBC operon. ArlR was previously suggested to regulate biofilm formation in an ica-dependent manner, activating icaADBC operon expression in S. epidermidis (39). Recently, it has also been reported that ArlRS can modulate icaADBC expression at the transcriptional level using northern blotting in S. aureus (5). However, the recognition sequence of ArlR was not identified, possibly due to the low binding affinity of unphosphorylated ArlR. We have successfully used BeF3− to mimic phosphorylation of ArlR and generate stable activated dimeric ArlR. To further validate the physiologic relevance of the phosphomimetic BeF3−-induced dimer, we studied its binding to the ica operon using electrophoretic mobility shift assay (EMSA). The ArlR wildtype monomeric protein binds the full promoter region between icaR and icaADBC with low affinity, as described previously (39). However, dimeric ArlR in complex with BeF3− and Mg2+ displayed significantly increased affinity, as indicated by EMSA (Figure 2A). We further applied DNase I footprinting assay to identify the DNA-binding motif of ArlR, using full-length ArlR in complex with BeF3− and magnesium. The binding of activated ArlR protects a 20-bp AT-rich inverted repeat (Figure 2B): ATNGYANNTTAANNTRCNAT, where N is any nucleotide and Y/R is a pyrimidine/purine, respectively (Figure 2B). The ArlR binding motif was located in the ica operon between the icaR and icaADBC loci, at around 50 bp upstream of the icaADBC start codon and 90 bp upstream from the icaR start codon on the other side (Supplementary Figure S1).
Figure 2.
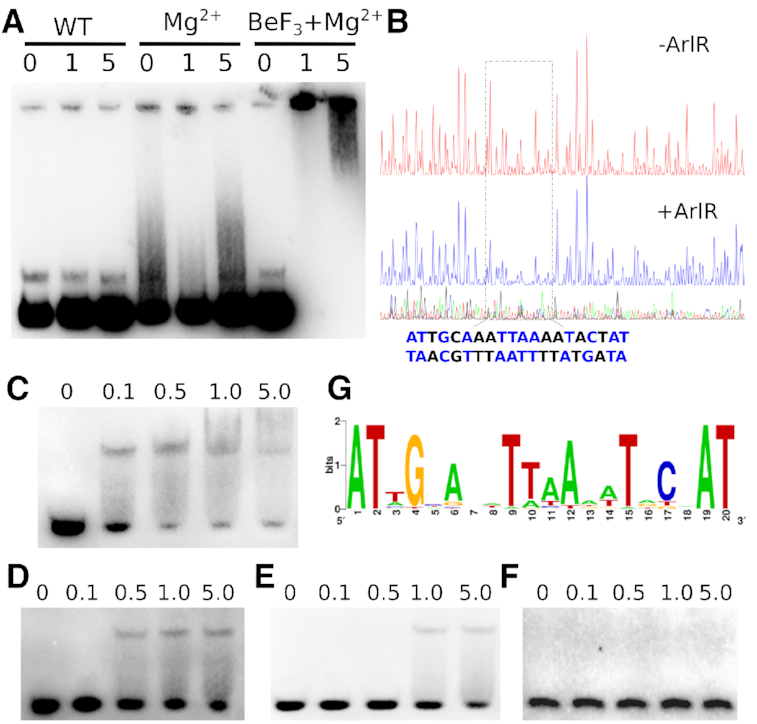
BeF3−-activated ArlR recognizes an imperfect inverted repeat in the ica operon. (A) Purified ArlR in complex with BeF3− and Mg2+ is activated and binds the icaADBC operon, as indicated by EMSA. 0 μM, 1 μM and 5 μM ArlR with or without added BeF3− and Mg2+ is indicated. (B) The activated ArlR recognizes the AT-rich imperfected inverted repeat binding box (ATNGYANNTTAANNTRCNAT) identified through DNase I footprinting. (C) EMSA experiment with the identified wildtype 24-bp ArlR-binding ica operon DNA sequence with concentration of 0 μM, 0.1 μM, 0.5 μM, 1 μM and 5 μM ArlR used in D, E, F panel as well. (D) EMSA experiment of activated ArlR with the identified ica operon DNA sequence with position 4 mutated. (E) EMSA experiment of activated ArlR with the identified recognition ica operon DNA sequence with positions 1 and 6 doubly mutated. (F) The combined mutations of positions 1, 4 and 6 completely abolish the binding of ArlR. (G) Bioinformatics searching of the S. aureus genome for similar DNA sequences suggests a consensus sequence (Supplementary Table S3), shown here as generated by the WebLogo server.
To further verify the identified ArlR-binding sequence within the ica operon, a short 24-bp DNA fragment including the 20-bp imperfect inverted repeat (with a 2-bp extension at both ends) was used for EMSA instead of the whole ica operon region. ArlR bound to the 24-bp fragment and caused a shift at an affinity comparable to that seen with the full ica operon region (Figure 2C). Mutation of position 4 or positions 1 and 6 in the identified sequence attenuated binding of activated ArlR (Figure 2D and E, respectively). Combined mutations of positions 1, 4 and 6 completely abolished the binding of activated ArlR to the DNA fragment (Figure 2F).
ArlR is reported to regulate over 114 genes involved in autolysis, cell division, growth, and pathogenesis (40), and multiple ArlR targeting sites have been speculated because of its broad regulation profile. We have performed bioinformatics searching on the whole S. aureus genome and identified more than 15 potential ArlR binding sites sharing sequence similarity with the identified imperfect inverted repeat ica operon DNA sequence (Supplementary Table S3). The consensus DNA sequence derived from whole genome searching indicate highly conserved A1, T2, G4, A6 and T9 positions, with positions 3 and 10 showing preferences for A or T, as shown using WebLogo (Figure 2G) (41). Many of the potential ArlR targeting sequence are located within or near the regulating regions of previously reported genes regulated by ArlR. Of particular interest, one of the top sequences identified was ATAGCATATTTATATGCAAT, which is within 40 bp of hypothetical protein SA0257 and probable ribokinase rbsK SA0258, genes shown to be downregulated in biofilm formation and close to other genes shown to be downregulated by ArlR (SA0252 and SA0253) (40,42).
Crystal structures of activated and inactive ArlR receiver domain suggest its activation mechanism
To further explore the activation mechanism of ArlR, we solved the crystal structure of activated ArlR receiver domain in complex with BeF3− and Mg2+ at 1.59 Å (Table 1). The ArlR receiver domain adopts a canonical α5β5 topology arranged as a central parallel β-sheet sandwiched by α1 and α5 on one side and α2, α3, and α4 on the other side (Figure 3A). Similar to other PhoB family members, there is a prominent dimerization interface comprised of α4-β5-α5, involving 21 hydrogen bonds and 22 salt bridges and burying a total surface area of 960 Å2, as determined by PDBPISA (Figure 3A, B, Supplementary Table S4) (43). Positively charged residues R111, R113 and R118 from α5 interact with negatively charged α4 residues D97, D92 and D96 across the dimerization interface. Interestingly, Y98, which is involved in the typical response regulator Y–T coupling activation mechanism, forms a hydrogen bond with R111 across the dimer interface through its backbone. R118 also forms hydrogen bonds across the dimer interface with S72 in the loop between α3 and β4 (Figure 3B). A detailed list of dimer-forming interactions is provided in Supplementary Table S3. ArlR receiver domain has an overall sequence identity of 36% to the well-characterized OmpR/PhoB family response regulator, PhoP (Figure 3C). Most of the residues involved in dimerization, S72, D96, D97, Y98, R111 and R118, are conserved between ArlR and PhoP.
Figure 3.
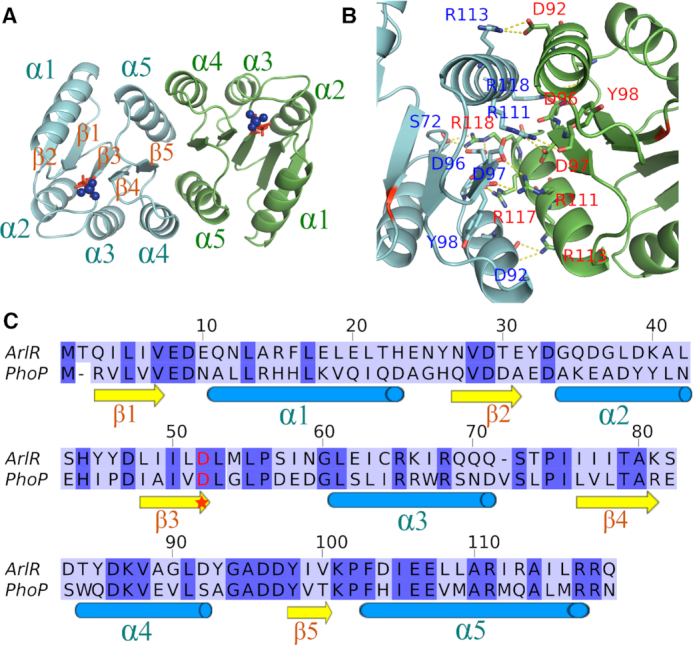
Crystal structure of ArlR receiver domain in complex with BeF3−. (A) Crystal structure of ArlR receiver domain dimer in complex with BeF3−. The ArlR receiver domain adopts a canonical α5β5 topology and exhibits a central parallel β-sheet surrounded by α1 and α5 on one side and α2, α3 and α4 on the other side. The phosphorylation site aspartate and BeF3− are shown in red sticks and blue spheres, respectively. (B) Dimerization interface of ArlR receiver domain involves the α4-β5-α5 motif, with a buried surface area of 960 Å2. A detailed interaction profile is shown in Supplementary Table S2. (C) Sequence alignment of ArlR receiver domain with the well-characterized response regulator PhoP. The conserved aspartate phosphorylation site is highlighted with a star.
The highly conserved aspartate residue D52 at the C-terminal end of β3 is the phosphorylation site (Figure 3C). The aspartate phosphorylation analogue BeF3− was found on the conserved D52 residue as expected. The phosphorylation of the aspartate residue in the response regulator domain regulates the activity of the effector DNA-binding domain. The atomic resolution crystal structure of ArlR receiver domain in complex with BeF3− and Mg2+ demonstrates that there are 7 residues from the β2-β5 strands and 2 water molecules involved in the complexation of BeF3− and Mg2+. BeF3− binds the OD1 atom of D52, and the paired Mg2+ ion coordinates with one fluoride atom from BeF3−, OD2 of D52, side chains of E8 and D9, the main chain carbonyl oxygen of M54 and two water molecules (Figure 4A). The other two fluoride atoms form hydrogen bonds with T79, A80, K101 and M54 (Figure 4A). These residues involved in BeF3− and Mg2+ binding are highly conserved in most response regulator family members (Figure 3C).
Figure 4.
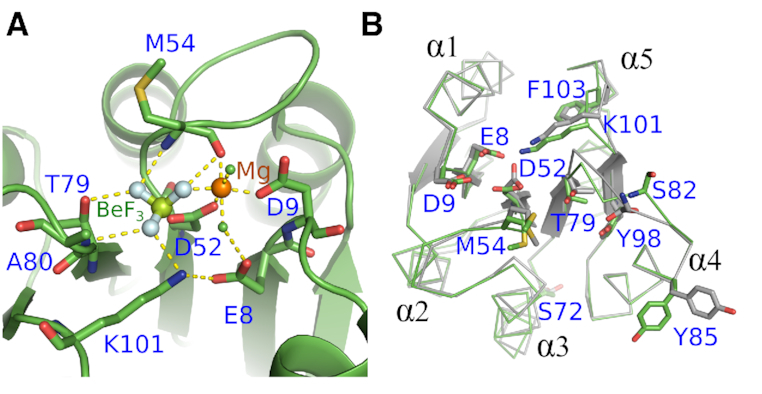
Binding motif of BeF3− and conformational changes upon ArlR activation. (A) Interactions between BeF3 (F: cyan sphere, Be: green sphere), Mg2+ (orange sphere), and ArlR. The conserved residues E8, D9, T79, K101 shown in stick colored with elements and main chain of M54, A80 form a binding pocket for BeF3− and Mg2+. (B) Structural alignment between ArlR in complex with BeF3− and the inactivated ArlR shown in ribbon_side_chain_helper mode. The residues involved in binding BeF3− and Mg2+ are shown in sticks. ArlR activation engages a significant conformational change in the β4–α4 and β5–α5-connecting loops. The canonical Y–T coupling residue Y98 of β5 remains in the same conformation before and after activation.
To assess the conformational changes between the active and inactive state of ArlR, we also solved the crystal structure of inactive ArlR receiver domain in the absence of BeF3− and Mg2+. Structural alignment of ArlR in the presence versus the absence of BeF3− and Mg2+ demonstrated an RMSD of 0.312 Å. Although inactive ArlR exists as monomer in solution, it crystallized as an active state-like dimer, similar to other members of this family (44,45). This occurs because, within the PhoB family, helices α4 and α5 possess a fair degree of flexibility. Phosphorylation shifts the equilibrium towards an active conformation, but an active-like conformation is also present as a minor species in the unphosphorylated form, and this is what tends to crystallize. Thus, the dominant inactive conformation, which is not conducive to dimerization, is not observed in the crystal structures. Nevertheless, it can be observed, by comparing the ArlR receiver domain in the absence and the presence of BeF3−/Mg2+ that BeF3−/Mg2+ binding pulls the loops connecting β4-α4 and connecting β5-α5 toward D52. Specifically, T79, which is located in β4–α4, and K101, located in β5–α5, directly complex BeF3−, causing conformational changes in their respective loops. In solution, it would be expected that these conformational changes be further transmitted to helices α4 and α5 to impact the dimerization surfaces in a phosphorylation-dependent manner. It was originally proposed in PhoP that the highly conserved threonine (T79 in PhoP) side chain on β5 is coupled to a conformational rearrangement of the adjacent aromatic Y98 residue located on β5, giving rise to a ‘Y–T’ coupling activation mechanism (26,45,46). However, this coupling was not observed in our ArlR crystal structures, suggesting that it may not be a feature universal to this family. Instead, what we observe is a conformational change in the β4-α4 and β5-α5-connecting loops, with the largest conformational changes to be found in K81, S82, D83 and Y86 in the β4-α4 connecting loop (Figure 4B).
Divalent cation binding of ArlR
A divalent cation is essential for the addition or removal of the phosphoryl group at D52 in the receiver domain. Most response regulator receiver domains prefer Mg2+, but other divalent ions such as Mn2+ and trivalent ions have been reported to promote reactions with even tighter binding, although without physiological validation (47,48). Crystals of apo ArlR receiver domain were produced in the presence of 0.2 M CaCl2 in the reservoir. Interestingly, the resulting structures show that, within a dimer of ArlR receiver domains, one monomer was found to have a Ca2+ ion bound, while the other monomer was free of cation. The calcium ion occupied the expected cation binding site liganded to four residues: D52, D9, E8 via a single water molecule, and the carbonyl group of M54. The binding of calcium caused a rotational switch of D52 resulting in conformational changes in K101 and M54 (Figure 5A). To investigate the divalent ion binding further, we crystallized the ArlR receiver domain in the presence of 10 mM Mg2+ without BeF3−. Mg2+ bound in a similar manner, but with six-ligand coordination (compared to the Ca2+-bound form, two additional water molecules are seen complexing the Mg2+ ion), in which the coordination with a BeF3− fluoride atom was replaced by a water molecule (Figure 5B). Compared to the metal ion-free state of ArlR receiver domain, the Mg2+-bound and Ca2+-bound ArlR receiver domain were both structurally very similar, having RMSD values of 0.3 Å (Figure 5C). Notably, however, the Mg2+-bound ArlR receiver domain more resembled the activated form even in the absence of BeF3−, whereas the Ca2+-bound more resembled the form with no metal ion bound (Figure 5D).
Figure 5.
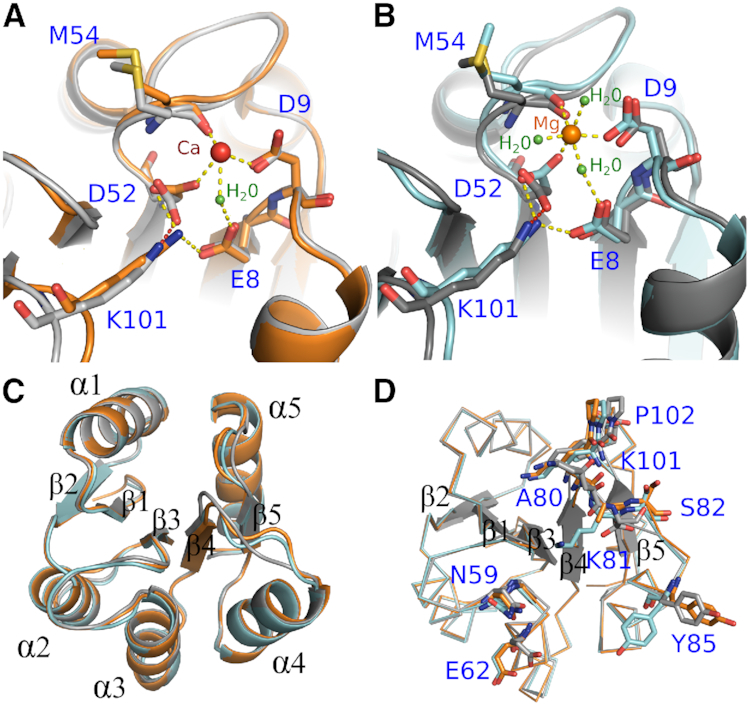
Divalent cation binding of ArlR. (A) Interactions between Ca2+ ion (red sphere) and ArlR. (B) Interactions between Mg2+ (orange sphere) and ArlR. Water molecules were shown as green sphere. (C) Structural alignment between ArlR in complex with Ca2+, Mg2+ and the uncomplexed inactivated form shown in cartoon and colored in orange, cyan and grey respectively. (D) Structural alignment of ArlR in complex with Ca2+, Mg2+ and inactivated form shown in ribbons highlighting interacting residues. Mg2+ but not Ca2+ can induce isomerization of Y85.
Crystal structure of the ArlR DNA-binding domain reveals domain swapping of the β1–β2 hairpin
Based on sequence homology, the ArlR DNA-binding domain was classified as OmpR/PhoB-type, having a winged helix DNA-binding domain. The ArlR DNA-binding domain has an overall sequence identity of around 40% to the well-characterized PhoB, with most of the conserved residues in the DNA-binding helix-turn-helix region (Supplementary Figure S2). Here, we solve the crystal structure of the ArlR DNA-binding effector domain. The ArlR effector domain consists of four N-terminal β strands (β1–β4), followed by a thre-helix-bundle (α1–α3) and a C-terminal β hairpin (β5–β6). However, compared to all other known OmpR/PhoB DNA-binding domain structures, which crystallized as monomers, the ArlR DNA-binding domain crystallized as a domain-swapped dimer (Figure 6A, B). Instead of a single anti-parallel N-terminal β sheet composed of four consecutive β strands, the N-terminal β1–β2 strands are swapped with the dimeric partner, so that the N-terminal β sheet is made up of β1–β2 strands from one monomer and β3–β4 strands from the other monomer (Figure 6A, B).
Figure 6.
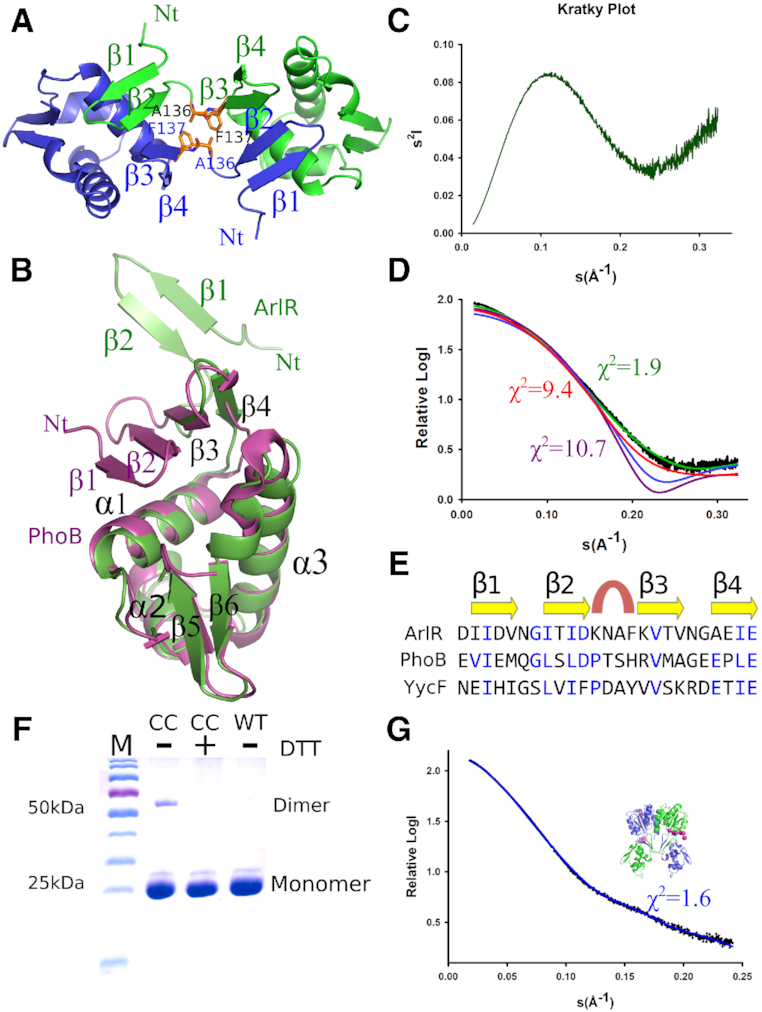
Crystal structure of the ArlR DNA-binding domain reveals domain swapping of the β1–β2 hairpin. (A) ArlR DNA-binding domain dimer generated from the symmetry unit cell. The β1–β2 hairpins of each monomer swap and form a connecting antiparallel β sheet. A136 and F137 residues are shown as sticks. (B) Structural alignment of ArlR DNA-binding domain (green) with the well characterized OmpR/ PhoB family protein PhoB (purple). In ArlR, the β1–β2 hairpin is dissociated from the rest of the domain, as compared with the monomeric β1–β2–β3–β4 antiparallel β sheet observed in PhoB. (C) The Kratky plot of the ArlR DNA-binding domain in solution by small angle X-ray scattering indicates a partly unfolded protein. (D) The ArlR DNA-binding domain small angle X-ray scattering experimental curve (black) is fit to curves generated from the crystal structure of ArlR (green) and PhoB (purple) DNA-binding domains, demonstrating a strong correlation between the crystal and solution structures of ArlR DNA-binding domain. The model of PhoB DNA-binding domain with an additional N-terminal 6-His-tag (red) demonstrates a X2 of 9.4. The β1–β2-truncated ArlR DBD model (blue) presents a smaller apparent size as compared to ArlR DBD, with a X2 value over 100 as generated from FoXS. (E) Structure-based sequence alignment of ArlR, PhoB and YycF generated from PROMALS3D. (F) Double cysteine mutant A136CF137C of ArlR full length recombinant protein presents a small population of dimer in SDS PAGE gel, and addition of DTT eliminates the small dimer population. (G) Small angle X-ray scattering experiment of ArlR in solution agrees with the full length ArlR model generated from the receiver domain and DNA-binding domain structure. The pink spheres represent the missing residues of the His tag in the crystal structure.
To validate the ArlR DNA-binding domain structure, we performed small angle X-ray scattering in solution. The Kratky plot is informative of the globularity and flexibility of a protein in solution. The curvature of the ArlR DNA-binding domain SAXS data was consistent with a globular domain with a partially unfolded region (Figure 6C). The Porod volume and SAXS MoW2 software indicated that the ArlR DNA-binding domain exists as a monomer in solution, consistent with the size exclusion chromatography data (Supplementary Table S2). However, the SAXS data are not consistent with a fully folded single compact domain, with a high X2 value of 10.7 when the experimental data is fit against the curve predicted for the PhoB DNA-binding domain (Figure 6D). Inclusion of an additional N-terminal 6-His-tag on the PhoB model slightly improved the X2 value to 9.4, indicating that the flexible His-tag on our ArlR construct could not account for the SAXS curve. Instead, the structure of a monomeric ArlR DNA-binding domain, with a β1–β2 hairpin extended away from the rest of the domain, demonstrated a compatible fit with the small angle x-ray scattering experiment data, with a X2 value of 1.9. The fit of the experimental ArlR DBD SAXS data to a β1–β2-truncated ArlR DBD model yields a X2 value over 100, as generated by FoXS (49). The hypothetical SAXS curve generated from the β1–β2-truncated ArlR DBD model is suggestive of a compact shape, similar to PhoB DBD, but demonstrated an apparent smaller size with Rg of 12.6 Å as compared to the full ArlR DBD of 18.7 Å, both of which are inconsistent with the experimental SAXS data for ArlR DBD. Taken together, the SAXS experiment supports dissociation of the β1–β2 strands in the ArlR DBD (Figure 6D). It is likely that ArlR DBD exists in an ensemble of states that includes partially folded monomer (unfolding of the N-terminal β1–β2 strands), giving rise to the observed SAXS data.
It is interesting to observe that the N-terminal β sheet of the ArlR DNA-binding domain is unstable with a propensity to domain swap (Figure 6A). A β1–β2–β3–β4 structural sequence alignment of multiple PhoB/OmpR family DNA-binding domain generated from PROMALS3D indicates conserved secondary structure elements with relatively low sequence identity (Figure 6E) (50). In the well-characterized PhoB/OmpR family DNA-binding domain structure, the 4 residues between the β2 and β3 strand comprise a tight β turn. PhoB and YycF have been shown to possess Type I β turns. However, the 4-residue sequence of ArlR at this position, KNAF, has a relatively low propensity for forming such a β turn structure, with lower turn propensities of each amino acid type at every position of the turn compared to PhoB: K (0.53), N (0.74), A (0.63), F (1.03) for ArlR (overall score of 0.25) versus P (1.28), T (0.82), S (1.49), H (1.13) for PhoB (overall score of 1.77) (51). Thus, it is possible that the main factor destabilizing the intramonomeric β-sheet in ArlR versus PhoB is the preference of the sequence between strands β2 and β3 in ArlR to form a structure other than a tight turn.
The program H-predictor calculates theoretical unfolding temperatures (ΔE/ΔL) at every position in a protein by treating each position as a putative hinge region for domain swapping, and then calculating the predicted change in enthalpy (ΔE) divided by the change in average minimal path (ΔL) (52). H-predictor analysis of ArlR unfolding temperatures (ΔE/ΔL) revealed that the 134KNAF137 sequence has a global minimum ΔE/ΔL value, which supports the notion that the KNAF sequence is a potential hinge region for domain swapping (Supplementary Figure S3).
To further support the domain swapping of the β1–β2 hairpin observed in the ArlR DNA-binding domain, we engineered double cysteine point mutations, A136C and F137C with its distance favorable for cysteine disulfide bridge engineering. These residues form intermolecular contacts in the domain-swapped dimer (Figure 6A). It is known that following purification of ArlR directly from an affinity column, there exists a small population of active ArlR able to bind to DNA without addition of BeF3−, which has been explained as being due to the response regulator accepting phosphoryl groups from intermediary metabolites such as acetyl phosphate and carbamoyl phosphate acting as phosphor-donors (39,53). Consistent with this low basal level of ArlR activation and dimer formation, the A136C-F137C double mutant form of ArlR full-length protein is observed to form a small population of disulfide-linked dimer, and the addition of DTT eliminated this dimer population (Figure 6F).
The small angle X-ray scattering data for the full length ArlR in complex with BeF3− and Mg2+ agrees with the rigid model generated from the receiver domain and domain swapped DNA-binding domain, with the missing N-terminal tag residues modeled as spheres with CORAL and SASREF program in the ATSAS package (Figure 6G) (37).
Functional characterization of ArlR mutants involved in its activation and recognition
We applied a series of point mutations to the receiver domain and DNA-binding domain to determine the functional relevance of these residues in vivo by monitoring the impact of mutations on icaA transcriptional level. The key mutations include D96A/D97A, Y98A, R111A, R113A, R117A/R118A, all designed to disrupt ArlR dimerization as confirmed by the size exclusion chromatography profiles of the recombinant mutants ArlR purification (Supplementary Figure S4). All of these mutants also demonstrated a significant decrease in icaA transcription, confirming that our structural models are consistent with those of other response regulators (33). Mutations in conserved residues in the DNA-binding domain, including R196A, Y216A and the minor groove anchoring residue R212A, were designed to disrupt recognition of target DNA, and all of these significantly reduced the transcriptional level of icaA (Figure 7). Interestingly, the deletion of the β1–β2 hairpin linking the receiver domain and the effector domain in ArlR also demonstrates a strong disruption of icaA transcription (Figure 7).
Figure 7.

Functional characterization of ArlR mutants targeting residues involved in its dimerization and DNA recognition. Quantitative PCR of key-residue mutants involved in ArlR dimerization and DNA-binding. Fold changes in various icaA transcripts in ArlR knockout and mutant strains relative to wildtype ArlR were calculated using the 2−ΔΔCt formula. All the data were generated with at least 3 biological replicas.
DISCUSSION
ArlR is classified as a member of the bacterial OmpR/PhoB response regulator family. Although its secondary structure elements and basic two-domain organization resemble that of the other family members, ArlR has a number of features that are unique and have not previously been observed in this family. First of all, the ArlR effector domain binds to target DNA sequences in the form of inverted repeats, whereas PhoB and most other homologs have been shown to bind to direct repeats. Generally speaking, however, winged helix-turn-helix DNA-binding domain response regulators have been reported to recognize either direct repeat or inverted repeat target sequences (54–58). DNA-binding domains that bind to inverted repeats tend to form symmetric dimers, we observe in the X-ray crystal structure of the domain-swapped DBD dimer, which also characterized in Mycobacterium tuberculosis of RegX3 (59).
The ArlR DNA-binding domain dimer is connected by a short KNAF sequence that is key to the domain swapping mechanism. This sequence is not favorable for the tight turn observed in other homologs, and it adopts a somewhat extended conformation that acts as a short connecting linker between monomers of the domain swapped dimer. The DNA-binding α3 helices that suggested to bind to the DNA major groove are perfectly spaced in the domain swapped dimer to bind to adjacent major grooves, corresponding to one helical B-DNA step or 10–11 nucleotides. Thus, dimerization of the receiver domain would potentially drive domain-swapped dimer formation of the DNA-binding domain with the domain orientation needed to bind the inverted repeat target sequence. However, in full-length ArlR, domain orientations would be constrained by the very short 4-residue 120PQKD linker between the receiver domain and DNA-binding domain. The inter-domain linkers from each monomer are brought into close proximity by dimerization of the receiver domain (see Figure 3A), and this would require a 60- to 90-degree twist of both DNA-binding domains along the A136-F137 connector sequence (see Figure 6A), necessitating a rearrangement of the A136-F137 dimeric interface. It is unknown whether this rearrangement would be more favorable in the context of a domain-swapped dimer or a non-swapped dimer that would place fewer constraints on the A136–F137 interface. The twist rotation necessary to bring the N-termini of the DBD domains into close proximity (see Figure 6A) also positions the α3 helices into the right orientation (facing the same way) to bind a DNA inverted repeat, suggesting that this is the actual geometry behind dimerization-driven DNA recognition, further evidence such as solution NMR structure of ArlR full length would valid our hypothesis.
ArlR activation of the receiver domain engages a significant conformational change in the β4–α4 and β5–α5-connecting loops, and possible isomerization of the neighboring aromatic Y85 from α4, which would lead to a re-orientation of helices α4 and α5 to promote dimerization. However, the classical Y–T coupling mechanism is not observed in ArlR, as Y98 remains in the same conformation in all crystal forms. ArlR may be part of a non-Y–T coupling activation mechanism subfamily of response regulators, similar to SdrG, for which it was proposed that a reorganization of several aromatic resides around a conserved motif is implicated in activation rather than Y–T coupling (60).
ArlR is a master regulator reported to globally regulate over hundred genes expression involved in autolysis, cell division, growth, and pathogenesis (40). However the direct targets of ArlR have not been identified, possibly due to the difficulty associated with preparing activated ArlR. The ArlR binding motif we have identified in the ica operon is the only ArlR binding site identified thus far in S. aureus, and it is distinct from the proposed binding motif involved in multidrug resistance transporter NorA modulation (61). This strongly supports the role of ArlR in regulating biofilm formation in an ica-dependent manner (62,63). The successful identification of an ArlR binding site within the ica operon has also suggested a consensus motif for binding that will enable the identification of other ArlR-modulated genes. This will help to characterize the ArlR regulation network and to elucidate its multi-faceted role in biofilm formation and pathogenesis.
In conclusion, we demonstrate that ArlR functions in a manner similar to other two-domain response regulators involving a distinct activation mechanism, with phosphorylation driving dimerization of the effector domain. The ArlRS two-component regulatory system plays a unique role in promoting the pathogenicity of S. aureus, making it an interesting potential target in the development of new antimicrobial strategies.
DATA AVAILABILITY
Structure factors and atomic coordinates have been deposited in the Protein Data Bank under accession codes: 6IS1 (ArlR receiver domain in complex with BeF3− and Mg2+), 6IS2 (ArlR receiver domain in complex with Mg2+), 6IS3 (ArlR receiver domain) and 6IS4 (ArlR DNA-binding domain).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the staff from the BL18U1/BL19U1 beamline and Dr Na Li from BL19U2 beamline for technique support during data collection at the National Center for Protein Sciences Shanghai (NCPSS) at Shanghai Synchrotron Radiation Facility.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31870132, 81741088, 31500051, 81772745 to Y.W.]; NSERC (Natural Sciences and Engineering Research Council of Canada) Discovery [RGPIN-2015-06664 to P.M.H.]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Tong S.Y.C., Davis J.S., Eichenberger E., Holland T.L., Fowler V.G.. Staphylococcus aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin. Microbiol. Rev. 2015; 28:603–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pendleton J.N., Gorman S.P., Gilmore B.F., Pendleton J.N., Gilmore B.F.. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti. Infect. Ther. 2013; 11:297–308. [DOI] [PubMed] [Google Scholar]
- 3. Malachowa N., Deleo F.R.. Mobile genetic elements of Staphylococcus aureus. Cell. Mol. Life Sci. 2010; 67:3057–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wertheim H.F.L., Melles D.C., Vos M.C., Van Leeuwen W., Van Belkum A., Verbrugh H.A., Nouwen J.L.. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 2005; 5:751–762. [DOI] [PubMed] [Google Scholar]
- 5. Burgui S., Gil C., Solano C., Lasa I., Valle J.. A systematic evaluation of the two-component systems network reveals that ArlRS is a key regulator of catheter colonization by Staphylococcus aureus. Front. Microbiol. 2018; 9:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stock A.M., Robinson V.L., Goudreau P.N.. Two-Component signal transduction. Annu. Rev. Biochem. 2000; 69:183–215. [DOI] [PubMed] [Google Scholar]
- 7. Groisman E.A. Feedback control of two-component regulatory systems. Annu. Rev. Microbiol. 2016; 70:103–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Capra E.J., Laub MT. The evolution of two-component signal transduction systems. Annu. Rev. Microbiol. 2012; 66:325–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ortet P., Whitworth D.E., Santaella C., Achouak W., Barakat M.. P2CS: updates of the prokaryotic two-component systems database. Nucleic Acids Res. 2015; 43:D536–D541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu Q., Yeo W.S., Bae T.. The SaeRS two-component system of Staphylococcus aureus. Genes (Basel). 2016; 7:1–20. [Google Scholar]
- 11. Kawada-Matsuo M., Yoshida Y., Nakamura N., Komatsuzawa H.. Role of two-component systems in the resistance of Staphylococcus aureus to antibacterial agents. Virulence. 2011; 2:427–430. [DOI] [PubMed] [Google Scholar]
- 12. Kinkel T.L., Roux C.M., Dunman P.M., Fang F.C.. The Staphylococcus aureus SrrAB two-component system promotes resistance to nitrosative stress and hypoxia. MBio. 2013; 4:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kolar S.L., Nagarajan V., Oszmiana A., Rivera F.E., Miller H.K., Davenport J.E., Riordan J.T., Potempa J., Barber D.S., Koziel J. et al.. NsaRS is a cell-envelope-stress-sensing two-component system of Staphylococcus aureus. Microbiology. 2011; 157:2206–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hall J.W., Yang J., Guo H., Ji Y.. The AirSR two-component system contributes to Staphylococcus aureus survival in human blood and transcriptionally regulates sspABC operon. Front. Microbiol. 2015; 6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Watanabe T., Okada A., Gotoh Y., Utsumi R.. Inhibitors targeting two-component signal transduction. Adv. Exp. Med. Biol. 2008; 631:229–236. [DOI] [PubMed] [Google Scholar]
- 16. Gotoh Y., Eguchi Y., Watanabe T., Okamoto S., Doi A., Utsumi R.. Two-component signal transduction as potential drug targets in pathogenic bacteria. Curr. Opin. Microbiol. 2010; 13:232–239. [DOI] [PubMed] [Google Scholar]
- 17. Memmi G., Nair D.R., Cheung A.. Role of ArlRS in Autolysis in Methicillin-Sensitive and Methicillin- Resistant Staphylococcus aureus Strains. J. Bacteriol. 2012; 12:759–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hooper D.C. A new Two-Component regulatory system involved in adhesion, Autolysis, and extracellular proteolytic activity of staphylococcus aureus. J. Bacteriol. 2000; 182:3955–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walker J.N., Crosby H.A., Spaulding A.R., Salgado-Pabón W., Malone C.L., Rosenthal C.B., Schlievert P.M., Boyd J.M., Horswill A.R.. The Staphylococcus aureus ArlRS Two-Component system is a novel regulator of agglutination and pathogenesis. PLoS Pathog. 2013; 9:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luong T.T., Lee C.Y.. The arl locus positively regulates Staphylococcus aureus type 5 capsule via an mgrA -dependent pathway. Microbiology. 2006; 152:3123–3131. [DOI] [PubMed] [Google Scholar]
- 21. Wen Y., Ouyang Z., Devreese B., He W., Shao Y., Lu W., Zheng F.. Crystal structure of master biofilm regulator CsgD regulatory domain reveals an atypical receiver domain. Protein Sci. 2017; 26:2073–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bourret R.B. Receiver domain structure and function in response regulator proteins. Curr. Opin. Microbiol. 2010; 13:142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao R., Mack T.R., Stock A.M.. Bacterial response regulators: versatile regulatory strategies from common domains. Trends Biochem. Sci. 2007; 32:225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y., Cen X.F., Zhao G.P., Wang J.. Characterization of a New GlnR binding box in the promoter of amtB in Streptomyces coelicolor inferred a PhoP/GlnR competitive binding mechanism for transcriptional regulation of amtB. J. Bacteriol. 2012; 194:5237–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cho H., Wang W., Kim R., Yokota H., Damo S., Kim S.H., Wemmer D., Kustu S., Yan D.. BeF(3)(-) acts as a phosphate analog in proteins phosphorylated on aspartate: structure of a BeF(3)(-) complex with phosphoserine phosphatase. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:8525–8530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li Y.C., Chang C.K., Chang C.F., Cheng Y.H., Fang P.J., Yu T., Chen S.C., Li Y.C., Hsiao C.D., Huang T.H.. Structural dynamics of the two-component response regulator RstA in recognition of promoter DNA element. Nucleic. Acids. Res. 2014; 42:8777–8788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kabsch W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McCoy A.J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 2006; 63:32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barbieri C.M., Mack T.R., Robinson V.L., Miller M.T., Stock A.M.. Regulation of response regulator autophosphorylation through interdomain contacts. J. Biol. Chem. 2010; 285:32325–32335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 31. Adams P.D., Afonine P. V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al.. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schrödinger L. 2015; The PyMOL Molecular Graphics System, Version∼1.8.
- 33. Wen Y., Ouyang Z., Yu Y., Zhou X., Pei Y., Devreese B., Higgins P.G., Zheng F.. Mechanistic insight into how multidrug resistant Acinetobacter baumannii response regulator AdeR recognizes an intercistronic region. Nucleic Acids Res. 2017; 45:9773–9787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bae T., Schneewind O.. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid. 2006; 55:58–63. [DOI] [PubMed] [Google Scholar]
- 35. Livak K.J., Schmittgen T.D.. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 36. Fischer H., De Oliveira Neto M., Napolitano H.B., Polikarpov I., Craievich A.F.. Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J. Appl. Crystallogr. 2010; 43:101–109. [Google Scholar]
- 37. Petoukhov M.V., Franke D., Shkumatov A.V., Tria G., Kikhney A.G., Gajda M., Gorba C., Mertens H.D.T., Konarev P.V., Svergun D.I.. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012; 45:342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Svergun D., Barberato C., Koch M.H.J.. CRYSOL - a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. J. Appl. Crystallogr. 1995; 28:768–773. [Google Scholar]
- 39. Wu Y., Wang J., Xu T., Liu J., Yu W., Lou Q., Zhu T., He N., Qu D., Ben H. et al.. The Two-Component signal transduction system arlrs regulates staphylococcus epidermidis biofilm formation in an ica-Dependent Manner. PLoS One. 2012; 7:e40041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liang X., Zheng L., Landwehr C., Lunsford D., Holmes D.. Global regulation of gene expression by ArlRS, a Two-Component signal transduction regulatory system of staphylococcus aureus. J. Bacteriol. 2005; 187:5486–5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Crooks G., Hon G., Chandonia J., Brenner S.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beenken K.E., Dunman P.M., Mcaleese F., Macapagal D., Murphy E., Projan S.J., Blevins J.S., Smeltzer M.S.. Global Gene Expression in Staphylococcus aureus Biofilms. J. Bacteriol. 2004; 186:4665–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Krissinel E., Henrick K.. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007; 372:774–797. [DOI] [PubMed] [Google Scholar]
- 44. Toro-Roman A., Wu T., Stock A.M.. A common dimerization interface in bacterial response regulators KdpE and TorR. Protein Sci. 2005; 14:3077–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bachhawat P., Stock A.M.. Crystal structures of the receiver domain of the response regulator PhoP from Escherichia coli in the absence and presence of the phosphoryl analog beryllofiuoride. J. Bacteriol. 2007; 189:5987–5995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang S. Cai H. Bacterial Two-Component Systems: Structures and Signaling Mechanisms. Protein Phosphorylation in Human Health. 2012; InTech. [Google Scholar]
- 47. Needham J. V, Chen T.Y., Falke J.J.. Novel ion specificity of a carboxylate cluster Mg (II) Binding Site: Strong charge selectivity and weak size selectivity. Biochemistry. 1993; 32:3363–3367. [DOI] [PubMed] [Google Scholar]
- 48. Febs S., Lukat G.S., Stock A.M., Stock J.B.. Divalent metal ion binding to the CheY Protein and its significance to phosphotransfer in bacterial chemotaxis. Biochemistry. 1990; 29:5436–5442. [DOI] [PubMed] [Google Scholar]
- 49. Schneidman-duhovny D., Hammel M., Tainer J.A., Sali A., Angle S., Saxs X.S.. FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 2016; 44:424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pei J., Kim B.H., Grishin N.V.. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic. Acids. Res. 2008; 36:2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shen Y., Bax A.. Identification of helix capping and β-turn motifs from NMR chemical shifts. J. Biomol. NMR. 2012; 52:211–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ding F., Prutzman K.C., Campbell S.L., Dokholyan N.V.. Topological determinants of protein domain swapping. Structure. 2006; 14:5–14. [DOI] [PubMed] [Google Scholar]
- 53. Lukat G.S., McCleary W.R., Stock A.M., Stock J.B.. Phosphorylation of bacterial response regulator proteins by low molecular weight phospho-donors. Proc. Natl. Acad. Sci. U.S.A. 2006; 89:718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Edayathumangalam R., Wu R., Garcia R., Wang Y., Wang W., Kreinbring C.A., Bach A., Liao J., Stone T.A., Terwilliger T.C. et al.. Crystal structure of Bacillus subtilis GabR, an autorepressor and transcriptional activator of gabT. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:17820–17825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ryan P.L., Friendship R.M., Raeside J.I.. Impaired estrogen production by leydig cells of the naturally retained testis in unilaterally cryptorchid boars and stallions. J. Androl. 1986; 7:100–104. [DOI] [PubMed] [Google Scholar]
- 56. He X., Wang L., Wang S.. Structural basis of DNA sequence recognition by the response regulator PhoP in Mycobacterium tuberculosis. Sci. Rep. 2016; 6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Martínez-Hackert E., Stock A.M.. The DNA-binding domain of OmpR: Crystal structure of a winged helix transcription factor. Structure. 1997; 5:109–124. [DOI] [PubMed] [Google Scholar]
- 58. Maris A.E., Sawaya M.R., Kaczor-Grzeskowiak M., Jarvis M.R., Bearson S.M.D., Kopka M.L., Schröder I., Gunsalus R.P., Dickerson R.E.. Dimerization allows dna target site recognition by the narl response regulator. Nat. Struct. Biol. 2002; 9:771–778. [DOI] [PubMed] [Google Scholar]
- 59. King-scott J., Nowak E., Mylonas E., Panjikar S., Roessle M., Svergun D.I., Tucker P.A.. The structure of a Full-length response regulator from mycobacterium tuberculosis in a stabilized Three-dimensional Domain-swapped, Activated State *. J. Biol. Chem. 2007; 282:37717–37729. [DOI] [PubMed] [Google Scholar]
- 60. Campagne S., Dintner S., Gottschlich L., Thibault M., Bortfeld-Miller M., Kaczmarczyk A., Francez-Charlot A., Allain F.H.T., Vorholt J.A.. Role of the PFXFATG[G/Y] Motif in the activation of SdrG, a Response regulator involved in the alphaproteobacterial general stress response. Structure. 2016; 24:1237–1247. [DOI] [PubMed] [Google Scholar]
- 61. Fournier B., Aras R., Hooper D.C.. Expression of the multidrug resistance transporter nora from staphylococcus aureus is modified by a Two-Component regulatory system. J. Bacteriol. 2000; 182:664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Toledo-arana A., Merino N., Vergara-irigaray M., De M., Penade R.. Staphylococcus aureus develops an alternative, ica- Independent biofilm in the absence of the arlRS Two-Component System †. J. Bacteriol. 2005; 187:5318–5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wu Y., Liu J., Jiang J., Hu J., Xu T., Wang J., Qu D.. Microbial Pathogenesis Role of the two-component regulatory system arlRS in ica operon and aap positive but non-bio fi lm-forming Staphylococcus epidermidis isolates from hospitalized patients. Microb. Pathog. 2014; 76:89–98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Structure factors and atomic coordinates have been deposited in the Protein Data Bank under accession codes: 6IS1 (ArlR receiver domain in complex with BeF3− and Mg2+), 6IS2 (ArlR receiver domain in complex with Mg2+), 6IS3 (ArlR receiver domain) and 6IS4 (ArlR DNA-binding domain).