

Abstract
Several recent studies suggest that cancer stem cells (CSCs) are involved in intrinsic resistance to cancer treatment. Maintenance of quiescence is crucial for establishing resistance of CSCs to cancer therapeutics. F-box/WD repeat-containing protein 7 (FBXW7) is a ubiquitin ligase that regulates quiescence by targeting the c-MYC protein for ubiquitination. We previously reported that gefitinib-resistant persisters (GRPs) in EGFR-mutant non-small cell lung cancer (NSCLC) cells highly expressed octamer-binding transcription factor 4 (Oct-4) as well as the lung CSC marker CD133, and they exhibited distinctive features of the CSC phenotype. However, the role of FBXW7 in lung CSCs and their resistance to epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitors in NSCLC is not fully understood. In this study, we developed GRPs from the two NSCLC cell lines PC9 and HCC827, which express an EGFR exon 19 deletion mutation, by treatment with a high concentration of gefitinib. The GRPs from both PC9 and HCC827 cells expressed high levels of CD133 and FBXW7, but low levels of c-MYC. Cell cycle analysis demonstrated that the majority of GRPs existed in the G0/G1 phase. Knockdown of the FBXW7 gene significantly reduced the cell number of CD133-positive GRPs and reversed the cell population in the G0/G1-phase. We also found that FBXW7 expression in CD133-positive cells was increased and c-MYC expression was decreased in gefitinib-resistant tumors of PC9 cells in mice and in 9 out of 14 tumor specimens from EGFR-mutant NSCLC patients with acquired resistance to gefitinib. These findings suggest that FBXW7 plays a pivotal role in the maintenance of quiescence in gefitinib-resistant lung CSCs in EGFR mutation-positive NSCLC.
Keywords: FBXW7, quiescence, cancer stem cells, gefitinib resistance, NSCLC
INTRODUCTION
Cancer stem cells (CSCs), also known as tumor-initiating cells and stem-like cancer cells, are thought to constitute a minor subpopulation of cancer cells [1]. Accumulating evidence indicates that CSCs persist after cancer treatments such as chemotherapy, radiotherapy, and molecularly targeted therapy [2]. Recent studies revealed that CSCs are maintained in a non-proliferative state, referred to as quiescence, dormancy, or G0 phase, which is thought to render CSCs resistant to various cancer therapeutics [3].
Advanced non-small cell lung cancer (NSCLC) is the leading cause of cancer-related deaths globally [4]. Somatic mutations in the epidermal growth factor receptor (EGFR) gene, such as an in-frame deletion mutation in exon 19 and the point mutation L858R, are associated with a favorable response to the EGFR tyrosine kinase inhibitors (EGFR-TKIs) gefitinib and erlotinib [5]. However, primary or acquired resistance to EGFR-TKIs is a major obstacle to improving the prognosis of EGFR mutation-positive NSCLC patients. The mechanisms of resistance identified to date include the secondary mutation of EGFR T790M, amplification of MET, hepatocyte growth factor (HGF) overexpression, HER2 amplification or mutation, conversion to SCLC, and epithelial-mesenchymal transition (EMT) [6,7]. However, the mechanisms responsible for resistance to EGFR-TKIs are not well understood.
F-box/WD repeat-containing protein 7 (FBXW7), also known as FBW7, SEL-10, hCdc4, or hAgo, is a substrate recognition subunit of the SCF (Skp1-Cullin-F-box protein) ubiquitin ligase complex [8,9]. Several studies demonstrated that FBXW7 is involved in quiescence by degradation of c-MYC protein [10-12]. It has been reported that FBXW7 plays an important role in the maintenance of quiescence in leukemia-initiating cells (LICs) by reducing the level of c-MYC protein [10]. Furthermore, abrogation of quiescence in LICs by FBXW7 ablation increased sensitivity to the TKI imatinib [10]. Thus, targeting quiescence might be a promising strategy for effective control of CSCs.
We previously reported that gefitinib-resistant persisters (GRPs) in EGFR-mutant NSCLC cells showed elevated expression of stem cell genes including PROM1 (prominin-1, CD133), POU5F1 (octamer-binding transcription factor 4, Oct-4), SOX2, NANOG, CXCR4, and ALDH1A1 and characteristic features of CSCs [13,14]. In this study, we examined whether FBXW7 plays a crucial role in the maintenance of quiescence in gefitinib-resistant CSCs using an in vitro and in vivo GRP model with stem cell features. We also evaluated the cell cycle status by introducing a fluorescent ubiquitination-based cell cycle indicator (FUCCI)-expressing plasmid into GRPs. The biological role of FBXW7 for the maintenance of quiescence in gefitinib-resistant lung CSCs in EGFR-mutant NSCLC is discussed.
MATERIALS AND METHODS
Cell culture and reagents
In this study, we used two NSCLC cell lines, PC9 and HCC827, which harbor deletion mutations of EGFR exon 19 (ΔE746-A750) as previously depicted [15]. The reagents and condition of the culture are explained in the supplemental Materials and Methods.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
The qRT-PCR conditions and sequences of the primers applied for transcript detection are explained in the supplemental Materials and Methods.
RNA interference
Short interfering RNAs (siRNAs) inhibiting FBXW7 (stealth select RNAi siRNA), a negative control, and Lipofectamine RNAiMAX were purchased from Invitrogen (Carlsbad, CA, USA). The Lipofectamine RNAiMAX and RNAi duplex were mixed in Opti-MEM® I (Gibco, MA, USA). The details of this procedure are explained in the supplemental Materials and Methods.
Immunofluorescence
Cells were cultured either on Lab-Tek chamber II slides (Nunc, Rochester, NY, USA) or on 35 mm glass bottom dishes (Greiner Bio-One, Frickenhausen, Germany) with 1 µM gefitinib for 72 h, and the immunofluorescence of FBXW7, c-MYC, and CD133 was conducted as described in the supplemental Materials and Methods. The number of FBXW7-, c-MYC-, and CD133-positive cells was counted; the ratio of positive cells to the total cell number was calculated in five fields for each experiment.
FUCCI
pFucci-S/G2/M green and pFucci-G1 orange plasmids were purchased from Medical and Biological Laboratories (Nagoya, Japan). Fucci-S/G2/M green (mKO2-hCdt1) and Fucci-G1 orange (mAG-hGem) were amplified by PCR using LA Taq DNA Polymerase (TaKaRa Bio, Kyoto, Japan), and they were linked in frame by a T2A sequence [16]. Then, the Fucci-S/G2/M green-T2A-Fucci-G1 orange fusion gene was cloned into the lentiviral vector CSII-CMV (kindly provided by Dr. Miyoshi, RIKEN BioResource Center, Tsukuba, Japan), and the resulting plasmid was designated as CSII-CMV-FUCCI-S/G2/M green-G1 orange. The plasmid of CSII-CMV-FUCCI-S/G2/M green-G1 orange was mixed with packaging plasmids and transfected into 293T cells (Invitrogen). Lentiviral infection was carried out as previously depicted [17]. FUCCI-expressing positive cells were used for further experiments.
Mice
The NOD/Shi-scid/IL-2Rcnull (NOG) mice (7-week-old, female) were obtained from the Central Institute for Experimental Animals (Kanagawa, Japan). The mice were lodged as described in the supplemental Materials and Methods.
Establishment of gefitinib-resistant tumors (GRTs) in vivo
PC9 cells (1 × 105) were inoculated into NOG mice, and when tumors reached the size of 75 mm2 we started intraperitoneal injection to these mice with gefitinib. The tumors still remained after 14 days of gefitinib treatment, and we called these remaining tumors as GRTs. Fourteen days after gefitinib administration, tumors were taken as described in the supplemental Materials and Methods.
Immunohistochemistry
In this study, we used tumor specimens from NSCLC patients at Juntendo University Hospital before gefitinib treatment and after relapse. Immunohistochemistry for FBXW7, c-MYC, and CD133 was carried out as depicted in the supplemental Materials and Methods.
Ethics
All animal experiments were conducted in accordance with the Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Notice No. 71, 2006) and were approved by the Committee for Animal Experimentation of Juntendo University with the Approval No. 240182. Tumor specimens were obtained from NSCLC patients at Juntendo University Hospital before gefitinib treatment and after acquisition of resistance, with patient consent and under a protocol approved by the Institutional Review Board.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA). The two-tailed Student’s t-test and ANOVA were used to compare values. Significant differences between the means were statistically regarded if p < 0.05.
RESULTS
GRPs expressed high levels of FBXW7 and CD133 and low levels of c-MYC
We developed GRPs from two NSCLC cell lines, PC9 and HCC827, harboring a sensitive EGFR mutation by exposing cells to a high concentration of gefitinib. After 9 days, the vast majority of cells were dead, but a small population of viable cells remained. We called these remaining cells GRPs of PC9 and HCC827 (PC9-GRPs and HCC827-GRPs). We previously demonstrated using genomic DNA analysis that short tandem repeat (STR) profiles of the GRPs and parental cells were similar, and direct sequencing revealed that GRPs still harbored the EGFR exon 19 deletion mutation [13]. These results indicate that GRPs were not derived from contaminating cells. Furthermore, we previously revealed that GRPs also did not harbor either the secondary T790M mutation of EGFR or amplification of MET [13].
We then investigated the expression level of FBXW7 and the lung CSC marker CD133, as well as of c-MYC in parental cells and GRPs, 9 days after exposure to gefitinib. The mRNA expression levels of FBXW7 and PROM1 (CD133) were higher in GRPs of both PC9 and HCC827, as shown by qRT-PCR analysis (Figure 1A). We also evaluated the protein expression level of FBXW7 and c-MYC by immunofluorescence in GRPs of PC9 and HCC827 cells after exposing cells to gefitinib for 72 h. The immunofluorescence analysis showed increased FBXW7 expression and loss of c-MYC expression in PC9-GRPs and HCC827-GRPs (Figure 1B and C).
FIGURE 1.
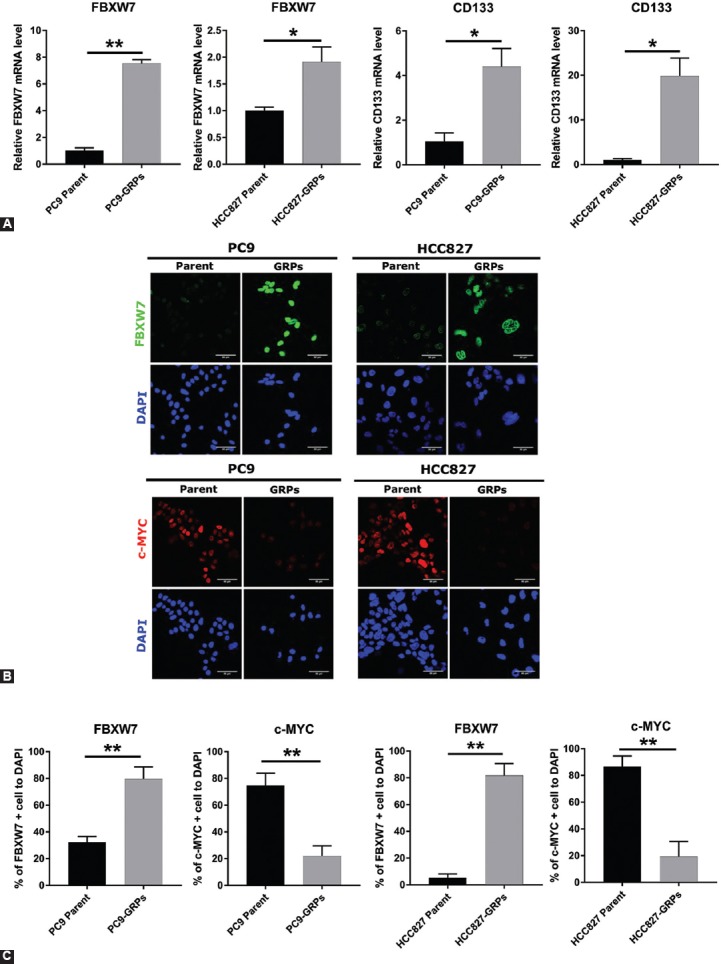
Gefitinib-resistant persisters (GRPs) of PC9 and HCC827 cells express high levels of F-box/WD repeat-containing protein 7 (FBXW7) and CD133 but a low level of c-MYC. (A) The mRNA expression of FBXW7 and PROM1 (CD133) in PC9 and HCC827 cells was analyzed by quantitative reverse transcription polymerase chain reaction (qRT-PCR) 9 days after exposure to gefitinib. Expression levels of FBXW7 and CD133 in parental cells and in GRPs of PC9 and HCC827 were compared. Data were normalized to actin expression. All values represent the average of triplicate experiments. Average values for each parental cell line were set to 1. *p < 0.05, **p < 0.01. (B) Immunofluorescence of FBXW7 and c-MYC in PC9 and HCC827 cells. Cells were cultured with 1 µM gefitinib on slides of Labo-Tek chamber for 72 h. Nuclei were stained with DAPI. (C) The ratio of FBXW7-positive or c-MYC-positive cells in PC-9 and HCC827 cells. Parental cells and GRPs from each cell line were compared. Average values were calculated from five fields in each sample. **p < 0.01; scale bar indicates 50 µm.
Knockdown of FBXW7 decreased CD133-positive GRPs and reversed c-MYC expression in vitro
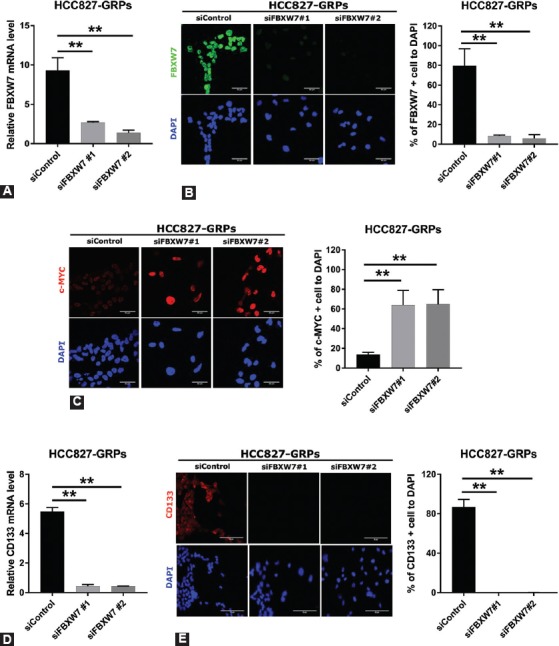
To investigate the role of FBXW7 in the maintenance of GRPs, FBXW7 was silenced using two different siRNAs (siRNA#1 and siRNA#2) in GRPs of PC9 and HCC827. The expression of FBXW7 was significantly suppressed by the two different siRNAs in GRPs of PC9 and HCC827 cells (Figure 2A, B and Figure S1A, B). Furthermore, the reduction in FBXW7 expression significantly reversed c-MYC protein expression (Figure 2C and Figure S1C). We also examined CD133-positive GRPs after inhibition of FBXW7. CD133 mRNA expression and the number of CD133-positive GRPs were significantly decreased after using the two siRNAs targeting FBXW7 in the GRPs of PC9 cells and HCC827 cells (Figure 2D, E and Figure S1D, E). These results collectively suggest that FBXW7 regulates and maintains CD133-positive GRPs of PC9 and HCC827 cells and mediates degradation of c-MYC at the protein level.
FIGURE 2.
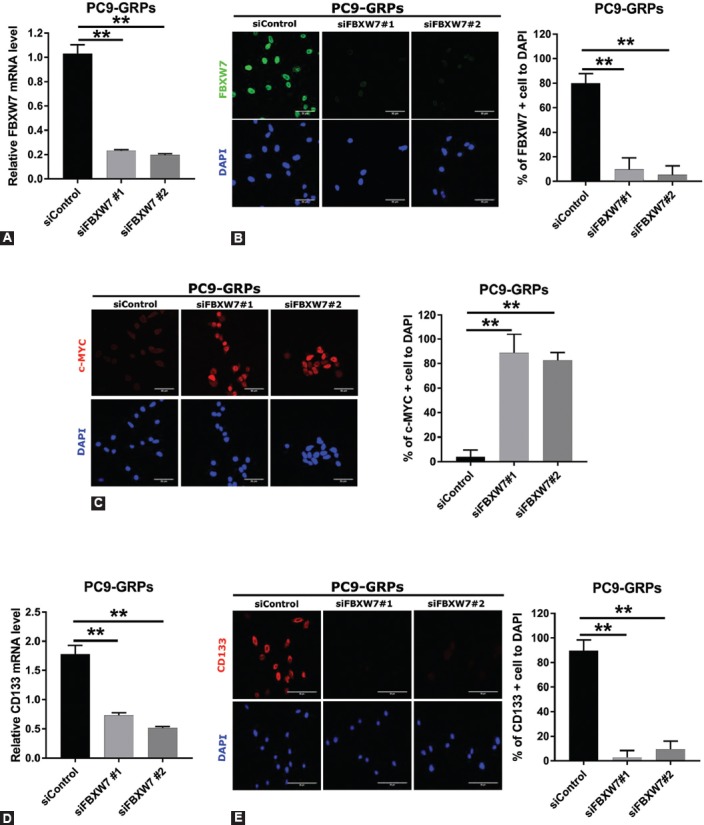
Silencing of F-box/WD repeat-containing protein 7 (FBXW7) significantly decreased the population of CD133-positive cells and reversed c-MYC expression in PC9 gefitinib-resistant persisters (GRPs). (A) FBXW7 expression was knocked down in PC9-GRPs with short interfering RNA (siRNA) followed by gefitinib treatment for 72 h and examined by quantitative reverse transcription polymerase chain reaction (qRT-PCR). Two specific siRNAs and one non-specific control siRNA were used, and representative data from the siRNA experiment are shown. Average values for cells treated with the control siRNA were set to 1. Average values were calculated from triplicate experiments. Error bars, SD. **p < 0.01. (B, left) FBXW7 protein expression knocked down with siRNA in PC9-GRPs cells after gefitinib treatment for 72 h was analyzed by immunofluorescence. Nuclei were stained with DAPI. (C, left) c-MYC expression was analyzed by immunofluorescence after silencing FBXW7 followed by gefitinib treatment for 72 h in PC9-GRPs cells. DAPI (blue) was used to stain nuclei. (B, right and C, right) The ratio of FBXW7-positive cells (B, right) or c-MYC-positive cells (C, right) in PC9-GRPs cells after silencing FBXW7. Average values were calculated from five fields in each sample. **p < 0.01, scale bar indicates 50 µm. (D) Knockdown of FBXW7 expression with siFBXW7 followed by gefitinib treatment for 72 h in PC9-GRPs cells significantly suppressed CD133 expression as determined by qRT-PCR. Average values and SDs were calculated from triplicate samples. Average values for cells treated with the control siRNA were set to 1. **p < 0.01. (E, left) PC9 cells were cultured on slides of Labo-Tek chamber, and FBXW7 expression was silenced with siFBXW7. Cells were treated with 1 µM gefitinib for 72 h. CD133-positive GRPs were significantly decreased. DAPI (blue) was used to stain nuclei. (E, right) The ratio of CD133-positive cells in PC9-GRPs after silencing FBXW7 and followed by gefitinib treatment for 72 h. Average values were calculated from five fields in each sample **p< 0.01, scale bar indicates 50 µm.
FIGURE 4.
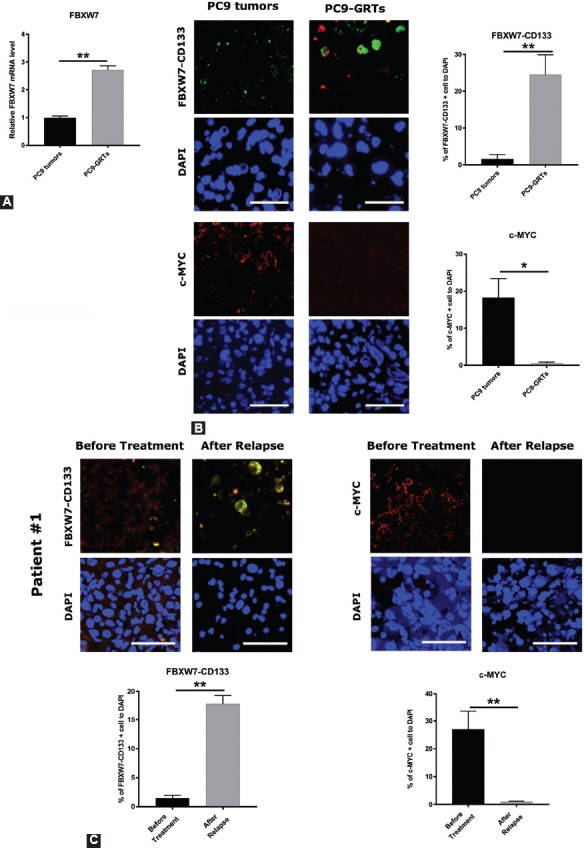
The expression of F-box/WD repeat-containing protein 7 (FBXW7) was higher and that of c-MYC was lower in CD133-positive cells in gefitinib-resistant tumors (GRT) of PC9 in vivo as well as in lung cancer specimens from NSCLC patients who had acquired resistance to gefitinib. (A) PC9 cells were injected into NOD/Shi-scid/IL-2Rcnull (NOG) mice subcutaneously followed by intraperitoneal injection (6 times/week) of 20 mg/kg of gefitinib or vehicle after the tumor volume reached 75 mm3. After 14 to 17 days of gefitinib treatment, the tumors still remained (the mean size of the tumor was 35.3 mm3). Tumors were taken, and quantitative reverse transcription polymerase chain reaction (qRT-PCR) was carried out with FBXW7 primers, in PC9 tumors and PC9-GRT. Data were normalized to actin expression and represent mean ± SD. **p <0.01. (B, left) The expression levels of FBXW7 in CD133-positive cells and c-MYC in PC9 tumors and PC9-GRTs were examined by immunohistochemistry. (B, right) The ratio of FBXW7- and CD133-positive cells and c-MYC-positive cells. Cell nuclei were stained with DAPI (blue). Images were obtained using a ZEISS LSM 780 system. Five pictures were taken from each group and the number of cells expressing FBXW7 and c-MYC in CD133-positive cells was counted *p < 0.05, **p < 0.01. Scale bar indicates 25 µm. (C, top) The expression of FBXW7 in CD133-positive cells and c-MYC in pairs of tumor specimens prior gefitinib treatment and specimens with acquired resistance to gefitinib from NSCLC patients was examined by immunohistochemistry. (C, bottom) The ratio of FBXW7 in CD133-positive cells and c-MYC-positive cells. Cell nuclei were stained with DAPI (blue). Images were obtained using a ZEISS LSM 780 system. Five pictures were taken from each group, and the number of CD133-positive cells that expressed FBXW7 and the number of cells that expressed c-MYC was counted. *p < 0.05, **p < 0.01. Scale bar indicates 25 µm.
The majority of GRPs exist in G0/G1 phase, and FBXW7 is implicated in the quiescence of GRPs
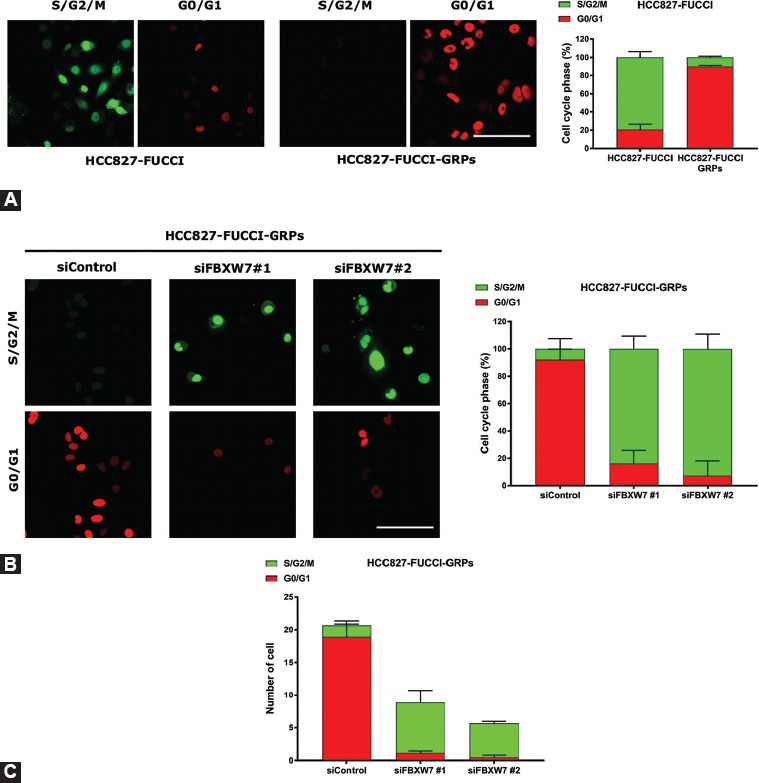
Using a lentiviral transfection procedure, we introduced a FUCCI vector plasmid into PC9 and HCC827 cells to develop FUCCI-expressing PC9 cells (PC9-FUCCI) and FUCCI-expressing HCC827 cells (HCC827-FUCCI), to examine the cell cycle status. The transfection efficiency was confirmed by the appearance of fluorescent color in both cell types, using fluorescence microscopy. To clarify whether the GRPs were in a quiescent state, PC9-FUCCI and HCC827-FUCCI cells were treated with or without a high concentration of gefitinib (1 µM) for 72 h and were subjected to cell cycle analysis. The cells in G0/G1 or S/G2/M phases appeared red and green, respectively [18]. To determine whether the same cells expressed CD133 and FBXW7 in GRPs, we conducted double immunofluorescence staining. We found that GRPs of PC9-FUCCI and GRPs of HCC827-FUCCI expressed both FBXW7 and CD133 in the quiescent state [G0/G1 phase] (Figure S2). Cell cycle analysis demonstrated that the vast majority of cells were in G0/G1 phase after gefitinib treatment as compared with untreated cells (Figure 3A, S3A). To determine whether the upregulation of FBXW7 could maintain the quiescence of GRPs, we knocked down FBXW7 in PC9-FUCCI and HCC827-FUCCI by transfecting two specific siRNAs, followed by gefitinib treatment for 72 h. We found that most FBXW7-silenced cells were in S/G2/M phase, whereas control cells were still in G0/G1 phase (Figure 3B and Figure S3B). The number of GRPs was significantly decreased following knockdown of FBXW7 in FUCCI-expressing PC9 and HCC827 cells (Figure 3C and Figure S3C). Taken together, these findings suggest that GRPs largely exist in G0/G1 phase and that FBXW7 plays a pivotal role in the quiescence of GRPs.
FIGURE 3.
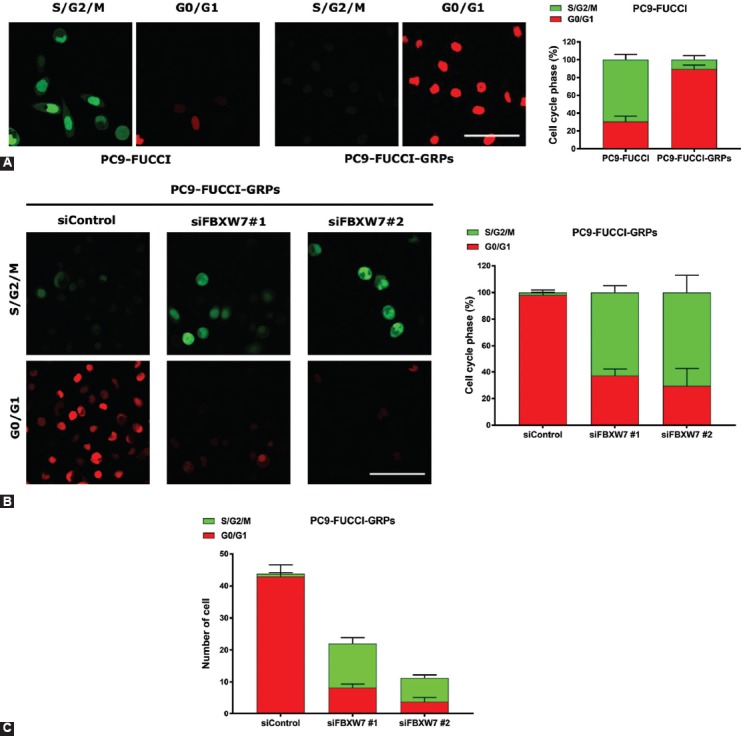
The majority of gefitinib-resistant persisters (GRPs) of PC9 fluorescent ubiquitination-based cell cycle indicator (FUCCI) exist in a quiescent state, maintained by F-box/WD repeat-containing protein 7 (FBXW7). Stable FUCCI-expressing cells were made by introducing FUCCI vector plasmid into PC9. (A, left) PC9-FUCCI cells were seeded in 35 mm glass bottom dishes, followed by treatment with or without gefitinib and subsequently subjected to cell cycle analysis. The cells in G0/G1 or S/G2/M phases appear red and green, respectively. (A, right) Histograms demonstrate the cell cycle phase distribution before and after gefitinib treatment. (B, left) The cell cycle profile of PC9-FUCCI was analyzed after knockdown of FBXW7 using siRNA transfection and gefitinib treatment for 72 h. (B, right) Histograms show the distribution of cells in G0/G1 phase and S/G2/M phase after knocking down FBXW7 under gefitinib treatment. Five pictures were taken from each group, and the cell number in G0/G1 phase as well as in S/G2/M phase was counted and divided by the total number of cells. (C) The number of GRPs was decreased after knockdown of FBXW7 in PC9-FUCCI. Five pictures were taken from each group and the number of cells in G0/G1 phase as well as in S/G2/M phase was counted. Scale bar indicates 50 µm.
FBXW7 expression in CD133-positive cells was increased and c-MYC expression was decreased in GRTs in vivo as well as in tumor specimens from NSCLC patients with acquired resistance to gefitinib
To evaluate in vivo the expression of FBXW7 in the CD133-positive cells and c-MYC in the GRTs of NSCLC cells, we inoculated 1 × 105 PC9 cells into NOG mice. After the tumor size reached 75 mm3, we injected intraperitoneally 20 mg/kg of gefitinib or vehicle to the mice. Following 14 to 17 days of gefitinib treatment, the tumors still remained (the mean size of the tumor was 35.3 mm3); these tumors were considered GRTs [14]. We previously revealed that the GRTs showed high expression of Oct-4 as well as of the lung CSC marker CD133 at both mRNA and protein levels [14]. Moreover, neither the T790M mutation of EGFR nor amplification of the MET gene was found in PC9-GRTs, as previously described [14]. As shown in Figure 4A, PC9-GRTs displayed elevated FBXW7 expression at mRNA level, according to qRT-PCR analysis. The immunohistochemical analysis showed upregulation of FBXW7 expression in the CD133-positive cells and downregulation of c-MYC expression in GRTs as compared with control tumors (Figure 4B). We also examined the expression of FBXW7 in CD133-positive cells as well as of c-MYC in pairs of tumor specimens prior gefitinib treatment and specimens with acquired resistance to gefitinib from 14 NSCLC patients, which lacked the T790M mutation. The immunohistochemical analysis demonstrated that the FBXW7 expression level in CD133-positive cells was higher and that of c-MYC was lower in 9 out of the 14 gefitinib-resistant tumor specimens from NSCLC patients that had the EGFR mutation as compared with the tumor specimens collected before the treatment (Figure 4C).
DISCUSSION
In this study, we developed GRPs by exposing PC9 and HCC827 cells harboring an activating EGFR mutation to a high concentration of gefitinib. GRPs expressed a high level of FBXW7 and CD133 as well as a low level of c-MYC in vitro. The cell cycle analysis revealed that the majority of GRPs were in G0/G1 phase. GRPs expressed both FBXW7 and CD133 in the quiescent state, and the knockdown of the FBXW7 gene in GRPs significantly decreased the CD133-positive cell population after gefitinib exposure and reversed the cell population in G0/G1 phase. Furthermore, we demonstrated in vivo that the GRTs of PC9 cells that remained after gefitinib treatment in mice also expressed a high level of FBXW7 in CD133-positive cells and a low level of c-MYC protein. FBXW7 expression was increased in CD133-positive cells while c-MYC expression was decreased in tumor specimens from EGFR mutation-positive NSCLC patients with acquired resistance to gefitinib. These findings indicate that FBXW7 plays a crucial role in the maintenance of quiescence in GRPs in NSCLC with an activating EGFR mutation.
We previously showed that GRPs have high expression levels of stem cell genes, including PROM1 (CD133), POU5F1 (Oct-4), SOX2, NANOG, CXCR4, and ALDH1A1 and that they have distinct CSC phenotypes [13]. GRPs showed a high ability for the formation of sphere in vitro and tumorigenicity in vivo [13]. In this study, we analyzed the cell cycle status using FUCCI-expressing PC9 cells and FUCCI-expressing HCC827 cells. Our cell cycle analysis revealed that the vast majority of GRPs from PC9-FUCCI and HCC827-FUCCI were in G0/G1 phase, while the majority of untreated cells were in S/G2/M phase. These findings strongly indicate that GRPs are very quiescent, exist in G0/G1 phase, and have characteristic features of CSC phenotype.
FBXW7 is the substrate recognition subunit of the SCF ubiquitin ligase complex that is responsible for the degradation of many proteins, including c-MYC, through the ubiquitylation process [9,19]. FBXW7 has been shown to play a role in maintaining quiescence by degradation of c-MYC protein. Silencing of FBXW7 in quiescent prostate cancer cells resulted in cell cycle reentry via c-MYC protein accumulation [11]. Another study demonstrated that FBXW7 expression was upregulated in CSCs from colorectal cancer and that silencing of FBXW7 significantly increased the chemosensitivity of the colorectal CSCs and led to the upregulation of c-MYC expression [12]. This evidence is consistent with our data that FBXW7 expression was upregulated in GRPs in NSCLC and that the silencing of FBXW7 resulted in the upregulation of c-MYC expression. We also found that FBXW7 expression was higher in CD133-positive cells and that c-MYC was lower in our in vivo GRT model after gefitinib treatment in mice. In addition, we demonstrated that FBXW7 expression in CD133-positive cells was increased and c-MYC expression was decreased in tumor specimens from NSCLC patients with acquired resistance to gefitinib as compared with tumor specimens collected before treatment. These results are consistent with a previous study that showed that high FBXW7 expression significantly correlates with low c-MYC expression in specimens of resected liver metastases from colorectal cancer after chemotherapy [12].
LICs of chronic myeloid leukemia (CML) have been shown to be resistant to the TKI imatinib, and quiescence in LICs of CML is thought to contribute to imatinib resistance [20]. FBXW7 is required for the maintenance of quiescence in LICs and the abrogation of quiescence in LICs by FBXW7 ablation sensitized LICs to imatinib [10]. In our study, the knockdown of FBXW7 significantly reduced the number of CD133-positive GRPs and reversed the cell population in G0/G1 phase, indicating that FBXW7 plays a pivotal role in the maintenance and quiescence of GRPs.
Recently, it has been reported that silencing of FBXW7 induces gefitinib resistance in NSCLC cell lines through the regulation of the EMT and mTOR pathway [21]. The loss of FBXW7 leads to resistance to EGFR and ALK inhibitors in NSCLC cell lines through the stabilization of MCL-1 [22]. However, these studies investigated the role of FBXW7 in the overall population of differentiated NSCLC cells. In our study, we focused on a small population of these cells with CSC properties, i.e., CD133-positive GRPs. Our present study suggests a different role for FBXW7 in the differentiated cells as compared with CSCs or tumor-initiating cells in gefitinib resistance of NSCLC with an activating EGFR mutation. However, further studies are necessary to clarify this finding.
Collectively, our study identifies a role for FBXW7 in the maintenance and quiescence of GRPs. Targeting FBXW7-mediated quiescence may represent a promising strategy to treat gefitinib-resistant lung CSCs in NSCLC with an EGFR mutation.
ACKNOWLEDGMENTS
This work was supported by JSPS KAKENHI Grant Number 15K09230 (Kazuhisa Takahashi). This study was also supported by a grant for cross-disciplinary collaboration and a grant from the Institute for Environmental and Gender-specific Medicine, Juntendo University.
SUPPLEMENTAL DATA
SUPPLEMENTAL MATERIALS AND METHODS
Cell culture and reagents
Two NSCLC cell lines, PC9 and HCC827, which express EGFR exon 19 deletion mutations (ΔE746-A750), were used in this study. PC9 cells were established at the Tokyo Medical University (Tokyo, Japan) as previously described [15] and were kindly provided by Dr. Kazuto Nishio (Department of Genome Biology, School of Medicine; Kinki University, Osaka). HCC827 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). Cell lines were verified to be mycoplasma free. Cells were cultured in RPMI-1640 medium (Wako, Osaka, Japan) supplemented with 10% fetal bovine serum (FBS), penicillin, and streptomycin (100 U/mL and 100 µg/mL, respectively) and were grown in a humidified 5% CO2 atmosphere at 37°C in an incubator, in which the oxygen tension was held at 21% (normoxia). Gefitinib was purchased from JS Research Chemicals Trading (Wedel, Germany).
Quantitative reverse transcription PCR (qRT-PCR)
Total RNA was extracted from cell lines using the mirVana miRNA Isolation kit (Ambion, Austin, TX, USA). cDNA was created from 1 µg of RNA using the ReverTra cDNA synthesis kit (Toyobo, Osaka, Japan) according to the manufacturer’s protocol. Quantitative (qPCR) was carried out using the Fast SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA) as previously described [13]. The following program was used: holding at 95°C for 20 s and amplification through 40 cycles (denaturation 95°C for 3 s, annealing, and extension at 60°C for 30 s), with concurrent melt curve analysis. Actin served as an internal control.
The following primers were used:
CD133
Forward, 5’-GGCCCAGTACAACACTACCAA-3’
Reverse, 5’-CGCCTCCTAGCACTGAATTGATA-3’
FBXW7
Forward, 5’-CGACGCCGAATTACATCTGTC-3’
Reverse, 5’-CGTTGAAACTGGGGTTCTATCA-3’
Actin
Forward, 5’-CTCTTCCAGCCTTCCTTCCT-3’
Reverse, 5’-AGCACTGTGTTGGCGTACAG-3’
RNA interference
Short interfering RNAs (siRNAs) targeting FBXW7 (Stealth Select RNAi siRNA) were custom synthesized by Invitrogen. A negative control was also purchased from Invitrogen. To exclude off-target effects, PC9 cells and HCC827 cells were transfected with two different specific siRNAs and one non-specific control using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. The cells were detached and diluted in complete growth medium without antibiotics and then plated in each of the wells. RNAi duplex and Lipofectamine RNAiMAX were mixed in Opti-MEM® I (Gibco) reduced serum medium and incubated for 15 min at room temperature. RNAi duplex-Lipofectamine™ RNAiMAX complexes were added to the wells containing cells. The cells were then incubated for 24 h at 37°C followed by gefitinib treatment for 72 h. The sequences of the siRNA against FBXW7 were as follows:
Stealth FBXW7 oligo #1: 5’-GAGACUUCAUUUCAUUGCUCCCUAA-3’;
Stealth FBXW7 oligo #2: 5’-AGUUGGCACUCUAUGUGCUUUCAUU-3’.
Immunofluorescence
Cells were cultured with 1 µM gefitinib for 72 h, fixed with 4% paraformaldehyde for 5 min, and permeabilized with 0.1% Triton X-100 for 5 min. After blocking with 10% goat serum in phosphate-buffered saline (PBS) for 1 h at room temperature, cells were incubated at 4°C overnight with the primary antibody: FBXW7 (Abcam, Cambridge, United Kingdom), c-MYC (Cell Signaling Technology, Danvers, Massachusetts, USA), and CD133 (Miltenyi Biotec, Bergisch Gladbach, Germany). Protein was visualized by incubation with secondary antibody labeled with Alexa Fluor 594 goat anti-mouse IgG, Alexa Fluor 488 goat anti-mouse IgG, and Alexa Fluor 647 goat anti-mouse and anti-rabbit IgG (Invitrogen). Slides were mounted using Vectashield Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA, USA). The images were obtained with a confocal microscope (ZEISS LSM 780, Oberkochen, Germany) outfitted with 488 nm (argon), 561 nm (red DPSS), and 633 (He/Ne) using ×40 water objective and with ZEN 2 black edition software (ZEISS). Alternatively, an Axioplan 2 imaging system was used (ZEISS, Oberkochen, Germany) with AxioVision software (ZEISS). Images used for comparisons of different cells and/or treatments were acquired with the same instrument settings and exposure times and were processed similarly. The number of FBXW7, c-MYC, and CD133-positive cells was counted; the ratio of positive cells was calculated in five fields for each experiment. As for transient knockdown of FBXW7, PC9 and HCC827 were grown on Lab-Tek chamber II slides (Nunc, Rochester, NY, USA) and transiently knocked down using reverse siRNA transfection and treated with 1 µM gefitinib for 72 h, and the subsequent steps were the same as described above. For the double staining of FBXW7/CD133 in PC9-FUCCI and HCC827 FUCCI, cells were grown on 35 mm glass bottom dishes (Greiner Bio-One), permeabilized with 0.2% tween 20 for 20 min, and FBXW7 protein was visualized by incubation with secondary antibody labeled with Alexa Fluor 450 goat anti-mouse IgG (Invitrogen). DAPI staining was not used in this experiment. The subsequent steps were the same as described above.
FUCCI
pFucci-S/G2/M green and pFucci-G1 orange plasmid were purchased from MBL (Nagoya, Japan). Fucci-S/G2/M green (mKO2-hCdt1) and Fucci-G1 orange (mAG-hGem) were amplified by PCR using LA Taq DNA Polymerase (TaKaRa Bio, Kyoto, Japan), and they were linked in-frame by a T2A sequence [16]. Then, the Fucci-S/G2/M green-T2A-Fucci-G1 orange fusion gene was cloned into the lentiviral vector CSII-CMV (kindly provided by Dr. Miyoshi, RIKEN BioResource Center, Tsukuba, Japan), and the resulting plasmid was designated as CSII-CMV-FUCCI-S/G2/M green-G1 orange. The expressing plasmid of CSII-CMV-FUCCI-S/G2/M green-G1 orange was mixed with packaging plasmids and transfected into 293T cells (Invitrogen). The supernatant medium was collected 2 days after transduction and filtered through a 0.45-µm membrane (Millipore, Massachusetts, USA). Then, PC9 and HCC827 cells were infected using 6 µg/mL of polybrene as previously described [17]. The successfully transfected cells were identified using confocal microscope (ZEIS LSM 780). FUCCI-expressing positive cells were used for further experiments.
Mice
Seven-week-old female NOD/Shi-scid/IL-2Rcnull (NOG) mice were obtained from the Central Institute for Experimental Animals (Kanagawa, Japan). All mice were shipped to the Juntendo University and were maintained under pathogen-free conditions. The mice were housed in a room under controlled temperature (25°C), humidity, and lighting (12-h light/dark cycle). Ad libitum access to food and tap water was allowed throughout the study period.
Establishment of GRTs in vivo
We implanted 1 × 105 PC9 cells into NOG mice, and once tumors reached the size of 75 mm2, we began treatment of these mice with gefitinib (20 mg/kg) by intraperitoneal injection (6 times/week). After 14 days of gefitinib treatment, the tumors remained, and these tumors were referred to as GRTs. Tumors were collected 14 days after gefitinib treatment and were subjected to FBXW7 expression analysis by qRT-PCR and expression of FBXW7 and c-MYC by immunohistochemistry.
Immunohistochemistry
Paraffin-fixed GRTs samples and lung cancer specimens were subject to the following steps: Deparaffinization, antigen retrieval, blocking, and immunohistochemical staining with FBXW7 antibody at a 1:1000 dilution (Abcam, Cambridge, United Kingdom) and with c-MYC antibody at a 1:200 dilution (Abcam). Slides were incubated at 4°C overnight with primary antibody. Protein was visualized by incubation with secondary antibody labeled with Alexa Fluor 594 goat anti-mouse IgG (Invitrogen) and Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen). Slides were mounted using Vectashield Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA, USA). The number of both FBXW7- and CD133- as well as c-MYC-positive cells was counted; the ratio of positive cells was calculated in five fields for each experiment. Images were obtained using a confocal microscope (ZEIS LSM 780) with ×40 water objective, and with ZEN 2 black edition software (ZEISS).
FIGURE S1.
Silencing of F-box/WD repeat-containing protein 7 (FBXW7) significantly decreased the population of CD133-positive cells and reversed c-MYC expression in HCC827 gefitinib-resistant persisters (GRPs). (A) FBXW7 expression was knocked down in HCC827 GRPs with short interfering RNA (siRNA) followed by gefitinib treatment for 72 h and examined by quantitative reverse transcription polymerase chain reaction (qRT-PCR). Two specific siRNAs and one non-specific control were used, and data representative of the siRNA experiment are shown. Average values for cells treated with the control siRNA were set to 1. Average values were calculated from triplicate experiments. Error bars, SD. **p < 0.01. (B, left) FBXW7 expression at protein level was knocked down with siRNA in HCC827-GRPs and analyzed by immunofluorescence after gefitinib treatment for 72 h. Nuclei were stained with DAPI. (C, left) c-MYC expression was analyzed by immunofluorescence after silencing of FBXW7 and gefitinib treatment for 72 h in HCC827-GRPs. Nuclei were stained with DAPI. (B, right and C, right) The ratio of FBXW7-positive cells (B, right) or c-MYC-positive cells (C, right) in HCC827 GRP cells following the silencing of FBXW7. Average values were calculated from five fields in each sample. **p < 0.01, scale bar indicates 50 µm. (D) Knockdown of FBXW7 expression with siFBXW7 followed by gefitinib treatment for 72 h in HCC827-GRPs significantly suppressed CD133 expression as determined by qRT-PCR. Average values and SDs were calculated from triplicate samples. Average values for cells treated with the control siRNA were set to 1. **p < 0.01. (E, left) HCC827 cells were cultured on Labo-Tek chamber slides and FBXW7 expression was silenced with siFBXW7, after which cells were treated with 1 µM gefitinib for 72 h. The number of CD133-positive GRPs was significantly decreased. Nuclei were stained with DAPI (blue). (E, right) The ratio of CD133-positive cells in HCC827 GRPs following FBXW7 silencing and gefitinib treatment for 72 h. Average values were calculated from five fields in each sample. **p < 0.01, scale bar indicates 50 µm.
FIGURE S2.
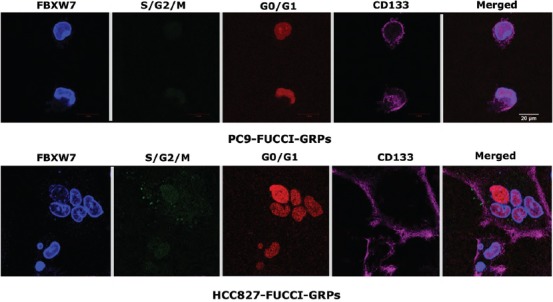
. Both F-box/WD repeat-containing protein 7 (FBXW7) and CD133 proteins were highly expressed in gefitinib-resistant persisters (GRPs) of PC9 fluorescent ubiquitination-based cell cycle indicator (FUCCI) and HCC827 FUCCI. The fluorescence image of GRPs showed red color, indicating that GRPs existed in G0/G1 phase. Cells were seeded in 35 mm glass bottom dishes, followed by treatment with or without 1 µM gefitinib for 72 h. Scale bar indicates 20 µm.
FIGURE S3.
The majority of gefitinib-resistant persisters (GRPs) of HCC827 fluorescent ubiquitination-based cell cycle indicator (FUCCI) existed in a quiescent state and were maintained by F-box/WD repeat-containing protein 7 (FBXW7). Stable FUCCI-expressing cells were made by introducing FUCCI vector plasmid into HCC827. (A, left) HCC827 FUCCI cells were seeded in 35 mm glass bottom dishes, followed by treatment with or without gefitinib and subsequently were subjected to cell cycle analysis. The cells in G0/G1 or S/G2/M phases appear red and green, respectively. (A, right) Histograms demonstrate the distribution of the cell cycle phases before and after treatment with gefitinib. (B, left) The cell cycle profile of HCC827 FUCCI was analyzed following the knockdown of FBXW7 using reverse short interfering RNA (siRNA) transfection and gefitinib treatment for 72 h. (B, right) Histograms show the distribution of G0/G1 phase and S/G2/M phase after knockdown of FBXW7 under gefitinib treatment. Five images were taken from each group, and the cell population in G0/G1 phase as well as S/G2/M phase was counted and divided by the total number of cells. (C) The number of GRPs was decreased following the knockdown of FBXW7 in HCC827 FUCCI. Five images were taken from each group and the number of cells in G0/G1 phase as well as S/G2/M phase was counted. Scale bar indicates 50 µm.
DECLARATION OF INTERESTS
K.T. (Kazuhisa Takahashi) received research funding from Chugai Pharm, Ono Pharma, Taiho Pharm, Nippon Boehringer Ingelheim, AstraZeneca, Pfizer, MSD, and Lilly Japan. The remaining authors declare no conflict of interests.
REFERENCES
- 1.Clevers H. The cancer stem cell:Premises, promises and challenges. Nat Med. 2011;17(3):313–9. doi: 10.1038/nm.2304. https://doi.org/10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- 2.Yoshida GJ, Saya H. Therapeutic strategies targeting cancer stem cells. Cancer Sci. 2016;107(1):5–11. doi: 10.1111/cas.12817. https://doi.org/10.1111/cas.12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alarcón T, Jensen HJ. Quiescence:A mechanism for escaping the effects of drug on cell populations. J R Soc Interface. 2011;8(54):99–106. doi: 10.1098/rsif.2010.0130. https://doi.org/10.1098/rsif.2010.0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siegel RL, Miller KD, Jemal A. Cancer statistics 2018. CA Cancer J Clin. 2018;68(1):7–30. doi: 10.3322/caac.21442. https://doi.org/10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 5.Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10(11):760–74. doi: 10.1038/nrc2947. https://doi.org/10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang K, Yuan Q. Current mechanism of acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitors and updated therapy strategies in human nonsmall cell lung cancer. J Cancer Res Ther. 2016;12(2):131–7. doi: 10.4103/0973-1482.200613. https://doi.org/10.4103/0973-1482.200613. [DOI] [PubMed] [Google Scholar]
- 7.Wu SG, Shih JY. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol Cancer. 2018;17(1):38. doi: 10.1186/s12943-018-0777-1. https://doi.org/10.1186/s12943-018-0777-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Welcker M, Clurman BE. FBW7 ubiquitin ligase:A tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8(2):83–93. doi: 10.1038/nrc2290. https://doi.org/10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 9.Nakayama KI, Nakayama K. Ubiquitin ligases:Cell-cycle control and cancer. Nat Rev Cancer. 2006;6(5):369–81. doi: 10.1038/nrc1881. https://doi.org/10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 10.Takeishi S, Matsumoto A, Onoyama I, Naka K, Hirao A, Nakayama KI, et al. Ablation of FBXW7 eliminates leukemia-initiating cells by preventing quiescence. Cancer Cell. 2013;23(3):347–61. doi: 10.1016/j.ccr.2013.01.026. https://doi.org/10.1016/j.ccr.2013.01.026. [DOI] [PubMed] [Google Scholar]
- 11.Xi Z, Yao M, Li Y, Xie C, Holst J, Liu T, et al. Guttiferone K impedes cell cycle re-entry of quiescent prostate cancer cells via stabilization of FBXW7 and subsequent c-MYC degradation. Cell Death Dis. 2016;7(6):e2252. doi: 10.1038/cddis.2016.123. https://doi.org/10.1038/cddis.2016.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Izumi D, Ishimoto T, Miyake K, Eto T, Arima K, Kiyozumi Y, et al. Colorectal cancer stem cells acquire chemoresistance through the upregulation of F-box/WD repeat-containing protein 7 and the consequent degradation of c-myc. Stem Cells. 2017;35(9):2027–36. doi: 10.1002/stem.2668. https://doi.org/10.1002/stem.2668. [DOI] [PubMed] [Google Scholar]
- 13.Murakami A, Takahashi F, Nurwidya F, Kobayashi I, Minakata K, Hashimoto M, et al. Hypoxia increases gefitinib-resistant lung cancer stem cells through the activation of insulin-like growth factor 1 receptor. PLoS One. 2014;9(s1):e8645–9. doi: 10.1371/journal.pone.0086459. https://doi.org/10.1371/journal.pone.0086459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kobayashi I, Takahashi F, Nurwidya F, Nara T, Hashimoto M, Murakami A, et al. Oct4 plays a crucial role in the maintenance of gefitinib-resistant lung cancer stem cells. Biochem Biophys Res Commun. 2016;473(1):125–32. doi: 10.1016/j.bbrc.2016.03.064. https://doi.org/10.1016/j.bbrc.2016.03.064. [DOI] [PubMed] [Google Scholar]
- 15.Minakata K, Takahashi F, Nara T, Hashimoto M, Tajima K, Murakami A, et al. Hypoxia induces gefitinib resistance in non-small-cell lung cancer with both mutant and wild-type epidermal growth factor receptors. Cancer Sci. 2012;103(11):1946–54. doi: 10.1111/j.1349-7006.2012.02408.x. https://doi.org/10.1111/j.1349-7006.2012.02408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shao L, Feng W, Sun Y, Bai H, Liu J, Currie C, et al. Generation of iPS cells using defined factors linked via the self-cleaving 2A sequences in a single open reading frame. Cell Res. 2009;19(3):296–306. doi: 10.1038/cr.2009.20. https://doi.org/10.1038/cr.2009.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takahashi F, Chiba N, Tajima K, Hayashida T, Shimada T, Takahashi M, et al. Breast tumor progression induced by loss of BTG2 expression is inhibited by targeted therapy with the erbB/HER inhibitor lapatinib. Oncogene. 2011;30(27):3084–95. doi: 10.1038/onc.2011.24. https://doi.org/10.1038/onc.2011.24. [DOI] [PubMed] [Google Scholar]
- 18.Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132(3):487–98. doi: 10.1016/j.cell.2007.12.033. https://doi.org/10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- 19.Takeishi S, Nakayama KI. To wake up cancer stem cells, or to let them sleep, that is the question. Cancer Sci. 2016;107(7):875–81. doi: 10.1111/cas.12958. https://doi.org/10.1111/cas.12958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holtz M, Forman SJ, Bhatia R. Growth factor stimulation reduces residual quiescent chronic myelogenous leukemia progenitors remaining after imatinib treatment. Cancer Res. 2007;67(3):1113–20. doi: 10.1158/0008-5472.CAN-06-2014. https://doi.org/10.1158/0008-5472.can-06-2014. [DOI] [PubMed] [Google Scholar]
- 21.Xiao Y, Yin C, Wang Y, Lv H, Wang W, Huang Y, et al. FBXW7 deletion contributes to lung tumor development and confers resistance to gefitinib therapy. Mol Oncol. 2018;12:883–95. doi: 10.1002/1878-0261.12200. https://doi.org/10.1002/1878-0261.12200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ye M, Zhang Y, Zhang X, Zhang J, Jing P, Cao L, et al. Targeting FBW7 as a strategy to overcome resistance to targeted therapy in non-small cell lung cancer. Cancer Res. 2017;77(13):3527–39. doi: 10.1158/0008-5472.CAN-16-3470. https://doi.org/10.1158/0008-5472.can-16-3470. [DOI] [PubMed] [Google Scholar]