

Abstract
The ability to write DNA code from scratch will allow for the discovery of new and interesting chemistries as well as allowing the rewiring of cell signal pathways. Herein, we have utilized synthetic evolution artificial intelligence (SYN-AI) to intelligently design a set of 14-3-3 docking genes. SYN-AI engineers synthetic genes utilizing a parental gene as an evolution template. Wherein, evolution is fast-forwarded by transforming template gene sequences to DNA secondary and tertiary codes based upon gene hierarchical structural levels. The DNA secondary code allows identification of genomic building blocks across an orthologous sequence space comprising multiple genomes. Where, the DNA tertiary code allows engineering of supersecondary structures. SYN-AI constructed a library of 10 million genes that was reduced to three structurally functional 14-3-3 docking genes by applying natural selection protocols. Synthetic protein identity was verified utilizing Clustal Omega sequence alignments and Phylogeny.fr phylogenetic analysis. Wherein, we were able to confirm the three-dimensional structure utilizing I-TASSER and protein–ligand interactions utilizing COACH and Cofactor. The conservation of allosteric communications was confirmed utilizing elastic and anisotropic network models. Whereby, we utilized elNemo and ANM2.1 to confirm conservation of the 14-3-3 ζ amphipathic groove. Notably, to the best of our knowledge, we report the first 14-3-3 docking genes to be written from scratch.
1. Introduction
The ability to write DNA code from scratch is a primary goal in the area of synthetic biology. Wherein, intelligent gene design will allow researchers to address a broad range of scientific conundrums such as the rewiring of signal pathways and the intelligent design of small genomes. Thusly, allowing for the engineering of novel organisms and the potential for new and interesting chemistries that may include degradation of nonbiodegradable products. Saliently, these technologies will open frontiers in medicine allowing the design of novel drug receptors for the discovery of new cancer and disease treatments. The possibilities that such a technology can offer are seemingly endless and essential for our current technological challenges. It is worth mentioning that over the past decade, there have been multiple attempts at the modest ambition of de novo protein engineering. Wherein, the science was limited to a range of mutagenesis techniques that often resulted in nonfunctional proteins. Although, there has been considerable progress, the ability to intelligently design fully functional genes from scratch has been elusive.
In the current study, we focus on the intelligent gene design utilizing synthetic evolution artificial intelligence (SYN-AI), an AI that accelerates the evolution process by performing a domain shuffling-like mechanism similar to the “Domain Lego” principle.1−3 Evolutional acceleration is achieved by transforming gene sequences into DNA secondary (DSEC) and tertiary codes (DTER) based on gene hierarchical structure levels. We assume that modern genes have a common ancestor that partitioned over time via DNA crossovers, and that genetic diversity occurred by processes such as gene duplication, inversion, insertion, and deletion.4,5 Our assumption of a common ancestor is in agreement with the “Universal Ancestor” and LUCA “Last Universal Common Ancestor” models.6,7 Saliently, the DSEC allows for the identification of short highly conserved sequences occurring across multiple genomes referred to herein as genomic building blocks (GBBs). Based upon “The Fundamental Theory of the Evolution Force”, these sequences are genetic artifacts formed during the evolution process.71 The DTER allows partitioning of genes at the supersecondary structure level. Whereby, synthetic genes are engineered by walking the DTER followed by random selection and ligation of supersecondary structures. Thusly, the exchange of information is analogous to the swapping of genomic building blocks in a game of Legos.
To fast forward the evolution process, SYN-AI forms an expanse orthologous sequence space followed by an exponential number of DNA crossovers within the genomic alphabet comprising the DNA secondary code. SYN-AI identifies genomic building block formation across genomes by analysis of evolution force associated with DNA crossovers. Whereby, evolution force is a compulsion acting at the matter–energy interface that accomplishes genetic diversity while simultaneously conserving architecture and function.71 In the current study, we utilize SYN-AI to intelligently design a set of 14-3-3 docking genes. These genes are responsible for regulating protein–protein interactions (PPI) in cell signal pathways. Saliently, docking protein interactions can have a profound effect on the target protein, altering its localization, stability, conformation, phosphorylation state, activity, and/or molecular interactions.9 Thusly, the ability to write 14-3-3 docking genes from scratch will allow the rewiring of cell signal pathways. In addition to acting as key components of cell signal pathways, 14-3-3 docking proteins play a role in cell growth and development, cancer cell signaling,9,10 cellular metabolism, and organelle communication.11 Whereby, 14-3-3 proteins have been shown to interact with key signaling components such as the insulin-like growth receptor,57−59 PI3K,60,61 cdc25 phosphatase,62−64 and bad.65−67
In the current study, we intelligently designed a set of 14-3-3 docking genes utilizing SYN-AI. Wherein, a truncated B. taurus 14-3-3 ζ docking gene was utilized as a template for gene engineering. Genomic building blocks were identified across multiple genomes by partitioning the parental docking gene into a DNA secondary code and simulating evolution by performing an exponential number of DNA crossovers within the genomic alphabet forming the DSEC, Figure 1 (top). Whereby, DNA crossover partners were randomly selected across an orthologous sequence space. Synthetic supersecondary structures were engineered by targeting evolution within the genomic alphabet comprising the DNA tertiary code, Figure 1 (middle). In all, SYN-AI generated a library of 10 million genes by walking the DTER, followed by random selection and ligation of synthetic supersecondary structures, Figure 1 (bottom). This expanse gene library was reduced to three genes utilizing natural selection protocols. Synthetic 14-3-3 docking genes were confirmed utilizing the Clustal Omega multiple sequence alignment tool and by phylogenetic analysis utilizing Phylogeny.fr. We were able to confirm the three-dimensional structure of synthetic proteins utilizing I-TASSER as well as verify conservation of small-molecule and fusicoccin binding sites utilizing Cofactor and Coach. The conservation of the 14-3-3 ζ amphipathic groove57 as well as allosteric interactions were confirmed utilizing the elastic network model. Whereby, we utilized ElNemo and ANM2.1 to analyze normal modes, root-mean-square deviation (RMSD), and deformation energies. Notably, based upon the aforementioned, we confirm the intelligent design of a set of novel 14-3-3 docking proteins utilizing the synthetic evolution artificial intelligence.
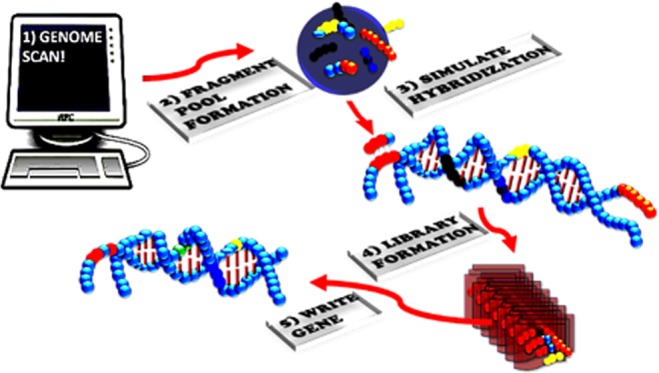
Figure 1.

SYN-AI mechanism. SYN-AI partitions the parental gene into a DNA secondary code (DSEC) and fast forwards the evolution process by performing an exponential number of DNA crossovers within each genomic alphabet comprising the DSEC, (Top). DNA crossovers characterized by the highest magnitude of evolution force are selected and stored in libraries. Subsequently, genomic building block (GBB) libraries are subjected to natural selection. Following natural selection, synthetic supersecondary structures are formed by random selection and ligation of GBBs from appropriate genomic alphabet libraries, (Middle). Synthetic genes are engineered by walking the DNA tertiary code (DTER) followed by random selection and ligation of synthetic supersecondary structures. Gene libraries are restricted to functional 14-3-3 docking genes by a natural selection process (Bottom).
2. Results
SYN-AI identified genomic building block formation by performing an exponential number of DNA crossovers within the genomic alphabet forming the DSEC followed by the analysis of the magnitude of the evolution force associated with the aforementioned. Whereby, evolution force was analyzed over single and multidimensional planes of evolution formed as functions of the four evolution engines, (i) evolution conservation, (ii) wobble, (iii) DNA binding state, and (iv) periodicity according to “The Fundamental Theory of the Evolution Force” and as described in ref (71). In addition, genomic building blocks were identified by applying natural selection protocols that limited selection to evolutionarily conserved DNA crossovers utilizing pattern recognition filters and that limited selection to sequences comprised of naturally occurring mutations utilizing Blosum80 mutation frequency-based algorithms. Subsequently, SYN-AI-engineered synthetic 14-3-3 docking genes by walking the DNA tertiary code and randomly selecting and ligating synthetic supersecondary structures formed by Domain Legos shuffling of genomic building blocks. Whereby, SYN-AI limited selection to structurally functional 14-3-3 docking genes by application of natural selection protocols.
Evolution force was analyzed utilizing the rotation model, as described in71 and as illustrated in Figure 2. Wherein, genomic building blocks appeared as low-density occurrences displaying (+) molecular wobble and high magnitude moments of inertia about the evolutional axis. In identifying genomic building blocks, SYN-AI analyzed evolution force across single and multidimensional planes formed by the four evolution engines. Evolution force distribution across a one-dimensional evolution plane is illustrated in Figure 2. Where, evolution force associated with genomic building block formation was analyzed within genomic alphabets (1–4) of the parental bovine 14-3-3 docking gene DNA secondary code.
Figure 2.
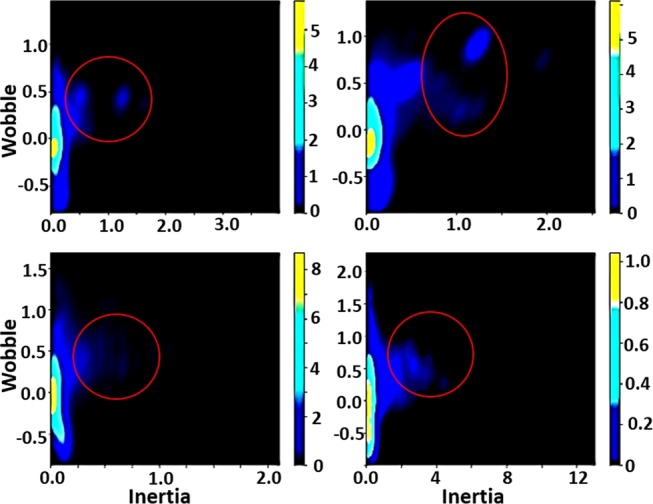
Analysis of evolution force utilizing the rotation model. Evolution force was evaluated in genomic alphabets (1–4) of the parental bovine brain 14-3-3 docking gene DNA secondary code. Evolution force was evaluated as described in ref (71).
Protein identity was assessed by aligning synthetic docking genes with the truncated B. taurus 14-3-3 ζ docking gene utilizing the Clustal Omega multiple sequence alignment tool.12 Synthetic proteins displayed 58.5 to 71.5 percent identity to the parental sequence characterized by stretches of high sequence identity between residues 100 and 130 and residues 154 and 180, Figure 3A. Phylogenetic analysis was performed utilizing Phylogeny.fr.13,14 Saliently, SYN-AI-1 and SYN-AI-2 diverged from the parental bovine gene forming novel 14-3-3 gene families. SYN-AI-3 was most closely related to the parental bovine 14-3-3 docking gene. However, each of the synthetic genes displayed significant branch distance from the parental gene, Figure 3B. We also performed phylogeny.fr blast, Figure 4. Notably, each synthetic protein was characterized by distinctive phylogenetic relationships. Whereby, SYN-AI-1 was most closely related to 14-3-3 protein zeta/delta of Ophiophagus hannah and Anolis carolinensis. The aforementioned phylogenetic relationships were characterized by score: 281 bits (718), expect: 7e-73, identities: 150/213 (70%), and positives: 165/213 (77%). SYN-AI-2 also displayed close identity to the aforementioned but with greater divergence. Wherein, the relationship was characterized by alignment score: 266 bits (679), expect: 2e-68, identities: 142/212 (66%), and positives: 156/212 (73%). Saliently, SYN-AI-3 exhibited close phylogenetic relationship to protein zeta/delta of Ovis aries and the parental B. taurus 14-3-3 ζ docking protein characterized by an alignment score: 281 bits (719), expect: 5e-73, identities: 149/213 (69%), and positives: 169/213 (79%).
Figure 3.
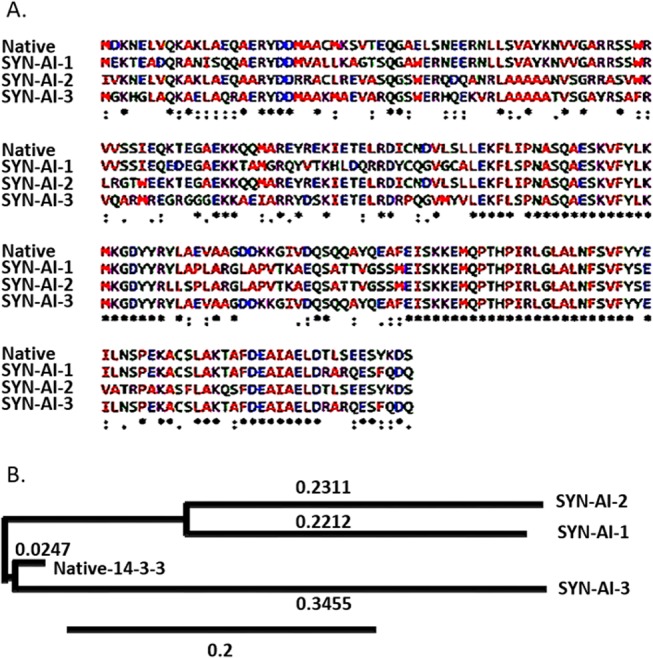
Sequence alignment and phylogenetic analysis of synthetic 14-3-3 docking proteins. Synthetic 14-3-3 docking proteins were aligned to truncated parental 14-3-3 ζ docking protein utilizing the Clustal Omega multiple sequence alignment tool, (A). Phylogenetic relationships between synthetic 14-3-3 docking proteins were compared to the parental utilizing Phylogeny.fr. (B).
Figure 4.
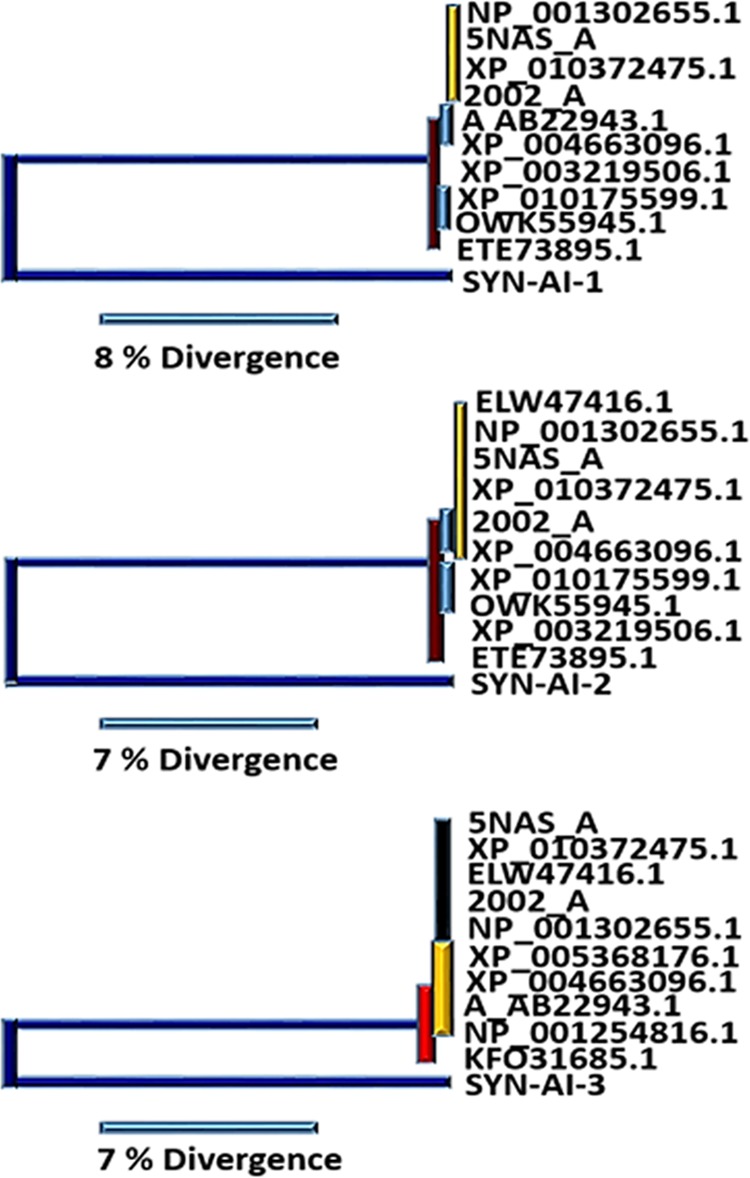
Phylogenetic analysis of synthetic proteins. Phylogenetic relationships characterizing synthetic 14-3-3 docking genes engineered utilizing SYN-AI were analyzed by performing a Phylogeny.fr blast. Phylogenetic tree depicting synthetic protein SYN-AI-1 (Top), SYN-AI-2 (Middle), and SYN-AI-3 (Bottom).
Three-dimensional structure of the synthetic protein was analyzed utilizing the I-TASSER suite, Zhang Laboratory University of Michigan. Structural analysis revealed that synthetic proteins conserved the 14-3-3 ζ architecture and surface as well as conserving volume of the ligand-binding site, Figure 5. SYN-AI-3 folded at the highest confidence characterized by a C-score of 1.54 and a resolution of 2.5 ± 1.9 Å. However, I-TASSER structural predictions of all three synthetic proteins were very reliable wherein synthetic proteins folded at an average C-score of 1.51. Saliently, the truncated parental 14-3-3 ζ docking protein folded at a similar confidence score of 1.55 with a TM-score of 0.93 ± 0.06 and a RMSD of 2.5 ± 1.9 Å. In addition, SYN-AI-1 and SYN-AI-3 were characterized by a TM-score of 0.93 ± 0.06 compared to SYN-AI-2 that was characterized by a TM-Score of 0.92 ± 0.06. Further, SYN-AI-1 and SYN-AI-3 were predicted at an RMSD of 2.5 ± 1.9 Å wherein SYN-AI-2 was predicted at an RMSD of 2.6 ± 1.9 Å.
Figure 5.
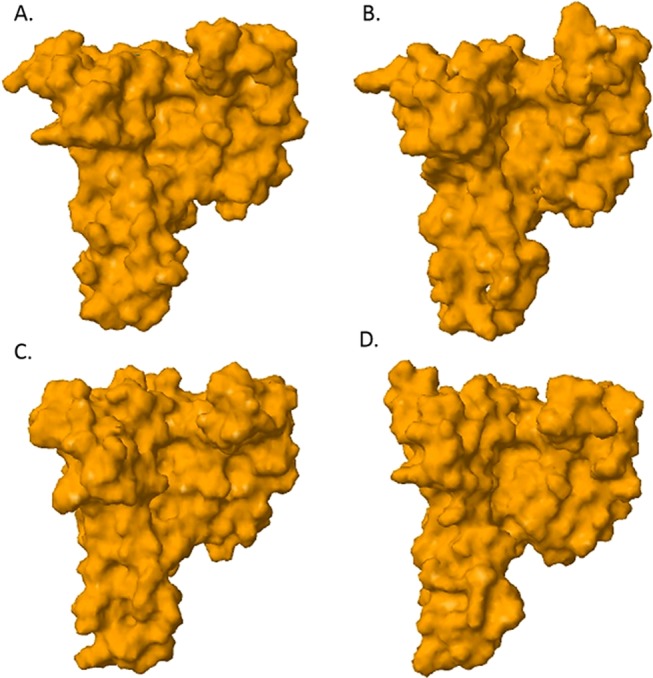
Three-dimensional structure predictions. The I-TASSER Suite, Zhang Laboratory University of Michigan was utilized to analyze the three-dimensional structure. A molecular surface image of the truncated parental 14-3-3 ζ docking protein predicted at a confidence of 1.55 and a resolution of 2.5 ± 1.9 Å (A). A molecular surface image of SYN-AI-3 predicted at a confidence of 1.54 and a resolution of 2.5 ± 1.9 Å (B). A molecular surface image of SYN-AI-1 predicted at a confidence of 1.51 and a resolution of 2.5 ± 1.9 Å (C). SYN-AI-2 predicted at a confidence of 1.48 and a resolution 2.6 ± 1.9 Å (D).
Synthetic protein–ligand-binding interactions were analyzed utilizing Cofactor and Coach. Notably, SYN-AI successfully conserved fungal toxin fusicoccin complex (FC) binding within the BS03 site of SYN-AI-3, Figure 6 (top left). The aforementioned was predicted at a confidence score of 0.40 and a resolution of 2.74 Å. Wherein, FC binding within parental bovine 14-3-3 docking protein was predicted at a slightly higher confidence of 0.45. The aforementioned analysis revealed that fusicoccin ligand–residue interactions were conserved with exception of N42 → V42 and V46 → A46 mutations. Fusicoccin formed hydrogen bonding interactions with residues V42, A46, K120, M121, P165, I166, and D213 and Van der Waals interaction with F117 of the synthetic protein amphipathic groove Figure 6 (Top Right). Whereby, the FC complex formed bonding interactions with the residue V42 at a distance of 0.285 nm, A46 at 0.375 nm, F117 at 0.223 nm, K120 at 0.357 nm, M121 at 0.306 nm, P165 at 0.332 nm, I166 at 0.300 nm, and residue D213 at 0.353 nm. Compared to the parental 14-3-3 docking protein, where fusicoccin formed bonding interactions with the residue N42 at a distance of 0.41 nm, V46 at 0.353 nm, F117 at 0.267 nm, K120 at 0.322 nm, M121 at 0.333 nm, P165 at 0.332 nm, I166 at 0.285 nm, and residue D213 at a distance of 0.42 nm. Notably, the FC complex ligand-binding site was fully conserved in SYN-AI-1, Figure 7. Where, fusicoccin formed bonding interactions with the residue N42 at a distance of 0.354 nm, V46 at 0.27 nm, F117 at 0.213 nm, K120 at 0.333 nm, M121 at 0.287 nm, P165 at 0.33 nm, I166 at 0.379 nm, and D213 at a distance of 0.386 nm. Comparatively, synthetic protein SYN-AI-2 conserved the fusicoccin ligand-binding site except V46 → A46 mutation, Figure 7. Whereby, fusicoccin formed bonding interactions with the residue N42 at a distance of 0.222 nm, A46 at 0.445 nm, F117 at 0.282 nm, K120 at 0.320 nm, M121 at 0.344 nm, P165 at 0.329 nm, I166 at 0.301 nm, and D213 at a distance of 0.343 nm.
Figure 6.
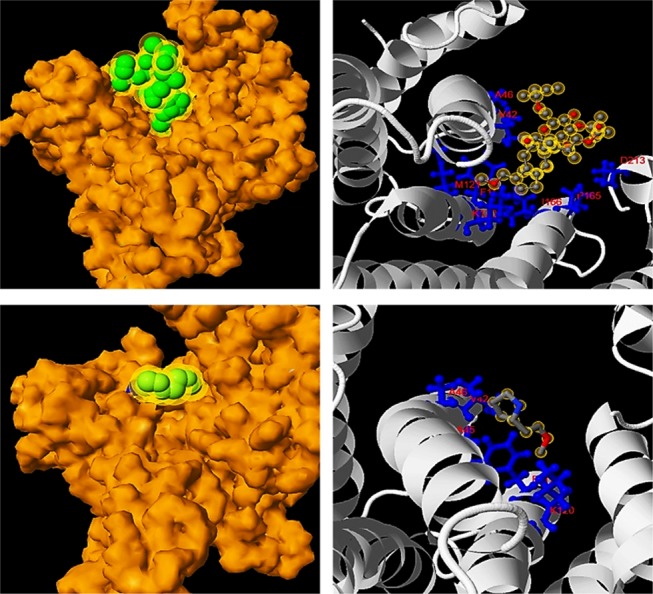
Analysis of protein–ligand interactions. Ligand and small-molecule interactions were analyzed utilizing Cofactor and Coach. Van der Waals surface image of fusicoccin binding at a resolution of 2.74 Å, (Top Left). Ligand–residue interactions between fusicoccin and SYN-AI-3 (Top Right). Van der Waals surface image of the small-molecule (2S)-(2)-methoxyethyl pyrrolidine at a resolution of 1.7 Å, (Bottom Left). Ligand–residue interactions between SYN-AI-3 and (2S)-(2)-methoxyethyl pyrrolidine (Bottom Right).
Figure 7.
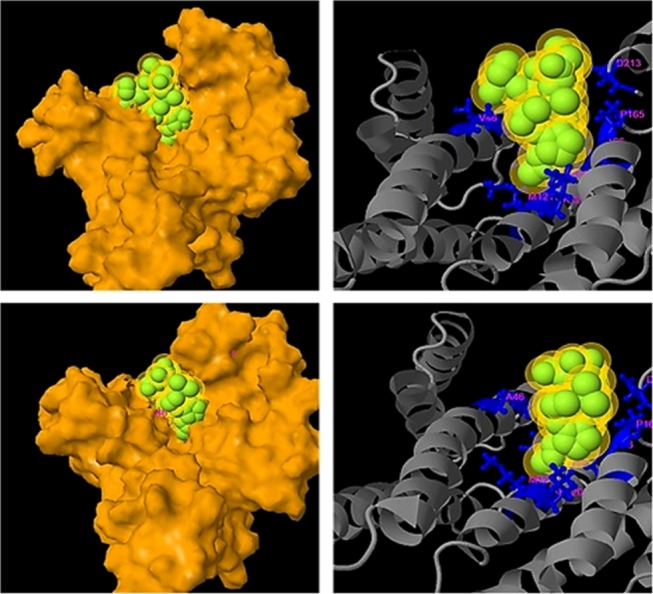
Analysis of ligand-binding interactions. Synthetic protein–ligand-binding interactions were analyzed utilizing Cofactor and Coach. A molecular surface image of fusicoccin SYN-AI-1 ligand-binding reaction predicted at a C-score of 0.46 (Top Left). SYN-AI-1 fusicoccin ligand–residue interactions (Top Right). A molecular surface image of fusicoccin SYN-AI-2 ligand-binding interaction predicted at a C-score of 0.41 (Bottom Left). SYN-AI-2 fusicoccin ligand–residue interactions (Bottom Right).
In addition to conserving FC complex ligand–residue interactions, synthetic evolution artificial intelligence conserved small molecule (2S)-2-(2-methoxyethyl) pyrrolidine ligand binding within the BS02 site, Figure 6 (Bottom). Wherein, Cofactor and Coach predicted the binding of the small molecule within the SYN-AI-3 BS02 site at a resolution of 1.7 Å. Our analysis revealed that the SYN-AI-3 BS02 ligand-binding site comprised N42 → V42, S45 → A45, and V46 → A46 mutations. Further, the analysis of ligand–residue interactions within the SYN-AI-3 BS02 site revealed that (2S)-2-(2-methoxyethyl) pyrrolidine interacts with the residue V42 at a distance of 0.264 nm, A45 at 327 nm, A46 at 0.391 nm, F117 at 0.252 nm, and the residue K120 at a distance of 0.397 nm. Comparatively, (2S)-2-(2-methoxyethyl) binding within the parental bovine 14-3-3 docking protein BS02 site was characterized by the interaction of residues N42 at 0.244 nm, S45 at 0.305 nm, V46 at 0.268 nm, F117 at 0.282 nm, and the residue K120 at a distance of 0.366 nm. Notably, synthetic protein SYN-AI-1 displayed full conservation of the BS02 ligand-binding site. Whereby, (2S)-2-(2-methoxyethyl) pyrrolidine interacted with the residue N42 at a distance of 0.261 nm, S45 at 0.273 nm, V46 at 0.270 nm, F117 at 0.250 nm, and the residue K120 at a distance of 0.385 nm.
Notably, synthetic evolution artificial intelligence successfully conserved protein–protein interactions, as corroborated by Coach and Cofactor analyses of BS01 and BS02 ligand-binding sites. Whereby, test probe I (TMLNLVSGRRR) was occupied and was deeply buried within BS01 and BS02 ligand-binding sites, Figure 8 (Top Left). Test probe I ligand interaction was predicted at 2.74 Å with a C-score of 0.31. Whereby, the probe interacted with residues H38, K41, V42, A45, A46, R56, R60, F117, K120, R127, Y128, P165, I166, G169, L172, N173, V176, and E180 of the synthetic protein amphipathic grove, Figure 8 (Top right). Test probe II (VTYSG) binding within the BS01 site was predicted at a C-score of 0.82 and at a resolution of 2.3 Å, Figure 8 (Bottom Left). Whereby, probe II interacted with residues K49, R56, R60, K120, R127, Y128, L172, V176, and E180 of the amphipathic grove, Figure 8 (Bottom Right). Saliently, synthetic evolution artificial intelligence accomplished full conservation of BS01 ligand-binding residues within synthetic protein SYN-AI-3 except a K49 → A49 mutation as well as an additional G169 residue contact. Comparatively, synthetic protein SYN-AI-1 displayed full conservation of ligand–residue interactions with the addition of the G169 residue contact. Whereby, SYN-AI-2 comprised K49 → A49 and R56 → A56 mutations in addition to the G169 contact.
Figure 8.
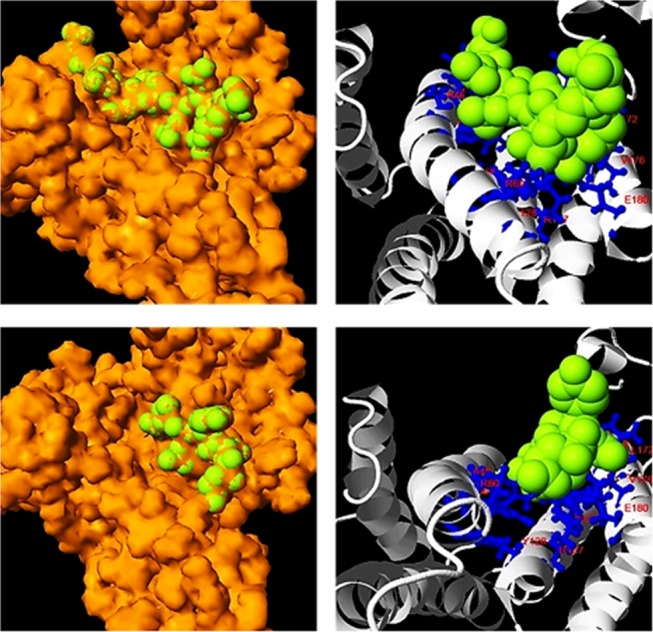
Analysis of protein–protein interaction sites. Protein interactions were analyzed utilizing Cofactor and Coach. Van der Waals surface image of probe I (TMLNLVSGRRR) buried within BS01 and BS02 ligand-binding sites, predicted at 2.74 Å (Top Left). Probe I ligand–residue interactions (Top Right). Van der Waals surface image of probe II (VTYSG) buried within the BS01 site, predicted at 2.3 Å (Bottom Left). Probe II ligand–residue interactions (Bottom Right).
To demonstrate SYN-AI’s ability to conserve protein allosteric effects, we analyzed parental and SYN-AI engineered 14-3-3 ζ docking proteins utilizing the elastic network model. Whereby, truncated parental and synthetic structures were predicted utilizing I-TASSER. ElNemo68,69 was utilized to perform the normal mode analysis, Figure 9. Normal mode analysis resulted in 107 modes, of which five (7–11) were low-frequency modes indicating a role in ligand binding and intraprotein communication. Saliently, Cα strains within synthetic proteins were similar to those occurring within the parental docking protein, as shown in Figure 9A. Whereby, mode 7 was characterized by a mean residue sample variance of σ2 = 2.62 × 10–4. Residue root mean square deviations of synthetic proteins also closely overlapped the parental 14-3-3 ζ docking protein, Figure 9B. When comparing RMSD of parental 14-3-3 ζ docking protein to synthetic proteins, there exists a miniscule variance of σ2 = 1.115 × 10–4 Å. Normal mode analysis also revealed that the frequency and collectivity of synthetic protein modes closely mirrored those of the parental 14-3-3 ζ docking protein, Figure 9C,D. The frequency of synthetic modes was characterized by a variance of σ2 = 0.202 from the parental docking protein. Wherein, synthetic proteins were characterized by a variance in the collectivity of σ2 = 9.593 × 10–3 from parental 14-3-3 ζ. The anisotropic network model ANM2.170 was utilized to analyze energy deformation and solvent accessibility. Synthetic protein energy deformation peak pattern and strength were analogous to that of the parental protein with a strong deformation peak ranging from residues 43–106 as well as residues 127–148, Figure 10A–D. Notably, energy deformation peaks correlate well with the location of the 14-3-3 ζ amphipathic groove as well as ligand-binding residues predicted by I-TASSER. Wherein, the strong peak at residue 127 suggests a significant role of the Van der Waal interaction with arginine in ligand-binding, in both the parental and synthetic proteins. Synthetic protein solvent accessibility also overlapped with that of the parental protein with little variation per residue, as illustrated in Figure 10E.
Figure 9.

Analysis of allosteric interactions. ElNemo was utilized to perform normal mode analysis of parental 14-3-3 ζ and synthetic proteins SYN-AI-1, SYN-AI-2, and SYN-AI-3. Whereby, we analyzed carbon α strain (A), root mean square deviation (B), mode frequency (C), mode collectivity (D).
Figure 10.

Analysis of amphipathic groove. Conservation of the 14-3-3 ζ amphipathic groove was confirmed by the anisotropic model, as described in (70). Whereby, ANM2.1 was utilized to analyze energy deformations occurring in the parental 14-3-3 ζ docking protein (A), SYN-AI-1 (B), SYN-AI-2, (C) and synthetic protein SYN-AI-3 (D). To further verify the conservation of the amphipathic groove, ANM2.1 was also utilized to predict parental and synthetic 14-3-3 ζ docking protein solvent accessibility (E).
3. Discussion
In the current study, we utilized SYN-AI to design a set of 14-3-3 docking proteins utilizing B. taurus 14-3-3 ζ docking protein as a parental template. Notably, SYN-AI is not a rational design technology but simulates evolution by evaluating evolution force associated with genomic building block formation and subsequently builds genes from scratch by randomly assembling the aforementioned. Our approach anticipates the evolution process by simulating DNA shuffling. Thusly, SYN-AI is advantageous in engineering functional genes, as anticipatory evolution has been shown to generate functional proteins with high efficacy.74 Evolution force was solved by overlapping gene sequences occurring over an orthologue sequence space with that of the parental template. Thereby, SYN-AI is able to analyze the evolutional character of DNA crossovers going back to “LUCA”. It is worth mentioning that our technology is limited by its dependence on the availability of PDB structural data. In the current study, crystal data was available for 228 of 245 B. taurus 14-3-3 ζ residues. Further, these crystal data contained gaps wherein the STRIDE structure-based analysis was able to generate structural data for 213 of the 228 residues. Thusly, SYN-AI protein engineering was limited to the available empirical data, whereby we showed the potential of our technology for gene and signal pathway engineering by synthesizing a set of truncated 14-3-3 docking proteins of 214 residues.
According to “The Fundamental theory of the Evolution Force”, we were able to analyze evolution force associated with genomic building block formation based upon four evolution engines, (i) evolution conservation, (ii) wobble, (iii) DNA binding state, and (iv) periodicity. Molecular biologists have long utilized evolution conservation as a tool when selecting mutable gene regions. Where, it has been assumed that highly evolutionarily conserved residues are critical to protein function.15−23 Thusly, SYN-AI is based upon the hypothesis that evolution conservation is an artifact of the evolution force. Classically, wobble has been defined by genetic diversity in the third codon with the conservation of the residue sequence.24−30 However, in fingerprinting the evolution force we expanded the definition of wobble to the acquisition of genetic diversity with the conservation of architecture. Thusly, allowing wobble to be viewed at all structural levels. For instance, supersecondary structures such as helices, turns, and β sheets are expressed in genetically diverse species yet retain basic architecture and function. A further example of wobble at a higher structural level is that of bipedal animals. Although the aforementioned are genetically diverse, anatomical structure and the architecture are reserved across species. Also when constructing SYN-AI, we assumed that evolution force interacts at the matter–energy interface via DNA crossovers. Thusly, we were able to evaluate the interaction of the evolution force at the matter–energy interface by analyzing DNA binding states. Wherein, DNA binding states measure DNA crossover selectivity in respect to the recombinant pool and are a function of Gibb’s free energy associated with DNA base stacking interactions.8,31−33 The final evolution engine we considered was periodicity. We assumed that nature has a tendency to repeat successful structures that promote the survival of an organism. Thereby, evolution force at the molecular level is a function of sequence periodicity.34−36
In the current study, we have demonstrated that genomic building blocks can be identified across multiple genomes by analyzing evolution force and exploited to write genes from scratch. Wherein, by transforming a parental gene template into DNA secondary and tertiary codes based on hierarchical structure levels, SYN-AI was able to fast-forward the evolution process. The aforementioned allowed the identification of genomic building blocks by characterization of evolution force and allowed for intelligent gene design by a Legos-like swapping of genetic material in agreement with the Domain Lego principle.1−3 Notably, SYN-AI generated synthetic proteins displaying high sequence identity to naturally occurring 14-3-3 docking proteins suggesting that the AI successfully simulated the evolution process allowing divergence from the parental 14-3-3 ζ docking gene while evolutionarily conserving 14-3-3 global architecture. We were able to corroborate the aforementioned by performing phylogeny.fr blast and established that each synthetic protein comprised of diverse phylogenetic relationships as characterized by diverse phylogenetic trees. Synthetic proteins also displayed significant branch distance from one another further confirming that SYN-AI fast-forwarded the evolution process. Wherein, each synthetic protein diverged into a different evolutional pathway. Saliently, despite performing ∼300 million DNA crossovers within genomic alphabet comprising the 14-3-3 DNA secondary code, Clustal Omega sequence alignments of synthetic proteins were characterized by stretches of high sequence identity wherein no genetic diversity was accomplished by SYN-AI. Notably, the aforementioned corroborates that our natural selection protocols implemented into SYN-AI successfully conserved slow evolving regions of genes. Saliently, the resistance of these regions to mutation suggests that they are essential to cellular function.37−39
In addition to fast-forwarding the evolution process, SYN-AI successfully conserved the 14-3-3 docking protein architecture. Whereby, I-TASSER three-dimensional structural analysis revealed that global 14-3-3 docking protein architecture was conserved in all three synthetic proteins. Based on molecular dynamics simulation data in conjunction with previously discussed phylogenetic data, we validate our hypothesis of evolution force effects on genomic building block formation by proof-of-concept. Further corroborating our proof-of-concept, SYN-AI evolutionarily conserved ligand-binding sites and ligand–residue interactions. Notably, analysis by Coach and Cofactor40 confirmed that SYN-AI conserved small-molecule binding as demonstrated by the conservation of (2S)-2-(2-methoxyethyl) pyrrolidine binding within the BS02 site. While conserving small-molecule binding, synthetic evolution accomplished significant modification of the SYN-AI-3 BS02 ligand-binding site. As characterized by mutation of three of the five residues participating in the binding of (2S)-2-(2-methoxyethyl) pyrrolidine. SYN-AI successfully altered positioning and conformation of the molecule within the binding site. Contrary to SYN-AI-3, synthetic protein SYN-AI-1 was characterized by full conservation of the BS02 binding site. Wherein, the artificial intelligence preserved parental ligand–residue interactions. Saliently, conformation and positioning of (2S)-2-(2-methoxyethyl) pyrrolidine within the SYN-AI-1 BS02 ligand-binding site was altered due to changes in binding groove volume. As a result of the modification of global protein architecture due to the evolution process. The aforementioned is corroborated by phylogenetic analysis. Wherein, SYN-AI-1 is significantly diverged from parental bovine brain 14-3-3 docking protein in comparison to synthetic protein SYN-AI-3. Notably, the ability of synthetic evolution artificial intelligence to evolutionarily conserve small molecule binding sites while altering conformation and binding affinities of ligands is significant as small molecules stabilize and inhibit 14-3-3 protein–protein interactions that play a role in neurodegenerative diseases and cancer.41
Contrary to the surprising level of divergence achieved in the BS02 ligand-binding site, SYN-AI evolutionarily conserved amphipathic interfaces and ligand-binding residues within the BS01 and BS03 ligand-binding sites. Whereby, Coach and cofactor confirmed the conservation of fusicoccin binding within all three synthetic proteins engineered utilizing synthetic evolution artificial intelligence. Fusicoccin ligand–residue interactions were also conserved with exception of N42 → V42 and V46 → A46 mutations. In agreement with our previous experiments, modifications in global protein architecture altered the conformation of fusicoccin within the amphipathic groove signifying that fusicoccin exhibited an altered binding affinity for the ligand-binding site. Notably, SYN-AI’s ability to evolutionarily conserve the FC complex binding site is significant in that the complex is responsible for activating H+ pumping across the plasma membrane.42 In plants, the interaction of the FC complex with the 14-3-3 docking protein activates KAT1 channels and is responsible for cell growth by regulating diffusion through K+ channels.43 Saliently, the FC 14-3-3 complex also regulates defense responses in tomato plants.44 In addition to conservation of the BS02 and BS03 sites, analysis of protein probe localizations confirmed the conservation of the BS01 ligand-binding site with exception of a K49 → A49 mutation and an additional G169 residue contact. The aforementioned corroborates evolutional conservation of protein–protein interactions in synthetic proteins.
Notably, altered positioning and conformation of small molecules, protein probes, and the FC complex within synthetic protein BS01, BS02, and BS03 sites substantiate our assumption that synthetic proteins designed in this study display the potential for altered PPI. The aforementioned is significant in that the interaction of globular domains of protein interaction partners within the 14-3-3 amphipathic grove regulates stress signaling proteins such as ERK, MAPK, JNK, and p38 MAPK as well as growth and cell cycle regulators raf, PI3K, and cdc25 phosphatase.45−49,61−67 We confirmed the conservation of these ligand-binding interactions as well as confirmed the conservation of intraprotein communication by analyzing allosteric effects utilizing the elastic network model. Whereby, normal mode analysis of parental and synthetic 14-3-3 ζ docking proteins was performed utilizing ElNemo and ANM2.1. The analysis of mode 7 indicated that there existed little variance in Cα strain occurring in parental 14-3-3 ζ and synthetic proteins. These data suggest that synthetic proteins are energetically stable and corroborate the validity of I-TASSER structure prediction. The aforementioned are corroborated by RMSD results, wherein synthetic proteins exhibited little variance from the parental 14-3-3 ζ protein. Thusly, SYN-AI was able to evolve protein sequence and local structures without disrupting the global protein architecture. Notably, the aforementioned was accomplished while conserving mode frequency and collectivity suggesting that there exist little variance in residue potential energies during structural transitioning from parental to synthetic 14-3-3 ζ proteins. This is prominent due to the many ligand and signal pathway interactions occurring within the 14-3-3 ζ amphipathic groove, as corroborated by the presence of 107 modes indicated by ElNemo. Our data suggests that SYN-AI conserved allosteric interactions regulating signal transduction pathways. The conservation of the 14-3-3 ζ amphipathic groove is corroborated by solvent accessibility data indicating only infinitesimal differences in protein hydrophobicity. Notably, we demonstrate the conservation of the binding of the R18 protein peptide within the amphipathic groove as well as the conservation of a “hug and squeeze” mechanism. Where, the left and right torso of the 14-3-3 monomer flex closed and secured the R18 protein peptide in the amphipathic groove. The open configuration is shown in Figure 11A,C and the closed configuration in Figure 11B,D. Notably, we also successfully dimerized the SYN-AI-1 monomer and engineered a functional 14-3-3 ζ dimer, while maintaining the “bend and flex” mechanism present in the WT dimer, as illustrated in Figure 12. Whereby, we show the synthetic 14-3-3 ζ dimer in both the open and closed positions. Modes 7–11 were also conserved in SYN-AI-generated dimmers. We have demonstrated that SYN-AI conserved allosteric communications in the monomeric form of the 14-3-3 ζ protein as well as low-frequency vibrations occurring in the 14-3-3 ζ dimeric form, thusly validating the identification of genomic building blocks by characterization of evolution force.
Figure 11.
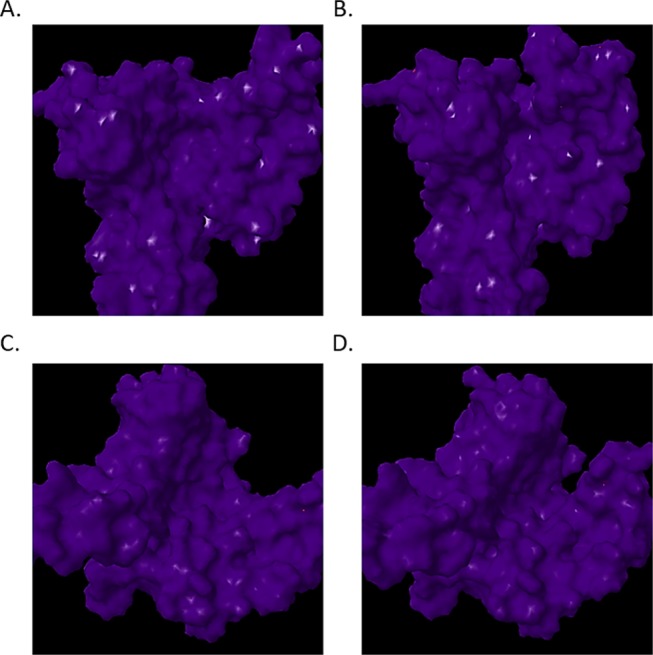
Hug and squeeze mechanism. The elastic network model was utilized to perform normal mode analysis. Low-frequency vibrations occurring within parental and synthetic 14-3-3 ζ docking proteins were analyzed utilizing elNemo. Frontal view of mode 7 open configuration (A), frontal view of mode 7 closed configuration (B), view of the open configuration down amphipathic groove (C), view of the closed configuration down amphipathic groove (D).
Figure 12.
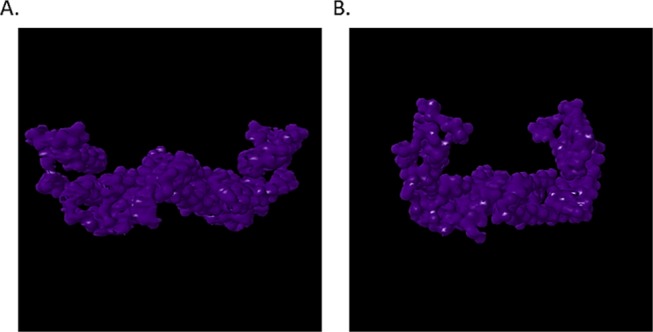
Synthetic 14-3-3 ζ dimer formation. Dimerization of the SYN-AI-1 14-3-3 monomer was predicted utilizing COTH,73 Zhang Laboratory University of Michigan. The normal mode analysis was performed utilizing ElNemo and graphics generated utilizing the Jena3D Viewer. The open configuration of the Mode 7 bend and flex mechanism is illustrated in (A), whereby the closed configuration is shown in (B).
Saliently, our study relies heavily on the accuracy of computational structure prediction. However, our results are corroborated by the parental 14-3-3 ζ structure reported in ref (57). Whereby, as an experimental control, we overlapped the native 14-3-3 ζ crystal structure with the I-TASSER-predicted PDB structure. As illustrated in Figure 13, the I-TASSER 14-3-3 ζ structure prediction is identical to the reported crystal structure. Wherein, we utilized the SuperPose server72 to overlap the PDB structure reported in ref (57) to the I-TASSER-predicted structure, with only minor deviations present in the coiled coil regions. It is worth mentioning that this level of accuracy is not guaranteed for all subsequent predictions performed in this study. However, the claims presented herein are supported by sufficient scientific data to corroborate the experimental controls and I-TASSER structure prediction of synthetic proteins. Wherein, we can confidently conclude that SYN-AI successfully engineered a set of functional 14-3-3 ζ docking genes from scratch.
Figure 13.
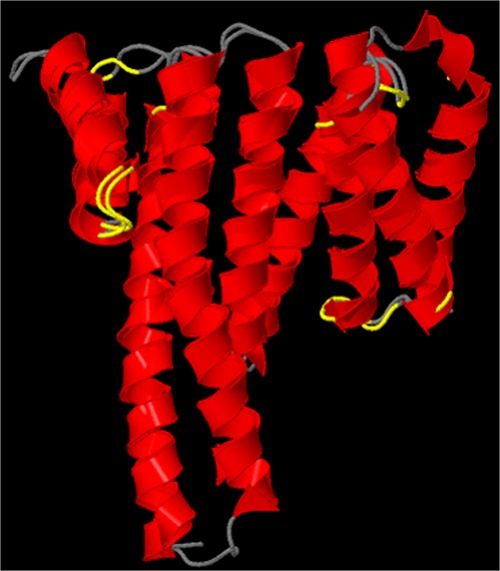
Superimposition of experimental and predicted 14-3-3 ζ structures. The SuperPose server was utilized to compare the native B. taurus 14-3-3 crystal structure reported in57 to the I-TASSER-predicted structure. The native crystal structure covered residues 1–228 with two gaps of 5 and 7 amino acids, respectively. Superimposition of the parental and synthetic proteins was performed with a minimum sequence similarity of 25% identity, a similarity cutoff of RMSD of 2.0 Å, a dissimilarity cutoff of RMSD of 3.0 Å, and a dissimilar subdomain cutoff of 7 residues.
According to Wang, mutations to residues L172, V176, and L220 drastically interrupted the interaction of 14-3-3 ζ with raf1 kinase, thusly effecting the promotion of prosurvival by the docking protein. Although our study does not focus on the role of 14-3-3 ζ on apoptosis, L220 is located in the C′ terminal, thusly is truncated in SYN-AI engineered docking proteins. In considering effects of mutation on protein interaction, we note that in vivo 14-3-3 ζ forms both a tetrameric complex comprised of a 14-3-3 ζ homodimer and two serotonin N-acetyl transferase (AANAT) monomers as well forms an octameric biological complex with AANAT. Of the two complexes, inter-residue C-α strains are lower in the octameric complex, suggesting that it is more thermodynamically stable and more likely to occur in nature. The identical overlap of SYN-AI-engineered and parental 14-3-3 ζ docking proteins allowed us to investigate the effects of C′-terminal truncation by mutating rotamers of the parental 14-3-3 ζ serotonin N-acetyl transferase octameric complex utilizing UCSF Chimera.75,76 Rotamers were mutated to those characterizing SYN-AI-1, and residues 215−228 deleted from chains A through D of the biological complex. To obtain a good structure, energy minimization was performed on each chain of the octamer as reported in the Methods section. When analyzing hydrogen bonding, we find that the native octameric complex is characterized by 40 hydrogen bond interactions with conserved [RRHTLP] residues 28–33 of ovine AANAT. The AANAT motif is similar to canonical motifs [RSXpSXP; RXY/FXpSXP].75 Notably, the SYN-AI-1 ζ biological complex was characterized by 55 hydrogen bond interactions with the [RRHTLP] motif, thusly displayed increased binding affinity. As illustrated in Figure S1A, native B. taurus 14-3-3 ζ C-terminal residues 215–228 (cyan) contribute two hydrogen bonds (circled white) between asparagine 224 and histidine 30 of the [RRHTLP] motif (green) within each 14-3-3 ζ AANAT ligand interaction. These hydrogen bond interactions are lost in the SYN-AI-1 octameric complex. However, N-acetyl transferase is secured by alternate hydrogen bond formations between the [RRHTLP] motif and SYN-AI-1 characterized by hydrogen bond interactions between proline 33 and lysine 49, TPO 31 and arginine 56, arginine 29 and glutamate 180, as well as glycine 214 of SYN-AI-1 and arginine 89 of serotonin N-acetyl transferase, Figure S1B. The increase in binding affinity in SYN-AI engineered 14-3-3 ζ opposed to the loss of function reported in Fu76 is due to Fu intentionally performing point mutations characterized by dissimilar residues, thusly disallowing alternate hydrogen bond formations, as acceptor atoms were conformationally blocked by the C′-terminal motif. By doing so, Fu was able to block the promotion of prosurvival by 14-3-3 ζ and demonstrate the potential of mutated lines for treating cancer. Contrarily, SYN-AI deleted the entire C′-terminal helix allowing for conformational shift of serotonin N-acetyl transferase within the binding pocket that led to alternative hydrogen bond formations with adjacent α-helices located within the amphipathic groove (Figure S1B, Table S1). Thusly, allowing stable but modified hydrogen bonding between the [RRHTLP] motif of serotonin N-acetyl transferase and SYN-AI-1 monomers. Modifications were not restricted to hydrogen bonding interactions occurring with the [RRHTLP] motif but were seen globally throughout the biological complex as AANAT formed 218 hydrogen bond interactions within the biological complex Figure S2, compared to 145 hydrogen bond interactions within the native 14-3-3 ζ N-acetyl transferase complex. It is unfeasible to analyze the effects of synthetic evolution over 130 14-3-3 ζ protein interaction partners. However, it is safe to assume that the effects of synthetic evolution are pathway sensitive and depend upon the protein interaction partner.
4. Conclusions
Based upon our findings, SYN-AI was able to engineer genes from scratch by identifying evolution force associated with genomic building block formation as well as by applying natural selection protocols that mimic those that occur in nature. The evidence reported herein suggests that synthetic evolution methodologies are excellent tools for the intelligent design of genes and should offer an alternative to rational design approaches. Notably, SYN-AI technology may be expanded allowing for the design of genomes at a very high resolution compared to current technologies that are based on the exchange of very large segments of genomic DNA. SYN-AI’s ability to write DNA code from scratch at high resolution opens an endless potential for scientific exploration and gene design that may be applied to the evolution of any gene, dependent on the availability of PDB structural data. The ability to write DNA code at a high resolution also allows the rewiring of cell signal pathways. Saliently, SYN-AI synthetic evolution technology explores multiple evolution pathways based on researcher’s experimental parameters, whereby each pathway results in the formation of an alternate gene or gene family dependent on the mutability of the sequence space. As SYN-AI technology simulates evolution, outcomes also rely on randomness, whereby under identical experimental parameters, there exists a possibility of exploring a diverse evolution pathway. Thusly, SYN-AI offers an excellent opportunity for the discovery of new chemistries that have potential applications in the treatment of cancer and other diseases as well as allow for the design of industrial genes.
5. Methods
5.1. High-Performance Computing
SYN-AI was performed utilizing the Stampede 2 supercomputer located at the Texas Advanced Computing Center, University of Texas. Experiments were performed in the normal mode utilizing SKX compute nodes comprising 48 cores on two sockets with a processor base frequency of 2.10 GHz and a max turbo frequency of 3.70 GHz. Each SKX node comprises 192 GB RAM at 2.67 GHz with 32 KB L1 data cache per core, 1 MB L2 per core, and 33 MB L3 per socket. Each socket can cache up to 57 MB with local storage of 144/tmp partition on a 200 GB SSD.
5.2. Simulating Evolution
5.2.1. Identification and Isolation of Genomic Building Blocks
SYN-AI analyzed evolution force associated with genomic building block formation across an orthologous sequence space comprising genes occurring at a homology threshold of >80 percent identity to parental bovine brain 14-3-3 docking gene. The orthologous sequence space comprised 2.5 × 106 bp of genetic material. Evolution force was analyzed by transforming the bovine brain 14-3-3 docking gene into a DSEC and performing 3 × 108 DNA crossovers within genomic alphabets. DNA hybridization partners were randomly selected across orthologous sequence space. Evolution at the matter–energy interface was simulated by performing DNA hybridizations in a buffering solution of 3 mM Mg2+ and 1.2 mM dNTP at 328.15° kelvin.8 Gibb’s free energy was calculated according to Owczarzy50 and a penalty assessed for DNA base pair mismatches.
Evolutionarily fit DNA crossovers were selected by applying natural selection protocols. Neural networks limited selection to DNA crossovers based upon Gibb’s free energy. Genomic building blocks were passed through pattern recognition filters that removed sequences displaying low sequence homology to the parental bovine brain 14-3-3 docking gene. The selection was limited to DNA crossover instances comprising evolutionarily favored mutations by quantum-normalized Blosum80 mutation frequency-based neural networks. Natural selection was further accomplished by limiting selection to sequences characterized by (+) molecular wobble vectors. Subsequently, genomic building block libraries were constructed by quantum-normalized neural networks that limited selection to DNA crossovers characterized by high magnitude of evolution. Evolution force was enumerated over single and multidimensional evolution planes, as described in ref (71).
5.2.2. Evaluation of Evolution Force Associated with Genomic Building Block Formation
Evolution force associated with genomic building block formation was solved utilizing the rotation model, as described in ref (71). Evolution force τϵ was solved as a function of inertial moments Ic about the evolution axis and molecular wobble, eq 1. Whereby, inertial vector Ic is a function of evolution conservation ϵ and variance r from a recombinant pool comprising a total 3 × 108 DNA crossovers, eq 2. Evolution conservation ϵ was solved as a function of DNA and protein evolution vectors (ϵDNAc, ϵPro), eq 3, and molecular wobble ωm likewise solved as a function of evolution vectors (ϵDNAc, ϵPro), eq 4. The aforementioned evolution vectors are functions of DNA and protein similarity vectors (Xi, Xj) weighted by the recombinant pool, eqs 5 and 6. Whereby, evolution weights (Wd, Wp) describe similarity of the recombinant pool to the parental sequence in respect to DNA and protein primary sequence. Evolution weights are a function of mean DNA μsDNA and mean protein μs evolution vectors, eqs 7 and 8. Wherein, μsDNA and μs were solved by summation of genomic building block (GBB) similarity functions (Xi/n, Xj/n) occurring across the orthologue sequence space (sspacer) divided by the total number of DNA crossovers N. Where, sspacer comprised 2.5 × 106 bp of the genetic material.
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
| 8 |
5.2.3. Engineering Synthetic Supersecondary Structures
Parental supersecondary structures were identified by partitioning the bovine brain 14-3-3 gene into a DNA tertiary code followed by analysis utilizing STRIDE51 knowledge-based secondary structure algorithms. Evolution was performed by ligation of genomic building blocks randomly selected from genomic alphabet libraries encompassing 5′ and 3′ terminals of parental structures. A cleaving algorithm was utilized to remove 5′ and 3′ prime overhangs from synthetic supersecondary structures. Natural selection was performed by limiting selection to naturally occurring mutations utilizing Blosum80 mutation frequency algorithms. SYN-AI also accomplished natural selection by imposing a secondary structure homology threshold >90 percent identity, wherein synthetic sequences were aligned with parental 14-3-3 secondary sequences. A standalone version of PSIPRED 4.052 was utilized to evaluate secondary structure. Synthetic supersecondary structures were stored in DTER libraries for writing DNA code.
5.2.4. Writing DNA Code from Scratch
DNA code was written from scratch by walking the DTER followed by random selection and ligation of synthetic supersecondary structures stored in genomic alphabet libraries. SYN-AI constructed a library of 1 × 107 genes that was passed through a set of neural networks that evaluated the closeness of synthetic protein structural states to native states. Wherein, SYN-AI set a minimal closeness threshold of >90 percent identity according to.71 A subsequent selection limited supersecondary structures to those characterized by naturally occurring mutations. The aforementioned was performed utilizing BLOSUM8053,54 mutation frequency algorithms. A further round of natural selection restricted selection to synthetic 14-3-3 docking proteins characterized by mean secondary structure identities within the top quantile. Structurally conserved and functional 14-3-3 docking proteins were selected by a final natural selection protocol that evaluated the closeness of protein active sites and hydrophobic interfaces to the parental bovine 14-3-3 docking protein. Wherein, selection was limited to synthetic proteins characterized by closeness thresholds of >90 percent identity.
5.2.5. Analysis of Synthetic Proteins
Sequence homology of synthetic proteins to parental bovine 14-3-3 docking protein was analyzed utilizing the Clustal Omega multiple sequence alignment tool.12 Wherein, phylogenetic analysis was performed utilizing Phylogeny.fr. Furthermore, we analyzed synthetic protein three-dimensional structure utilizing the I-TASSER suite.55 Wherein, protein–protein interaction and ligand-binding sites were analyzed utilizing Cofactor and Coach.56
5.2.6. Building SYN-AI-1 Biological Complex
SYN-AI-1 was overlapped with the native 14-3-3 ζ monomer utilizing the Superpose server. Upon the conformation of nearly identical overlap, the SYN-AI-1 N-acetyl transferase octameric complex was built utilizing UCSF Chimera76 with Biological Complex 2 reported in ref (75) as the template. Rotamers were mutated to those characterizing SYN-AI-1 and C′-terminal residues 215–228 deleted from chains A–D. Energy minimization of each chain within the complex was performed utilizing AMBER78 with 100 steepest decent steps with a step size of 0.02 Å and 10 conjugant gradient steps at a step size of 0.02 Å. The process was repeated until a good structure was obtained. Energy minimization was performed utilizing the steric method. Charges were added to standard residues utilizing the Gasteiger method and computed utilizing ANTECHAMBER.77 Upon obtaining a good structure, energy minimization was performed upon the entire biological complex utilizing the hydrogen bond method with 400 steepest decent steps and 20 conjugant gradient steps. Ions were removed from the structure, and the structure solvated utilizing AMBER with a shell of 12.0 Å applying the hydrogen bond and TIP3PBOX method. Hydrogen bond interactions with residues 28–33 of the serotonin N-acetyl transferase [RRHTLP] motif were analyzed with relaxed constraints of 4.0 Å and 20 degrees. Clashes and contacts were identified with Van der Waals overlap distances of greater or equal to 6.0 Å, with a correction of −5.0 Å for potential hydrogen bonding pairs. Overlaps and clashes within four bonding pairs were excluded.
Acknowledgments
This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562. The authors acknowledge the Texas Advanced Computing Center (TACC) at the University of Texas at Austin for providing HPC resources that contributed to research results reported within this paper. URL: http://www.tacc.utexas.edu
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b03100.
UCSF chimera images of B. taurus 14-3-3 ζ and SYN-AI-1 ζ serotonin N-acetyl transferase biological complex hydrogen bond formations within conserved RRHTLP motif, UCSF chimera images of SYN-AI-1 ζ hydrogen bond formation with serotonin N-acetyl transferase, table of hydrogen bond formations within conserved RRHTLP motif of B. taurus 14-3-3 ζ serotonin N-acetyl transferase biological complex, table of hydrogen bond formations within conserved RRHTLP motif of SYN-AI-1 ζ serotonin N-acetyl transferase biological complex (PDF)
The author declares no competing financial interest.
Supplementary Material
References
- Bork P.; Doolittle R. Drosophilia kelch motif is derived from a common enzyme fold. J. Mol. Biol. 1994, 236, 1277–1282. 10.1016/0022-2836(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Doolittle R. The multiplicity of domains in proteins. Annu. Rev. Biochem. 1995, 64, 287–314. 10.1146/annurev.bi.64.070195.001443. [DOI] [PubMed] [Google Scholar]
- Henikoff S.; Greene E. A.; Pietrokovski S.; Bork P.; Attwood T. K.; Hood L. Gene families: the taxonomy of protein paralogs and chimeras. Science 1997, 278, 609–614. 10.1126/science.278.5338.609. [DOI] [PubMed] [Google Scholar]
- Schubert I. Chromosome evolution. Curr. Opin. Plant Biol. 2007, 10, 109–115. 10.1016/j.pbi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Zhang J. Evolution by gene duplication: an update. Trends Ecol. Evol. 2003, 18, 292–298. 10.1016/S0169-5347(03)00033-8. [DOI] [Google Scholar]
- Woese C. The universal ancestor. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6854–6859. 10.1073/pnas.95.12.6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glansdorf N.; Xu Y.; Labedan B. The Last Universal Common Ancestor: emergence, constitution and genetic legacy of an elusive forerunner. Biology 2008, 3, 29 10.1186/1745-6150-3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis L. Engineering cellulosic bioreactors by template assisted DNA shuffling and in vitro recombination (TADSir). Biosystems 2014, 124, 95–104. 10.1016/j.biosystems.2014.06.007. [DOI] [PubMed] [Google Scholar]
- Freeman A.; Morrison D. 14-3-3 Proteins: Diverse functions in cell proliferation and cancer progression. Semin. Cell Dev. Biol. 2011, 22, 681–687. 10.1016/j.semcdb.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermeking H. The 14-3-3 cancer connection. Nat. Rev. Cancer 2003, 3, 931–942. 10.1038/nrc1230. [DOI] [PubMed] [Google Scholar]
- Kleppe R.; Martinez A.; Døskelanda S.; Haavik J. The 14-3-3 proteins in regulation of cellular metabolism. Semin. Cell Dev. Biol. 2011, 22, 713–719. 10.1016/j.semcdb.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Sievers F.; Wilm A.; Dineen D.; Gibson T. J.; Karplus K.; Li W.; Lopez R.; McWilliam H.; Remmert M.; Söding J.; Thompson J. D.; Higgins D. G. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539–544. 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dereeper A.; Guignon V.; Blanc G.; Audic S.; Buffet S.; Chevenet F.; Dufayard J.-F.; Guindon S.; Lefort V.; Lescot M.; Claverie J.-M.; Gascuel O. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, 465–469. 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dereeper A.; Audic S.; Claverie J.-M.; Blanc G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol. Biol. 2010, 10, 8–14. 10.1186/1471-2148-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boffelli D.; McAuliffe J.; Ovcharenko D.; Lewis K. D.; Ovcharenko I. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science 2003, 299, 1391–1394. 10.1126/science.1081331. [DOI] [PubMed] [Google Scholar]
- Capra J.; Singh M.; John A. Predicting functionally important residues from sequence conservation. Bioinformatics 2007, 23, 1875–1882. 10.1093/bioinformatics/btm270. [DOI] [PubMed] [Google Scholar]
- Capra J.; Laskowshi R.; Thornton J.; Singh M.; Funkhouser T. Predicting protein ligand sites by combining sequence conservation and 3D structure. PLoS Comput. Biol. 2009, 5, e1000585 10.1371/journal.pcbi.1000585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper G.; Brown C. Qualifying the relationship between sequence conservation and molecular function. Genome Res. 2008, 18, 201–205. 10.1101/gr.7205808. [DOI] [PubMed] [Google Scholar]
- Hardison R. Comparative Genomics. PLoS Biol. 2003, 2, 156–160. 10.1371/journal.pbio.0000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingles-Prieto A.; Ibarra-Molero B.; Delgado-Delgado A.; Perez-Jimenez R.; Fernandez J.; Gaucher E.; Sanchez-Ruiz J.; Gavira J. Conservation of protein structure over four billion years. Structure 2013, 21, 1690–1697. 10.1016/j.str.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrie D.; Petrov D. Comparative population genomics: power and principles for the inference of functionality. Trends Genet. 2014, 30, 133–139. 10.1016/j.tig.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting C. Biological function in the twilight zone of sequence conversion. BMC Biol. 2017, 71. 10.1186/s12915-017-0411-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhold N.; Sander O.; Dominques F. S.; Sommer L. Local function conservation in sequence and structure Space. PLoS Comput. Biol. 2008, 4, e1000105 10.1371/journal.pcbi.1000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulmer M. Coevolution of codon usage and transfer RNA abundance. Nature 1987, 325, 728–730. 10.1038/325728a0. [DOI] [PubMed] [Google Scholar]
- Bulmer M. The selection-mutation-drift theory of synonymous codon usage. Genetics 1991, 129, 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. Codon – anticodon pairing: The wobble hypothesis. J. Mol. Biol. 1966, 19, 548–555. 10.1016/S0022-2836(66)80022-0. [DOI] [PubMed] [Google Scholar]
- Diwan D.; Agashe D. Wobbling forth and drifting back: The Evolutionary History and Impact of Bacterial tRNA Modifications. Mol. Biol. Evol. 2018, 35, 2046–2059. 10.1093/molbev/msy110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y.; Li Q. Mutation and selection on the wobble nucleotide in tRNA anticodons in marine bivalve mitochondrial genomes. PLoS One 2011, 6, e16147 10.1371/journal.pone0016147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong K. L.; Wong J. T. Anticodon and wobble evolution. Gene 2004, 333, 169–177. 10.1016/j.gene.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Xia X. Mutation and selection on the anticodon of tRNA genes in vertebrate mitochondrial genomes. Gene 2005, 345, 13–20. 10.1016/j.gene.2004.11.019. [DOI] [PubMed] [Google Scholar]
- Boger D.; Fink B.; Brunette S.; Winston T.; Hedrick M. A simple, high-resolution method for establishing DNA binding affinity and sequence selectivity. J. Am. Chem. Soc. 2001, 123, 5878–5891. 10.1021/ja010041a. [DOI] [PubMed] [Google Scholar]
- Moore G.; Maranas C.; Lutz S.; Benkovic J. Predicting crossover generation in DNA shuffling. Theor. Biol. 2000, 219, 9–17. 10.1016/S0022-5193(02)93102-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore G.; Maranas C. Predicting out-of-sequence reassembly in DNA shuffling. J. Theor. Biol. 2002, 219, 9–17. 10.1016/S0022-5193(02)93102-4. [DOI] [PubMed] [Google Scholar]
- Bellesia G.; Jewett A.; Shea J. Sequence periodicity and secondary structure propensity in model proteins. Protein Sci. 2009, 19, 141–154. 10.1002/pro.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H.; Buckwalter B.; Shieh H.; Hecht M. Periodicity of polar and nonpolar amino acids is the major determinant of secondary structure in self-assembling oligomeric proteins. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 6349–6353. 10.1073/pnas.92.14.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonov H.; Arkin I. A periodicity analysis of transmembrane helices. Bioinformatics 2005, 21, 2604–2610. 10.1093/bioinformatics/bti369. [DOI] [PubMed] [Google Scholar]
- Crochet P. A.; Desmarais E. Slow rate of evolution in the mitochondrial control region of gulls (Aves: Laridae). Mol. Biol. Evol. 2000, 17, 1797–1806. 10.1093/oxfordjournals.molbev.a026280. [DOI] [PubMed] [Google Scholar]
- Dötsch A.; Klawonn F.; Jarek M.; Scharfe M.; Blöcker H.; Häussler S. Evolutionary conservation of essential and highly expressed genes in Pseudomonas aeruginosa. BMC Genomics 2010, 11, 234–245. 10.1186/1471-2164-11-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.; Roberts R.; Kasif S. Identification of genes with fast-evolving regions in microbial genomes. Nucleic Acids Res. 2004, 32, 6347–6357. 10.1093/nar/gkh935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Roy A.; Zhang Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. 10.1093/bioinformatics/btt447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan A.; Ottman C.; Fournier A. E. 14-3-3 adaptor protein-protein interactions as therapeutic targets for CNS diseases. Pharmacol Res. 2017, 125, 114–121. 10.1016/j.phrs.2017.09.007. [DOI] [PubMed] [Google Scholar]
- Bunney T. D.; De Boer A. H.; Levin M. Fusicoccin signaling reveals 14-3-3 protein function as a novel step in left-right patterning during amphibian embryogenesis. Development 2003, 130, 4847–58. 10.1242/dev.00698. [DOI] [PubMed] [Google Scholar]
- Saponaro A.; Pooro A.; Chaves-Sanjuan A.; Nordini M.; Rauh O.; Thiel G.; Moroni A. Fusicoccin activates KAT1 channels by stabilizing the interaction with 14-3-3 proteins. Plant Cell 2017, 29, 2570–2580. 10.1105/tpc.17.00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts M.; Bowles D. Fusicoccin, 14-3-3 proteins, and defense responses in tomato plants. Plant Physiol. 1999, 119, 1243–1250. 10.1104/pp.119.4.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapp C.; Portt L.; Khoury C.; Sheibani C.; Norman G.; Ebner P.; Eid R.; Vali H.; Mandato C. A.; Madeo F. 14-3-3 Protects against stress-induced apoptosis. Cell Death Dis. 2012, 3, e348 10.1038/cddis.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong S.; Kang S.; Gu T.; Kardar S.; Fu H.; Lonial S.; Khoury H.; Khuri F.; Chen J. 14–3-3 integrates prosurvival signals mediated by the AKT and MAPK pathways in ZNF198-FGFR1–transformed hematopoietic cells. Blood 2007, 110, 360–369. 10.1182/blood-2006-12-065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanger G.; Widmann C.; Porteri A. C.; Sather S.; Johnson G. L.; Vaillancourt R. R. 14-3-3 Proteins interact with specific MEK kinases. J. Biol. Chem. 1998, 273, 3476–3483. 10.1074/jbc.273.6.3476. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan V.; Martinez J. 14-3-3γ Induces oncogenic transformation by stimulating MAP kinase and PI3K Signaling. PLoS One 2010, 5, e11433 10.1371/journal.pone.0011433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing H.; Zhang S.; Weinheimer C.; Kovacs A.; Muslin A. 14-3-3 proteins block apoptosis and differentially regulate MAPK cascades. EMBO J. 2000, 19, 349–358. 10.1093/emboj/19.3.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owczarzy R.; Moreira B.; You Y.; Behike A.; Walder J. Predicting stability of DNA duplexes in solutions containing magnesium and monovalent cations. Biochemistry 2008, 19, 5336–5353. 10.1021/bi702363u. [DOI] [PubMed] [Google Scholar]
- Frishman D.; Argos P. Knowledge-based protein secondary structure assignment. Proteins 1995, 23, 566–579. 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- McGuffin L.; Bryson K.; Jones D. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- Henikoff S.; Henikoff J. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 10915–10919. 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomii K.; Kanehisa M. Analysis of amino acid indices and mutation matrices for sequence comparison and structure prediction of proteins. Protein Eng., Des. Sel. 1996, 9, 27–36. 10.1093/protein/9.1.27. [DOI] [PubMed] [Google Scholar]
- Yang J.; Renxiang Y.; Roy A.; Xu D.; Poisson J.; Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat. Methods 2015, 12, 7–8. 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Roy A.; Zhang Y. BioLiP: a semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 2013, 41, 1096–1103. 10.1093/nar/gks966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petosa C.; Masters S. C.; Bankston L. A.; Pohli J.; Wang B.; Fu H.; Liddington R. C. 14-3-3z binds a phosphorylated raf peptide and an unphosphorylated peptide via its conserved amphipathic groove. J. Biol. Chem. 1998, 273, 16305–16310. 10.1074/jbc.273.26.16305. [DOI] [PubMed] [Google Scholar]
- Spence S. L.; Dey B. R.; Terry C.; Albert P.; Nissley P.; Furlanetto R. W. Interaction of 14-3-3 proteins with the insulin-like growth factor I receptor (IGFIR): evidence for a role of 14-3-3 proteins in IGFIR signaling. Biochem. Biophys. Res. Commun. 2003, 312, 1060–1066. 10.1016/j.bbrc.2003.11.043. [DOI] [PubMed] [Google Scholar]
- Furlanetto R. W.; Dey B. B.; Lopaczynski W.; Nissley S. P. 14-3-3 Proteins interact with the insulin-like growth factor receptor but not the insulin receptor. Biochem. J. 1997, 327, 765–771. 10.1042/bj3270765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal C. L.; Xu J.; Li P.; Mori S.; Yang J.; Neal N. N.; Zhou X.; Wyszomierski S. L.; Yu D. Overexpression of 14-3-3ζ in cancer cells activates PI3K via binding the p85 regulatory subunit. Oncogene 2011, 31, 897–906. 10.1038/onc.2011.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J. J.; Okamoto Y.; Coblitz B.; Li M.; Qiu Y.; Shikano S. PI3K/Akt signalling-mediated protein surface expression sensed by 14-3-3 interacting motif. FEBS J. 2009, 276, 5547–5558. 10.1111/j.1742-4658.2009.07241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin D. S.; Galaktionov K.; Beach D. 14-3-3 proteins associate with cdc25 phosphatases. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 7892–96. 10.1073/pnas.92.17.7892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardino A. K.; Yaffe M. B. 14-3-3 proteins as signaling integration points for cell cycle control and apoptosis. Semin. Cell Dev. Biol. 2011, 22, 688–695. 10.1016/j.semcdb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J.; Cui C.; Liu Y.; Jin M.; Wu D.; Liu C.; Wang E.; Yu B. The Role of 14-3-3ε Interaction with Phosphorylated Cdc25B at Its Ser321 in the Release of the Mouse Oocyte from Prophase I Arrest. PLoS One 2013, e53633 10.1371/journal.pone.0053633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters S. C.; Yang H.; Datta S. R.; Greenberg M. E.; Fu H. 14-3-3 Inhibits bad-induced cell death through interaction with serine-136. Mol. Pharmacol. 2001, 60, 1325–1331. 10.1124/mol.60.6.1325. [DOI] [PubMed] [Google Scholar]
- Mackintosh C. Dynamic interactions between 14-3-3 proteins and phosphoproteins regulate diverse cellular processes. Biochem. J. 2004, 381, 329–342. 10.1042/BJ20031332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington K. L.; Chan T. Y.; Torres M. P.; Andersen J. L. The dynamic and stress-adaptive signaling hub of 14-3-3: emerging mechanisms of regulation and context-dependent protein–protein interactions. Oncogene 2018, 37, 5587–5604. 10.1038/s41388-018-0348-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhre K.; Sanejouand Y. H. ElNemo: a normal mode web-server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004, 32, 610–614. 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhre K.; Sanejouand Y. H. On the potential of normal mode analysis for solving difficult molecular replacement problems. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 796–799. 10.1107/S0907444904001982. [DOI] [PubMed] [Google Scholar]
- Eyal E.; Lum G.; Bahar I. The anisotropic Network Model web server at 2015 (ANM 2.0). Bioinformatics 2015, 31, 1487–1489. 10.1093/bioinformatics/btu847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis L. K.; Carson L.; Uppu R.; Fares A.; Godson O. Fundamental Theory of the Evolution Force: Gene engineering utilizing synthetic evolution artificial intelligence (SYN-AI). bioRxiv 2019, 585042. [Google Scholar]
- Maiti R.; Domselaar G. H.; Zhang H.; Wishart D. S. SuperPose: a simple server for sophisticated structural superposition. Nucleic Acids Res. 2004, 1, 590–594. 10.1093/nar/gkh477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Zhang Y. Protein-protein complex structure prediction by multimeric threading and template recombination. Structure 2011, 19, 955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacher J. M.; Reiss B. D.; Ellington A. D. Anticipatory evolution and DNA shuffling. Genome Biol. 2002, 3, reviews1021-1 10.1186/gb-2002-3-8-reviews1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obsil T.; Ghirlando R.; Klein D. C.; Ganguly S.; Dyda F. Crystal structure of the 14-3-3 zeta:Serotonin N-acetyltransferase complex. Cell 2001, 105, 257–267. 10.1016/S0092-8674(01)00316-6. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wang W.; Kollman P. A.; Case D. A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graphics Modell. 2006, 25, 247–260. 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Cadwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general AMBER force field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.