

Abstract
The G‐protein‐coupled receptor GPR132, also known as G2A, is activated by 9‐hydroxyoctadecadienoic acid (9‐HODE) and other oxidized fatty acids. Other suggested GPR132 agonists including lysophosphatidylcholine (LPC) have not been readily reproduced. Here, we identify N‐acylamides in particular N‐acylglycines, as lipid activators of GPR132 with comparable activity to 9‐HODE. The order‐of‐potency is N‐palmitoylglycine > 9‐HODE ≈ N‐linoleoylglycine > linoleamide > N‐oleoylglycine ≈ N‐stereoylglycine > N‐arachidonoylglycine > N‐docosehexanoylglycine. Physiological concentrations of N‐acylglycines in tissue are sufficient to activate GPR132. N‐linoleoylglycine and 9‐HODE also activate rat and mouse GPR132, despite limited sequence conservation to human. We describe pharmacological tools for GPR132, identified through drug screening. SKF‐95667 is a novel GPR132 agonist. SB‐583831 and SB‐583355 are peptidomimetic molecules containing core amino acids (glycine and phenylalanine, respectively), and structurally related to previously described ligands. A telmisartan analog, GSK1820795A, antagonizes the actions of N‐acylamides at GPR132. The synthetic cannabinoid CP‐55 940 also activates GPR132. Molecular docking to a homology model suggested a site for lipid binding, predicting the acyl side‐chain to extend into the membrane bilayer between TM4 and TM5 of GPR132. Small‐molecule ligands are envisaged to occupy a “classical” site encapsulated in the 7TM bundle. Structure‐directed mutagenesis indicates a critical role for arginine at position 203 in transmembrane domain 5 to mediate GPR132 activation by N‐acylamides. Our data suggest distinct modes of binding for small‐molecule and lipid agonists to the GPR132 receptor. Antagonists, such as those described here, will be vital to understand the physiological role of this long‐studied target.
Keywords: G protein‐coupled receptor, G2A, GPR132, N‐acylglycine, N‐linoleoylglycine, N‐palmitoylglycine
Abbreviations
- 9‐HODE
9‐hydroxyoctadecadienoic acid
- LPC
lysophosphatidylcholine
- GPCRs
G‐protein‐coupled receptors
- AEA
arachidonoyl ethanolamide
- NAGly
N‐arachidonylglycine
1. INTRODUCTION
N‐acylamides, also known as lipoamines, are a family of diverse naturally occurring lipids derived from the conjugation of saturated, unsaturated, or hydroxylated fatty acids to amines such as dopamine, ethanolamine, or amino acids (or simply to amide, as in linoleamide). Recent lipidomic studies confirm the widespread occurrence of N‐acylamides in mammalian tissue, including brain.1 The family includes the endocannabinoid N‐arachidonoyl ethanolamide (AEA, also known as anandamide), and the cannabinoid system is the best understood signalling pathway in the class, with cognate G‐protein‐coupled receptors (GPCRs) CB1 and CB2 and well‐elucidated pathways for synthesis and degradation of AEA.2 Other N‐acylamides have demonstrated biological effects, but generally the molecular targets or receptors mediating these effects are not yet clear.3
N‐acyl amino acids have received particular attention in recent years, with the demonstration that they occur in mammalian tissues at concentrations comparable to other lipid signalling mediators. Moreover several N‐acyl amino acids are biologically active (see Ref. 4 for review). A human biosynthetic enzyme for N‐acylglycines, GLYATL2 (glycine N‐acyltransferase‐like 2, Ref.5), and N‐acyl amide synthase genes genes in gut bacteria6, 7 further emphasizes their importance. N‐arachidonylglycine (NAGly), the first‐discovered N‐acyl amino acid, is reported to activate the orphan GPCR, GPR188, 9, 10 and the lysophosphatidic acid receptor LPA5.11 NAGly concentrations detected in brain are remarkably high and may exceed AEA.12 Literature suggests NAGly may be involved in nociception, inflammation, and regulation of ocular pressure.4 Physiological processes for NAGly beyond GPCR signalling have also been proposed, including modulation of transporters, ion channels, and enzymes, especially the AEA‐degrading enzyme, fatty acid amide hydrolase (FAAH, Ref.4).
Here we show that N‐acylglycines activate another orphan family A GPCR, namely GPR132 (originally called G2A). The most potent N‐acylglycine activators of GPR132 are N‐palmitoylglcine (NPGly) and N‐linoleoylglcine (NLGly) which have mono‐unsaturated or saturated acyl side‐chains. NAGly has weak agonist activity on GPR132 and is unlikely to be a physiologically relevant ligand. NPGly and NLGly bear structural similarity to the oxidized fatty acid, 9‐HODE, the candidate endogenous GPR132 ligand.13, 14, 15 Administered orally, NLGly reduces leukocyte migration in a mouse peritonitis model.16 In vivo and in vitro, NLGly can stimulate production of prostaglandin 15‐deoxy‐Δ12,14‐PGJ2, an inflammation‐resolving eicosanoid, from macrophages and macrophage‐like mouse RAW cells, respectively.16 Biological activity of N‐acylglycines and oxidized fatty‐acids coincides with GPR132 location, in lymphocytes,17, 18 monocyte‐lineage cells including macrophages19 and keratinocytes.20 GPR132 is implicated in diverse functions including nociception,21, 22 positioning of macrophages at sites of inflamation,19 haematopoiesis,23 sensing oxidative stress,20 regulating macrophage responses in a tumor microenvironment,24 and microglial colonization from periphery into developing brain (in zebrafish25). However, understanding of GPR132 function has been hampered because multiple other ligands have been published to activate GPR132, and selective pharmacological tools for functional studies have not been available.
To address this, we adopted a chemical biology approach. First we identified synthetic drug‐like agonists to permit validation of cellular expression systems for GPR132. To screen a set of bioactive lipids, we employed yeast‐based assays to monitor GPR132 in isolation from other potentially confounding GPCRs, revealing N‐acylglycines as agonists. Small‐molecule antagonists of GPR132 block activation by N‐acylglycine. Rat and mouse GPR132 orthologs also act as receptors for N‐acylglycines and oxidized fatty‐acids. Structural modelling suggests that binding of N‐acylglycines to GPR132 is analogous to lipid binding to other GPCRs, with lipid entry through the transmembrane region. During our studies, Cohen et al described two further N‐acyl amino acids derived from the gut microbiota which also activate GPR132.6, 7 Our data, together with that of Cohen et al, support a GPR132 signalling axis in the immune system with dual N‐acylglycine and oxidized fatty‐acid ligands, which may play distinct roles in different tissues and physiological contexts.
2. METHODS
2.1. Chemicals
N‐acyl amino acids and linoleamide were obtained from Cayman Chemical (Ann Arbor, MI, USA). SKF‐95667, SB‐583355, SB‐583831, GSK1820795A, and telmisartan were prepared synthetically.
2.2. Mammalian cell culture
Cell lines were maintained at 37°C in 5% CO2. DiscoverX PathHunter (CHO) cells were maintained using 10% heat‐inactivated fetal bovine serum (HI‐FBS) in Dulbecco's minimal essential media (DMEM):Ham's F‐12 (1:1) with 2 mmol/L L‐glutamine. Selection was maintained using G418 (800 μg/mL) and hydromycin‐B (300 μg/mL). Rat basophilic leukemia (RBL) cells were maintained in DMEM F12 supplemented with 10% HI‐FBS at 37°C and 5% CO2. RBL cells stably expressing human GPR132a (RBL‐GPR132) were supplemented with G418 (500 µg/mL).
GeneBLAzer™ T‐REx™‐G2A(GPR132)‐NFAT‐bla FreeStyle™ 293F cells were maintained according to manufacturer's instructions (ThermoFisher Scientific).
2.3. Ca2+ microfluorimetry
A digital epifluorescence imaging system (Perkin‐Elmer) incorporating an Olympus BX50WI microscope was used to measure changes in intracellular Ca2+ levels in individual cells. Cells grown overnight to ~80% confluency on poly‐D‐lysine‐treated glass coverslips (0.9 mm) were serum starved in 0.2% FBS. Cells were incubated for 50 minutes with the Ca2+ sensitive dye Fura 2‐AM (6 μmol/L) in HEPES buffered saline (HBS: NaCl 135 mmol/L, HEPES 10 mmol/L, KCl 5 mmol/L, CaCl2 1.8 mmol/L, MgCl2 1 mmol/L, D‐glucose 25 mmol/L, pH 7.4). For experimentation, cells were perfused with HBS at a rate of ~2 mL/min (28‐30°C) and ratiometric images (350/380 nm) were collected at 5 second intervals using MetaFluor software (Universal Imaging Corporation). Data from multiple individual cells within a field were collated from three independent experiments for analysis.
2.4. β‐arrestin association assay
hGPR132a and mGPR132 were separately cloned into DiscoverX Prolink vectors and transfected into HEK293 cells which stably express β‐arrestin2‐β‐gal‐EA fusion protein. Cells were plated in 384‐well white‐opaque bottom (12 000 cells/well in 20 μL) in F‐12 medium containing 10% FBS. After 4 hours, media was removed and cells were serum starved in 0.2% FBS overnight. Test ligand diluted in F12 media (2 μL per well; 90 minutes, 37°C) followed by DiscoverX reagent solution (12 μL per well; 30 minutes, 37°C) were added and plates read using an Envision luminescence plate reader.
2.5. NFAT reporter gene assay
To identify GSK1820795A, 2256 compounds were dispensed into black clear‐bottom 384‐well plates (Greiner) as solutions in 100% DMSO (40 nL/well; final concentration 1 µmol/L), using an Echo accoustic dispenser (Labcyte). GeneBLAzer™ T‐REx™‐G2A(GPR132)‐NFAT‐bla FreeStyle™ 293F cells were thawed, resuspended in 90% DMEM; 10% dialyzed FBS; 0.1 mmol/L NEAA; 25 mmol/L HEPES (pH 7.3); 100 U/mL Penicilin; 100 µg/mL Streptomycin, and added to wells (10 000 cells/well in 40 µL). Media was supplemented with 1 ng/mL doxycycline to induce expression of GPR132, resulting in high GPR132 constitutive activity. After incubtion (18 hours at 37°C; 5% CO2), LiveBLAzer™‐FRET β‐galactosidase substrate (CCF‐AM) prepared according to manufacturer's instructions (ThermoFisher Scientific, Loughborough, UK) was added (8 µL/well; room temperature for 2 hours). Inverse agonists including GSK1820795A were identified from the 460/530 nm emission ratio (Envision, Perkin Elmer) by reference to doxycycline‐treated and untreated controls. Half‐maximal inhibition (pIC50) values were determined in similar experiments, except that cells were supplemented with SKF‐95667 (1 µmol/L).
2.6. Angiotensin AT1 antagonist assay
Chinese Hamster Overy (CHO) cells stably expressing human angiotensin AT1 and containing luciferase under an NFAT‐responsive promoter (NFAT‐hAT1‐CHO) were used. Briefly, cells are incubated with 30 nmol/L angiotensin II for 18 hours, in the presence of test compound. Luciferase activity was quantified using Bright‐Glo™ Luciferase reagent (Promega), measured on the Lumistar Galaxy (BMG Labtech).
2.7. Activation of the yeast pheromone‐response pathway
Modified yeast used to express mammalian GPCRs and measure GPCR activation have been described previously.26 GPR132 orthologs were cloned into pRS306GPD for chromosomal integration. Human GPR132a (hGPR132a) was integrated into MMY24 (Gpa1/Gαi3) to generate YIG95. GPR132b (hGPR132b) isoform and rat GPR132 (rGPR132) were also integrated into MMY24. Mouse GPR132 (mGPR132) was integrated into MMY23 (Gpa1/Gαi1). To test for ligand specificity, yeast with chromosomally integrated human CB2 (in MMY23) and human adenosine A2A (in MMY23) receptors were used. Mutant hGPR132‐pRS306GPD plasmids were generated by DC Biosciences (Dundee, UK) and integrated into MMY24. Yeast expressing GPR68 (OGR1) were generated using p426GPD vector27 transfected into MMY14 (Gpa1/Gαq5). To measure agonist activity, GPCR‐expressing yeast were plated at 0.02 OD600nm/mL in black 384‐well microtiter plates in the presence of test compound for 24 hours at 30°C. Fluorescein production from FDGlu was quantified using an Envision plate reader.
2.8. Statistical analysis
Half‐maximal activities (pEC50 and pIC50) were calculated by subjecting technical replicates within an experiment to 4‐parameter logistic fitting, using PRISM. Values for pEC50 and pIC50 are presented as mean ± standard deviation across separate experiments (n = experiment days). For calcium assay, data are presented as mean ± SE of ratiometric fluorescent units. Two‐tailed unpaired Welch t‐test was used to compare the response of 10 μmol/L ligand in parental RBL cells vs hGPR132a‐RBL cells. Data are presented as non‐normalized fluorescence counts, or normalized to the maximum response of a reference full agonist (NPGly or NLGly for yeast assays; 9‐HODE or SB‐583831 for β‐arrestin association assays).
3. RESULTS
At the outset of this project, we had access to the GPR132 agonist, SKF‐95667 (Figure 1A). SKF‐95667 originates from a collaboration between SmithKline Beecham and CADUS.28 In this collaboration, synthetic surrogate agonists were identified at orphan GPCRs, by subjecting putative GPCRs to compound screening using the CADUS modified yeast (Saccharomyces cerevisiae) as expression host. Here we confirmed that SKF‐95667 activates the mating‐pheromone response pathway of yeast expressing human splice variant GPR132a (hGPR132a). Yeast strains used in this study are similar to those originally developed by CADUS, but include a chimeric Gα subunit to optimize communication between mammalian GPCR and downstream yeast signalling proteins.29 YIG95 yeast, which constitutively express hGPR132a and the Gpa1‐Gαi3 chimera, when treated with SKF‐95667 showed a specific gene‐reporter response (Figure S1). Half‐maximal (pEC50) concentrations and maximal responses relative to a reference agonist (%Emax) for SKF‐95667 and other compounds tested in this study are shown in Table 1.
Figure 1.
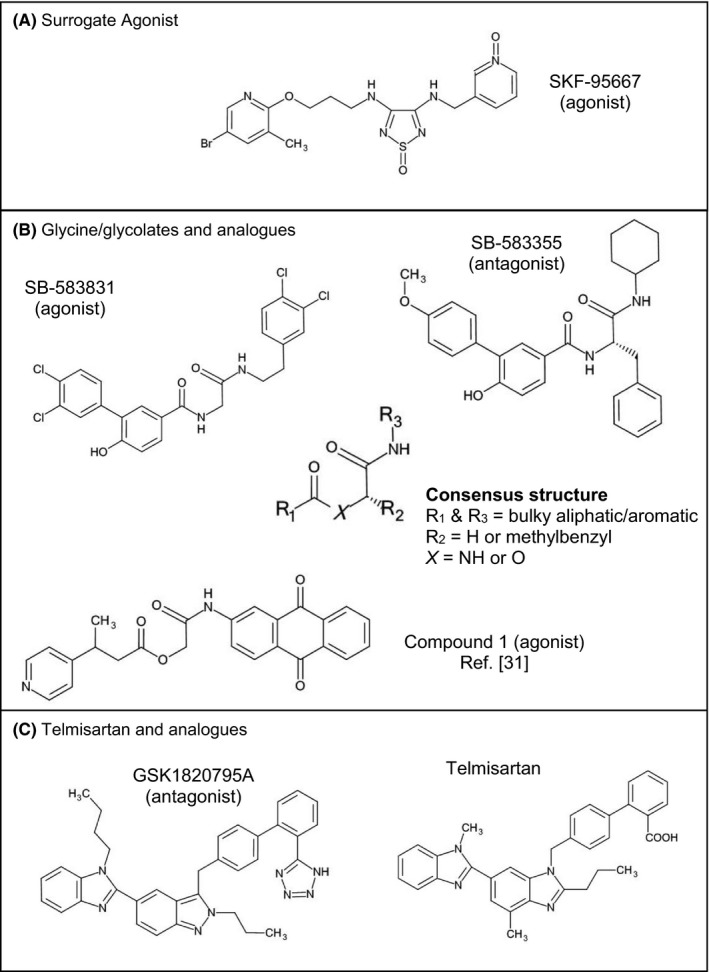
Structures of GPR132 ligands described in this study
Table 1.
Structure‐activity relationship of N‐acylamides at GPR132. Values are shown as mean ± SD
| Ligand | Receptor | |||||||
|---|---|---|---|---|---|---|---|---|
| Human GPR132a | Rat GPR132 | Mouse GPR132 | ||||||
| Yeast | β‐arrestin | Yeast | β‐arrestin | |||||
| pEC50 (n) | Emax | pEC50 (n) | Emax | pEC50 (n) | Emax | pEC50 (n) | Emax | |
| NPGly | 6.2 ± 0.16 (7) | 100 | <5 (3) | 14 | 7.0 ± 0.12 (4) | 53 ± 14 | <5 (2) | 11 |
| 9‐HODE | 5.9 ± 0.14 (4) | 91 ± 8 | 5.5 ± 0.13 (4) | 100 | 6.2 ± 0.12 (5) | 100 | 5.6 ± 0.23 (5) | 97 ± 6 |
| NLGly | 5.8 ± 0.08 (3) | 84 ± 6 | 5.5 ± 0.23 (2) | 38 ± 4 | 6.5 ± 0.36 (4) | 68 ± 7 | 5.3 (1) | 40 |
| Linoleamide | 5.8 ± 0.01 (3) | 79 ± 11 | 5.5 (1) | 22 | 5.9 ± 0.21 (2) | 37 ± 5 | <5 (1) | IA |
| NOGly | 5.1 ± 0.24 (3) | 95 ± 6 | 5.6 ± 0.18 (2) | 29 ± 15 | NT | NT | 5.3 ± 0.05 (2) | 44 ± 10 |
| NSGly | 5.1 ± 0.08 (3) | 80 ± 4 | 5.4 (1) | 23 | NT | NT | <5 (1) | IA |
| NAGly | <4.5 (3) | 38 | IA (2) | IA | 5.3 ± 0.09 (4) | 87 ± 12 | NT | NT |
| NHGly | <5 (4) | IA | NT | NT | NT | NT | NT | NT |
| NDGly | <4.4 (4) | 33 | <5 (2) | IA | NT | NT | IA (1) | IA |
| NOSer | 5.8 ± 0.07 (4) | 21 ± 5 | NT | NT | NT | NT | NT | NT |
| NATyr | <4 (3) | IA | NT | NT | <4 (3) | IA | NT | NT |
| 2‐AG | <5 (4) | IA | NT | NT | IA (4) | IA | NT | NT |
| Linoleic acid | <4 (3) | IA | <5 (1) | IA | NT | NT | IA (1) | IA |
| LPC | <4 (3) | IA | IA (3) | IA | NT | NT | IA (2) | IA |
| SKF‐95667 | 6.6 ± 0.26 (4) | 99 ± 3 | 5.8 ± 0.32 (2) | 281 ± 81 | IA (3) | IA | 5.8 (1) | 320 |
| SB‐583831 | 7.7 ± 0.32 (4) | 95 ± 3 | 7.3 ± 0.17 (5) | 100 | IA (4) | IA | 7.5 ± 0.3 (2) | 100 |
| CP‐55,940 | 5.5 ± 0.13 (3) | 102 ± 3 | NT | NT | <5.5 (3) | IA | NT | NT |
Number of independent experiment occasions is shown in parentheses (n). Typically four technical replcates were conducted on each experiment occasion. Values based on limited data (n ≤ 2) require further experimentation to confirm. For yeast assays, raw data was normalized to the effect of NPGly (hGPR132a) or 9‐HODE (rGPR132a). For β‐arrestin assays, raw data was normalized to the effect of SB‐583831. NT = Not Tested. For inactive (IA) compounds, top concentration tested is shown (for example, pEC50 < 4 denotes a compound tested up to 100 µmol/L for which no conentration‐response curve could be fitted). For compounds which showed evidence of activity at higher test concentrations, but where no pEC50 could be fitted, Emax (%) is included. Note SKF‐95667 behaved as a superagonist (>100% Emax) at both hGPR132a and mGPR132 in β‐arrestin assays, and did not activate rGPR132.
Obinata et al showed that 10 μmol/L 9‐HODE induced Ca2+ transients in CHO cells overexpressing hGPR132a.13 Using yeast YIG95, we confirmed 9‐HODE as an agonist of hGPR132a, causing a concentration‐dependent response (pEC50 = 5.9 ± 0.14; Figure 2A). To further corroborate the ligand pairing, we used two mammalian hosts. Perfusion of rat basophilic leukemia cells expressing hGPR132a (RBL‐hGPR132a) with 10 μmol/L 9‐HODE induced intracellular Ca2+ release (Figure 3A). No response was detected in parental RBL cells (Figure 3C). We also used Chinese hamster ovary cells expressing a C‐terminally‐tagged version of hGPR132a (CHO‐hGPR132aPL), in the PathHunter assay. 9‐HODE induced association between hGPR132aPL and β‐arrestin (Figure 4A; pEC50 = 5.4 ± 0.16), similar to results reported by others.15 Together, these data confirm 9‐HODE as an agonist of hGPR132a.
Figure 2.
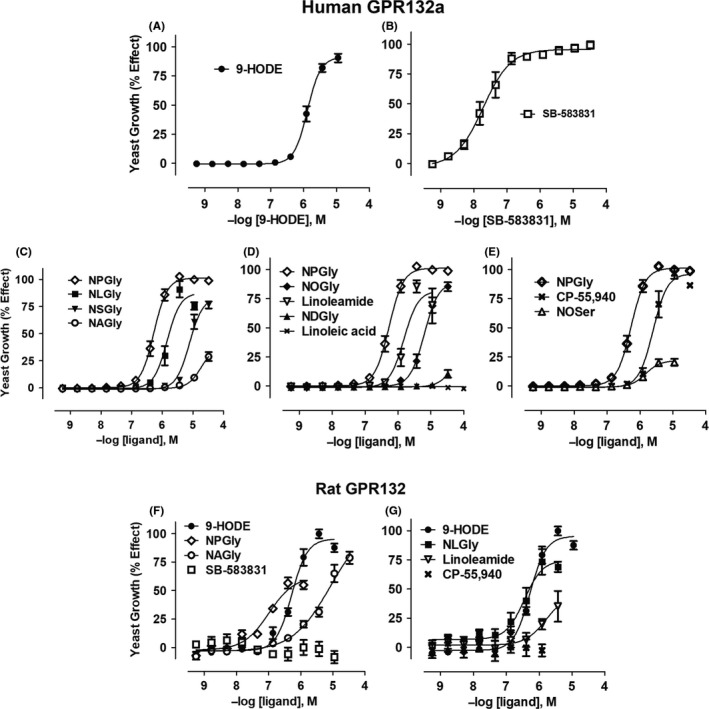
Agonist activation of yeast cells expressing human or rat GPR132. Yeast cells containing a gene‐reporter linked to the pheromone response pathway and engineered to express human GPR132a were treated as follows: Oxidized fatty acid 9‐HODE13 (A); the glycine‐containing ligand, SB‐583831 (C); N‐palmitoylglycine (NPGly; C); N‐linoleoylglycine (NLGly; C); N‐stereoylglycine (NSGly; C); N‐arachidonoylglycine (NAGly; C); N‐oleoylglycine (NOGly; D); linoleamide (D); linoleic acid (D): N‐oleoylserine (NOSer; E) and CP‐55,940 (E). Similarly, yeast expressing rat GPR132 were treated with 9‐HODE (F); N‐palmitoylglycine (NPGly; F); N‐arachidonoylglycine (NAGly; F); SB‐583831 (F); N‐linoleoylglycine (NLGly; G); linoleamide (G) and CP‐55,940 (G). Ligand responses were normalized to the effect of NPGly (for hGPR132a) or 9‐HODE (for rGPR132). Data are presented as mean ± SEM (from n = 3 to 7 independent experiments per ligand; four technical replicates were conducted for each condition). N‐acylamide ligands caused inhibition of yeast cell growth at concentrations >10 µmol/L or >30 µmol/L, and these were therefore used as the top threshold test concentrations curve‐fitting
Figure 3.
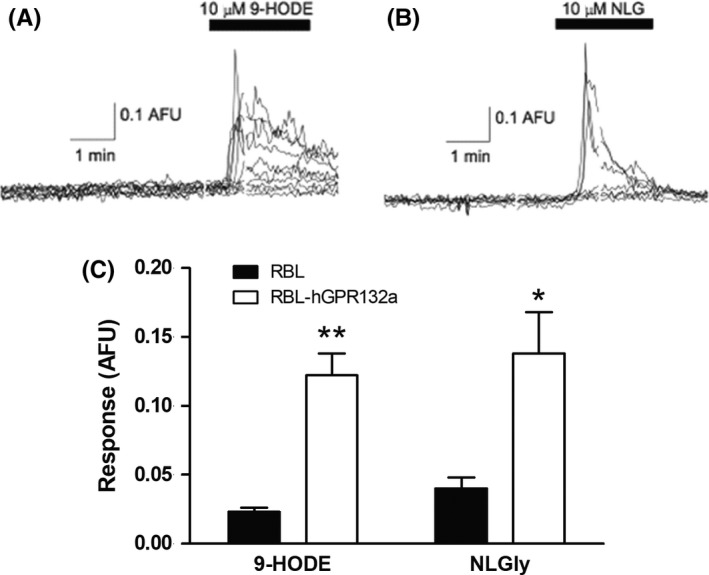
9‐HODE and NLGly induce calcium transients in Rat Basophilic Leukemia (RBL) cells overexpressing hGPR132a. Representative transient calcium release measurements taken from microfluorimetry experiments showing effect of (A) 9‐HODE and (B) NLGly (both 10 µmol/L) on RBL‐hGPR132a cells. Each trace represents an individual cell. (C) NLGly (10 µmol/L) induced Ca2+ transients in RBL‐hGPR132a cells (0.138 ± 0.03 ratio units, n = 17 cells), significantly greater than observed in parental RBL cells (0.040 ± 0.008 ratio units, n = 14 cells, t (17.78) = 2.88, P = .01). Similarly, 9‐HODE (10 µmol/L) induced Ca2+ in RBL‐hGPR132a cells (mean, peak response: 0.122 ± 0.016 ratio units, n = 46 cells), with little or no response detected in parental RBL cells (0.023 ± 0.003 ratio units, n = 27 cells; t (48.39) = 6.27; P = <.0001). *P < .05 **P < .01
Figure 4.
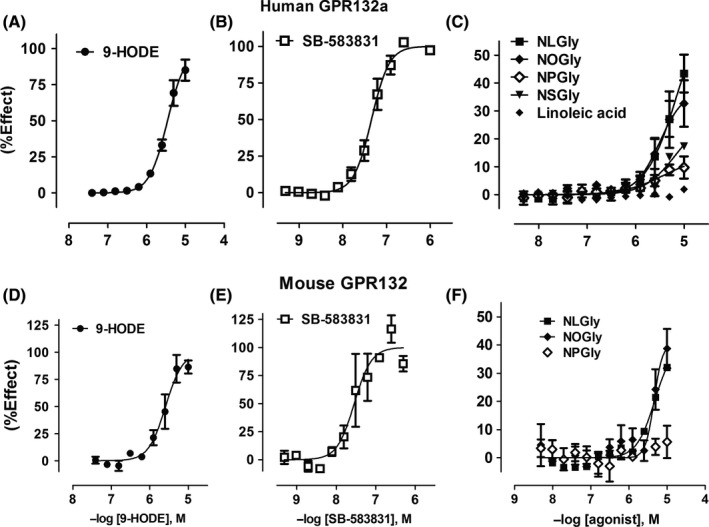
N‐acylglycines induce association of human and mouse GPR132 with β‐arrestin. CHO cells stably expressing C‐terminally tagged human (A‐C) or mouse GPR132 (D‐F) (CHO‐hGPR132aPL or CHO‐mGPR132PL) were challenged with agonists as follows: 9‐HODE (A and D); SB‐583831 (B and E); linoleic acid (C) and N‐acylglycines (C and F). N‐acylglycines tested were N‐linoleoylglycine (NLGly), N‐oleoylglycine (NOGly), N‐palmitoylglycine (NPGly), and N‐stereoylglycine (NSGly). PathHunter assay was used to measure receptor‐β‐arrestin interaction. Data were normalized to the maximal effect of SB‐583831 (100%) in each experiment, and are presented as mean ± SEM (from n = 2 to 7 independent tests per ligand with 2 technical replicates per experiment)
RBL‐hGPR132a cells were used to screen 6465 chemically diverse compounds, measuring intracellular Ca2+ mobilization using the fluorescence imaging plate reader (FLIPR). Among active agonists, SB‐583831 was chosen based on its potency and similarity to previously described agonists.30 SB‐583831 activated YIG95 (Figure 2B; pEC50 = 7.7 ± 0.32) whereas yeast expressing either GPR68 or CB2 were not activated up to 100 μmol/L (Figure S1). SB‐583831 also induced association between hGPR132aPL and β‐arrestin (Figure 4B; pEC50 = 7.3 ± 0.17). SB‐583831 contains a glycine core linked via amide bonds to aliphatic or aromatic appendage groups (Figure 1B), resembling hGPR132a agonists described previously.30 Compound 1 disclosed by Shehata et al (Figure 1B) contains hydroxyacetate (glycolate) isosteric to glycine in SB‐583831. SB‐583831 is >1 log unit more potent than compound 1 (pEC50 = 7.3 compared to 5.5 for compound 1,30 in β‐arrestin association assays). Appendage groups of SB‐583831 and compound 1 also differ, so it is not clear whether glycine‐ or glycolate‐containing structures are preferred for GPR132 binding. To identify antagonists, CHO‐hGPR132aPL cells were incubated with test compound prior to challenge with SKF‐95667. SB‐583355 inhibited agonist‐induced hGPR132aPL association with β‐arrestin, showing preliminary evidence of a concentration‐dependent effect (pIC50 ≈ 7.0; n = 2; data not shown). To confirm that this effect was receptor‐specific, we showed that SB‐583355 inhibited SKF‐95667‐induced activation of YIG95 (pIC50 = 6.5 ± 0.23; n = 5; data not shown). SB‐583355 is similar to SB‐583831 but contains L‐phenylalanine in place of glycine (Figure 1B). Thus, GPR132 ligands with either agonist or antagonist activity occur in the same structural series. Exemplars contain amino acid (or isostere) linked to aromatic or aliphatic groups by amide or ester (consensus structure shown in Figure 1B).
Lipid GPCRs often have multiple endogenous cognate ligands. Cannabinoid receptor CB1, for example, is activated by lipids with ethanolamide and glycerol ester head groups (AEA and 2‐arachidonylglycerol, respectively2). To identify candidate physiologically relevant ligands of GPR132 beyond 9‐HODE, we screened a set of lipids, using YIG95 yeast to quantify agonist activity. N‐linoleoylglycine (NLGly) and linoleamide were identified as novel GPR132 agonists (Figure 2C and D). 9‐HODE was also a component of this lipid set and was identified alongside NLGly in the screen. Testing of further commercially available N‐acylglycines revealed N‐palmitoylglycine (NPGly; C16 side‐chain) to be the most potent N‐acylamide agonist of hGPR132a, among those tested (pEC50 = 6.2 ± 0.16, Figure 2C). In the yeast assay, 9‐HODE, SB‐583831, NPGly and CP‐55,940 (see below) all behaved as full agonists of hGPR132a (Figure 2A‐E). N‐hexanoylglycine (NHGly; C6 side‐chain) was inactive (Table 1), indicating a minimum side‐chain length amongst unsaturated N‐acylglycines between C6 and C16, for agonist activity. Linoleic acid had no detectable agonist activity up to 100 µmol/L (Figure 2D). We also tested lysophosphatidylcholine (LPC), since this proinflammatory lipid was originally described to activate GPR132. We could detect no agonist or antagonist activity of LPC in any of the test systems used (Table 1), consistent with published findings.13, 15 Lipid order‐of‐potency (based on yeast pEC50 values for hGPR132a activation) was NPGly > 9‐HODE ≈ NLGly ≈ linoleamide > N‐oleoylglycine (NOGly) ≈ N‐stereoylglycine (NSGly) > N‐arachidonoylglycine (NAGly) > N‐docosahexanoylglycine (NDGly) (Figure 2A‐E).
We tested the effect of replacing glycine with other head groups. N‐oleoylserine (NOSer) activated hGPR132a more potently than NOGly (Figure 2E; pEC50 = 5.8 ± 0.07 and 5.1 ± 0.24, respectively). However, NOSer exhibited partial agonism (Emax = 21±5%; Figure 2E) compared with the reference agonist SB‐583831, in contrast to N‐acylglycine agonists which typically had Emax ≥ 80% (Table 1). N‐arachidonyltyrosine (NATyr) was inactive, suggesting a steric limit on the N‐acylamide head group for GPR132 binding (Table 1). 2‐arachidonylglycerol (2‐AG) was inactive on hGPR132a (Table 1). Several N‐acylamides including 9‐HODE, NLGly and linoleamide produced bell shaped responses in the hGPR132a yeast assay (Figure 2). In these cases, data at higher concentrations where response was depressed were excluded from curve fitting. The assay is based on yeast growth, and this signal depression was attributed to toxicity because control receptors were also inhibited in the same concentration range (Figure S1). Yeast expressing splice variant hGPR132b31 were activated by N‐acylglycines with similar pharmacology to hGPR132a (Figure S2).
To examine specificity of N‐acylamides for GPR132, we tested three other family‐A GPCRs expressed in yeast. GPR68 sequence is related to GPR132, but GPR68 responds to acid pH (protons) and no endogenous lipids are known to activate GPR68.32 CB2 binds N‐acylamide ligands (AEA and 2‐arachidonylglycerol) but has low sequence similarity to GPR132. Adenosine A2a has neither sequence similarity nor comparable ligands to GPR132.33 In general, the N‐acylamides were inactive at GPR68, CB2 and A2a, indicating they are specific agonists of GPR132 (Figure S1). Unexpectedly, NAGly showed weak partial agonist activity on CB2 (pEC50 = 5.6 ± 0.3, Emax = 17±2%, n = 3; Figure S1). NAGly does not bind CB2 when tested up to 10 µmol/L,34 but our data raise the possibility of an interaction between NAGly and CB2 in higher concentration ranges. Unexpectedly CP‐55,940 also activated hGPR132a (Figure 2E; pEC50 = 5.5 ± 0.13, Emax = 102 ± 3%), though more weakly than CB2 (Figure S1; pEC50 = 7.6 ± 0.27, Emax = 100%). CP‐55,940 is a synthetic analogue of the Cannabis sativa constituent, Δ9‐tetrahydrocannabinol. CP‐55 940 also modulates the lysophophaditylinositol receptor, GPR55.35, 36 GPR132 is the fourth lipid receptor (in addition to CB1, CB2, and GPR55) modulated by CP‐55 940.
To confirm the ligand pairing, we perfused RBL‐hGPR132a cells with 10 μmol/L NLGly, which induced intracellular Ca2+ release comparable to 9‐HODE (Figure 3B&C). NLGly induced association of hGPR132 with β‐arrestin (Figure 4C; pEC50 = 5.5 ± 0.2, Emax = 39±4%). NOGly had similar activity to NLGly whereas NPGly and NSGly were less active (Figure 4C). N‐acylglycines act as less efficacious or less potent agonists to induce hGPR132a association with β‐arrestin, compared with 9‐HODE, which acts as a full agonist. This is in contrast to G‐protein‐mediated signals (in RBL‐hGPR132a and yeast) where N‐acylglycines and 9‐HODE are equi‐efficacious (Figures 2 and 3). 9‐HODE and N‐acylglycines are thus potentially able to elicit differential cellular responses via the same receptor. During our studies, Cohen et al showed two structurally‐related N‐acyl amino acids to stimulate association of hGPR132 (G2A) with β‐arrestin. N‐acyl‐3‐hydroxypalmitoylglycine (commendamide)7 and N‐myristoylalanine6 are products of N‐acyl amide synthase enzymes of gut‐resident bacteria proposed to modulate host immune responses in via GPR132. Our data, together with that of Cohen et al, confirm N‐acyl amino acids as bona fide activators of hGPR132.
Family A GPCRs have varying degrees of homology across mammals. Assuming most physiological processes change little through mammalian evolution, specificity for cognate ligands is expected to be conserved even where GPCR sequence conservation is not high, due to selective pressure to retain affinity and activity. hGPR132 and mouse GPR132 (mGPR132) are 68% identical. Thus, GPR132 appears less well‐conserved than CB2, CB1, or GPR68 (respectively 83%, 97%, and 92% identical between human and mouse). No activation of mGPR132 by 9‐HODE was originally observed.14 Those authors concluded GPR132 may have distinct roles in human and rodent, and the late‐onset autoimmune syndrome in GPR132‐null mice37 may not be relevant to human. We developed yeast expressing rGPR132. 9‐HODE, NPGly, NAGly (Figure 2F) and also NLGly and linoleamide (Figure 2G) activated rGPR132, with potencies comparable to those observed at hGPR132 (Table 1). We fused a C‐terminal ProLink tag to mGPR132. In HEK293 cells expressing EA‐β‐arrestin, 9‐HODE (Figure 4D), SB‐583831 (Figure 4E), NLGly and NOGly (Figure 4F) induced association between mGPR132PL and β‐arrestin. Ligands inactive at hGPR132 (NHGly, linoleic acid, and LPC) were inactive at mGPR132 and rGPR132 (Table 1). In conclusion, the pharmacological profile of N‐acylglycines and oxidized free fatty acids is conserved across mammalian GPR132 orthologs.
N‐acylglycines and SB‐583831 share a glycine moiety (Figure 1B). To determine whether these internal glycines bind a common GPR132 site, we conducted antagonist Schild analysis. GSK1820795A had been identified as an hGPR132 antagonist (see Methods). GSK1820795A is an analogue of telmisartan, the dual angiotensin AT1 antagonist and PPARγ modulator (Figure 1C). AT1 antagonism by GSK1820795A and telmisartan are comparable (not shown; pIC50 = 8.2 ± 0.13; n = 3 and pIC50 = 8.6 ± 0.3; n = 28, respectively). GSK1820795A blocks agonist‐induced activation of hGPR132 (Figure 5A‐D). GSK1820795A did not antagonize free fatty acid receptor FFA2 (Figure 5E), confirming specificity for GPR132. GSK1820795A blocked agonist‐induced association of hGPR132 with β‐arrestin (not shown; pIC50 = 7.8 ± 0.22; n = 13), and we have preliminary evidence that GSK1820795A blocks rGPR132 (n = 2; not shown). Maximum asymptotes (Emax) for NPGly, NLGly and linoleamide (Figure 5A‐C) were reduced by increasing concentrations of GSK1820795A with minimal change to pEC50, typical of non‐competitve antagonism. GSK1820795A caused rightward shifts of SB‐583831 agonist‐response curves with minimal change to Emax (Figure 5D), in contrast to N‐acylamides. Schild regression showed linearity (Figure S3B). GSK1820795A was ≈10‐fold more potent than telmisartan as an antagonist of hGPR132 (Figure S3). It was unclear whether GSK1820795A acted as a competitive antagonist of SB‐583831, since Hill slope differed from unity (nH = 1.9 for both GSK1820795A and telmisartan; Figure S3B). Extending antagonist preincubation time from 5 to 45 minutes had no effect on Hill slope (not shown), suggesting non‐unity Hill slopes were not caused by insufficient binding equilibration. We concluded that N‐acylglycines and SB‐583831 have distinct modes of binding to GPR132.
Figure 5.
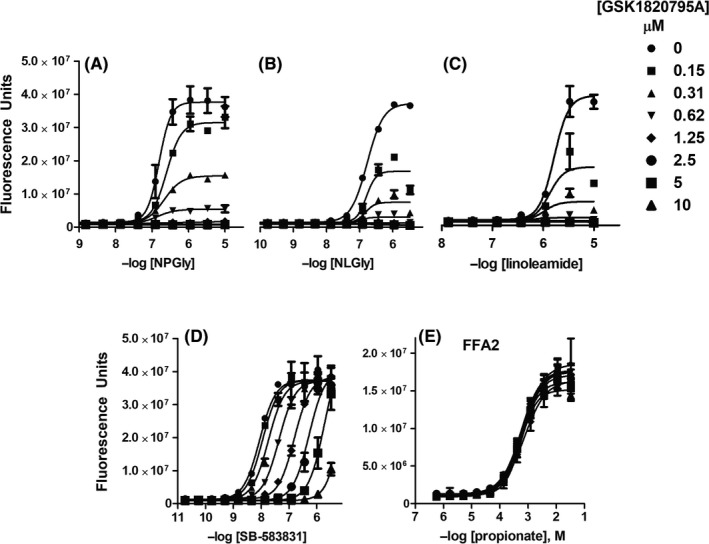
GSK1820795A is a selective antagonist at hGPR132a. Telmisartan analogue GSK1820795A was identified as a GPR132 antagonist through high‐throughput screening. Responses of yeast expressing hGPR132a to agonists NPGly (A), NLGly (B), linoleamide (C), and SB‐583831 (D) were blocked by GSK1820795A in a concentration‐dependent manner. Maximal effects NPGly, NLGly, and linoleamide (A‐C) were depressed by GSK1820795A with little effect on EC50. In contrast, GSK1820795A caused rightward shifts of concentration‐response curves to SB‐583831 without reducing maximal responses. The effect of telmisartan on hGPR132a and Schild analysis are presented in Figure S3. Agonist responses of yeast expressing GPR43/FFA3 to propionate were not significantly affected by GSK1820795A (E). Data points show mean ± SD of four technical replicates (non‐normalized fluorescent counts) from a representative experiment
To probe further the interaction of GPR132 with lipids, we predicted a 3D pose for N‐acylglycine and 9‐HODE binding, and tested this by mutagenesis. A 3D homology model of hGPR132 from GPCRdb was used.38 Due to the flexible acyl side‐chain of both ligands, we adopted an “anchor and grow” approach (see Figure S6). 9‐HODE is similar to ligands of FFA1. Carboxylate‐binding residues in FFA1 have been established by mutagenesis39 and crystallization.40, 41 We hypothesized that lipid heads groups bind GPR132 in an equivalent pocket, so we focused on this region as the initial anchoring site (residues 4.57, 5.39, 5.42, 6.51, and 6.55 by Ballesteros‐Weinstein numbering). Meta‐analysis of the ternary complex between FFA1, fatty‐acid mimetic MK‐8666 and allosteric ligand AP8 further supported importance of these residues (Figure S4). “Anchor and grow” docking yielded superimposable and reproducible low‐energy‐scoring binding poses for 9‐HODE and NPGly. Growth trees with the best grid scores are shown in Figures S6 and S7. The lowest‐energy binding poses for 9‐HODE and NPGly are highly similar (Figure 6). At neutral pH, negatively charged lipid headgroups are predicted to stabilize a charge network involving Y200 (5.39), Y258 (6.51), K265 (6.58), and K183 (ECL2). We noted a potential interaction between R203 (5.42) and the alkyl side chain of both 9‐HODE and NPGly. This residue is conserved across GPR132 orthologs. In published GPCR structures, residue 5.42 often lines the “classical” ligand‐binding pocket. Residue 5.42 tends to contribute more to agonist than antagonist binding (Figure S5). We mutated R203 (5.42) and adjacent residues Y199 (5.38) and Y200 (5.39) of hGPR132a, and also K183 in ECL2. Agonist potencies for a panel of ligands is shown in Figure 7 and Table S1. Mutant R203A (5.42) abolished responses to NPGly and NLGly (Figure 7C and D). These effects were unlikely to be due to reduced expression of hGPR132a, since R203A responded to CP‐55,940 with a potentiated response (wild‐type: pEC50 = 5.64 ± 0.08; R203A: pEC50 = 7.04 ± 0.06; Figure 7B and Table S1). These observations are consistent with our binding pose, and the hypothesis that R203(5.42) contributes significant energy to N‐acylglycine binding. Strikingly, SB‐583831 had no agonist activity but appeared to act as an inverse agonist on R203A, inhibiting basal signaling (Figure 7A; pIC50 = 7.7 ± 0.2; n = 3). Mutants K183A (ECL2) and Y199A (5.38) retained agonist responses, suggesting neither residue contributes to ligand binding or G‐protein activation. Responses of K183A (ECL2) and Y199A (5.38) to N‐acylglycines were somewhat potentiated compared with wild‐type (Figure 7C and D), which could be due to increased receptor expression. Mutant Y200A (5.39) responded to SB‐583831 with similar pEC50 as wild‐type, showing that Y200A is expressed and retains the ability to respond to agonists (Figure 7A). N‐acylglycines activated Y200A but with substantially reduced Emax compared with wild‐type (Figure 7C and D). Activation by CP‐55,940 was abolished in Y200A (Figure 7B). Residue 5.39 in the free‐fatty acid receptors (FFA1‐3) is arginine, forming the critical salt‐bridge to carboxylate‐containing agonists.39, 41 The effect of Y200A on activation of hGPR132a by N‐acylglycines supports the hypothesis that there are similarities in the mode of binding of lipid agonists between GPR132, FFA1‐3, and other lipid‐binding Family‐A GPCRs. In summary, modelling identified residues in GPR132 important in binding N‐acylglycines and other novel agonists examined in this study. Combined with the effects of antagonist GSK1820795A (Figure 5), our data differentiate between GPR132 agonists with flexible acyl side‐chains (N‐acylamides) and more rigid small‐molecule surrogate agonists such as SB‐583831.
Figure 6.
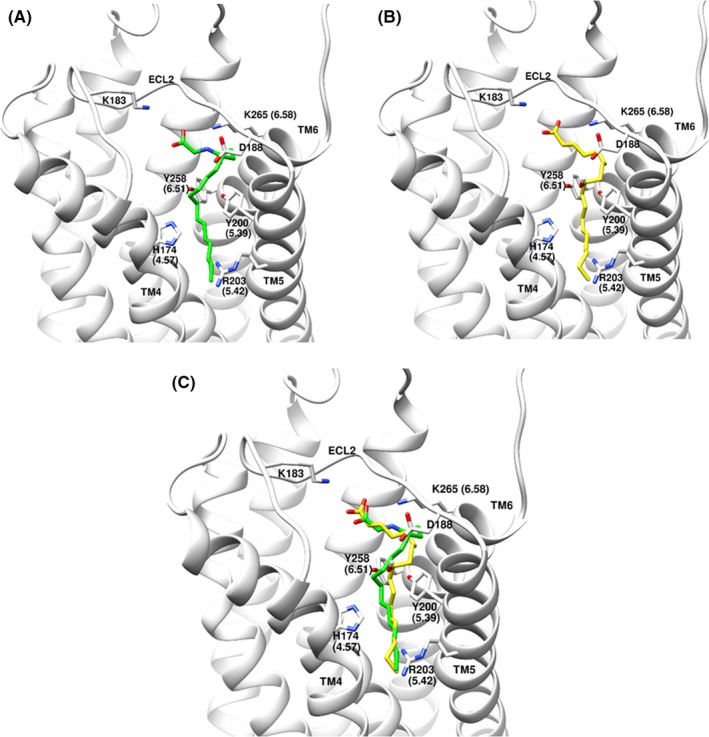
Proposed binding poses of NPGly and 9‐HODE docked to GPR132 homology model, showing suggested hydrophobic interactions of acyl side‐chains perpendicular to the guanidinium plane of R203(5.42): (A) NPGly; (B) 9‐HODE; (C) overlay of 9‐HODE and NPGly
Figure 7.
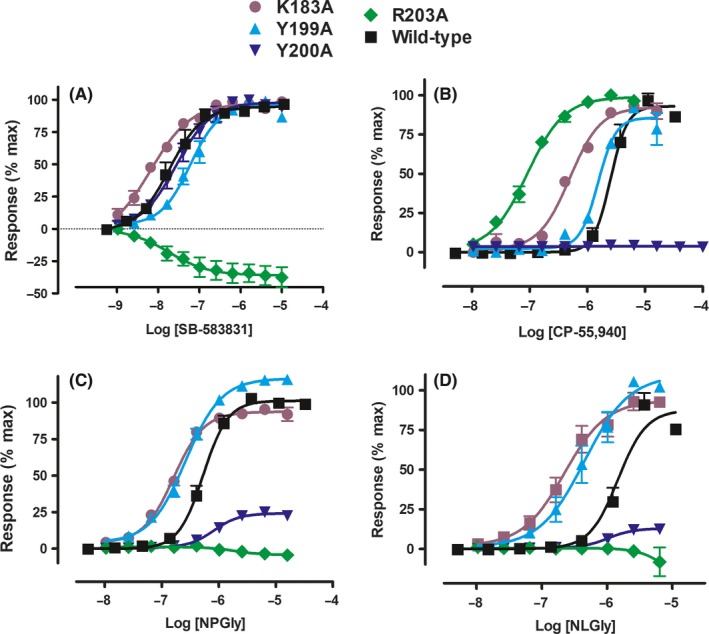
Yeast assay showing the effect of hGPR132a residue mutagenesis on the agonist potency of ligands: (A) SB‐583831, (B) CP‐55,940, (C) NPGly, (D) NLGly. hGPR132a sequences are represented as follows: K183A (purple), Y199A (cyan), Y200A (blue), R203A (green), and wild‐type (black). Response data for each mutant were normalized to effects of the corresponding ligand at wild‐type hGPR132a. SB‐583831 did not activate mutant R203A but showed an inverse agonist concentration‐response, inhibiting the elevated basal signal (elevated basal is due to receptor constitutive activity). Data are presented as mean ± SEM of n = 3 independent experiments
4. DISCUSSION
Here we show that endogenously‐occurring N‐acylamides activate the orphan receptor, GPR132. To gain confidence in this ligand pairing, we identified small‐molecule agonists and antagonists of GPR132, also disclosed here. This chemical biology approach, starting with the surrogate agonist, SKF‐95667, allowed validation of multiple host systems for functional GPR132 expression, before screens of endogenously‐occurring lipids were conducted. N‐acylamides trigger intracellular Ca2+ release in RBL‐hGPR132a cells, induce association between hGPR132a and β‐arrestin, and activate the G‐protein‐linked pheromone response pathway of yeast cells expressing hGPR132a. GSK1820795A blocks activation of yeast cells expressing hGPR132a by N‐acylamides. Activation of rat and mouse GPR132 by N‐acylamides further supports the ligand pairing. We corroborate 9‐HODE as a candidate endogenous agonist for hGPR132.14, 15 Our data are consistent with activation of Gαi pathways by GPR132.13 Chimeric Gpa1‐Gαi3 supported hGPR132 signalling in yeast, and Gαi activation in RBL cells has been shown to trigger Ca2+ mobilization.42 We show for the first time that rGPR132 and mGPR132 are also activated by 9‐HODE. Broadly, 9‐HODE and N‐acylglycines have comparable effects on GPR132, which is unsurprising given their similar structures.
N‐acylserine (NOSer; Figure 2) and N‐acylalanine6 also activate GPR132, though N‐acylglycines appeared most efficacious. Moreover, a specific biosynthetic enzyme for N‐acylglycines, GLYATL2, has been characterized suggesting biological relevance.5 Few N‐acyl derivatives of larger amino acids are commercially available. N‐arachidonyltyrosine was inactive, but arachidonate is not an optimal side‐chain for GPR132 activation. A full understanding of SAR will require a systematic evaluation across N‐acyl amino acids with a common, optimal acyl groups for GPR132 activation. We observed no activation of GPR132 by free linoleic acid up to 100 µmol/L in either yeast or β‐arrestin assocation assays, in agreement with,13 though not.15 Amidation of linoleic acid to linoleamide confers agonist activity (Figure 2). This contrasts with FFA receptors, where fatty acid amidation abolishes agonism.43 We also disclose SB‐583831 and SB‐583355 as peptidomimetic GPR132 agonist and antagonist. These are based on amino acid templates (glycine in SB‐583831; L‐phenylalanine in SB‐583355) with structural similarity to previously described agonists.30
Yeast is suited to GPCR deorphanization because no potentially confounding GPCRs are encoded in the yeast genome (the pheromone receptor, Ste2p, is deleted). Inadvertent upregulation of endogenous mammalian GPCRs is a common pitfall of orphan GPCR studies, and may explain why initial deorphanizations are not always reproduced. Other GPCRs reportedly activated by N‐acylglycines (LPA5 and GPR18) could not be responsible for effects observed at GPR132 in yeast. The pharmacological profile of GPR18 is controversial: multiple groups report NAGly activity,8, 9, 10 other publications do not reproduce this.15, 44 The recent identification of synthetic GPR18 ligands should allow resolution of authentic ligand specificity, as has been the case for other controversial GPCRs.45, 46 We observed evidence of weak activation of yeast expressing CB2 by NAGly. NAGly did not bind CB2 up to 10 µmol/L,34 but the possibility of an interaction between CB2 and NAGly at higher concentration merits further investigation.
LPC and lactate have been published as GPR132 ligands, but have not gained widespread acceptance (one publication was retracted47). GPR132 was also reported as a sensor of extracellular pH,48 but other authors were unable to replicate these observations.32, 49 GPR4, GPR65, and GPR68, whose pH‐sensing activity is well‐established, have significant similarity to each other. In contrast, GPR132 is more distantly related, showing homology also to purinergic receptors,50 and three of the four basic amino acids involved in pH‐sensing by GPR4, GPR65, and GPR68 are non‐basic residues in GPR132.50 Discovery of 9‐HODE allowed direct comparison of the effect of acidic pH to a molecular agonist. 9‐HODE was approximately 10‐fold more efficacious than acidic pH.13 Considering these factors, and the challenges in discriminating specific effects of pH on receptors from the effects of pH on host cells, we present here data generated in buffered conditions (constant pH), to show pH‐independent agonist effects.
We postulated lipids to enter GPR132 via a crevice between TM4 and TM5 domains and to occupy similar poses. Acyl groups extend through the TM4/TM5 crevice into the lipid bilayer, accommodating larger acyl groups (C20 in 11‐HETE13). Acyl chains are predicted to interact with R203 (5.42) via hydrophobic interactions perpendicular to planar guanidinium.51 R203A (5.42) mutation eliminated the activity of NPGly, NLGly and NOGly, consistent with our model. Phenotypes of mutations discriminated agonists into three classes, which may correspond to different binding modes to GPR132. R203A (5.42) eliminated activity of N‐acylglycines, whereas CP‐55,940 retained its agonist effect. This is consistent with crystal structures of AM11542 and AM841 in CB1, which showed that structurally equivalent 5.43 in CB1 is not essential for interaction with Δ9‐tetrahydrocannabinol derivatives.52 R203A (5.42) retained affinity for SB‐583831 but efficacy was lost and it behaved as an inverse agonist. Similar studies on β2 adrenoceptor showed the potential importance of S203 (5.42) rotameric conformation in mediating inverse versus full agonism of ligands.53 Mutant Y200A (5.39) had no effect on SB‐583831 but substantially reduced reponses to N‐acylglycines and abolished responses to CP55940. Previous studies have shown residue 5.39 to influence binding of CP‐55,940 to CB1.54 Together, our data raise the possibility of a comparable pocket on two lipid receptors, CB1 and hGPR132a, able to accommodate CP‐55,940. A limitation of our functional assay data is that it does not distinguish mutational effects on efficacy and affinity. It will be informative to analyze mutations using probes to directly quantify affinity at GPR132, and reliable antibodies to measure protein expression, when such tools become available. An alternative interpretation is that R203 (5.42) forms a salt‐bridge to carboxylate and Y200 (5.39) forms a carboxylate‐stabilizing interaction, as observed in the FFA1‐TAK‐875 structure.41 Confidence in the structure of GPR132 and pose of bound ligands will require crystallography or other biophysical methods.
How does the identification of N‐acylglycines as ligands for GPR132 advance understanding of this multi‐functional GPCR? In general, N‐acylglycines are anti‐inflammatory 3, 12 and anti‐nociceptive,12 whilst 9‐HODE is proinflammatory 20 and pro‐nociceptive.21 These differences could relate to preferential activation of separate pathways downstream of GPR132. N‐acylglycines induced weaker association of ß‐arrestin with GPR132 compared to 9‐HODE, whereas NPGly activated GPR132‐expresssing yeast more strongly than 9‐HODE. However, the situation is complicated because 9‐HODE also activates PPARγ, a known modifier of macrophage activation.55, 56 N‐acylglycines also are unlikely to be selective for GPR132.4 Both 9‐HODE and N‐acylamides (such as AEA) interact with TRPV1.57, 58 We have not tested the panel of N‐acylglycines at TRPV1, but the GPR132 antagonist SB‐583355 is inactive at TRPV1 (tested at a single concentration; 12.5 µmol/L) so might allow confirmation of GPR132‐mediated lipid effects in tissue. Structure‐activity relationship (SAR) for N‐acyl amino acid stimulation of PGJ production by mouse RAW cells16 resembles, at least in part, order‐of‐potency at GPR132, with NLGly being more active than NAGly. Thus, RAW cell activation may be mediated by GPR132. However, selective blockade will be required to conclusively attribute GPR132‐mediated effects of N‐acylglycines and oxidized fatty acids in primary cells, and in vivo. Oxidized fatty acids are produced in skin and other tissues under stress conditions, such as exposure to UV light. Hattori et al found that skin keratinocytes under oxidative stress upregulate GPR132, and 9‐HODE treatment causes GPR132‐mediated intracellular Ca2+ release in these cells.20 The identification of N‐acylglycines as GPR132 ligands also points to functions in barrier tissues, since skin fibroblasts express high levels of GLYATL2.5 N‐acylglycines from skin fibroblasts may signal to GPR132 on local keratinocytes, or to infiltrating or tissue‐resident macrophages. At sites of zymosan‐induced inflammation, GPR132 functions to position infiltrating macrophages into proinflammatory microenvironments, facilitating M1‐like differentiation and production of proinflammatory mediators.19 N‐acylglycines signalling to GPR132 in dermis may operate in a similar way.
In conclusion, GPR132 is activated by two lipid classes: oxidized fatty acids exemplified by 9‐HODE and 11‐HETE, and N‐acyl amino acids, exemplified by NPGly and NLGly. Oxidized fatty acids are associated with pathological states such as oxidative stress. N‐acylglycines are produced by dedicated biosynthetic pathways under non‐pathological conditions. Knowledge of the endogenous ligands of GPR132 will be crucial to fully understand the immunomodulatory roles of this GPCR.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
AUTHOR CONTRIBUTIONS
JRF, MXC, JH, SJD, AJI, and AJB participated in research design. JRF, SU, and AJB conducted experiments. JRF, SU, AJI, and AJB performed data analysis. JRF, JH, SJD, AJI, and AJB wrote or contributed to the writing of manuscript.
Supporting information
ACKNOWLEDGEMENTS
The authors thank Ben Moore, Charlotte Beresford, Mark Chen, Pam Thomas, Cara Purvis, Shaohui Wang, Yannick Saintillan, Nicolas Ancellin, Nicola Bevan, and past members of the GPR132 program (all GSK) for their contributions to this work.
Foster JR, Ueno S, Chen MX, et al. N‐Palmitoylglycine and other N‐acylamides activate the lipid receptor G2A/GPR132. Pharmacol Res Perspect. 2019;e00542 10.1002/prp2.542
REFERENCES
- 1. Leishman E, Mackie K, Luquet S, Bradshaw HB. Lipidomics profile of a NAPE‐PLD KO mouse provides evidence of a broader role of this enzyme in lipid metabolism in the brain. Biochim Biophys Acta. 2016;1861:491‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Iannotti FA, Di Marzo V, Petrosino S. Endocannabinoids and endocannabinoid‐related mediators: targets, metabolism and role in neurological disorders. Prog Lipid Res. 2016;62:107‐128. [DOI] [PubMed] [Google Scholar]
- 3. Burstein SH, Adams JK, Bradshaw HB, et al. Potential anti‐inflammatory actions of the elmiric (lipoamino) acids. Bioorg Med Chem. 2007;15:3345‐3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burstein SH. N‐acyl amino acids (Elmiric Acids): endogenous signaling molecules with therapeutic potential. Mol Pharmacol. 2018;93:228‐238. [DOI] [PubMed] [Google Scholar]
- 5. Waluk DP, Schultz N, Hunt MC. Identification of glycine N‐acyltransferase‐like 2 (GLYATL2) as a transferase that produces N‐acyl glycines in humans. FASEB J. 2010;24:2795‐2803. [DOI] [PubMed] [Google Scholar]
- 6. Cohen LJ, Esterhazy D, Kim SH, et al. Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature. 2017;549:48‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cohen LJ, Kang HS, Chu J, et al. Functional metagenomic discovery of bacterial effectors in the human microbiome and isolation of commendamide, a GPCR G2A/132 agonist. Proc Natl Acad Sci USA. 2015;112:E4825‐E4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McHugh D, Page J, Dunn E, Bradshaw HB. Delta(9) ‐Tetrahydrocannabinol and N‐arachidonyl glycine are full agonists at GPR18 receptors and induce migration in human endometrial HEC‐1B cells. Br J Pharmacol. 2012;165:2414‐2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Console‐Bram L, Brailoiu E, Brailoiu GC, Sharir H, Abood ME. Activation of GPR18 by cannabinoid compounds: a tale of biased agonism. Br J Pharmacol. 2014;171:3908‐3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kohno M, Hasegawa H, Inoue A, et al. Identification of N‐arachidonylglycine as the endogenous ligand for orphan G‐protein‐coupled receptor GPR18. Biochem Biophys Res Commun. 2006;347:827‐832. [DOI] [PubMed] [Google Scholar]
- 11. Oh DY, Yoon JM, Moon MJ, et al. Identification of farnesyl pyrophosphate and N‐arachidonylglycine as endogenous ligands for GPR92. J Biol Chem. 2008;283:21054‐21064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang SM, Bisogno T, Petros TJ, et al. Identification of a new class of molecules, the arachidonyl amino acids, and characterization of one member that inhibits pain. J Biol Chem. 2001;276:42639‐42644. [DOI] [PubMed] [Google Scholar]
- 13. Obinata H, Hattori T, Nakane S, Tatei K, Izumi T. Identification of 9‐hydroxyoctadecadienoic acid and other oxidized free fatty acids as ligands of the g protein‐coupled receptor G2A. J Biol Chem. 2005;280:40676‐40683. [DOI] [PubMed] [Google Scholar]
- 14. Obinata H, Izumi T. G2A as a receptor for oxidized free fatty acids. Prostaglandins Other Lipid Mediat. 2009;89:66‐72. [DOI] [PubMed] [Google Scholar]
- 15. Yin H, Chu A, Li W, et al. Lipid G protein‐coupled receptor ligand identification using beta‐arrestin PathHunter assay. J Biol Chem. 2009;284:12328‐12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burstein S, McQuain C, Salmonsen R, Seicol B. N‐Amino acid linoleoyl conjugates: anti‐inflammatory activities. Bioorg Med Chem Lett. 2012;22:872‐875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bond J, Domaschenz R, Roman‐Trufero M, et al. Direct interaction of Ikaros and Foxp1 modulates expression of the G protein‐coupled receptor G2A in B‐lymphocytes and acute lymphoblastic leukemia. Oncotarget. 2016;7:65923‐65936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weng Z, Fluckiger AC, Nisitani S, et al. A DNA damage and stress inducible G protein‐coupled receptor blocks cells in G2/M. Proc Natl Acad Sci USA. 1998;95:12334‐12339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kern K, Schafer SMG, Cohnen J, et al. The G2A receptor controls polarization of macrophage by determining their localization within the inflamed tissue. Front Immunol. 2018;9:2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hattori T, Obinata H, Ogawa A, et al. G2A plays proinflammatory roles in human keratinocytes under oxidative stress as a receptor for 9‐hydroxyoctadecadienoic acid. J Invest Dermatol. 2008;128:1123‐1133. [DOI] [PubMed] [Google Scholar]
- 21. Hohmann SW, Angioni C, Tunaru S, et al. The G2A receptor (GPR132) contributes to oxaliplatin‐induced mechanical pain hypersensitivity. Sci Rep. 2017;7:446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Su YS, Huang YF, Wong J, Lee CW, Hsieh WS, Sun WH. G2A as a threshold regulator of inflammatory hyperalgesia modulates chronic hyperalgesia. J Mol Neurosci. 2018;64:39‐50. [DOI] [PubMed] [Google Scholar]
- 23. Lahvic JL, Ammerman M, Li P, et al. Specific oxylipins enhance vertebrate hematopoiesis via the receptor GPR132. Proc Natl Acad Sci USA. 2018;115:9252‐9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen P, Zuo H, Xiong H, et al. Gpr132 sensing of lactate mediates tumor‐macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci USA. 2017;114:580‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu J, Wang T, Wu Y, Jin W, Wen Z. Microglia colonization of developing zebrafish midbrain is promoted by apoptotic neuron and lysophosphatidylcholine. Dev Cell. 2016;38:214‐222. [DOI] [PubMed] [Google Scholar]
- 26. Dowell SJ, Brown AJ. Yeast assays for G protein‐coupled receptors. Methods Mol Biol. 2009;552:213‐229. [DOI] [PubMed] [Google Scholar]
- 27. Mumberg D, Muller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995;156:119‐122. [DOI] [PubMed] [Google Scholar]
- 28. Chambers JK, Macdonald LE, Sarau HM, et al. A G protein‐coupled receptor for UDP‐glucose. J Biol Chem. 2000;275:10767‐10771. [DOI] [PubMed] [Google Scholar]
- 29. Dowell SJ, Brown AJ. Yeast assays for G‐protein‐coupled receptors. Receptors Channels. 2002;8:343‐352. [PubMed] [Google Scholar]
- 30. Shehata M, Christensen H, Isberg V, et al. Identification of the first surrogate agonists for the G protein‐coupled receptor GPR132. Rsc Adv. 2015;5:48551‐48557. [Google Scholar]
- 31. Ogawa A, Obinata H, Hattori T, et al. Identification and analysis of two splice variants of human G2A generated by alternative splicing. J Pharmacol Exp Ther. 2010;332:469‐478. [DOI] [PubMed] [Google Scholar]
- 32. Ludwig MG, Vanek M, Guerini D, et al. Proton‐sensing G‐protein‐coupled receptors. Nature. 2003;425:93‐98. [DOI] [PubMed] [Google Scholar]
- 33. Brown AJ, Dyos SL, Whiteway MS, et al. Functional coupling of mammalian receptors to the yeast mating pathway using novel yeast/mammalian G protein α‐subunit chimeras. Yeast. 2000;16:11‐22. [DOI] [PubMed] [Google Scholar]
- 34. Sheskin T, Hanus L, Slager J, Vogel Z, Mechoulam R. Structural requirements for binding of anandamide‐type compounds to the brain cannabinoid receptor. J Med Chem. 1997;40:659‐667. [DOI] [PubMed] [Google Scholar]
- 35. Kapur A, Zhao P, Sharir H, et al. Atypical responsiveness of the orphan receptor GPR55 to cannabinoid ligands. J Biol Chem. 2009;284:29817‐29827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brown AJ, Daniels DA, Kassim M, et al. Pharmacology of GPR55 in yeast and identification of GSK494581A as a mixed‐activity glycine transporter subtype 1 inhibitor and GPR55 agonist. J Pharmacol Exp Ther. 2011;337:236‐246. [DOI] [PubMed] [Google Scholar]
- 37. Le LQ, Kabarowski JH, Weng Z, et al. Mice lacking the orphan G protein‐coupled receptor G2A develop a late‐onset autoimmune syndrome. Immunity. 2001;14:561‐571. [DOI] [PubMed] [Google Scholar]
- 38. Pandy‐Szekeres G, Munk C, Tsonkov TM, et al. GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res. 2018;46:D440‐D446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stoddart LA, Smith NJ, Jenkins L, Brown AJ, Milligan G. Conserved polar residues in transmembrane domains V, VI, and VII of free fatty acid receptor 2 and free fatty acid receptor 3 are required for the binding and function of short chain fatty acids. J Biol Chem. 2008;283:32913‐32924. [DOI] [PubMed] [Google Scholar]
- 40. Lu J, Byrne N, Wang J, et al. Structural basis for the cooperative allosteric activation of the free fatty acid receptor GPR40. Nat Struct Mol Biol. 2017;24:570‐577. [DOI] [PubMed] [Google Scholar]
- 41. Srivastava A, Yano J, Hirozane Y, et al. High‐resolution structure of the human GPR40 receptor bound to allosteric agonist TAK‐875. Nature. 2014;513:124‐127. [DOI] [PubMed] [Google Scholar]
- 42. Brown AJ, Goldsworthy SM, Barnes AA, et al. The Orphan G protein‐coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem. 2003;278:11312‐11319. [DOI] [PubMed] [Google Scholar]
- 43. Brown AJ, Jupe S, Briscoe CP. A family of fatty acid binding receptors. DNA Cell Biol. 2005;24:54‐61. [DOI] [PubMed] [Google Scholar]
- 44. Lu VB, Puhl HL 3rd, Ikeda SR. N‐Arachidonyl glycine does not activate G protein‐coupled receptor 18 signaling via canonical pathways. Mol Pharmacol. 2013;83:267‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Simon K, Merten N, Schroder R, et al. The orphan receptor GPR17 is unresponsive to uracil nucleotides and cysteinyl leukotrienes. Mol Pharmacol. 2017;91:518‐532. [DOI] [PubMed] [Google Scholar]
- 46. Schoeder CT, Kaleta M, Mahardhika AB, et al. Structure‐activity relationships of imidazothiazinones and analogs as antagonists of the cannabinoid‐activated orphan G protein‐coupled receptor GPR18. Eur J Med Chem. 2018;155:381‐397. [DOI] [PubMed] [Google Scholar]
- 47. Witte ON, Kabarowski JH, Xu Y, Le LQ, Zhu K. Retraction. Science. 2005;307:206. [DOI] [PubMed] [Google Scholar]
- 48. Murakami N, Yokomizo T, Okuno T, Shimizu T. G2A is a proton‐sensing G‐protein‐coupled receptor antagonized by lysophosphatidylcholine. J Biol Chem. 2004;279:42484‐42491. [DOI] [PubMed] [Google Scholar]
- 49. Radu CG, Nijagal A, McLaughlin J, Wang L, Witte ON. Differential proton sensitivity of related G protein‐coupled receptors T cell death‐associated gene 8 and G2A expressed in immune cells. Proc Natl Acad Sci USA. 2005;102:1632‐1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Seuwen K, Ludwig MG, Wolf RM. Receptors for protons or lipid messengers or both? J Recept Signal Transduct Res. 2006;26:599‐610. [DOI] [PubMed] [Google Scholar]
- 51. Armstrong CT, Mason PE, Anderson JL, Dempsey CE. Arginine side chain interactions and the role of arginine as a gating charge carrier in voltage sensitive ion channels. Sci Rep. 2016;6:21759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hua T, Vemuri K, Pu M, et al. Crystal structure of the human cannabinoid receptor CB1 . Cell. 2016;167:750–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Warne T, Tate CG. The importance of interactions with helix 5 in determining the efficacy of β‐adrenoceptor ligands. Biochem Soc Trans. 2013;41:159‐165. [DOI] [PubMed] [Google Scholar]
- 54. McAllister SD, Rizvi G, Anavi‐Goffer S, et al. An aromatic microdomain at the cannabinoid CB(1) receptor constitutes an agonist/inverse agonist binding region. J Med Chem. 2003;46:5139‐5152. [DOI] [PubMed] [Google Scholar]
- 55. Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93:229‐240. [DOI] [PubMed] [Google Scholar]
- 56. Chawla A. Control of macrophage activation and function by PPARs. Circ Res. 2010;106:1559‐1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. De Petrocellis L, Schiano Moriello A, Imperatore R, Cristino L, Starowicz K, Di Marzo V. A re‐evaluation of 9‐HODE activity at TRPV1 channels in comparison with anandamide: enantioselectivity and effects at other TRP channels and in sensory neurons. Br J Pharmacol. 2012;167:1643‐1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hargreaves KM, Ruparel S. Role of oxidized lipids and TRP channels in orofacial pain and inflammation. J Dent Res. 2016;95:1117‐1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials