

Single-molecule photoswitchable emitters are demonstrated in the near-infrared based on single-wall carbon nanotube hybrids.
Abstract
The design of single-molecule photoswitchable emitters was the first milestone toward the advent of single-molecule localization microscopy, setting a new paradigm in the field of optical imaging. Several photoswitchable emitters have been developed, but they all fluoresce in the visible or far-red ranges, missing the desirable near-infrared window where biological tissues are most transparent. Moreover, photocontrol of individual emitters in the near-infrared would be highly desirable for elementary optical molecular switches or information storage elements since most communication data transfer protocols are established in this spectral range. Here, we introduce a type of hybrid nanomaterials consisting of single-wall carbon nanotubes covalently functionalized with photoswitching molecules that are used to control the intrinsic luminescence of the single nanotubes in the near-infrared (beyond 1 μm). Through the control of photoswitching, we demonstrate super-localization imaging of nanotubes unresolved by diffraction-limited microscopy.
INTRODUCTION
Over the past decade, super-resolution microscopy revolutionized fluorescence microscopy by delivering optical images with resolutions below the diffraction limit down to nanometer scales. Most of the super-resolution approaches, such as stimulated emission depletion microscopy (1), photoactivated localization microscopy (PALM) (2), or stochastic optical reconstruction microscopy (STORM) (3) [and related techniques (4)], are based on controlling the emission properties of fluorescent molecules to switch the emitters between on- and off-emission states. For instance, the advent of single-molecule localization microscopy (e.g., PALM and STORM), which is based on the super-localization of single molecules, was directly related to the conception of photoswitchable fluorescent emitters having photocontrollable blinking properties at the single-molecule level (5–7). To access the optical window of biological tissues, i.e., where tissue scattering and absorption are minimal and thus should allow localization microscopy at depth within biological tissue, the use of near-infrared (NIR) nanoprobes, with an emission wavelength of λ > 1 μm, will be crucial (8, 9). However, the available red-shifted dyes do not exhibit strong emission properties at the single-molecule level, and photoactivable emitters in this spectral range do not yet exist. The lack of appropriate emitters for implementing super-resolution techniques is even more detrimental in the NIR than in visible light: The limit of diffraction, an upper boundary for resolution in conventional far-field microscopy, increases linearly with wavelength [1.22 ∙ λ/(2NA)], e.g., ~450 nm for λ = 1065 nm with high numerical aperture (NA) objectives.
Single-wall carbon nanotubes (CNTs) display strong optical resonances in the NIR (10), and the discovery of their NIR luminescence (11) soon paved the way for their use as imaging probes in living cells (12) and whole animals (13, 14). They display exceptional luminescence signal stabilities in aqueous environments [~tens of minutes (15, 16)], superior to most other fluorescent nanoprobes used in single-molecule experiments for biological applications. NIR luminescent CNTs have thus proven to be unparalleled single-molecule probes due to their brightness and photostability for intracellular single-molecule tracking in cultured cells (17–19). Using small CNTs (L < 300 nm), it was previously shown that localization precision of ~50 nm, which corresponds to ~λ/20, could be achieved (16, 18). Their emission spectral range was also a key for long-term single-molecule tracking at depth in living brain tissue to uncover the extracellular dimensions of the brain at nanometer resolutions. Blinking photoluminescent CNTs have already been observed in acidic environments or through charge transfer near surfaces, which allowed achieving super-resolution imaging of CNTs emission sites (20, 21) or quenching sites (22–24). However, in these reports, photoblinking was observed without control of the blinking rate or efficiency. For future application of CNTs as NIR single molecules in localization microscopy, the first building block is thus still missing. Toward this aim, here, we report the design, experimental characterization, and modeling through simulations of photoswitchable CNTs having controlled blinking properties in the NIR (1065 nm) at the single-nanotube level.
RESULTS
Our approach is based on spiropyran-merocyanine (SP-MC) molecules covalently attached to CNTs through a nitrene-based cycloaddition reaction (fig. S1A) (25). The process noticeably preserves the conjugation of the sp2 network and ensures the CNTs to remain fluorescent upon covalent functionalization (up to 4% density of functional groups, fig. S2A). The unique functionalization yields a fully conjugated SP-CNT hybrid as demonstrated by the behavior of the π-electrons after conversion from SP to MC: In previous noncovalent approaches, the π-electron remained confined to the MC molecule, giving rise to the charge distribution that yields the strong MC dipole moment and its characteristic visible absorption band (26). In our covalent approach, there is no interruption of the conjugation between SP and nanotubes. The π-electron released by the switch after isomerization to MC conjugates over the extended CNT, uplifting the position of its Fermi level (25).
Illuminating a solution of SP-CNTs with an ultraviolet (UV) lamp induced a ~50% loss of luminescence intensity within a few seconds (fig. S2, A and B). This effect was previously shown to be fully reversible; it is due to the photoisomerization of SP-MC molecules, which modulates charge transfer to the CNT (25). The excitons are insensitive to the presence of the functional group in the SP state due to the π-preserving character of the functionalization. Upon UV illumination, the MC configuration is favored, and the subsequent charge transfer from the MC to the CNT induces nonradiative recombination of the exciton (i.e., photoluminescence partially quenched; fig. S2).
We will first reveal the origin of this incomplete loss of fluorescence upon UV illumination by performing a single-molecule study. We prepared (10,2) CNTs carrying randomly distributed SP-MC molecules [~1 per 100 carbon atoms (25)]. We imaged individual CNTs spin-coated on a microscope glass coverslip excited in a wide-field configuration using a circularly polarized 730 nm laser line (Ti:Sa) for resonant excitation on the second-order transition (S22). Photoluminescence of the (10,2) CNTs, which occurs at ~1065 nm, was imaged using an NIR indium gallium arsenide (InGaAs) camera (Fig. 1, A and B, and Materials and Methods section). Single (10,2) CNTs showed bright and stable photoluminescence when illuminated via the resonant S22 excitation (Fig. 1, C and D, black curves). We then added a lamp UV illumination (centered at 387 nm, with a bandwidth of 11 nm) and observed an overall decrease in intensity associated with independent blinking events (Fig. 1C, violet curves, and movie S1). This behavior is the origin for the incomplete loss of luminescence at the ensemble level. Averaging 79 nanotube intensity profiles (Fig. 1D) exhibits a dynamic that is similar to the isomerization observed through absorption spectroscopy on SP-CNTs in solution (fig. S1C). The 40% loss in intensity in Fig. 1D is comparable to the ~50% reduction measured for the (10,2) nanotube on bulk SP-CNT samples (fig. S2). We conclude that the photoblinking of the individual CNTs is responsible of the incomplete loss of photoluminescence observed at the ensemble level. Note that the single-nanotube photoluminescence was recovered over a few tens of seconds when the UV illumination was turned off (see movie S1). This fast MC to SP back-isomerization is most likely stimulated by the continuous 730 nm illumination during the recordings, since full luminescence recovery of nanotube solutions kept in the dark was observed previously over hours (25).
Fig. 1. UV irradiation induces luminescence intermittency on CNTs functionalized with SP-MC.
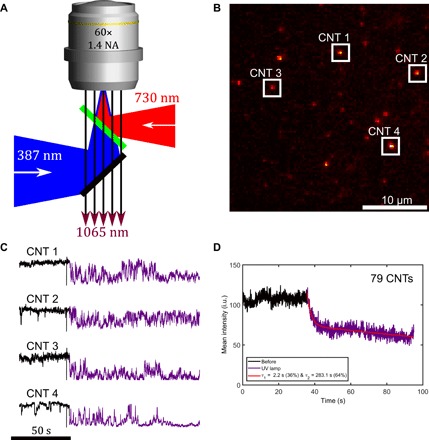
(A) Schema representing the excitation and emission fluorescence setup with UV illumination. (B) Wide-field NIR image of (10,2) CNTs spin-coated on polyvinylpyrrolidone (PVP) illuminated with a 730 nm circular polarized laser (~10 kW/cm2). (C) Luminescence time traces showing that UV irradiation induces emission intermittency and reduces the mean intensity. Imaging rate = 20 Hz. (D) Time evolution of the average of 79 CNTs before and during UV irradiation. i.u., intensity units.
Monte Carlo simulation of the photoblinking behavior of single CNTs
To better understand the blinking behavior of the nanotube under UV illumination, we developed a model for the complete photophysical processes in individual CNTs, taking into account the photogeneration of excitons, their spatial diffusion along the CNT backbones, and their radiative or nonradiative recombination (Fig. 2, A and B). This model bears similarities with previous work investigating how permanent quenching defects can affect the CNT luminescence properties (27), but here, the fast photoinduced quenching dynamics due to SP-MC isomerizations is critical and has to be additionally taken into account. We use the reported experimental value ld = 180 nm (28) for the exciton diffusion length in sodium cholate–stabilized (10,2) CNTs, and we consider that SP-MC molecules are randomly distributed along the nanotube. We further model the transition probability rate transfer statistics, between the SP and the MC conformations, by a two-state Markov process. We assume that all SP-MC molecules follow the same rate transfer probability and independent transitions. We define tSP and tMC as the average residence times in the SP and the MC states respectively, i.e., the time a molecule stays in the respective conformation, and the ratio between the residence time φ = tSP/tMC. Residence times should not be confused with the isomerization times required by the single SP-MC molecules to switch from one state to the other. The isomerization times occur on a much faster time scale (~hundreds of picoseconds) and are assumed to happen instantly. The generation rate of excitons is assumed to be constant along the nanotube. The probability for an exciton created at x0 to recombine at position x is given by c(x, x0, ld) = 1/(2ld) ∙ e−∣x − x0∣/ld. During its diffusion process, the exciton recombines nonradiatively if it encounters a molecule in the MC conformation or in the end of the CNT [which also acts as a photoluminescence quencher (29)]. Otherwise, the exciton will recombine radiatively, emitting a photon. Unless stated otherwise, the linear density of SP-MC molecules, NSM, is one per nanometer of nanotube (25). Diffusion and recombination are repeated for every exciton generated, and the state of each SP-MC molecules is updated at the end of each integration time (50 ms to match experimental observations). Figure 2 (C and D) presents examples of intensity time traces generated by this simulation considering a CNT length of L = 300 nm, respectively, for φ = 100 with varying tSP and for tSP = 50 s with varying φ.
Fig. 2. Simulation modeling of the excitonic photophysical processes generating the luminescence.
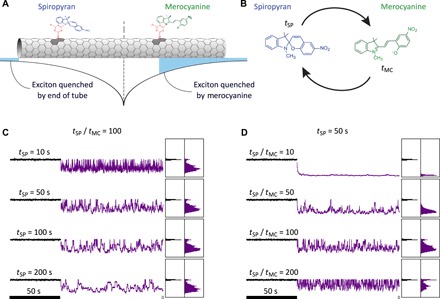
(A) Schematic representation of the nanotube functionalized with SP-MC molecules. An exciton generated in the middle of the nanotube and its probability of recombining are shown. Only excitons that recombine before encountering a merocyanine molecule or the end of the nanotube emit a photon. (B) The SP-MC group corresponds to a two-state model with transition rates defined as aSP = 1/tSP and aMC = 1/tMC. Simulated time traces are presented for φ =aMC/aSP = 100 with varying tSP (C) and for tMC = 50 s with varying φ (D). Simulated measurement rate = 20 Hz. Nanotube length and diffusion lengths were L = 300 nm, ld = 180 nm, and NSM = 1 per nm. The exciton generation rate was 133 nm−1 s−1.
An overall glance at the time traces suggests that replicating the experimental data requires tSP ≫ tMC. This can be explained by the fact that even if the large majority of the SP-MC molecules are in the SP configuration, the probability that a photogenerated exciton encounters an MC is high because of the high number of SP-MC molecules present on the nanotube along the exciton diffusion range (ld~φ/NSM). Only a few switching events are thus needed to quench excitons. In contrast, tMC~tSP (i.e., ld ≫ φ/NSM) markedly increases the quenching probability during the exciton diffusion process, resulting in full quenching instead of blinking.
From the simulations, we identify two parameters that allow determination of tSP and tMC from the experimental measurement of the blinking behavior of individual CNTs: (i) the mean intensity ratio between the stable state (before UV illumination, all molecules in SP form) and the blinking state (UV illumination, mixture of SP and MC forms) and (ii) the temporal autocorrelation function (ACF) of the blinking time traces. The mean intensity ratio for a given CNT length increases with φ and tends to 1 for large φ (Fig. 3A). The luminescence intensity ratio during UV irradiation is dependent on the residence times only through their ratio φ = tSP/tMC (fig. S4). An analytical solution of the intensity ratio is also obtained (Materials and Methods) and displayed on Fig. 3A for L = 300 nm and varying φ. This shows that φ can be analytically determined from the experimental intensity ratio knowing L, ld, and NSM with an error less than 12% for L = 300 ± 50 nm (Fig. 3A).
Fig. 3. Characterization of the luminescence temporal time traces.
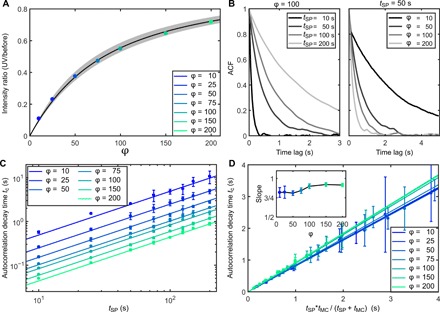
(A) Graph showing the influence of the ratio φ = aMC/aSP for tMC = 50 s and ld = 180 nm on the ratio of the average luminescence intensities before and during the UV irradiation. The gray shaded area corresponds to the error on the estimation of φ for L = 300 ± 50 nm. (B) Examples of temporal ACFs for the time traces presented in Fig. 2 (C and D) for φ = 100 with varying tSP and for tMC = 50 s with varying φ. (C) Graph summarizing the impact of varying φ and tSP on the temporal autocorrelation decay constant. For Fig. 3 (A to C), L = 300 nm, ld = 180 nm, and NSM = 1 per nm. (D) Graph summarizing the impact of varying φ and tSP on the temporal autocorrelation decay constant plotted versus . The slope is also shown in the inset as a function of φ. Values correspond to mean ± SD.
The autocorrelation decay time, tc, increases with tSP for a given φ and decreases with φ for a given tSP (Fig. 3B). This can be explained by interpreting tSP as a scaling factor at a given φ. The plateaus in the intensity time traces increase with tSP. Conversely, for constant tSP, an increase in φ induces faster varying events and decreases the autocorrelation time constant. From Fig. 3C, it can be observed that for a given nanotube length and linear defect density, the intensity ratio and the autocorrelation decay time fully describe the systems and provide estimates of the residence times of the SP and the MC states (tSP and tMC).
The temporal ACFs of the photoluminescence during blinking events are directly linked to the linear density of quenching molecules and the residence times in SP and MC states (Materials and Methods). Temporal time traces can thus be simulated using our model for different sets of parameters varying tSP and tMC and consequently φ (Fig. 3, C and D). The diffusion length, ld, couples the SP-MC states of adjacent molecules because the fate of the excitons depends on all quenching sites encountered along their path. For this reason, we find that the autocorrelation decay time tc < (tSP ∙ tMC)/(tSP + tMC) (Fig. 3D, inset). The temporal ACF of a single perfect fluorophore undergoing photointermittency between an on state and an off state (e.g., excited state and triplet state) would have given an autocorrelation decay time tc = (ton ∙ toff)/(ton + toff) (Materials and Methods).
Analysis of the experimental blinking traces for the determinations of tSP and tMC
The simulations described above indicate that the intensity time traces of a single CNT (Fig. 4A) provide φ using the ratio between the mean intensity when the nanotube is irradiated with UV light and the mean intensity before the UV irradiation. From the traces of 18 nanotubes, we obtain a mean value of φ = 80 ± 18 (mean±SEM) (Fig. 4, A and C). We next calculate the temporal ACFs (Fig. 4B) of the intensity time traces of each nanotube and obtain a mean decay time constant tC = 0.5 ± 0.1 s (mean ± SEM) (Fig. 4D). From the temporal autocorrelation time constants and using the slope obtained in the simulation data presented in Fig. 3D, we estimate the ratio (tSP ∙ tMC)/(tSP + tMC) = 0.4 ± 0.1 (mean ± SEM). Combining with the knowledge of φ = tSP/tMC, this yields the values for tSP = 46 ± 11 s (mean ± SEM) (Fig. 4E) and tMC = 0.6 ± 0.1 s (mean ± SEM) (Fig. 4F) from the experimental data.
Fig. 4. Estimation of the switching rate dynamics of the SP-MC molecules from the intensity time trace of individual CNTs.
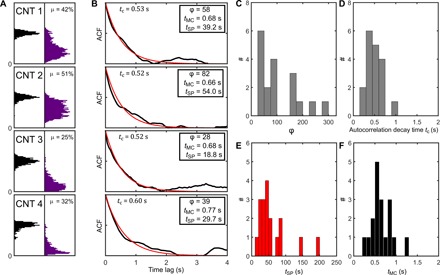
(A) Four normalized intensity histograms and the corresponding temporal ACFs (B) for the CNTs shown in Fig. 1. (C and D) The histograms of the intensity ratio φ and tc, respectively, obtained from individual carbon nanotube luminescence time traces. Knowing φ and tc, the values of tSP (E) and tMC (F) can be estimated. Values correspond to mean ± SEM.
Super-localization imaging of individual photoswitchable nanotubes
Using a forward-backward nonlinear filtering technique (30) on single-nanotube temporal intensity profiles, blinking events and consequently intensity plateaus are identified. Subtracting the average image of consecutive plateaus provides images of the single emitting sites. By fitting a two-dimensional Gaussian to these images, super-localization of individual blinking sites is obtained in a similar manner as used in super-resolution localization microscopy (4). The localization precision of single molecules can be estimated from (31). It depends on the strength of fluorescence signal (Si), the background noise (Br), the pixel size of the camera (a = 0.49 μm per pixel), and the width of the Gaussian point-spread function (σ). The localization precision is then given by , where , with , and , where G is the gain of the camera (G = 20e−/ADU where ADU means analog-to-digital unit). For this analysis, only plateaus longer than tC ≈ 0.5 s (i.e., 10 frames) are considered, and images from plateaus are averaged to increase the localization precision. Blinking detections with >7700 intensity units (corresponding to 385 photons) are used to generate super-localization of individual CNTs to guarantee a localization precision of <22 nm, corresponding to a full width at half maximum (FWHM) of ~50 nm (i.e., to ~λ/20). For display in Fig. 5 (A and B), single CNT detections are convolved with a two-dimensional Gaussian of ωFWHM = 50 nm to take into account the localization precision of single emitters. Given the short length of the nanotube (~300 nm) and the exciton diffusion length, only few switching events are sufficient to super-localize the nanotube centroid with a precision well beyond the emission wavelength (1064 nm) (Fig. 5A). For future applications, the use of bright ultrashort nanotubes (20) could potentially further enhance the localization precision.
Fig. 5. Super-localization and super-resolution imaging of single CNTs using photocontrolled luminescence intermittency.
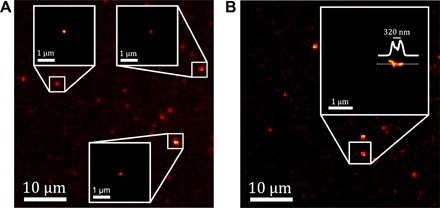
Wide-field NIR zoomed images of (10,2) CNTs spin-coated on PVP illuminated with a 730 nm circular polarized laser (~10 kW/cm2). The zoomed regions of interests correspond to reconstructed super-localization of individual CNTs (A) and super-resolved image of closely located nanotubes (B) using intensity transitions from all the blinking steps in the acquired movies (20 Hz). The super-resolved image shows different nanotube segments ~320 nm apart that could not be resolved in the wide-field image. For display, each localization is convolved with a Gaussian having a ωFWHM = 50 nm.
On occasion, owing to the blinking statistics of the photoswitchable CNTs, we can reveal the presence of distinct CNT segments that cannot be resolved initially. In Fig. 5B, different CNT segments are super-resolved 320 nm away from each other corresponding to λ/3.3. These super-resolved images provide the proof of principle that photoswitchable CNTs will be suitable for localization super-resolution microscopy applications in the NIR.
DISCUSSION
In this work, we show that photoswitchable CNTs can be created by conjugating SP-MC molecules onto the CNTs via triazine linkers. Single-nanotube experiments are presented to measure the blinking dynamics of the photoswitchable CNTs. Monte Carlo simulations of two-state Markov process, taking into account the spatiotemporal exciton dynamics occurring in CNTs, fully reproduced the experimental results. Combining simulations with the knowledge of tSP and tMC provides the means to create photoinduced blinking CNTs having arbitrary dynamics by varying the density of functionalization or illumination. It should thus be possible to tune these parameters to generate photoswitchable CNTs having blinking rates optimized for super-resolution imaging of densely labeled structures. The control of the emission properties of single CNTs using light is the first building block toward super-resolution studies in the NIR of biological samples (4) using CNTs. For this goal, future development will involve encapsulating CNTs in biocompatible surfactants (13, 32) and bioconjugation for specific labeling of cellular structures (33–35). Although low-dose UV illumination can, in principle, photoactivate molecules in vivo (36), it would be beneficial to perform CNT photoswitching in NIR (e.g., using two-photon excitation) to fully exploit their NIR photololuminescence. Photoswitchable CNTs might also find applications in the field of information science as elementary optical molecular switches or information storage elements operating in the NIR.
MATERIALS AND METHODS
SP-CNT preparation and characterization
High-pressure carbon monoxide synthetic method (HiPCO) single-walled CNTs were purchased from Unidym (0.8 to 1.2 nm in diameter and median length of 300 nm; batch no. SP0295). 2,4,6-Trichloro-1,3,5-triazine (cyanuric chloride or triazine), 2,3,3-trimethylindolenine, and 5-nitrosalicylaldehyde were purchased from Sigma-Aldrich. Sodium azide and N-methyl-2-pyrrolidone were purchased from Merck. Solvents and materials were used as they received and without further purifications. The synthesis of the SP-CNTs proceeded in two steps following (25): attachment of the triazine moieties onto the tubes (Trz-CNTs) and subsequent growth of the SP-MC switching moiety on the tubes (SP-CNTs).
Synthesis of Trz-CNTs
CNTs (1 g) were dispersed in N-methyl-2-pyrrolidone (150 ml) and sonicated for 0.5 to 1 hour. The dispersion was stirred for 1 to 2 hours at 25°C and cooled down to 0°C. 2,4,6-1,3,5-Trichloro-triazine (10 g, 54 mmol) was dissolved in N-methyl-2-pyrrolidone (50 ml), and the obtained solution was slowly added to the CNTs dispersion at 0°C. Sodium azide (1.76 g, 27 mmol) was added to the mixture and stirred for 2 hours at 0°C followed by 12 hours stirring at 70°C for . The product was purified by centrifugation redispersed in water and different organic solvents (acetone, toluene, and chloroform), and lyophilized for storage and characterization. The triazine functionalization was characterized by elemental analysis, x-ray photoelectron spectroscopy (XPS), thermogravimetric analysis, Raman, and infrared spectroscopy [see (25) for details].
Synthesis of SP-CNTs
SP-CNTs were synthesized in a multistep synthetic process (25). The indole segment was attached to the surface of CNTs-Trz by a nucleophilic reaction between the chlorine atoms of the triazine groups and 2,3,3-trimethylindolenine. CNT-Trz (0.2 g) was added to N-methyl-2-pyrrolidone (150 ml) and sonicated for 1 hour. A solution of 2,3,3-trimethylindolenine in N-methyl-2-pyrrolidone (2 ml, 12.47 mmol/10 ml) was added to this mixture at 0°C and stirred for 1 hour. After sonication at 25°C for 1 hour, the temperature of reaction was raised to 65°C and the mixture was stirred under nitrogen atmosphere for 4 days. The product (CNT-indole) was purified by centrifugation and redispersion in water and organic solvents such as acetone, chloroform, and tetrahydrofuran. CNT-indole was changed to CNT-indolene by dispersing it in a saturated aqueous solution of NaOH and sonicated for 30 min. The mixture was stirred at room temperature for 5 hours and then purified by repeated washing with water and centrifugation. SP-CNT was synthesized by adding 5-nitrosalicylaldehyde (2.5 g, 1.19 mmol) to a well-sonicated and degassed dispersion of CNT-indolene (0.1 g) in dry ethanol (70 ml) at 25°C. After sonication at 25°C with 35 kHz for 2 hours and stirring at 70°C for 12 hours, the solvent was evaporated and the mixture was redispersed in ethanol, chloroform, water, toluene, and acetone and collected by centrifugation at 5000 rpm for 5 min. CNT functionalization by SP was confirmed by elemental analysis, XPS, Raman scattering, UV/visible, infrared, and photoluminescence spectroscopy as described in detail in (25) (see also fig. S2).
Switching of SP-CNTs in solution and preparation of single SP-CNT samples
To monitor the switching behavior of the hybrids in suspension, we prepared a bulk solution by dissolving the SP-CNTs in water (density of tubes, 0.127 g/liter) and by adding sodium cholate (1 weight %). After tip sonication (Bandelin, SONOPULS HD 2070, 1 hour at 16 W) and centrifugation (Hettich Mikro 220R centrifuge, 30,000g for 1 hour), we collected the supernatant for optical characterization. Two-dimensional excitation-emission spectroscopy of the bulk solution was performed with a Nanolog spectrofluorometer from Horiba (xenon lamp source and liquid nitrogen–cooled InGaAs detector). Kinetic absorption measurements of the bulk suspensions under UV illumination were performed with a spectrophotometer from Thermo Fisher Scientific coupled with a handheld UV lamp emitting at 365 nm as a UV light source.
NIR single-nanotube imaging
Single-nanotube photoluminescence imaging was performed with an inverted microscope equipped with an NA of 1.40 and 60× objective. A volume of 10-μl drop of SP-CNT suspension was spin-coated on polyvinylpyrrolidone-coated glass coverslips to have isolated nanotubes. The excitation source consisted of a tunable Ti:Sa laser emitting at a wavelength of 730 nm to preferentially excite (10,2) CNTs at the resonance excitation on the second-order transition (S22). The excitation intensity was kept at 10 kW/cm2 with circularly polarized light. CNTs were detected by an InGaAs camera (Xenics XEVA 1.7-320 TE3) at 20 frames/s with a pixel size of 0.49 μm. A band-pass filter Z1064/10x (Chroma) was used to detect the (10,2) CNT–emitted fluorescence. An epifluorescence white light excitation illumination with FF01-387/11 (Semrock) was used for the UV illumination.
Modeling of the mean photoluminescence intensity ratio during blinking events
To model the mean photoluminescence intensity probability during the blinking processes (normalized to the luminescence without defects), we derived an analytical expression for the mean intensity of a segment of arbitrary length X. It corresponds to the probability of an exciton recombining before reaching the end of the nanotube and gives . Assuming that excitons are generated uniformly along the nanotube segment allows to estimate the average intensity of a nanotube segment of arbitrary length X by integrating p(x0, ld, X) over the nanotube segment
| (1) |
The integrated intensity from a nanotube with n quenching sites is given by the sum of the intensity of each (n + 1) nanotubes segments. The intensity of each segment is given by Eq. 1 and fig. S3A. The mean normalized intensity for a nanotube of length L = 300 nm can then be calculated (fig. S3B). This curve was obtained by numerically generating 50,000 random configurations having n quenchers. The relation is well approximated by int(L, ld, n) = 1/(1 + a(ld/L) ∙ n), where a(ld/L) depends only on the length ratio ld/L, and relates the probability of an exciton to encounter a quencher [a(ld/L) = c1 ∙ (ld/L)c2/(1 + c3 ∙ (ld/L)c2)], where c1, c2, and c3 are constants (fig. S3C). The average luminescence intensity of a nanotube of length L with diffusion length ld having NSM SP-MC molecules is then obtained by weighing each int(L, ld, n) by the probability of having exactly n SP-MC molecules in the MC state on the nanotube at a given time: Poi(NSM/φ, n) = (NSM/φ)n ∙ e(−NSM/φ)/n!, where NSM/φ is the average number of n SP-MC molecules in the MC state and φ = tSP/tMC. The final normalized intensity is then given by
| (2) |
and displayed on Fig. 3A for L = 300 ± 50 nm and varying φ. From Eq. 2, the ratio φ can thus be determined knowing L, ld, and NSM with an error ∣∆φ∣/φ < 12% for L = 300 ± 50 nm, error on ∣∆φ∣/φ < 13 % for NSM = 1 ± 0.1 per nm, and ∣∆φ∣/φ < 10 % for ld = 200 ± 50 nm (Fig. 3A and fig. S5).
Monte Carlo simulations of temporal luminescence intensity profiles
Nanotubes of length L with SP-MC linear density of NSM were simulated. The SP-MC molecules were randomly distributed along the nanotube length. The positions of the SP-MC molecules were fixed for each simulated nanotube. Each of them was simulated to undergo transition between two states SP and MC with residence times tSP and tMC, respectively. Initially, all SP-MC molecules are in the SP conformation. The generation probability for excitons is assumed uniform along the nanotube, and the probability for the exciton, created at x0, to recombine at a position x is given by c(x, x0, ld) = 1/(2ld) ∙ e−∣x−x0∣/ld, where ld is the diffusion length of the excitons. If the exciton encounters a molecule in the MC conformation or the end of the tube during the diffusion process, it recombines nonradiatively; otherwise, it recombines in a radiative way and emits a photon. This process is repeated for every exciton generated, and the state of each SP-MC molecules was updated at the end of each integration time. The exciton generation rate is set to 133 exciton nm−1 s−1. A minimum of five nanotubes were simulated for each set of parameters (L, ld, NSM, tSP, and tMC).
Temporal autocorrelation of the intensity time trace
The temporal statistics of a single fluorophore undergoing photointermittency between an on and off state (e.g., excited state and triplet state) correspond to a two-state Markovian model. Temporal ACF of the fluorophore emission defines an autocorrelation decay time given by tc = (ton ∙ toff)/(ton + toff), where 1/ton is the decay rate from the on state to the off state and 1/toff is the decay rate from the off state to the on state. Having more than one fluorophore undergoing the same blinking statistics will not modify tc, assuming that their transitions from one state to another are independent. Using our simulation, we also investigated the role of each parameter on tc. As expected, decreasing the number of quenching sites that each exciton encounters increases tc up to the limit of (tSP ∙ tMC)/(tSP + tMC) (e.g., ↓ ld,↓NSM, or ↑φ) (Fig. 3D, inset).
Supplementary Material
Acknowledgments
Funding: This work was supported by CNRS, the Agence Nationale de la Recherche (ANR-14-OHRI-0001-01 and ANR-16-CE29-0011-03), IdEx Bordeaux (ANR-10-IDEX-03-02), Conseil Régional d’Aquitaine (2011-1603009), and the France-BioImaging national infrastructure (ANR-10-INBS-04-01). A.G.G. acknowledges financial support from the Fondation pour la Recherche Médicale, the Fonds recherche du Québec–Nature et technologies, and a Chercheur-Boursier Award from the Fonds de recherche du Québec–Santé. A.S., M.A., R.H., and S.R. acknowledge the Focus Area Nanoscale of Freie Universität Berlin for financial support. Author contributions: A.G.G. and L.C. conceived the experiment and analyzed the data. A.G.G., M.G., and L.C. developed the model and the Monte Carlo simulations. A.S., M.A., R.H., and S.R. synthesized and characterized the SP-CNTs. A.G.G., A.S., S.R., and L.C. wrote the manuscript. L.C. and S.R. supervised the project. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaax1166/DC1
Movie S1. Movie showing the photoluminescence of single CNTs.
Fig. S1. SP-MC nanotube hybrid.
Fig. S2. SP-CNT luminescence in solution.
Fig. S3. Theoretical modeling of the luminescence intensity of a nanotube with quencher molecules.
Fig. S4. Influence of the residence times on the intensity ratio.
Fig. S5. Error estimation of the parameter theoretical φ.
REFERENCES AND NOTES
- 1.Hell S. W., Wichmann J., Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 19, 780–782 (1994). [DOI] [PubMed] [Google Scholar]
- 2.Betzig E., Patterson G. H., Sougrat R., Lindwasser O. W., Olenych S., Bonifacino J. S., Davidson M. W., Lippincott-Schwartz J., Hess H. F., Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Rust M. J., Bates M., Zhuang X., Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy ( STORM ). Nat. Methods 3, 793–7795 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Godin A. G., Lounis B., Cognet L., Super-resolution microscopy approaches for live cell imaging. Biophys. J. 107, 1777–1784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dickson R. M., Cubitt A. B., Tsien R. Y., Moerner W. E., On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature 388, 355–358 (1997). [DOI] [PubMed] [Google Scholar]
- 6.Patterson G. H., Lippincott-Schwartz J., A Photoactivatable GFP for selective photolabeling of proteins and cells. Science 297, 1873–1877 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Bates M., Blosser T. R., Zhuang X., Short-range spectroscopic ruler based on a single-molecule optical switch. Phys. Rev. Lett. 94, 108101 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong G., Antaris A. L., Dai H., Near-infrared fluorophores for biomedical imaging. Nat. Biomed. Eng. 1, 0010 (2017). [Google Scholar]
- 9.Danné N., Godin A. G., Gao Z., Varela J. A., Groc L., Lounis B., Cognet L., Comparative analysis of photoluminescence and upconversion emission from individual carbon nanotubes for bioimaging applications. ACS Photonics 5, 359–364 (2018). [Google Scholar]
- 10.Bachilo S. M., Strano M. S., Kittrell C., Hauge R. H., Smalley R. E., Weisman R. B., Structure-assigned optical spectra of single-walled carbon nanotubes. Science 298, 2361–2366 (2002). [DOI] [PubMed] [Google Scholar]
- 11.O’Connell M. J., Bachilo S. M., Huffman C. B., Moore V. C., Strano M. S., Haroz E. H., Rialon K. L., Boul P. J., Noon W. H., Kittrell C., Ma J., Hauge R. H., Weisman R. B., Smalley R. E., Band gap fluorescence from individual single-walled carbon nanotubes. Science 297, 593–596 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Cherukuri P., Gannon C. J., Leeuw T. K., Schmidt H. K., Smalley R. E., Curley S. A., Weisman R. B., Mammalian pharmacokinetics of carbon nanotubes using intrinsic near-infrared fluorescence. Proc. Natl. Acad. Sci. U.S.A. 103, 18882–18886 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Welsher K., Liu Z., Sherlock S. P., Robinson J. T., Chen Z., Daranciang D., Dai H., A route to brightly fluorescent carbon nanotubes for near-infrared imaging in mice. Nat. Nanotechnol. 4, 773–780 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong G., Diao S., Chang J., Antaris A. L., Chen C., Zhang B., Zhao S., Atochin D. N., Huang P. L., Andreasson K. I., Kuo C. J., Dai H., Through-skull fluorescence imaging of the brain in a new near-infrared window. Nat. Photonics 8, 723–730 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cognet L., Tsyboulski D. A., Rocha J.-D. R., Doyle C. D., Tour J. M., Weisman R. B., Stepwise quenching of exciton fluorescence in carbon nanotubes by single-molecule reactions. Science 316, 1465–1468 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Godin A. G., Varela J. A., Gao Z., Danné N., Dupuis J. P., Lounis B., Groc L., Cognet L., Single-nanotube tracking reveals the nanoscale organization of the extracellular space in the live brain. Nat. Nanotechnol. 12, 238–243 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Reuel N. F., Dupont A., Thouvenin O., Lamb D. C., Strano M. S., Three-dimensional tracking of carbon nanotubes within living cells. ACS Nano 6, 5420–5428 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Fakhri N., Wessel A. D., Willms C., Pasquali M., Klopfenstein D. R., MacKintosh F. C., Schmidt C. F., High-resolution mapping of intracellular fluctuations using carbon nanotubes. Science 344, 1031–1035 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Roxbury D., Jena P. V., Williams R. M., Enyedi B., Niethammer P., Marcet S., Verhaegen M., Blais-Ouellette S., Heller D. A., Hyperspectral microscopy of near-infrared fluorescence enables 17-chirality carbon nanotube imaging. Sci. Rep. 5, 14167 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Danné N., Kim M., Godin A. G., Kwon H., Gao Z., Wu X., Hartmann N. F., Doorn S. K., Lounis B., Wang Y., Cognet L., Ultrashort carbon nanotubes that fluoresce brightly in the near-infrared. ACS Nano 12, 6059–6065 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Kim Y., Velizhanin K. A., He X., Sarpkaya I., Yomogida Y., Tanaka T., Kataura H., Doorn S. K., Htoon H., Photoluminescence intensity fluctuations and temperature-dependent decay dynamics of individual carbon nanotube sp3 defects. J. Phys. Chem. Lett. 10, 1423–1430 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Cognet L., Tsyboulski D. A., Weisman R. B., Subdiffraction far-field imaging of luminescent single-walled carbon nanotubes. Nano Lett. 8, 749–753 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Sen F., Boghossian A. A., Sen S., Ulissi Z. W., Zhang J., Strano M. S., Observation of oscillatory surface reactions of ribofl avin, trolox, and singlet oxygen using single carbon nanotube fluorescence spectroscopy. ACS Nano 6, 10632–10645 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Pan J., Cha T.-G., Li F., Chen H., Bragg N. A., Choi J. H., Visible/near-infrared subdiffraction imaging reveals the stochastic nature of DNA walkers. Sci. Adv. 3, e1601600 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Setaro A., Adeli M., Glaeske M., Przyrembel D., Bisswanger T., Gordeev G., Maschietto F., Faghani A., Paulus B., Weinelt M., Arenal R., Haag R., Reich S., Preserving π-conjugation in covalently functionalized carbon nanotubes for optoelectronic applications. Nat. Commun. 8, 14281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Setaro A., Bluemmel P., Maity C., Hecht S., Reich S., Non-covalent functionalization of individual nanotubes with spiropyran-based molecular switches. Adv. Funct. Mater. 22, 2425–2431 (2012). [Google Scholar]
- 27.Harrah D. M., Swan A. K., The role of length and defects on optical quantum efficiency and exciton decay dynamics in single-walled carbon nanotubes. ACS Nano 5, 647–655 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Siitonen A. J., Tsyboulski D. A., Bachilo S. M., Weisman R. B., Surfactant-dependent exciton mobility in single-walled carbon nanotubes studied by single-molecule reactions. Nano Lett. 10, 1595–1599 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Oudjedi L., Parra-Vasquez A. N. G., Godin A. G., Cognet L., Lounis B., Metrological investigation of the (6,5) carbon nanotube absorption cross section. J. Phys. Chem. Lett. 4, 1460–1464 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Chung S. H., Kennedy R. A., Forward-backward non-linear filtering technique for extracting small biological signals from noise. J. Neurosci. Methods 40, 71–86 (1991). [DOI] [PubMed] [Google Scholar]
- 31.Mortensen K. I., Churchman L. S., Spudich J. A., Flyvbjerg H., Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat. Methods 7, 377–381 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao Z., Danné N., Godin A. G., Lounis B., Cognet L., Evaluation of different single-walled carbon nanotube surface coatings for single-particle tracking applications in biological environments. Nanomaterials 7, 393 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong G., Diao S., Antaris A. L., Dai H., Carbon nanomaterials for biological imaging and nanomedicinal therapy. Chem. Rev. 115, 10816–10906 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Kruss S., Hilmer A. J., Zhang J., Reuel N. F., Mu B., Strano M. S., Carbon nanotubes as optical biomedical sensors. Adv. Drug Deliv. Rev. 65, 1933–1950 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Mann F. A., Lv Z., Großhans J., Opazo F., Kruss S., Nanobody conjugated nanotubes for targeted near-infrared in vivo imaging and sensing. Angew. Chem. Int. Ed. 58, 11469–11473 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Dreier J., Castello M., Coceano G., Cáceres R., Plastino J., Vicidomini G., Testa I., Smart scanning for low-illumination and fast RESOLFT nanoscopy in vivo. Nat. Commun. 10, 556 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaax1166/DC1
Movie S1. Movie showing the photoluminescence of single CNTs.
Fig. S1. SP-MC nanotube hybrid.
Fig. S2. SP-CNT luminescence in solution.
Fig. S3. Theoretical modeling of the luminescence intensity of a nanotube with quencher molecules.
Fig. S4. Influence of the residence times on the intensity ratio.
Fig. S5. Error estimation of the parameter theoretical φ.