

Abstract
Background: White matter (WM) microstructure may vary significantly in pediatric‐onset (PO) and adult‐onset (AO) patients with multiple sclerosis (MS), a difference that could be explained by the effects of an inherent plasticity in the affected pediatric brains early in the disease, and a phenomenon that does not occur later in life. This hypothesis would support the observation that disease progression is much slower in POMS compared to AOMS patients. Objectives: To examine WM microstructure in the brain of adults with POMS and AOMS, using tract based spatial statistics (TBSS) analysis of diffusion‐tensor imaging (DTI). Methods: Adults with relapsing‐remitting (RR) POMS, who were diagnosed before age of 18 years (n = 16), were compared with age‐matched (AOA, n = 23) and disease duration‐matched (AOD, n = 22) RR patients who developed MS after the age of 18 years. Scans were analyzed using the FSL software package (Oxford, UK) and statistics were performed using TBSS to evaluate WM microstructure between groups based on the mean fractional anisotropy (FA) values obtained from the DTI. Results: Widespread cortical and deep WM area differences characterized by increased FA values were seen in the AOAMS compared with POMS group (P < 0.05, TFCE corrected). Significantly increased FA values of posterior WM areas were detected in the AODMS compared with POMS group (P < 0.05, TFCE corrected). Conclusion: Increased FA values in WM areas of the AOMS compared with the POMS patients suggest that diffuse WM microstructure changes are more attributable to age of onset than a simple function of disease duration and age. Hum Brain Mapp 35:53–60, 2014. © 2012 Wiley Periodicals, Inc.
Keywords: multiple sclerosis, diffusion‐tensor‐imaging, tract‐based spatial statistics (TBSS), pediatric‐onset, adult‐onset
INTRODUCTION
Multiple sclerosis (MS) is a chronic inflammatory and neurodegenerative autoimmune disease of the central nervous system (CNS). Pediatric‐onset (PO) MS constitutes roughly 2–5% of all MS patients and is defined by onset under 18 years of age [Chitnis et al., 2009; Krupp et al., 2007; Langer‐Gould et al., 2011; Renoux et al., 2007]. It has been reported that clinical features of POMS differ from adult‐onset (AO) MS patients [Yeh et al., 2011]. POMS patients tend to present with more frequent relapses [Gorman et al., 2009], while recovering faster from these events [Ruggieri et al., 2004]. One of the most notable differences in POMS patients compared with AOMS is the slower time to irreversible disability, when controlled for disease duration [Simone et al., 2002; Yeh et al., 2011].
The underlying factors that might influence the evolution of disease progression in POMS vs. AOMS have been the subject of recent neuroimaging research [Banwell et al., 2007; Chabas and Pelletier, 2009; Waubant et al., 2009]. Prior studies have examined the neuroarchitecture of the CNS in POMS [Banwell et al., 2007; Yeh et al., 2009] and AOMS [Poloni et al., 2011] using conventional MRI techniques. It has become widely accepted that inflammatory MRI measures are poorly related to clinical outcomes in patients with AOMS [Barkhof, 1999; Zivadinov and Leist, 2005]. The clinical‐MRI disconnect is even more dramatic when patients with POMS are examined [Banwell et al., 2007; Chabas and Pelletier, 2009; Chabas et al., 2008b]. For example, in children <11 years old, large lesions early in the disease may resolve completely on subsequent scans, a phenomenon rarely observed even in teenagers and much less frequently in patients with AOMS [Chabas et al., 2008a].
To overcome the limits of conventional MRI in predicting clinical outcomes, more sophisticated nonconventional imaging methods were applied to the study of AOMS [Poloni et al., 2011]. However, application of nonconventional MRI techniques is especially needed in patients with POMS, in order to better understand how the neuroarchitecture and neuroplasticity of the CNS interacts with the MS disease process.
Diffusion tensor imaging (DTI) is a nonconventional MRI technique that utilizes the microscopic random motion of water over short distances to allow visualization in vivo of the pathological changes that occur in the CNS [Rovaris et al., 2005]. Commonly analyzed parameters of the DTI are mean diffusivity (MD) and fractional anisotropy (FA), which reflect the magnitude of diffusion and the preferential directionality of water diffusion along the white matter (WM) tracts, respectively [Le Bihan et al., 2001]. These types of MRI outcomes have allowed examination of brain structure differences among MS subpopulations [Roosendaal et al., 2009]. A preliminary study in which 10 pediatric MS patients were compared using DTI to an age‐ and sex‐matched control group, found that WM tracts of pediatric MS showed increased mean apparent diffusion coefficient (ADC) values and lower mean FA values compared to healthy controls [Vishwas et al., 2010]. Another study demonstrated significantly reduced FA in a variety of normal‐appearing WM regions in 33 POMS subjects when compared to a group of age‐matched healthy controls [Bethune et al., 2011]. However, to date little is known regarding differences in WM microstructure between patients with POMS and AOMS. Recently, it has been suggested a preservation of brain adaptive properties in POMS patients may explain clinically favorable mid‐term clinical outcomes [Rocca et al., 2010].
Against this background, we sought to examine the WM pathways in POMS patients as compared to their age‐ and disease duration‐matched AOMS counterparts, and to determine whether this population of patients has a unique ability for preservation of WM microstructure that may help us understand the age of onset influence on the evolution of disease progression.
MATERIALS AND METHODS
Subjects
In this study, a group of POMS patients (n = 16) were analyzed against two cohorts of AOMS patients matched individually for age (AOA, n = 23) and for disease duration (AOD, n = 22). The DTI exams from the POMS group and two AOMS comparator groups were obtained from a preexisting dataset, as part of a >800 MS patients prospective clinical surveillance study [Yeh et al., 2009]. For this study, the following inclusion criteria were applied: presence of a 3T MRI scan, DTI sequence acquired with identical scanning protocol and relapsing‐remitting (RR) disease course [Lublin and Reingold, 1996]. In total, 61 subjects were randomly selected among the entire dataset who fulfilled these criteria (Table 1). The POMS group consisted of 16 adults who had age of onset <18 years (mean age was 38.2 ± 10.6 and mean disease duration was 21.5 years ± 10.3 years). The AOAMS group was selected by including patients with age of onset >18 and <45 years and disease duration <7 years. This AOAMS group consisted of 23 subjects who had a mean age of 40 ± 5 and mean disease duration of 5.3 ± 1.6). The AODMS group consisted of 22 subjects who had a mean disease duration similar to the POMS group (mean age 52.6 ± 7.3 and mean disease duration was 25.1 ± 3.4). The study protocol was approved by the Institutional Review Board.
Table 1.
Demographic and clinical characteristics of the study groups
| POMS (n = 16) | AOAMS (n = 23) | AODMS (n = 22) | P‐value | |
|---|---|---|---|---|
| Females, n (%) | 15 (93.8) | 19 (82.6) | 19 (86.4) | 0.94 |
| Age in years, mean (SD) | 38.2 (10.6) | 40 (5) | 52.6 (7.3) | 0.005 |
| Age of onset in years, mean (SD) | 16.7 (1.2) 16 | 34.7 (3.2)a | 27.5 (6.4) | <0.0001 |
| Disease duration in years, mean (SD) | 21.5 (10.3)a | 5.3 (1.6) | 25.1 (3.4) | <0.0001 |
| EDSS, mean (SD) median | 3.2 (2.0) 2.8b | 2.0 (0.9) 2.0 | 3.1 1.8 (2.5) | 0.56 |
| RRMS disease course, n (%) | 16 (100) | 23 (100) | 22 (100) | 0.396 |
MS, multiple sclerosis; PO, pediatric onset; AOA, adult onset age‐matched; AOD; adult onset disease duration‐matched; SD, standard deviation; EDSS, Expanded Disability Status Scale; RR, relapsing‐remitting; n, number; %, percentage. The statistical differences between the three groups were calculated using the chi‐square test, analysis of covariance and Krukal‐Walis test. The statistical differences between the POMS and AOAMS groups were calculated using the chi‐square test, Student's t‐test and Mann‐Whitney test.
P < 0.001,
P < 0.01, and
P < 0.05 between the POMS and AOAMS groups are reported in relative boxes.
MRI Acquisition
All subjects were examined on a 3T GE Signa Excite HD 12.0 Twin Speed 8‐channel scanner (General Electric, Milwaukee, WI), with a maximum slew rate of 150T/m/s and maximum gradient amplitude in each orthogonal plane of 50 mT/m (zoom mode) using 8‐channel head coil. All scans were prescribed parallel to the subcallosal line in an axial‐oblique orientation. One average was used for all sequences. All sequences were obtained as follows, except as noted below: 256 × 192 matrix (freq × phase), field‐of‐view (FOV) of 25.6 cm × 19.2 cm (256 × 256 matrix with Phase FOV = 0.75), for an in‐plane resolution of 1 mm × 1 mm, with 48 3‐mm slices collected with no gap. The following sequences were acquired: 2D dual fast spin‐echo (FSE) proton density PD and T2‐weighted image (PD/T2‐WI) with a first and second Echo Time (TE) of 9 and 98 ms, a Repetition Time (TR) of 5,300 ms, Flip Angle (FA) of 90o, and an ETL = 14); Fluid‐Attenuated Inversion‐Recovery (FLAIR; TE/Inversion Time (TI)/TR = 120/2,100/8,500 ms; FA = 90°; ETL = 24); 3D high resolution (HIRES) T1‐WI using a fast spoiled gradient echo (FSPGR) sequence with magnetization‐prepared inversion recovery (IR) pulse (TE/TI/TR = 2.8/900/5.9 ms, FA = 10); and spin echo (SE) T1‐WI (TE/TR = 16/600 ms, FA = 90°). The 3D HIRES IR‐FSPGR, unlike the 2D sequences, was acquired with 184 1mm thick locations, resulting in isotropic resolution.
A 2D DTI sequence was acquired with TE/TR = 73/8,200 ms, 96 × 96 matrix, 32 × 24 cm FOV (due to pFOV = 0.75), FA = 90°; ETL = 1, slice thickness 3 mm with no gap, resulting in an in‐plane resolution size of 3.33 mm × 3.33 mm. DTI‐specific parameters include b‐value of 800, 15 directions, and 1 repetition. DTI was acquired using GE's ASSET parallel imaging, with an acceleration factor of 2.
MRI Analysis
Image analysis was performed at the Buffalo Neuroimaging Analysis Center, State University of New York. All image processing and manipulation was done using the FMRIB Software Library (FSL, http://www.fmrib.ox.ac.uk/fsl) on Linux‐based systems. Motion and eddy‐current distortion were corrected using the FMRIB's Diffusion Toolbox (FDT) in all images. The FDT was also used to assign the diffusion tensor for each voxel, from which FA, radial, and axial diffusivity were derived in a voxelwise manner [Smith et al., 2004].
Creating the diffusion tensor
First, the b0 image (with no diffusion weighting) was extracted and deskulled. The first of these steps was performed using a linear rigid body registration with 6° of freedom in which the b0 image was registered to the high resolution 3D HIRES IR‐FSPGR, with accompanying matrix created. The brain extraction tool (BET) was run on this image and an output mask was generated. The matrix from the first step was then inverted. The brain mask was placed into the same space as the b0 image using the inverted matrix from the previous step. The brain mask (in b0 image space) was then used to create a brain‐only b0 image. After this data prealignment was completed, the diffusion tensor was fit and calculated using the previously described method, in order to determine FA values while simultaneously correcting for the concentration of free water in each voxel [Pasternak et al., 2009]. Quality control was then performed to ensure that the tensor FA's of each scan had values between 0 and 1.0, as expected.
Tract‐based spatial statistics
The Tract‐Based Spatial Statistics (TBSS) module preprocessesed the FA imaged by slightly eroding the images and zeroing the end slices to remove likely outliers from the tensor fitting. Next, all FA images were aligned to a 1 × 1 × 1 mm standard space using the FMRIB58_FA as the target image. The FA images from each subject in standard space were then averaged and the resulting mean FA image was skeletonized. This 4D mean FA skeletonized image was then thresholded at 0.2 to include only voxels indicative of WM. For each subject's coregistered FA image, the maximum FA value perpendicular to each voxel of the skeleton was projected onto this skeleton (further detail can be found at the FMRIB website listed above) [Smith et al., 2007]. In addition, we performed a similar analysis for mean diffusivity (MD) values, bringing them into the same space and projecting them onto the FA‐identified skeleton.
Lesion volume and atrophy analyses
T2‐ and T1‐lesion volumes (LVs) were measured using a semiautomated edge detection contouring‐thresholding technique previously described [Zivadinov et al., 2001]. To control for lesion presence during subsequent processing steps, all lesion masks were also brought into the target analysis space. Source FLAIR images were coregistered to b0 images, and for each subject the resulting matrix was composited with the subject‐specific FA‐to‐target‐space warp. This final transformation was applied to each lesion mask, and all resulting images were composited into a single 4D lesion image.
Statistical Parametric Mapping (SPM5, Institute of Neurology, Queens Square, London, UK) was used for the global and tissue‐specific atrophy measures [Ashburner and Friston, 2000]. Because the presence of abnormal T1 hypointensities can cause tissue misclassification when performing automated segmentation, we used a dilation‐based inpainting method to correct all areas of focal abnormality before proceeding with analysis, as previously described [Yeh et al., 2009]. The brain parenchymal fraction (BPF), a measure of normalized brain volume, was computed by dividing the total brain parenchyma by total intracranial volume. Similarly, GM fraction (GMF) and WM fraction (WMF) were obtained by dividing each of these variables by total intracranial volume.
Statistical Analysis
The randomise tool was utilized after design matrix and contrast files were created using the General Linear Model (GLM) function (FSL Package). The randomise command combines GLM testing with permutation inference statistics and allows for differences in FA and MD between groups to be analyzed in a voxel‐wise fashion. The Threshold‐Free Cluster Enhancement (TFCE) option was used in randomise, as it is generally more profound and avoids the necessity for the use of arbitrary cluster‐forming thresholding [Smith and Nichols, 2009]. TBSS was employed for data analysis with a corrected cluster size significance interval of P < 0.05 to correct for multiple comparisons [Smith et al., 2007]. In addition, lesion maps were included as voxel‐wise binary regressors to ensure that differences detected were not simply due to lesion presence. Differences in demographic, clinical and MRI characteristics between study groups were assessed using PASW Statistics, version 18.0 (IBM, Somers, NY).
RESULTS
Demographic, Clinical, and MRI Characteristics
Tables 1 and 2 show demographic, clinical and MRI characteristics of the included study groups. No sex differences were observed between the study groups. As expected, the POMS and AODMS groups had higher EDSS than the AOAMS group. All patients were on disease‐modifying therapy at the time of examination, and there were no differences between the study groups regarding type of treatment (65% of the patients were using inteferon‐beta, 25% glatiramer acetate, and 10% were on combination therapies).
Table 2.
MRI characteristics of the study groups
| Characteristic | POMS (n = 16) | AOAMS (n = 23) | AODMS (n = 22) | P‐value |
|---|---|---|---|---|
| T2‐LV, mean (SD) | 17.9 (17.8) | 10 (9) | 15.7 (14.5) | 0.198 |
| T1‐LV, mean (SD) | 2.7 (5.2) | 2.2 (2.8) | 2.5 (2.8) | 0.930 |
| BPF, mean (SD) | 0.801 (0.03) | 0.827 (0.03) | 0.795 (0.02) | <0.0001 |
| GMF, mean (SD) | 0.410 (0.03) | 0.434 (0.03) | 0.395 (0.02) | <0.0001 |
| WMF, mean (SD) | 0.392 (0.02) | 0.393 (0.02) | 0.397 (0.02) | 0.583 |
MS, multiple sclerosis; PO, pediatric onset; AOA, adult onset age‐matched; AOD; adult onset disease duration matched; SD, standard deviation; BPF, brain parenchymal fraction; GMF, gray matter fraction; WMF, white matter fraction; LV, lesion volume. The lesion volume measures are expressed in milliliters and brain volume measures in fractions. The statistical differences between the groups were calculated using analysis of covariance in which age and disease duration were used as covariates.
No differences were observed for T2‐ and T1‐LV between the groups; however, the AODMS and POMS patients had significantly lower BPF and GMF compared with AOAMS, when corrected for age and disease duration (Table 2). AODMS patients showed lower GMF compared to POMS (P = 0.008), a difference that became non‐significant after age‐correction. No significant differences in WMF were detected.
TBSS Differences Between POMS and AOAMS Groups
When comparing WM microstructure in the AOAMS and POMS patients (AOAMS > POMS), the AOAMS group showed diffuse areas in both superficial and deep WM pathways, where the FA was significantly increased in the AOAMS patients (P < 0.05, TFCE corrected; Fig. 1A). The opposite experiment (POMS > AOAMS) did not show any WM area of increased FA values that was statistically significant (P < 0.05, TFCE corrected). When disease duration was used as a covariate, no significant WM areas of increased/decreased FA values were found for both comparisons. Similar results were found for MD (Fig. 2A).
Figure 1.
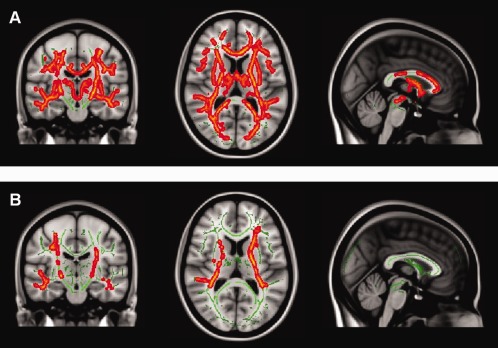
A: Adult‐onset age‐ matched MS (AOAMS) patients vs. pediatric‐onset MS (POMS) show significant areas of increased FA in the white matter (WM) of AOAMS when compared with POMS patients (P < 0.05, TFCE corrected). B: Adult‐onset disease duration‐matched MS (AODMS) patients vs. POMS show significant areas of increased FA in the WM of AODMS patients (P < 0.05, TFCE corrected).
Figure 2.
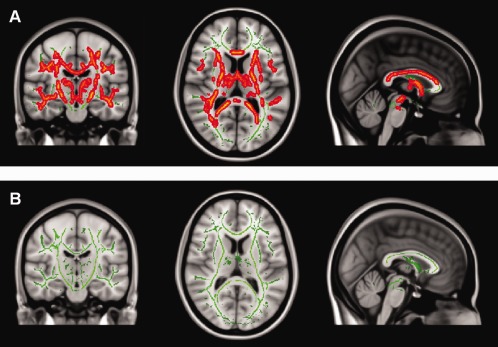
A: Pediatric‐onset MS (POMS) vs. adult‐onset age‐matched MS (AOAMS) patients show areas of significantly increased MD in the white matter (WM) of POMS when compared with AOAMS patients (P < 0.05, TFCE corrected). B: No statistically significant results are seen for POMS vs. AODMS (but uncorrected results show similar areas to the FA comparison).
TBSS Differences Between POMS and AODMS Groups
When comparing WM microstructure in AODMS and POMS patients (AODMS>POMS), the AODMS group showed diffuse areas in both posterior WM pathways in which the FA was significantly increased (P < 0.05, TFCE corrected; Fig. 1B). However, the opposite experiment did not yield detection of WM areas where POMS patients showed significantly increased FA values compared to AODMS. No statistically significant results were found for MD, but voxel‐wise uncorrected data followed a similar pattern to FA results (Fig. 2B).
DISCUSSION
The nature and progression of POMS compared to that of AOMS is still not fully understood despite recent advances in neuroimaging [Banwell et al., 2007; Chabas and Pelletier, 2009]. As we move into the future of MS treatment it has become increasingly more important to utilize more advanced, quantitative forms of MRI to elucidate better mechanisms for the diagnosis and treatment of POMS, as well as AOMS, and to determine what role age of onset may play in individual disease progression of MS patients.
The present study tried to elucidate the structural differences and similarities between the neuroarchitecture of the two MS groups. WM microstructure may vary significantly in POMS and AOMS patients, a difference that may be explained by the effects of inherent plasticity in the affected pediatric brains early in the disease, and a phenomenon that does not occur later in life. On the basis of this hypothesis, we would expect that the POMS would show significantly higher FA values, indicative of increased WM microstructure, compared to AOAMS patients, despite that they had significantly longer disease duration. However, since we were unsure of the findings and did not have prior evidence to confirm our hypothesis, we conducted all analyses in both directions (two‐tailed). In this case, widespread cortical and deep WM area differences characterized by increased FA values were seen in the AOAMS compared to POMS group (Fig. 1A). These findings suggest that the better WM microstructure in AOAMS compared with POMS patients may be related to the later age of onset.
One of our initial hypotheses was that WM microstructure could be preserved in POMS patients along tract‐specific areas, which could explain why there is slower disease progression in POMS patients in comparison to AOMS patients [Simone et al., 2002]. Interestingly, when controlled for disease duration, significance was lost when comparing FA values of WM areas between POMS and AOAMS groups. Whether possible compensatory changes in WM pathways allow POMS patients to maintain the functionality [Ruggieri et al., 2004] is to be further investigated, however, the findings from this study argue against the maintenance of WM pathway microstructure, as a possible mechanism to explain inherent plasticity in the affected pediatric brains.
Nevertheless, the compensatory hypothesis, despite a more destructive process identified in WM pathways in the POMS group, is supported by the findings from the present study indicating that AODMS patients had significantly increased FA values in posterior cortical WM tract‐specific areas compared to POMS patients with similar disease duration, but the same or less disability. These data suggest that brain injury at an early age may concede inherent plasticity in the affected pediatric brains that may maintain normal function for longer periods and slow down development of disability. Another possible explanation could be related to the time when mature WM microstructure was achieved. POMS were affected early in their lifetime, and it could be that they never acquired full myelination, although this is less likely, given the average age of 16 of the POMS group studied: the myelination process should be fully completed by 16. A comparison of WM microstructure in children/adolescents and adults, as measured by FA, should be further considered in order to fully explore this hypothesis.
The results from whole brain and tissue‐specific regional brain atrophy and lesion volume analyses in the current study do not help to explain how POMS patients have preserved function in comparison to AOMS patients. Whole brain atrophy was significantly worse in POMS patients compared to AOAMS patients, and no differences were observed between POMS and AODMS patients, despite significantly higher age (14.4 years) in the AODMS group. However, there was more severe GM atrophy in AODMS vs, POMS patients, a difference that became nonsignificant after correction for age. No differences between the groups were observed for lesion burden.
Therefore, in an effort to better understand the etiology of MS and more precisely how certain MS groups are affected by the debilitating neurodegenerative disease process, further studies are needed to address the issue of compensatory connections related to the age of onset, and whether these processes can explain the latency in disease progression among POMS patients [Rocca et al., 2010]. Functional studies, in combination with advanced structural MRI techniques—such as the DTI‐TBSS approach applied in this study—may help in this endeavor. Understanding the relationship of neurogenerative plasticity with respect to age‐ and disease duration can help us mitigate potential treatment strategies. The findings from this study suggest the pediatric brain is not necessarily using the classic WM tracts as a compensatory mechanism for diffuse injury, and this is in line with recent findings from our previous study using magnetization‐transfer imaging [Yeh et al., 2009]. It is also possible that immunoregulatory processes are at play in the disease state in which a less established or naively altered immune system in the POMS group may modulate cytokine responses differently than that of the more mature immune system in an AOMS patients, accounting for the variance in the disease course between these patient groups [Sosa and Forsthuber, 2011]. Additionally molecular mechanisms of neuroplasticity in a broader sense could be at work in POMS patients, rendering their brains more capable from a cellular perspective to initiate signaling and repair cascades, modulate gene regulation, and be more equipped to clear maligned cellular changes and reestablish a homeostatic state more effectively than in AOMS patients [Rocca et al., 2010].
There are certain limitations to this study, including retrospective comparison of MS patients who were part of the prospective clinical surveillance study [Yeh et al., 2009]. Another limit is the relatively small sample size and cross‐sectional rather than longitudinal design of the study. Future pediatric longitudinal studies should explore the interplay between GM and WM injury from the earliest clinical stages. In addition, we did not use spinal cord MRI to investigate whether differences in disability between POMS and AOMS patients could be due to disease in the spinal cord rather the in the brain. Treatment status also could play a role in interpretation of our findings; however, no differences between the study groups were found regarding type of treatment, although numerous changes in treatment status in the AODMS and POMS groups over 20+ years of the disease could have influenced study results. Another disadvantage may be related to the relatively low number of directions used to compute the DTI FA values. However, the DTI was applied as part of clinical protocol in this study.
In conclusion, increased FA values in WM areas of the AOAMS and AODMS vs. the POMS patients suggest that diffuse WM pathway microstructure changes are more attributable to age of onset than a simple function of disease duration and age, with worse FA corresponding to earlier onset. Therefore, it is unlikely that maintenance of WM microstructure is a key mechanism in the relative functional preservation often seen in POMS.
ACKNOWLEDGMENTS
The authors thank Christopher Magnano and Eve Salczynski for technical assistance in the preparation of this manuscript, and are also grateful to Ofer Pasternak for sharing his free water elimination code.
REFERENCES
- Ashburner J, Friston KJ (2000): Voxel‐based morphometry—The methods. Neuroimage 11(Part 1):805–821. [DOI] [PubMed] [Google Scholar]
- Banwell B, Shroff M, Ness JM, Jeffery D, Schwid S, Weinstock‐Guttman B (2007): MRI features of pediatric multiple sclerosis. Neurology 68(Suppl 2):S46–S53. [DOI] [PubMed] [Google Scholar]
- Barkhof F (1999): MRI in multiple sclerosis: Correlation with expanded disability status scale (EDSS). Mult Scler 5:283–286. [DOI] [PubMed] [Google Scholar]
- Bethune A, Tipu V, Sled JG, Narayanan S, Arnold DL, et al. (2011): Diffusion tensor imaging and cognitive speed in children with multiple sclerosis. J Neurol Sci 309:68–74. [DOI] [PubMed] [Google Scholar]
- Chabas D, Castillo‐Trivino T, Mowry EM, Strober JB, Glenn OA, Waubant E (2008a): Vanishing MS T2‐bright lesions before puberty: A distinct MRI phenotype? Neurology 71:1090–1093. [DOI] [PubMed] [Google Scholar]
- Chabas D, Pelletier D (2009): Sorting through the pediatric MS spectrum with brain MRI. Nat Rev Neurol 5:186–188. [DOI] [PubMed] [Google Scholar]
- Chabas D, Strober J, Waubant E (2008b): Pediatric multiple sclerosis. Curr Neurol Neurosci Rep 8:434–441. [DOI] [PubMed] [Google Scholar]
- Chitnis T, Glanz B, Jaffin S, Healy B (2009): Demographics of pediatric‐onset multiple sclerosis in an MS center population from the Northeastern United States. Mult Scler 15:627–631. [DOI] [PubMed] [Google Scholar]
- Gorman MP, Healy BC, Polgar‐Turcsanyi M, Chitnis T (2009): Increased relapse rate in pediatric‐onset compared with adult‐onset multiple sclerosis. Arch Neurol 66:54–59. [DOI] [PubMed] [Google Scholar]
- Krupp LB, Banwell B, Tenembaum S (2007): Consensus definitions proposed for pediatric multiple sclerosis and related disorders. Neurology 68(Suppl 2):S7–S12. [DOI] [PubMed] [Google Scholar]
- Langer‐Gould A, Zhang JL, Chung J, Yeung Y, Waubant E, Yao J (2011): Incidence of acquired CNS demyelinating syndromes in a multiethnic cohort of children. Neurology 77:1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bihan D, Mangin JF, Poupon C, Clark CA, Pappata S, Molko N, Chabriat H (2001): Diffusion tensor imaging: Concepts and applications. J Magn Reson Imaging 13:534–546. [DOI] [PubMed] [Google Scholar]
- Lublin FD, Reingold SC (1996): Defining the clinical course of multiple sclerosis: Results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 46:907–911. [DOI] [PubMed] [Google Scholar]
- Pasternak O, Sochen N, Gur Y, Intrator N, Assaf Y (2009).Free water elimination and mapping from diffusion MRI. Magn Reson Med 62:717–730. [DOI] [PubMed] [Google Scholar]
- Poloni G, Minagar A, Haacke EM, Zivadinov R (2011): Recent developments in imaging of multiple sclerosis. Neurologist 17:185–204. [DOI] [PubMed] [Google Scholar]
- Renoux C, Vukusic S, Mikaeloff Y, Edan G, Clanet M, Dubois B, Debouverie M, Brochet B, Lebrun‐Frenay C, Pelletier J, et al. (2007): Natural history of multiple sclerosis with childhood onset. N Engl J Med 356:2603–2613. [DOI] [PubMed] [Google Scholar]
- Rocca MA, Absinta M, Moiola L, Ghezzi A, Colombo B, Martinelli V, Comi G, Filippi M (2010): Functional and structural connectivity of the motor network in pediatric and adult‐onset relapsing‐remitting multiple sclerosis. Radiology 254:541–550. [DOI] [PubMed] [Google Scholar]
- Roosendaal SD, Geurts JJ, Vrenken H, Hulst HE, Cover KS, Castelijns JA, Pouwels PJ, Barkhof F (2009): Regional DTI differences in multiple sclerosis patients. Neuroimage 44:1397–1403. [DOI] [PubMed] [Google Scholar]
- Rovaris M, Gass A, Bammer R, Hickman SJ, Ciccarelli O, Miller DH, Filippi M (2005): Diffusion MRI in multiple sclerosis. Neurology 65:1526–1532. [DOI] [PubMed] [Google Scholar]
- Ruggieri M, Iannetti P, Polizzi A, Pavone L, Grimaldi LM (2004): Multiple sclerosis in children under 10 years of age. Neurol Sci 25 (Suppl 4):S326–S335. [DOI] [PubMed] [Google Scholar]
- Simone IL, Carrara D, Tortorella C, Liguori M, Lepore V, Pellegrini F, Bellacosa A, Ceccarelli A, Pavone I, Livrea P (2002): Course and prognosis in early‐onset MS: Comparison with adult‐onset forms. Neurology 59:1922–1928. [DOI] [PubMed] [Google Scholar]
- Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TE, Johansen‐Berg H, Bannister PR, De Luca M, Drobnjak I, Flitney DE, et al. (2004): Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 23 (Suppl 1):S208–S219. [DOI] [PubMed] [Google Scholar]
- Smith SM, Johansen‐Berg H, Jenkinson M, Rueckert D, Nichols TE, Miller KL, Robson MD, Jones DK, Klein JC, Bartsch AJ, et al. (2007): Acquisition and voxelwise analysis of multi‐subject diffusion data with tract‐based spatial statistics. Nat Protoc 2:499–503. [DOI] [PubMed] [Google Scholar]
- Smith SM, Nichols TE (2009): Threshold‐free cluster enhancement: Addressing problems of smoothing, threshold dependence and localisation in cluster inference. Neuroimage 44:83–98. [DOI] [PubMed] [Google Scholar]
- Sosa RA, Forsthuber TG (2011).The critical role of antigen‐presentation‐induced cytokine crosstalk in the central nervous system in multiple sclerosis and experimental autoimmune encephalomyelitis. J Interferon Cytokine Res 31:753–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishwas MS, Chitnis T, Pienaar R, Healy BC, Grant PE (2010): Tract‐based analysis of callosal, projection, and association pathways in pediatric patients with multiple sclerosis: A preliminary study. AJNR Am J Neuroradiol 31:121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waubant E, Chabas D, Okuda DT, Glenn O, Mowry E, Henry RG, Strober JB, Soares B, Wintermark M, Pelletier D (2009): Difference in disease burden and activity in pediatric patients on brain magnetic resonance imaging at time of multiple sclerosis onset vs. adults. Arch Neurol 66:967–971. [DOI] [PubMed] [Google Scholar]
- Yeh EA, Parrish JB, Weinstock‐Guttman B (2011): Disease progression in pediatric multiple sclerosis: Disparities between physical and neurocognitive outcomes. Expert Rev Neurother 11:433–440. [DOI] [PubMed] [Google Scholar]
- Yeh EA, Weinstock‐Guttman B, Ramanathan M, Ramasamy DP, Willis L, Cox JL, Zivadinov R (2009): Magnetic resonance imaging characteristics of children and adults with paediatric‐onset multiple sclerosis. Brain 132(Part 12):3392–3400. [DOI] [PubMed] [Google Scholar]
- Zivadinov R, Leist TP (2005): Clinical‐magnetic resonance imaging correlations in multiple sclerosis. J Neuroimaging 15(4 Suppl):10S–21S. [DOI] [PubMed] [Google Scholar]
- Zivadinov R, Rudick RA, De Masi R, Nasuelli D, Ukmar M, Pozzi‐Mucelli RS, Grop A, Cazzato G, Zorzon M (2001): Effects of IV methylprednisolone on brain atrophy in relapsing‐remitting MS. Neurology 57:1239–1247. [DOI] [PubMed] [Google Scholar]