

Abstract
The presence of brain atrophy and its progression in early Parkinson's disease (PD) are still a matter of debate, particularly in patients without cognitive impairment. The aim of this longitudinal study was to assess whether PD patients who remain cognitively intact develop progressive atrophic changes in the early stages of the disease. For this purpose, we employed high‐resolution T1‐weighted MR imaging to compare 22 drug‐naïve de novo PD patients without cognitive impairment to 17 age‐matched control subjects, both at baseline and at three‐year follow‐up. We used tensor‐based morphometry to explore the presence of atrophic changes at baseline and to compute yearly atrophy rates, after which we performed voxel‐wise group comparisons using threshold‐free cluster enhancement. At baseline, we did not observe significant differences in regional atrophy in PD patients with respect to control subjects. In contrast, PD patients showed significantly higher yearly atrophy rates in the prefrontal cortex, anterior cingulum, caudate nucleus, and thalamus when compared to control subjects. Our results indicate that even cognitively preserved PD patients show progressive cortical and subcortical atrophic changes in regions related to cognitive functions and that these changes are already detectable in the early stages of the disease. Hum Brain Mapp 35:3932–3944, 2014. © 2014 Wiley Periodicals, Inc.
Keywords: de novo PD, TBM, brain atrophy, longitudinal study, cognitive status
INTRODUCTION
Several structural magnetic resonance imaging (MRI) studies have consistently highlighted widespread cortical and subcortical atrophic changes in Parkinson's disease (PD) patients in advanced disease stages, and in particular in PD patients with dementia (PDD) (Beyer et al., 2007; Burton et al., 2004; Melzer et al., 2012; Nagano‐Saito et al., 2005; Summerfield et al., 2005; Weintraub et al., 2011). These results have been confirmed by pathological studies showing how the degenerative process (and signally the inclusion bodies pathology that is the hallmark of the disease) is first detectable in the brain stem, subsequently extends into the limbic system and finally progresses to increasingly involve the cerebral cortex (Braak et al., 2004).
There is increasing evidence that a substantial portion of PD patients (including those in the early, drug‐naïve stages of the disease) suffer from quantifiable cognitive deficits that do not meet the criteria for dementia (Aarsland et al., 2010; Poletti et al., 2012). These patients have been categorized as suffering from PD‐related mild cognitive impairment (PD‐MCI) (Litvan et al., 2012) and are considered at increased risk of developing dementia (Pedersen et al., 2013). MR studies in PD‐MCI patients have consistently revealed atrophic changes in a number of cortical regions. Such alterations were less extensive than those found in PDD patients, and the findings related to their precise regional distribution remain contradictory (Lee et al., 2012; Meltzer et al., 2012; Song et al., 2011; Weintraub et al., 2011).
In contrast, the presence and extent of atrophic changes in PD patients without cognitive impairment is still a matter of debate. While some cross‐sectional MR studies reported no significant atrophy in cognitively intact PD patients (Feldmann et al., 2008; Hattori et al., 2012; Meltzer et al., 2012; Weintraub et al., 2011), other authors described diverse patterns of cortical and subcortical atrophy in non‐demented PD patients (Beyer et al., 2007; Bouchard et al., 2008; Brenneis et al., 2003; Brück et al., 2004; Burton et al., 2004; Jubault et al., 2011; Lyoo et al., 2010; Nishio et al., 2010; Pereira et al., 2012; Song et al., 2011; Summerfield et al., 2005; Tam et al., 2005; Tinaz et al., 2011). The longitudinal/time‐dependent progression of regional brain atrophy in PD patients has been investigated in relatively few MR studies, with conflicting results. For instance, previous papers have described gray matter (GM) loss in limbic‐paralimbic structures in non‐demented PD patients and neocortical changes in PD patients (Ramírez‐Ruiz et al., 2005), widespread limbic, paralimbic, and neocortical grey‐matter loss in PD patients with visual hallucinations (Ibarretxe‐Bilbao et al., 2010), volume reduction in amygdala and temporal cortex in PD patients without severe hyposmia and (conversely) only sparse longitudinal volume changes in patients with hyposmia (Baba et al., 2012), progressive amigdalar atrophy and cortical thinning in frontotemporal regions in the early stages of the disease (Ibarretxe‐Bilbao et al., 2012), and greater progression of cortical thinning in frontal, limbic, and posterior cortical regions in advanced PD patients that converted to dementia with respect to non‐converters (Compta et al., 2013), whereas others reported no differences in regional brain atrophy rates between PD patients and healthy controls (Brenneis et al., 2003).
Also, some longitudinal MR studies employed measures of global atrophy in order to monitor the progression of the degenerative process in PD. Hu et al. (2001) found that annual brain volume loss was greater in patients with PD than in controls, whereas others studies reported no differences in global atrophy rates between controls and non‐demented patients with PDD (Burton et al., 2005; Paviour et al., 2006) but found significantly increased atrophy in patients with PD (Burton et al., 2005) with respect to non‐demented PD and controls or reported a higher rate of ventricular dilatation in PD patients that developed significant cognitive decline compared to PD patients who remained cognitively intact (Camicioli et al., 2011).
One possible explanation for the high variability in the findings described above could be the heterogeneous characteristics of PD patients under consideration. In particular, especially in less recent studies, non‐demented PD patients were often grouped into a single cohort which (a) also included advanced PD patients and (b) did not differentiate between cognitively intact and PD‐MCI patients. Therefore, in spite of the growing interest in evaluating patients in the initial stages of PD (warranted by a potentially greater responsiveness to putative neuroprotective and disease‐modifying treatment effects), the progression of structural brain changes in early, cognitively preserved PD patients remains uncertain.
Several methodological approaches have been applied to MR‐based investigation of brain atrophy in PD patients, including ROI‐based methods (Bouchard et al., 2008; Camicioli et al., 2003; Junque et al., 2005; Laakso et al., 1995), measurement of global atrophy (Burton et al., 2005; Camicioli et al., 2011; Hu et al., 2001), whole‐brain voxel‐wise techniques such as voxel‐based morphometry (VBM) (Beyer et al., 2007; Burton et al., 2004; Melzer et al., 2012; Nagano‐Saito et al., 2005; Nishio et al., 2010; Ramírez‐Ruiz et al., 2005; Song et al., 2011; Summerfield et al., 2005; Weintraub et al., 2011), and surface‐based methods that produce measures of cortical thickness and folding (Compta et al., 2013; Ibarretxe‐Bilbao et al., 2012; Jubault et al., 2011; Pereira et al., 2012; Tinaz et al., 2011; Zarei et al., 2013).
Tensor‐based morphometry (TBM) is a promising image processing technique that allows automated voxel‐wise analysis of brain tissue changes. In particular, local differences in brain tissue volume are evaluated by computing high‐dimensional nonlinear deformations to adjust the anatomy of each individual to match a custom‐built group‐average template and successively comparing maps of the Jacobian determinant (│J│) of the deformation fields in order to estimate the degree of tissue contraction/expansion at each location/voxel (Ashburner and Friston, 2003; Chung et al., 2001; Fox et al., 2001; Freeborough and Fox, 1998; Hua et al., 2009, 2011, 2013; Riddle et al., 2004; Studholme et al., 2001; Thompson et al., 2000). TBM has been proven to be an unbiased, robust, high‐throughput imaging marker in Alzheimer's disease (AD) and MCI and is particularly suited for longitudinal studies (Hua et al., 2013).
To the best of our knowledge, only few cross‐sectional studies employed TBM for the evaluation of PD. In a pilot report (Lu et al., 2009), PDD subjects showed cortical atrophic changes in more areas of the brain than PD‐MCI subjects (relative to healthy controls), while patients with normal cognition had only few areas of significant tissue loss. Borghammer et al. (2010) employed a TBM method based on a non‐linear deformation algorithm with a limited number of degrees of freedom (Klein et al., 2009). In this study, early PD patients showed an isolated cluster of GM loss in the right cerebellum with respect to controls. More recently, TBM has also been employed to detect tissue damage in an animal model of the disease (Westphal et al., 2013).
The aim of the present longitudinal study was to track the progression of regional brain atrophy in the early stages of the disease in cognitively intact PD patients. To this purpose, we employed an optimized TBM strategy based on high resolution T1‐weighted images in a cohort of newly diagnosed drug‐naïve (de novo) PD patients with preserved cognitive functioning at baseline and at three‐year follow‐up.
MATERIALS AND METHODS
Subjects
Thirty‐four (8 women and 26 men, mean age 63.1 ± 7.9 years) patients with de novo parkinsonian syndrome consecutively referred to a neurology unit for the diagnostic evaluation of PD over a 24‐month interval (from March 2004 to March 2006) were recruited in this prospective study. At baseline, clinical evaluation included history of disease‐related symptoms and signs, neurological examination and levodopa challenge test as a supportive criterion for the diagnosis of idiopathic PD. All patients were screened for cardiovascular autonomic dysfunction, which was considered as exclusion criterion. Severity of parkinsonism was evaluated by the Unified Parkinson's Disease Rating Scale (UPDRS) (Fahn et al., 1987) and the Hoehn–Yahr (HY) staging system (Hoehn and Yahr, 1967). All patients were required to satisfy the UK Brain Bank criteria for the diagnosis of PD (Gibb and Lees, 1988). We administered the Geriatric Depression Scale Short Form (GDS‐15: Sheik and Yesavage, 1986) to screen for depressive features in all PD patients, and diagnosis of depression was made according to DSM IV‐TR (Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text‐Revision) criteria (American Psychiatric Association, 2000). Patients also underwent a comprehensive neuropsychological assessment. Global cognitive status was assessed through the Mini Mental State Examination (Folstein et al., 1975). The battery of standardized neuropsychological tests included at least two tests within each of the five cognitive domains: attention and working memory, executive functions, language, memory, and visuospatial ability. We employed the Visual Search Test (Spinnler and Tognoni, 1987), the Digit Span (Orsini et al., 1987), and the Corsi Span (Spinnler and Tognoni, 1987) to assess attention and working memory, the Stroop Test (Caffarra et al., 2002a) and the Frontal Assessment Battery (Dubois et al., 2000) to evaluate executive functions, the Boston Naming Test short form (Kaplan et al., 1983), and the Phonemic and Semantic Fluency Test (Carlesimo et al., 1996) to assess language, the Rey Auditory Verbal Learning Task (Carlesimo et al., 1996) and Rey–Osterrieth Complex Figure Recall Test (Caffarra et al., 2002b) to evaluate memory and the Rey–Osterrieth Complex Figure Copy Test (Caffarra et al., 2002b), Benton Judgment of Line Orientation Test (Benton et al., 1978) and Raven Colored Progressive Matrices (Raven et al., 2003) to assess visuospatial ability. Due to the lack of specific criteria for evaluating MCI in PD at the time of inception of the current investigation, the presence of MCI was initially declared when a neuropsychological impairment was demonstrated within one or more cognitive domains; otherwise, patients were classified as cognitively preserved. In retrospective evaluation, the classification of our cohort members according to this criterion was seen to coincide with the assessment later performed using the recently proposed Movement Disorder Society Task Force diagnostic criteria for PD‐MCI (Litvan et al., 2012), which state that PD‐MCI may be diagnosed when a neuropsychological impairment is demonstrated by performances 1 to 2 standard deviations below appropriate norms in at least two tests of the same cognitive domain (PD‐MCI single domain) or in at least one test in two different cognitive domains (PD‐MCI multiple domains). The presence of depression or MCI at baseline or at follow‐up was considered as exclusion criterion.
Twenty healthy volunteers (8 women and 12 men, mean age 62.5 ± 9.1 years) with no history of familial or personal neurological diseases and normal neurological examination were recruited as controls. All subjects gave their written informed consent to participate in the study, which was approved by the Local Ethics Committee.
Study Design
One of the 34 de novo patients enrolled in the study was excluded after baseline clinical assessment because the cardiovascular autonomic test indicated autonomic dysfunction, and eight additional patients were excluded after cognitive assessment because of a MCI‐PD single domain diagnosis. The remaining 25 patients underwent baseline MRI scanning and began chronic dopaminergic treatment shortly thereafter.
Subsequently, patients were clinically evaluated every six months during the follow‐up. Two further patients were excluded from the study because of the lack of dopaminergic response and the appearance of atypical signs and/or symptoms during clinical follow‐up (for those two patients, respective diagnoses of possible Progressive Supranuclear Palsy and Multiple System Atrophy were made). At the end of the follow‐up, all subjects underwent a second comprehensive neuropsychological evaluation to ascertain possible development of cognitive impairment. One patient was excluded from the study because of the appearance of cognitive deficit classified as MCI‐PD single domain. Our cohort did not comprise any patients with a diagnosis of MCI multiple domain, neither at baseline, nor at follow‐up. Three control subjects did not consent to the follow‐up MR study. Overall, 22 patients (4 women and 18 men, mean age 61.5 ± 8.8) and 17 control subjects (8 women and 9 men, mean age 59.1 ± 8.5 years) completed the study and underwent a second MRI examination. The mean (± standard deviation) follow‐up time for patients and controls was 2.8 ± 0.6 (range 2–4) years and 3.9 ± 2.2 (range 2–7) years, respectively. Differences for age between PD patients and control subjects were not significant (P = 0.48, Mann‐Whitney U‐test). Clinical and neuropsychological data of the patients that completed the study are summarized in Table 1.
Table 1.
Clinical and neuropsychological data of PD patients at baseline and follow‐up
| Baseline assessment | Follow‐up assessment | |
|---|---|---|
| Disease duration (years) | 1.32 (0.80) | – |
| HY* | 1.25 (0.40) | 1.41 (0.43) |
| UPDRS II* | 6.3 (4.1) | 7.0 (3.5) |
| UPDRS III* | 10.5 (5.7) | 12.2 (5.0) |
| MMSE | 29.45 (0.73) | 28.77 (0.65) |
| Visual search | 48.28 (3.49) | 47.33 (2.55) |
| Digit span | 4.8 (1.1) | 4.6 (1.3) |
| Corsi span | 4.6 (0.9) | 4.2 (1.1) |
| Stroop interference: time | 17.00 (8.25) | 19.31 (9.50) |
| Stroop interference: errors | 0.39 (0.83) | 0.62 (1.14) |
| Frontal assessment battery | 16.37 (1.12) | 15.38 (1.40) |
| RAVLT immediate recall | 37.89 (6.80) | 37.70 (7.03) |
| RAVLT delayed recall | 7.30 (1.86) | 6.58 (1.98) |
| Rey figure immediate recall | 16.79 (4.25) | 13.98 (0.97) |
| Boston naming test short form | 26.93 (3.10) | 26.12 (3.37) |
| Semantic verbal fluency | 11.68 (2.37) | 11.67 (1.66) |
| Phonemic verbal fluency | 31.35 (6.22) | 31.30 (4.93) |
| CPM 47 | 31.35 (3.58) | 28.75 (4.06) |
| Rey–Osterrieth Complex Figure Copy Test | 32.71 (1.99) | 31.91 (2.34) |
| Benton JOL | 26.15 (2.66) | 25.05 (2.71) |
Mean (standard deviation) data are reported.
CPM 47 = Raven's Colored Progressive Matrices; HY = Hoehn and Yahr; JOL = Judgment of Line Orientation; MMSE = Mini Mental State Examination; PD = Parkinson's disease; RAVLT = Rey Auditory Verbal Learning Task; UPDRS = Unified Parkinson's Disease Rating Scale.
P < 0.05, General Linear Model, independent variables: HY, UPDRS II e UPDRS III, continuous between‐factors: age and follow‐up time, categorical between‐factors: gender, within‐factor: time.
MRI Data Acquisition
MRI was performed on a 1.5 T MR scanner system (Magnetom Symphony, Siemens, Erlangen‐Germany) with 30 mT/m maximum gradient strength and a standard quadrature birdcage head coil.
T1‐weighted MR images were acquired with an axial high resolution 3D sequence (Magnetization Prepared Rapid Gradient Echo, MPRAGE) with repetition time (TR) = 2500 ms, echo time (TE) = 3.7 ms, inversion time (TI) = 730 ms, flip angle = 15°, slice thickness = 1 mm, field of view (FOV) = 256 mm × 256 mm, matrix size = 256 × 256, number of excitations (NEX) = 1. The acquisition protocol also included axial T2 weighted images [fluid attenuated inversion recovery (FLAIR) sequence with TR = 9,000 ms, TE = 114 ms, TI = 2,500 ms, slice thickness = 4 mm, FOV = 230 mm × 230 mm, matrix size = 256 × 256, turbo factor = 21, NEX = 1, and turbo spin echo TSE sequence (TR = 4,730 ms, TE = 104 ms, slice thickness = 4 mm, FOV = 230 mm × 230 mm, matrix size = 256 × 256, turbo factor = 13, NEX = 2)].
Tensor‐Based Morphometry
In order to optimize TBM analysis, all registration procedures were based on variations of the SyN algorithm which, along with another registration strategy, has been shown to provide the most consistent high accuracy (Klein et al., 2009). We employed the Greedy SyN implementation of the SyN algorithm included in the ANTs package (Avants et al., 2011). All image registrations were initialized through a 12‐degree of freedom (DOF) affine transform, which was followed by a nonlinear diffeomorphic step, and employed neighborhood cross‐correlation as similarity metric. For improved accuracy with respect to the default package settings, in the second (nonlinear) registration step, we used four multiresolution levels with a maximum number of 200 iterations per level and smoothing resolutions of 3, 2, 1, and 0 mm per level (from coarsest to finest).
Custom T1 Template Construction
In order to generate an unbiased, population‐specific T1 template, we used baseline T1 images from all (number of subjects = 17) controls and an equal number of randomly selected patients. The template was generated using a procedure similar to that described in Avants et al. (2010). Briefly, after N4 bias field correction (Tustison et al., 2010), coregistrations of individual brain images were iteratively refined to create a group average, which is often referred to as an optimal average template. In particular, the algorithm works within the diffeomorphic space toward building an average shape and appearance brain by reducing dependence on the topological idiosyncracy of any individual brain. Within the ANTs package, the SyN tool is called to nonlinearly coregister all brain images to one another in an iterative manner for subsequent intensity averaging. The procedure is repeated recursively, thereby iteratively refining the co‐registration of the constituent images. Five global iterations were used to build the final template in this study. Creation of the custom template required approximately 420 hours of CPU time.
Image Registration
All N4‐corrected baseline images were registered to the unbiased template as described above. The voxel‐wise jacobian determinant of the nonlinear component of the warpfield (|J| baseline) was then computed for each subject. In particular, when the local volume in N4‐corrected image is greater/less than local volume in the unbiased template image the |J| baseline values are less/greater than 1, respectively.
Also, every N4‐corrected follow‐up image was registered (intra‐subject) to the N4‐corrected baseline image as described above, and the voxel‐wise jacobian determinant |J| longitudinal of the nonlinear component of the inverse warpfield (i.e., relative to the transformation which takes the baseline image into the space of the follow‐up image) was computed (resulting in |J| longitudinal being in baseline image space). The voxel‐wise yearly warp rate (WR) was then computed as WR = (|J| longitudinal − 1)/t, where t is time (in years) between baseline and follow‐up imaging. In particular, WR values less/greater than 0 indicate contraction/expansion of local tissue volume, respectively. WR is largely insensitive to differences both in follow‐up time and in spread around mean follow‐up time. Specifically, normalizing absolute volume changes by time and hence working with per‐year volume change yields a quantitative index (i.e., WR), which is statistically comparable across different follow‐up times both intra‐ and inter‐ subject groups. Finally, maps of voxel‐wise WR were transformed into custom template space by applying the transformations computed in the previous step (which take baseline images into custom template space).
Statistical Analysis
All voxel‐wise statistical analyses were performed within the framework of the general linear model (GLM) while controlling for age and gender as nuisance covariates. We employed a non‐parametric, permutation‐based inference approach which included full correction for multiple comparisons over space. In particular, P‐values were calculated and corrected for multiple comparisons using the “3D” parameter setting with threshold‐free cluster enhancement (TFCE), thereby avoiding the use of an arbitrary amount of spatial smoothing as well as threshold for the initial cluster‐formation, which can affect the sensitivity of statistical analysis and bias results (Smith and Nichols, 2009). For each comparison, we employed 50,000 permutation for increased accuracy, and corrected P‐values smaller than 0.05 were considered statistically significant. The comparisons we performed were (a) patients vs. controls at baseline (quantity of interest: |J| baseline), (b) follow‐up vs. baseline (quantity of interest: WR), and (c) within‐patient group correlation of WR with clinical/cognitive variables and their yearly changes. In (a) we tested the effect of group, in (b) we tested the effect of time (in each group) and of group x time interaction, and in (c) we tested the hypothesis of a regression slope being larger or smaller than zero. Resulting P‐value maps were transformed into MNI‐152 space by applying an affine, 12‐DOF transformation computed by registering the custom template to the MNI‐152 template. In order to avoid creating false P‐values, nearest neighbor interpolation was employed in this step.
RESULTS
Baseline Evaluation
No difference in local volume between patients and control subjects was revealed (i.e., the |J| baseline analysis showed no significant differences between PD patients and control subjects).
Longitudinal Evaluation
Control subjects: Baseline versus follow‐up
During the follow‐up period, control subjects developed atrophic changes (i.e., WR values were significantly smaller than 0) involving several white matter (WM) and GM regions (Fig. 1a) and showed cerebrospinal fluid (CSF) enlargement (i.e., WR values were significantly greater than 0; not shown). Atrophy clusters involved mainly WM and were more widespread in the frontal lobe. GM changes included paracentral lobule, precuneus, parietal superior gyrus, and anterior cingulum bilaterally, right frontal superior medial gyrus and left temporal middle gyrus, precentral gyrus, postcentral gyrus, middle cingulum, cuneus, and superior and middle occipital gyrus.
Figure 1.
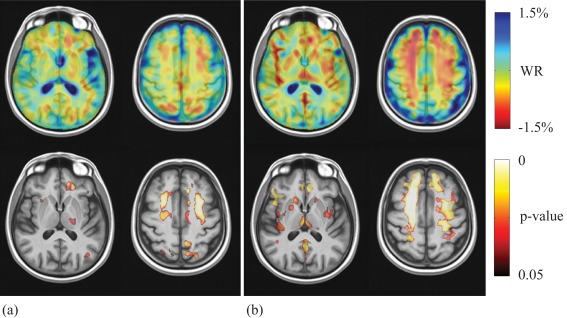
Top pane: Sample axial views of average WR maps in control subjects (a) and PD patients (b), where red indicates local atrophy and blue indicates local enlargement. Bottom pane: voxel‐wise corrected p‐value maps (threshold‐free cluster enhancement, TFCE) testing the null hypothesis of zero WR in control subjects (a) and PD patients (b) separately. Highlighted clusters indicate significant (p < 0.05) atrophic changes (i.e., WR significantly lower than zero) within the respective groups. All maps are overlayed on population‐specific template. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
PD patients: Baseline versus follow‐up
With respect to baseline examinations, PD patients at follow‐up displayed a number of clusters of reduced WM and GM volume (i.e., WR values were significantly less than 0), which were more widespread in WM and signally in the frontal lobe (Fig. 1b), and showed CSF enlargement (i.e. WR values were significantly greater than 0; not shown). GM involvement in PD patients was more widespread than in control subjects and included bilaterally the thalamus, caudate, putamen, superior and middle frontal gyrus, postcentral gyrus, anterior cingulum, insula, Rolandic operculum, Heschl gyrus, temporal middle and inferior gyrus, frontal inferior triangular gyrus, frontal inferior operculum, precuneus, frontal middle orbital gyrus, rectus gyrus and amygdala, right parahippocampal gyrus, olfactory gyrus and fusiform gyrus, and left precentral gyrus and posterior cingulum.
PD patients versus control subjects
With respect to control subjects, during the follow‐up period PD patients developed bilateral clusters of increased atrophy (i.e., WR values in PD patients were significantly lower than WR values in control subjects) in frontal superior and middle gyrus, anterior cingulum, caudate nucleus, and thalamus (Fig. 2). No other significant time (baseline vs. follow‐up) × group (PD patients vs. control subjects) interaction was revealed.
Figure 2.
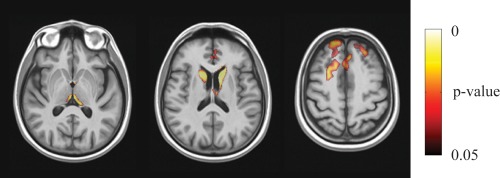
Voxel‐wise corrected p‐value maps (threshold‐free cluster enhancement, TFCE), testing the null hypothesis of zero differences in WR between in PD patients and control subjects. Highlighted clusters indicate significantly (p < 0.05) more pronounced mean atrophy in PD patients when compared to control subjects (i.e., WR in PD patients significantly lower than WR in control subjects). All maps are overlayed on population‐specific template. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Correlation analyses
In PD patients, no significant correlation between warp‐rates and motor or neuropsychological test scores or their average changes per year between baseline and follow‐up were identified.
DISCUSSION
TBM has been previously employed in few pilot, cross‐sectional PD studies both in patients (Borghammer et al., 2010; Lu et al., 2009) and in an animal model of the disease (Westphal et al., 2013). In this study, we employed TBM for the longitudinal evaluation of PD and focused on the progression of regional atrophy in a cohort of cognitively preserved de novo patients. Our main finding is that early PD patients who remain cognitively intact develop clusters of increased atrophy rates (when compared to control subjects) in cortical and subcortical regions related to cognitive functions.
Baseline Evaluation: De Novo PD Patients Versus Control Subjects
Our PD patients without cognitive impairment showed no detectable differences in atrophic changes with respect to controls subjects at baseline examination. Several studies have described inconsistent patterns of regional atrophy in the frontal (Burton et al., 2004; Jubault, et al., 2011; Lyoo et al., 2010; Nishio et al., 2010; Pereira et al., 2012; Tinaz et al., 2011), temporal (Beyer et al., 2007; Bouchard et al., 2008; Brück et al., 2004; Jubault et al., 2011; Lyoo et al., 2010; Nishio et al., 2010; Pereira et al., 2012; Summerfield et al., 2005; Tam et al., 2005), parietal (Jubault et al., 2011; Lyoo et al., 2010; Pereira et al., 2012; Tinaz et al., 2011), and occipital cortex (Pereira et al., 2012; Song et al., 2011; Tinaz et al., 2011), the cerebellum (Borghammer et al., 2010; Camicioli et al., 2009), and the striatum (Brenneis et al., 2003; Tinaz et al., 2011) of non‐demented PD patients. The majority of these reports did not distinguish between cognitively intact and PD‐MCI patients that do not fulfill the criteria for dementia, or included patients in moderate stages of the disease. In our opinion, the results of the present study reflect the early stage of PD in our patients, and they are in agreement with the findings of previous papers, which include an accurate distinction between patients with normal cognition and PD‐MCI patients (Hattori et al., 2012; Meltzer et al., 2012; Weintraub et al., 2011).
Longitudinal Evaluation: Baseline Versus Follow‐Up
We found that, during the follow‐up period, both PD patients and controls subjects developed atrophic changes that encompassed several WM and GM regions, although those changes were more widespread in WM and in particular in the frontal lobe. There is consensus that GM volume decreases linearly with age (Allen et al., 2005; Courchesne et al., 2000; Ge et al., 2002; Smith et al., 2007). In contrast, WM volume has been demonstrated to peak in the 40–50 year age range and subsequently to decline quickly in particular after 60 years of age (Allen et al., 2005; Barzokis et al., 2001; Courchesne et al., 2000; Ge et al., 2002; Guttman et al., 1998; Salat et al., 2009).
Further, MRI‐based studies have indicated that aging selectively affects different cortical regions, and frontal lobes have been described to show the largest volume reduction and WM changes (Allen et al., 2005; Grieve et al., 2005; Michielse et al., 2010; Raz et al., 2004). Our results essentially confirm an accelerated decrease in WM volume in the elderly population which is particularly pronounced in the frontal lobes.
Longitudinal Evaluation: PD Patients Versus Control Subjects
We found that, with respect to control subjects, cognitively intact PD patients displayed a higher atrophy rate bilaterally in the prefrontal cortex, anterior cingulum, head of caudate nucleus, and thalamus.
To the best of our knowledge, only one recent longitudinal MR study evaluated the progression of structural brain changes in early PD patients (Ibarretxe‐Bilbao et al., 2012) employing cortical thickness analysis, VBM, and volumetric measures of cortical and subcortical structures. The study found no significant VBM differences between PD patients and control subjects; however, when compared to controls, PD patients presented a faster rate of cortical thinning in bilateral frontotemporal regions, higher total GM and total cortical volume loss, and higher amygdalar atrophy after an average interval of 35 months. In the study by Ibarretxe‐Bilbao et al. (2012), the patient group had both a more severe clinical deficit (mean baseline UPDRS III 15.44 on treatment vs. 10.5 off treatment) and a longer disease duration (2.97 vs. 1.32 years at baseline) than our patient group. This could contribute to explaining why the cortical thickness changes described by Ibarretxe‐Bilbao et al. were more widespread than the ones presented in our whole brain TBM study.
In the following sections, areas of increased atrophy rates in PD patients with respect to control subjects are discussed in relation to their functions.
Prefrontal cortex
Atrophic changes in the prefrontal cortex of PD patients have been described in several cross‐sectional imaging studies (Beyer et al., 2007; Burton et al., 2004; Brück et al., 2004; Meltzer et al., 2012; Meppelink et al., 2011; Nagano‐Saito et al., 2005; Nishio et al., 2010; Pereira et al., 2012; Song et al., 2011; Tinaz et al., 2011), and prefrontal WM damage was reported in diffusion tensor imaging (DTI) studies (Karagulle‐Kendi et al., 2008; Rae et al., 2012). Further, decreased frontal lobe perfusion has been observed in Parkinson's disease without dementia (Antonini et al., 2001) and selective prefrontal hypometabolism has been detected in cognitively preserved PD (Hosokai et al., 2009). Neurodegeneration is expected to involve prefrontal cortex in Braak's stage 4/5. Although an exact clinical‐pathological correlation can only be established through pathological verification, considering the disease duration and the clinical scores it is likely that our patients were crossing this stage of the disease.
Structural changes in the frontal lobe have been hypothesized to contribute to the dysexecutive syndrome (Brück et al., 2004; Nagano‐Saito et al., 2005; Rae et al., 2012), the hallmark of cognitive impairment of early PD patients that has been attributed to functional derangement of specific prefrontal cortex‐striatal circuits (Williams‐Gray et al., 2007). Furthermore, pathological studies found a positive correlation between Lewy bodies load in the frontal cortex and cognitive impairment in PD (Mattila et al., 2000; Van den Berge et al., 2012).
Anterior cingulum
According to Braak's staging system, the anterior cingulate cortex is one of the cortical areas more precociously and severely affected by Lewy bodies pathology (Braak et al., 2004; Braak and Del Tredici, 2008), and atrophic changes in this region in cognitively impaired PD patients have been detected by previous cross‐sectional VBM studies (Nagano‐Saito et al., 2005; Summerfield et al., 2005), while in another study (Jubault et al., 2011) local surface area analysis (corticometry) showed anterior cingulate atrophy in a series of cognitively preserved PD patients. Furthermore, WM microstructural changes in the anterior cingulum of PD patients were reported in DTI studies (Gattellaro et al., 2009; Hattori et al., 2012; Kamagata et al., 2012).
Several studies have demonstrated the role of the cingulate cortex in attention and executive functioning, especially in inhibitory control and in the sensitivity to interference (Duncan and Owen, 2000; Fichtenholtz et al., 2004; Meindl et al., 2010; Silton et al., 2010), and a correlation between structural changes in the anterior cingulum and cognitive impairment in PD was suggested by pathological (Kövari et al., 2003) and imaging (Kamagata et al., 2012) studies.
Caudate nucleus
Both ROI‐based (Apostolova et al., 2010; Lisanby et al., 1993; Pitcher et al., 2012) and VBM (Brenneis et al., 2003; Burton et al., 2004; Melzer et al., 2012; Nagano‐Saito et al., 2005) cross sectional studies have revealed atrophic changes in the caudate nucleus of PD patients. The majority of this previous data concerned patients in advanced stages of the disease, and mostly dealt with demented subjects. However, Pitcher et al. (2012) found decreased caudate volumes, in particular of the head of the caudate nuclei, in both cognitively intact and impaired patients, although the changes were less conspicuous in the former than in the latter. In PD, the striatum is an elective site of Lewy body pathology in both neurons and astrocytes (Braak and Del Tredici, 2008); furthermore, pathological studies have described a decrease in dendritic spine density and dendrite atrophy in the caudate nuclei of PD patients, and these changes may contribute to a volume reduction (Stephens et al., 2005; Zaja‐Milatovic et al., 2005).
It is well known that the caudate nucleus is involved in a variety of cognitive processes and that it plays a regulatory role in different behavioral contexts, such as mood and motivation; lesions of this nucleus can cause a dysexecutive syndrome, learning difficulties, and affective disturbance (Cummings and Benson, 1984; Mendez et al., 1989; Richfield et al., 1987).
Moreover, reduced caudate dopaminergic function (Holthoff‐Detto et al., 1997; Jokinen et al., 2009) as well as caudate atrophy (Camicioli et al., 2009) have been associated with cognitive impairment in PD.
Thalamus
Atrophic changes in the thalamus have been described in a few previous VBM studies in PD patients (Burton et al., 2004; Hattori et al., 2012; Nagano‐Saito et al., 2005; Summerfield et al., 2005). Furthermore, in a ROI‐based study, PD patients in mild to moderate stages of the disease showed differences in shape, but not volume of the thalami, with respect to healthy subjects (McKeown et al., 2008).
Functional changes of thalamic neurons are traditionally related to motor symptoms of PD. According to the basal ganglia‐thalamocortical circuit model, activity changes in basal ganglia (which are due to the loss of dopaminergic regulation) are thought to cause a cortical hypo‐activity via a reduced excitatory outflow from the motor thalamus (Albin et al., 1989). This classic view assumes that there is no structural damage to the thalamus itself. However, more recently, several studies revealed that the thalamus is one of select targets of extranigral inclusion body pathology and that it undergoes significant neuronal loss in PD (Brooks and Halliday 2009; Halliday, 2009; Henderson et al., 2000; Rüb et al., 2002; Truong et al., 2009).
Furthermore, recent views posit that the thalamus is more than just a relay for information coming from the basal ganglia and instead that it actively modulates the function of both the basal ganglia and cortical neurons (Halliday, 2009; Sherman, 2007). Therefore, since pathological changes in PD also involve the non‐motor thalamus, it has been suggested that thalamic damage, while underlying motor symptoms, might also have an impact on cognitive functions and in particular on arousal, awareness, attention, memory, and dementia (Brooks and Halliday, 2009; Halliday, 2009; Kimura et al., 2004; Van der Werf et al., 2002).
Correlation Analyses
In our sample of PD patients, we found no significant correlation between TBM measures and clinical/cognitive variables or their yearly changes. This may reflect the relatively narrow range of scores in tests evaluating motor impairment and cognitive functions in our homogeneous sample of early de novo PD patients. In particular, we only selected patients who were cognitively intact at baseline and did not develop cognitive deficits during the follow‐up period, and this may contribute to explaining why we failed to find correlations between the rate of atrophic changes and cognitive measures. On the other hand, the lack of association between TBM data and motor scores or their yearly changes is not surprising, given the distribution of regional atrophy that predominantly involved cortical and subcortical structures belonging to cognitive circuits.
Inverse Consistency and Practical Implementation of TBM
Since our optimized TBM studies the Jacobian determinant of the deformation fields, accurate computation of intra‐subject transformations between baseline and follow‐up is crucial. In this context, one key property of TBM should be that the deformation field be inverse consistent. This means the computed deformations will be identical if the chronological order of the images is inverted (i.e., TBM results, in terms of regions with significant volume change, should be identical regardless if the deformation target is the baseline or the follow‐up image) (Hua et al., 2011). While analytically the SyN algorithm employed in our study is fully inverse consistent, a computer program which calculates deformations fields will only be able to work with finite precision, possibly creating small discrepancies in inverse consistency. We minimized this effect by employing double‐precision arithmetics for all our computations.
Also, our optimized TBM requires large computing times/resources, and in this study we circumvented this problem by running all registrations as well as permutation‐based inference on a parallel computing cluster with 512 Intel Xeon compute cores and 4GB RAM/core.
Possible Interactions Between Dopaminergic Treatment and Brain Morphometry
At follow‐up examination, all patients were receiving L‐dopa, dopamine‐agonists, or both. We are not aware of any proven effect of dopaminergic treatment on brain morphometry; however, a pilot VBM study reported midbrain volume increases in healthy controls after acute levodopa administration (Salgado‐Pineda et al., 2006). Furthermore, two recent MR studies by one group (Cerasa et al., 2011; 2013) found increased gray matter volume of the inferior frontal cortex in PD patients with levodopa‐induced dyskinesias when compared to non‐dyskinetic patients and raised an interesting debate regarding possible plastic effects of long term L‐dopa administration (Aron and Obeso, 2012; Vernon and Modo, 2012). For ethical reasons, a completely drug‐free longitudinal evaluation of PD patients is difficult if not impossible. Actually, since long‐duration symptomatic effects of dopaminergic drugs can persist for weeks or months (Hauser et al., 2000; Hauser and Holford, 2002; Olanow et al., 2009), the wash‐out of 12–24 h commonly prescribed before imaging studies probably is not fully adequate even in fMRI. In this context, it seems even more unlikely that hypothetical therapy‐induced plasticity effect could reverse so rapidly. We therefore decided not to suspend treatment before follow‐up MR examination.
CONCLUSIONS
Cognitively intact de novo PD patients show no significant GM or WM loss with respect to control subjects. However, already in these initial stages of the disease, these patients exhibit increased yearly atrophy rates in regions mainly related to cognitive functions, including prefrontal cortex, anterior cingulum, head of caudate nucleus, and thalamus. These structural changes could increase the susceptibility to developing cognitive impairment. In view of our findings, a longer term clinical follow‐up study is warranted in order to elucidate if the above changes can predict the onset of cognitive deficits.
REFERENCES
- Aarsland D, Bronnick K, Williams‐Gray C, Weintraub D, Marder K, Kulisevsky J, Burn D, Barone P, Pagonabarraga J, Allcock L, Santangelo G, Foltynie T, Janvin C, Larsen JP, Barker RA, Emre M (2010): Mild cognitive impairment in Parkinson's disease: A multicenter pooled analysis. Neurology 75:1062–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB (1989): The functional anatomy of basal ganglia disorders. Trends Neurosci 12:366–375. [DOI] [PubMed] [Google Scholar]
- Allen JS, Bruss J, Brown CK, Damasio H (2005): Normal neuroanatomical variation due to age: The major lobes and a parcellation of the temporal region. Neurobiol Aging 26:1245–1260. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association . 2000. Diagnostic and Statistical Manual of Mental Disorders, fourth edition, Text Revision. Washington, DC: American Psychiatric Publication, 943 p. [Google Scholar]
- Antonini A, De Notaris R, Benti R, De Gaspari D, Pezzoli G (2001): Perfusion ECD/SPECT in the characterization of cognitive deficits in Parkinson's disease. Neurol Sci 22:45–46. [DOI] [PubMed] [Google Scholar]
- Apostolova LG, Beyer M, Green AE, Hwang KS, Morra JH, Chou YY, Avedissian C, Aarsland D, Janvin CC, Larsen JP, Cummings JL, Thompson PM (2010): Hippocampal, caudate, and ventricular changes in Parkinson's disease with and without dementia. Mov Disord 25:687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aron AR, Obeso J (2012): Is executive control used to compensate for involuntary movements in levodopa‐induced dyskinesia? Mov Disord 27:339–340. [DOI] [PubMed] [Google Scholar]
- Ashburner J, Friston KJ (2003): Morphometry In: Frackowiak RSJ, Friston KJ, Frith C, Dolan R, Friston KJ, Price CJ, Zeki S, Ashburner J, and Penny WD, editors. Human Brain Function, 2nd ed. New York: Academic Press; pp 707–724. [Google Scholar]
- Avants BB, Yushkevich P, Pluta J, Minkoff D, Korczykowski M, Detre J, Gee JC (2010): The optimal template effect in hippocampus studies of diseased populations. Neuroimage 49:2457–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avants BB, Tustison NJ, Song G, Cook PA, Klein A, Gee JC (2011): A reproducible evaluation of ANTs similarity metric performance in brain image registration. Neuroimage 54:2033–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Kikuchi A, Hirayama K, Nishio Y, Hosokai Y, Kanno S, Hasegawa T, Sugeno N, Konno M, Suzuki K, Takahashi S, Fukuda H, Aoki M, Itoyama Y, Mori E, Takeda A (2012): Severe olfactory dysfunction is a prodromal symptom of dementia associated with Parkinson's disease: A 3‐year longitudinal study. Brain 135:161–169. [DOI] [PubMed] [Google Scholar]
- Barzokis G, Beckson M, Lu PH, Nuecherterlein KH, Edwards N, Mintz J (2001): Age related changes in frontal and temporal lobes in men. Arch Gen Psychiatry 58:461–465. [DOI] [PubMed] [Google Scholar]
- Benton AL, Varney NR, Hamsher KD (1978): Visuospatial judgement. A clinical test. Arch Neurol 35:364–367. [DOI] [PubMed] [Google Scholar]
- Beyer MK, Janvin CC, Larsen JP, Aarsland D (2007): A magnetic resonance imaging study of patients with Parkinson's disease with mild cognitive impairment and dementia using voxel‐based morphometry. J Neurol Neurosurg Psychiatry 78:254–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghammer P, Østergaard K, Cumming P, Gjedde A, Rodell A, Hall N, Chakravarty MM (2010): A deformation‐based morphometry study of patients with early‐stage Parkinson's disease. Eur J Neurol 17:314–320. [DOI] [PubMed] [Google Scholar]
- Bouchard TP, Malykhin N, Martin WR, Hanstock CC, Emery DJ, Fisher NJ, Camicioli RM (2008): Age and dementia‐associated atrophy predominates in the hippocampal head and amygdala in Parkinson's disease. Neurobiol Aging 29:1027–1039. [DOI] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K (2004): Stages in the development of Parkinson's disease‐related pathology. Cell Tissue Res 318:121–134. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K (2008): Nervous system pathology in sporadic Parkinson disease. Neurology 70:1916–1925. [DOI] [PubMed] [Google Scholar]
- Brenneis C, Seppi K, Schocke MF, Müller J, Luginger E, Bösch S, Löscher WN, Büchel C, Poewe W, Wenning GK (2003): Voxel‐based morphometry detects cortical atrophy in the Parkinson variant of multiple system atrophy. Mov Disord 18:1132–1138. [DOI] [PubMed] [Google Scholar]
- Brooks D, Halliday GM (2009): Intralaminar nuclei of the thalamus in Lewy body diseases. Brain Res Bull 78:97–104. [DOI] [PubMed] [Google Scholar]
- Brück A, Kurki T, Kaasinen V, Vahlberg T, Rinne JO (2004): Hippocampal and prefrontal atrophy in patients with early non‐demented Parkinson's disease is related to cognitive impairment. J Neurol Neurosurg Psychiatry 75:1467–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton EJ, McKeith IG, Burn DJ, Williams ED, O'Brien JT (2004): Cerebral atrophy in Parkinson's disease with and without dementia: a comparison with Alzheimer's disease, dementia with Lewy bodies and controls. Brain 127:791–800. [DOI] [PubMed] [Google Scholar]
- Burton EJ, McKeith IG, Burn DJ, O'Brien JT (2005): Brain atrophy rates in Parkinson's disease with and without dementia using serial magnetic resonance imaging. Mov Disord 20:1571–1576. [DOI] [PubMed] [Google Scholar]
- Caffarra P, Vezzadini G, Dieci F, Zonato F, Venneri A (2002a): A short version of the Stroop Test: normative data on Italian population. Nuova Rivista di Neurologia 12:111–115. [Google Scholar]
- Caffarra P, Vezzadini G, Dieci F, Zonato F, Venneri A (2002b): Rey–Osterreith Complex Figure: Normative values in an Italian population sample. Neurol Sci 22:443–447. [DOI] [PubMed] [Google Scholar]
- Camicioli R, Moore MM, Kinney A, Corbridge E, Glassberg K, Kaye JA (2003): Parkinson's disease is associated with hippocampal atrophy. Mov Disord 18:784–790. [DOI] [PubMed] [Google Scholar]
- Camicioli R, Gee M, Bouchard TP, Fisher NJ, Hanstock CC, Emery DJ, Martin WR (2009): Voxel‐based morphometry reveals extra‐nigral atrophy patterns associated with dopamine refractory cognitive and motor impairment in parkinsonism. Parkinson Relat Disord 15:187–195. [DOI] [PubMed] [Google Scholar]
- Camicioli R, Sabino J, Gee M, Bouchard T, Fisher N, Hanstock C, Emery D, Martin WR (2011): Ventricular dilatation and brain atrophy in patients with Parkinson's disease with incipient dementia. Mov Disord 26:1443–1450. [DOI] [PubMed] [Google Scholar]
- Carlesimo GA, Caltagirone C, Gainotti G (1996): The Mental Deterioration Battery: normative data, diagnostic reliability and qualitative analyses of cognitive impairment. The Group for the Standardization of the Mental Deterioration Battery. Eur Neurol 36:378–384. [DOI] [PubMed] [Google Scholar]
- Cerasa A, Messina D, Pugliese P, Morelli M, Lanza P, Salsone M, Novellino F, Nicoletti G, Arabia G, Quattrone A (2011): Increased prefrontal volume in PD with levodopa‐induced dyskinesias: A voxel‐based morphometry study. Mov Disord 26:807–812. [DOI] [PubMed] [Google Scholar]
- Cerasa A, Morelli M, Augimeri A, Salsone M, Novellino F, Gioia MC, Arabia G, Quattrone A (2013): Prefrontal thickening in PD with levodopa‐induced dyskinesias: new evidence from cortical thickness measurement. Parkinson Relat Disord 19:123–125. [DOI] [PubMed] [Google Scholar]
- Chung MK, Worsley KJ, Paus T, Cherif, C , Collins DL, Giedd JN, Rapoport J, Evans, AC (2001): A unified statistical approach to deformation‐based morphometry. Neuroimage 14:595–606. [DOI] [PubMed] [Google Scholar]
- Compta Y, Pereira JB, Ríos J, Ibarretxe‐Bilbao N, Junqué C, Bargalló N, Cámara A, Buongiorno M, Fernández M, Pont‐Sunyer C, Martí MJ (2013): Combined dementia‐risk biomarkers in Parkinson's disease: A prospective longitudinal study. Parkinson Relat Disord doi: 10.1016/j.parkreldis.2013.03.009. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Chisum H, Townsend J, Cowles A, Covington J, Egaas B, Harwood M, Hinds S, Press G (2000): Normal brain development and aging: quantitative analysis at in vivo MR imaging in healthy volunteers. Radiology 216:672–682. [DOI] [PubMed] [Google Scholar]
- Cummings JL, Benson F (1984): Subcortical dementia. Review of an emerging concept. Arch Neurol 41:874–879. [DOI] [PubMed] [Google Scholar]
- Dubois B, Slachevsky A, Litvan I, Pillon B (2000): The FAB: A Frontal Assessment Battery at bedside. Neurology 55:1621–1626. [DOI] [PubMed] [Google Scholar]
- Duncan J, Owen AM (2000): Common regions of the human frontal lobe recruited by diverse cognitive demands. Trends Neurosci 23:475–483. [DOI] [PubMed] [Google Scholar]
- Fahn S, Elton R, and the members of the Unified Parkinson's Disease rating Scale Development Committee (1987): Unified Parkinson's Disease Rating Scale In: Fahn S, Marsden CD, Calne D, editors. Recent Developments in Parkinson's Disease. New York: MacMillan; pp 153–163. [Google Scholar]
- Feldmann A, Illes Z, Kosztolanyi P, Illes E, Mike A, Kover F, Balas I, Kovacs N, Nagy F (2008): Morphometric changes of gray matter in Parkinson's disease with depression: A voxel‐based morphometry study. Mov Disord 23:42–46. [DOI] [PubMed] [Google Scholar]
- Fichtenholtz HM, Dean HL, Dillon DG, Yamasaki H, McCarthy G, LaBar KS (2004): Emotion‐attention network interactions during a visual oddball task. Brain Res Cogn Brain Res 20:67–80. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR (1975): Mini Mental State. A practical method for grading the cognitive state of patients for the clinician. J Psychiatry Res 12:189–198. [DOI] [PubMed] [Google Scholar]
- Fox NC, Crum WR, Scahill RI, Stevens JM, Janssen JC, Rossor MN (2001): Imaging of onset and progression of Alzheimer's disease with voxel‐compression mapping of serial magnetic resonance images. Lancet 358:201–205. [DOI] [PubMed] [Google Scholar]
- Freeborough, PA , Fox NC (1998): Modeling brain deformations in Alzheimer disease by fluid registration of serial 3D MR images. J Comput Assist Tomogr 22:838–843. [DOI] [PubMed] [Google Scholar]
- Gattellaro G, Minati L, Grisoli M, Mariani C, Carella F, Osio M, Ciceri E, Albanese A, Bruzzone MG (2009): White matter involvement in idiopathic Parkinson disease: A diffusion tensor imaging study. AJNR Am J Neuroradiol 30:1222–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Grossman RI, Babb JS, Rabin ML, Mannon LJ, Kolson DL (2002): Age‐related total gray matter and white matter changes in normal adult brain: Part I. Volumetric MR imaging analysis. Am J Neuroradiol 23:1327–1333. [PMC free article] [PubMed] [Google Scholar]
- Gibb WR, Lees AJ (1988): The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry 51:745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieve SM, Clark CR, Williams LM, Peduto AJ, Gordon E (2005): Preservation of limbic and paralimbic structures in aging. Hum Brain Mapp 25:391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman CRG, Jolesz FA, Kikinis R, Killiany RJ, Moss MB, Sandor T, Albert MS (1998): White matter changes with normal aging. Neurology 50:972–978. [DOI] [PubMed] [Google Scholar]
- Halliday GM (2009): Thalamic changes in Parkinson's disease. Parkinson Relat Disord 3(15 Suppl):S152–S155. [DOI] [PubMed] [Google Scholar]
- Hattori T, Orimo S, Aoki S, Ito K, Abe O, Amano A, Sato R, Sakai K, Mizusawa H (2012): Cognitive status correlates with white matter alteration in Parkinson's disease. Hum Brain Mapp 33:727–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser RA, Koller WC, Hubble JP, Malapira T, Busenbark K, Olanow CW (2000): Time course of loss of clinical benefit following withdrawal of levodopa‐carbidopa and bromocriptine in early Parkinson's disease. Mov Disord 15:485–489. [PubMed] [Google Scholar]
- Hauser RA, Holford NH (2002): Quantitative description of loss of clinical benefit following withdrawal of levodopa‐carbidopa and bromocriptine in early Parkinson's disease. Mov Disord 17:961–998. [DOI] [PubMed] [Google Scholar]
- Henderson JM, Carpenter K, Cartwright H, Halliday GM (2000): Degeneration of the thalamic caudal intralaminar nuclei in Parkinson's disease. Ann Neurol 47:345–352. [PubMed] [Google Scholar]
- Hoehn MM, Yahr MD (1967): Parkinsonism: onset, progression and mortality. Neurology 17:427–442. [DOI] [PubMed] [Google Scholar]
- Holthoff‐Detto VA, Kessler J, Herholz K, Bonner H, Pietrzyk U, Wurker M, Ghaemi M, Wienhard K, Wagner R, Heiss WD (1997): Functional effects of striatal dysfunction in Parkinson disease. Arch Neurol 54:145–150. [DOI] [PubMed] [Google Scholar]
- Hosokai Y, Nishio Y, Hirayama K, Takeda A, Ishioka T, Sawada Y, Suzuki K, Itoyama Y, Takahashi S, Fukuda H, Mori E (2009): Distinct patterns of cerebral glucose metabolism in Parkinson's disease with and without mild cognitive impairment. Mov Disord 23:854–862. [DOI] [PubMed] [Google Scholar]
- Hu MT, White SJ, Chaudhuri KR, Morris RG, Bydder GM, Brooks DJ (2001): Correlating rates of cerebral atrophy in Parkinson's disease with measures of cognitive decline. J Neural Transm 108:571–580. [DOI] [PubMed] [Google Scholar]
- Hua X, Lee S, Yanovsky I, Leow AD, Chou YY, Ho AJ, Gutman B, Toga AW, Jack CR Jr, Bernstein MA, Reiman EM, Harvey DJ, Kornak J, Schuff N, Alexander GE, Weiner MW, Thompson PM (2009): Optimizing power to track brain degeneration in Alzheimer's disease and mild cognitive impairment with tensor‐based morphometry: An ADNI study of 515 subjects. Neuroimage 48:668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Gutman B, Boyle CP, Rajagopalan P, Leow AD, Yanovsky I, Kumar AR, Toga AW, Jack CR Jr, Schuff N, Alexander GE, Chen K, Reiman EM, Weiner MW, Thompson PM (2011): Accurate measurement of brain changes in longitudinal MRI scans using tensor‐based morphometry. Neuroimage 57:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Hibar DP, Ching CR, Boyle CP, Rajagopalan P, Gutman BA, Leow AD, Toga AW, Jack CR Jr, Harvey D, Weiner MW, Thompson PM (2013): Alzheimer's Disease Neuroimaging Initiative. Unbiased tensor‐based morphometry: Improved robustness and sample size estimates for Alzheimer's disease clinical trials. Neuroimage 66:648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarretxe‐Bilbao N, Ramirez‐Ruiz B, Junque C, Marti MJ, Valldeoriola F, Bargallo N, Juanes S, Tolosa E (2010): Differential progression of brain atrophy in Parkinson's disease with and without visual hallucinations. J Neurol Neurosurg Psychiatry 81:650–657. [DOI] [PubMed] [Google Scholar]
- Ibarretxe‐Bilbao N, Junque C, Segura B, Baggio HC, Marti MJ, Valldeoriola F, Bargallo N, Tolosa E (2012): Progression of cortical thinning in early Parkinson's disease. Mov Disord 27:1746–1753. [DOI] [PubMed] [Google Scholar]
- Jokinen P, Brück A, Aalto S, Forsback S, Parkkola R, Rinne JO (2009): Impaired cognitive performance in Parkinson's disease is related to caudate dopaminergic hypofunction and hippocampal atrophy. Parkinson Relat Disord 15:88–93. [DOI] [PubMed] [Google Scholar]
- Jubault T, Gagnon JF, Karama S, Ptito A, Lafontaine AL, Evans AC, Monchi O (2011): Patterns of cortical thickness and surface area in early Parkinson's disease. Neuroimage 55:462–467. [DOI] [PubMed] [Google Scholar]
- Junque C, Ramirez‐Ruiz B, Tolosa E, Summerfield C, Martí MJ, Pastor P, Gómez‐Ansón B, Mercader JM (2005): Amygdalar and hippocampal MRI volumetric reductions in Parkinson's disease with dementia. Mov Disord 20:540–544. [DOI] [PubMed] [Google Scholar]
- Kamagata K, Motoi Y, Abe O, Shimoji K, Hori M, Nakanishi A, Sano T, Kuwatsuru R, Aoki S, Hattori N (2012): White matter alteration of the cingulum in Parkinson disease with and without dementia: evaluation by diffusion tensor tract‐specific analysis. Am J Neuroradiol 33:890–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan EF, Goodglass H, Wintraub S. 1983. The Boston Naming Test. Experimental Edition. Philadelphia, PA: Lea & Febiger. [Google Scholar]
- Karagulle‐Kendi AT, Lehericy S, Luciana M, Ugurbil K, Tuite P (2008): Altered diffusion in the frontal lobe in Parkinson disease. Am J Neuroradiol 29:501–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Minamimoto T, Matsumoto N, Hori Y (2004): Monitoring and switching of cortico‐basal ganglia loop functions by the thalamo‐striatal system. Neurosci Res 48:355–360. [DOI] [PubMed] [Google Scholar]
- Klein A, Andersson J, Ardekani BA, Ashburner J, Avants B, Chiang MC, Christensen GE, Collins DL, Gee J, Hellier P, Song JH, Jenkinson M, Lepage C, Rueckert D, Thompson P, Vercauteren T, Woods RP, Mann JJ, Parsey RV (2009): Evaluation of 14 nonlinear deformation algorithms applied to human brain MRI registration. Neuroimage 46:786–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kövari E, Gold G, Herrmann FR, Canuto A, Hof PR, Bouras C, Giannakopoulos P (2003): Lewy body densities in the entorhinal and anterior cingulate cortex predict cognitive deficits in Parkinson's disease. Acta Neuropathol (Berl) 106:83–88. [DOI] [PubMed] [Google Scholar]
- Laakso MP, Partanen K, Riekkinen P (1995): Hippocampal volumes in Alzheimer's disease, Parkinson's disease with and without dementia, and in vascular dementia: An MRI study. Neurology 46:678–681. [DOI] [PubMed] [Google Scholar]
- Lee JE, Cho KH, Kim M, Sohn YH, Lee PH (2012): The pattern of cortical atrophy in Parkinson's disease with mild cognitive impairment according to the timing of cognitive dysfunction. J Neurol 259:469–473. [DOI] [PubMed] [Google Scholar]
- Lisanby SH, McDonald WM, Massey EW, Doraiswamy PM, Rozear M, Boyko OB, Krishnan KR, Nemeroff C (1993): Diminished subcortical nuclei volumes in Parkinson's disease by MR imaging. J Neural Transm Suppl 40:13–21. [PubMed] [Google Scholar]
- Litvan I, Goldman JG, Tröster AI, Schmand BA, Weintraub D, Petersen RC, Mollenhauer B, Adler CH, Marder K, Williams‐Gray CH, Aarsland D, Kulisevsky J, Rodriguez‐Oroz MC, Burn DJ, Barker RA, Emre M (2012): Diagnostic criteria for mild cognitive impairment in Parkinson's disease: Movement Disorder Society Task Force guidelines. Mov Disord 27:349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PH, Apostolova LG, Green AE, Hwang K, Chung C, Thompson PM, Leow A, Lee G, Janvin CC, Larsen JP, Cummings JL, Aarsland D, Beyer MK (2009): A tensor‐based morphometry study of patients with cognitive impairment and dementia in Parkinson's disease. Proceedings of the International Conference on Alzheimer's Disease 2009. Alzheimer Dementia: J Alzheimer Assoc 5:P60.
- Lyoo CH, Ryu YH, Lee MS (2010): Topographical distribution of cerebral cortical thinning in patients with mild Parkinson's disease without dementia. Mov Disord 25:496–499. [DOI] [PubMed] [Google Scholar]
- Mattila PM, Rinne JO, Helenius H, Dickson DW, Röyttä M (2000): Alpha‐synuclein‐immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson's disease. Acta Neuropathol 100:285–290. [DOI] [PubMed] [Google Scholar]
- McKeown MJ, Uthama A, Abugharbieh R, Palmer S, Lewis M, Huang X (2008): Shape (but not volume) changes in the thalami in Parkinson disease. BMC Neurol 8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meindl T, Teipel S, Elmouden R, Mueller S, Koch W, Dietrich O, Coates U, Reiser M, Glaser C (2010): Test‐retest reproducibility of the default‐mode network in healthy individuals. Hum Brain Mapp 3:237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer TR, Watts R, MacAskill MR, Pitcher TL, Livingston L, Keenan RJ, Dalrymple‐Alford JC, Anderson TJ (2012): Grey matter atrophy in cognitively impaired Parkinson's disease. J Neurol Neurosurg Psychiatry 83:188–194. [DOI] [PubMed] [Google Scholar]
- Mendez MF, Adams NL, Lewandowski KS (1989): Neurobehavioral changes associated with caudate lesions. Neurology 39:349–354. [DOI] [PubMed] [Google Scholar]
- Meppelink AM, de Jong BM, Teune LK, van Laar T (2011): Regional cortical grey matter loss in Parkinson's disease without dementia is independent from visual hallucinations. Mov Disord 26:142–147. [DOI] [PubMed] [Google Scholar]
- Michielse S, Coupland N, Camicioli R, Carter R, Seres P, Sabino J, Malykhin N (2010): Selective effects of aging on brain white matter microstructure: A diffusion tensor imaging tractography study. Neuroimage 5:1190–1201. [DOI] [PubMed] [Google Scholar]
- Nagano‐Saito A, Washimi Y, Arahata Y, Kachi T, Lerch JP, Evans AC, Dagher A, Ito K (2005): Cerebral atrophy and its relation to cognitive impairment in Parkinson disease. Neurology 64:224–229. [DOI] [PubMed] [Google Scholar]
- Nishio Y, Hirayama K, Takeda A, Hosokai Y, Ishioka T, Suzuki K, Itoyama Y, Takahashi S, Mori E (2010): Corticolimbic gray matter loss in Parkinson's disease without dementia. Eur J Neurol 17:1090–1097. [DOI] [PubMed] [Google Scholar]
- Olanow WC, Stern MB, Sethi K (2009): The scientific and clinical basis for the treatment of Parkinson's disease. Neurology 72:S1–S136. [DOI] [PubMed] [Google Scholar]
- Orsini A, Grossi D, Capitani E, Laiacona M, Papagno C, Vallar G (1987): Verbal and spatial immediate memory span: normative data from 1355 adults and 1112 children. J Neurol Sci 8:539–548. [DOI] [PubMed] [Google Scholar]
- Paviour DC, Price SL, Jahanshahi M, Lees AJ, Fox NC (2006): Longitudinal MRI in progressive supranuclear palsy and multiple system atrophy: rates and regions of atrophy. Brain 129:1040–1049. [DOI] [PubMed] [Google Scholar]
- Pedersen KF, Larsen JP, Tysnes OB, Alves G (2013): Prognosis of mild cognitive impairment in early Parkinson's disease: The Norwegian Park West Study. JAMA Neurol 25:1–7. [DOI] [PubMed] [Google Scholar]
- Pereira JB, Ibarretxe‐Bilbao N, Marti MJ, Compta Y, Junqué C, Bargallo N, Tolosa E (2012): Assessment of cortical degeneration in patients with Parkinson's disease by voxel‐based morphometry, cortical folding, and cortical thickness. Hum Brain Mapp 33:2521–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher TL, Melzer TR, Macaskill MR, Graham CF, Livingston L, Keenan RJ, Watts R, Dalrymple‐Alford JC, Anderson TJ (2012): Reduced striatal volumes in Parkinson's disease: A magnetic resonance imaging study. Transl Neurodegener 1:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poletti M, Frosini D, Pagni C, Baldacci F, Nicoletti V, Tognoni G, Lucetti C, Del Dotto P, Ceravolo R, Bonuccelli U (2012): Mild cognitive impairments and cognitive‐motor relationships in newly diagnosed drug‐naive patients with Parkinson's disease. J Neurol Neurosurg Psychiatry 83:601–606. [DOI] [PubMed] [Google Scholar]
- Rae CL, Correia MM, Altena E, Hughes LE, Barker RA, Rowe JB (2012): White matter pathology in Parkinson's disease: the effect of imaging protocol differences and relevance to executive function. Neuroimage 62:1675–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez‐Ruiz B, Martí MJ, Tolosa E, Bartrés‐Faz D, Summerfield C, Salgado‐Pineda P, Gómez‐Ansón B, Junqué C (2005): Longitudinal evaluation of cerebral morphological changes in Parkinson's disease with and without dementia. J Neurol 252:1345–1352. [DOI] [PubMed] [Google Scholar]
- Raven J, Raven JC, Court JH. 2003. Manual for Raven's Progressive Matrices and Vocabulary scales: Section 1. General overview. San Antonio, TX: Harcourt Assessment. [Google Scholar]
- Raz N, Gunning‐Dixon F, Head D, Rodrigue KM, Williamson A, Acker JD (2004): Aging, sexual dimorphism, and hemispheric asymmetry of the cerebral cortex: Replicability of regional differences in volume. Neurobiol Aging 25:377–396. [DOI] [PubMed] [Google Scholar]
- Richfield EK, Twyman R, Berent S (1987): Neurological syndrome following bilateral damage to the head of the caudate nuclei. Ann Neurol 22:768–771. [DOI] [PubMed] [Google Scholar]
- Riddle WR, Li R, Fitzpatrick JM, DonLevy SC, Dawant BM, Price RR (2004): Characterizing changes in MR images with color‐coded Jacobians. Magn Reson Imaging 22:769–777. [DOI] [PubMed] [Google Scholar]
- Rüb U, Del Tredici K, Schultz C, Ghebremedhin E, de Vos RA, Jansen Steur E, Braak H (2002): Parkinson's disease: The thalamic components of the limbic loop are severely impaired by alpha‐synuclein immunopositive inclusion body pathology. Neurobiol Aging 23:245–254. [DOI] [PubMed] [Google Scholar]
- Salat DH, Greve DN, Pacheco JL, Quinn BT, Helmer KG, Buckner. L , Fischl B (2009): Regional white matter volume differences in nondemented aging and Alzheimer's disease. Neuroimage 44:1247–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado‐Pineda P, Delaveau P, Falcon C, Blin O (2006): Brain T1 intensity changes after levodopa administration in healthy subjects: a voxel‐based morphometry study. Br J Clin Pharmacol 62:546–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheik JI, Yesavage JA (1986): Geriatric Depression Scale (GDS): Recent evidence and development of a shorter version In: Clinical Gerontology. A Guide to Assessment and Intervention. New York: The Haworth Press; pp 165–173. [Google Scholar]
- Sherman SM (2007): The thalamus is more than just a relay. Curr Opin Neurobiol 17:417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silton RL, Helter W, Towers DN, Engels AS, Spielberg JM, Edgar JC, Sass SM, Stewart JL, Sutton BP, Banich MT, Miller GA (2010): The time course of activity in dorsolateral prefrontal cortex and anterior cingulate cortex during top‐down attentional control. Neuroimage 50:1292–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CD, Chebrolu H, Wekstein DR, Schmitt FA, Markesbery WR (2007): Age and gender effects on human brain anatomy: A voxel‐based morphometric study in healthy elderly. Neurobiol Aging 28:1075–1087. [DOI] [PubMed] [Google Scholar]
- Smith SM, Nichols TE (2009): Threshold‐free cluster enhancement: Addressing problems of smoothing, threshold dependence and localisation in cluster inference. Neuroimage 44:83–98. [DOI] [PubMed] [Google Scholar]
- Song SK, Lee JE, Park HJ, Sohn YH, Lee JD, Lee PH (2011): The pattern of cortical atrophy in patients with Parkinson's disease according to cognitive status. Mov Disord 26:289–96. [DOI] [PubMed] [Google Scholar]
- Spinnler H, Tognoni G (1987): Italian standardization and validation of neuropsychological tasks. Ital J Neurol Sci 6(Suppl 8):1–120. [PubMed] [Google Scholar]
- Stephens B, Mueller AJ, Shering AF, Hood SH, Taggart P, Arbuthnott GW, Bell JE, Kilford L, Kingsbury AE, Daniel SE, Ingham CA (2005): Evidence of a breakdown of corticostriatal connections in Parkinson's disease. Neuroscience 132:741–754. [DOI] [PubMed] [Google Scholar]
- Studholme C, Cardenas V, Schuff N, Rosen H, Miller B, Weiner M (2001): Detecting spatially consistent structural differences in Alzheimer's and fronto temporal dementia using deformation morphometry. Med Image Comput Computer Assist Interv (MICCAI) 41–48. [Google Scholar]
- Summerfield C, Junqué C, Tolosa E, Salgado‐Pineda P, Gómez‐Ansón B, Martí MJ, Pastor P, Ramírez‐Ruíz B, Mercader J (2005): Structural brain changes in Parkinson disease with dementia: A voxel‐based morphometry study. Arch Neurol 62:281–285. [DOI] [PubMed] [Google Scholar]
- Tam CW, Burton EJ, McKeith IG, Burn DJ, O'Brien JT (2005): Temporal lobe atrophy on MRI in Parkinson disease with dementia: a comparison with Alzheimer disease and dementia with Lewy bodies. Neurology 64:861–865. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Giedd JN, Woods RP, MacDonald D, Evans AC, Toga AW (2000): Growth patterns in the developing brain detected by using continuum mechanical tensor maps. Nature 404:190–193. [DOI] [PubMed] [Google Scholar]
- Tinaz S, Courtney MG, Stern CE (2011): Focal cortical and subcortical atrophy in early Parkinson's disease. Mov Disord 26:436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong L, Brooks D, Amaral F, Henderson JM, Halliday GM (2009): Relative preservation of thalamic centromedian nucleus in parkinsonian patients with dystonia. Mov Disord 24:2128–2135. [DOI] [PubMed] [Google Scholar]
- Tustison NJ, Avants BB, Cook PA, Zheng Y, Egan A, Yushkevich PA, Gee JC (2010): N4ITK: improved N3 bias correction. IEEE Trans Med Imag 29:1310–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub D, Doshi J, Koka D, Davatzikos C, Siderowf AD, Duda JE, Wolk DA, Moberg PJ, Xie SX, Clark CM (2011): Neurodegeneration across stages of cognitive decline in Parkinson disease. Arch Neurol 68:1562–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal R, Simmons C, Bernanos M, Wood T, Crum W, Duricki D, Vernon A, Williams S, Cash D (2013): Pharmacological MRI and tensor‐based morphometry in the 6‐OHDA rat model of Parkinson's disease. Proceedings of the 21st International Society for Magnetic Resonance in Medicine, p 1043.
- Williams‐Gray CH, Foltynie T, Brayne CE, Robbins TW, Barker RA (2007): Evolution of cognitive dysfunction in an incident Parkinson's disease cohort. Brain 130:1787–1798. [DOI] [PubMed] [Google Scholar]
- Van den Berge SA, Kevenaar JT, Sluijs JA, Hol EM (2012) Dementia in Parkinson's disease correlates with α‐synuclein pathology but not with cortical astrogliosis. Parkinson Dis 420957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Werf YD, Witter MP, Groenewegen HJ (2002): The intralaminar and midline nuclei of the thalamus. Anatomical and functional evidence for participation in processes of arousal and awareness. Brain Res Brain Res Rev 39:107–140. [DOI] [PubMed] [Google Scholar]
- Vernon AC, Modo M (2012): Do levodopa treatments modify the morphology of the parkinsonian brain? Mov Disord 27:166–167. [DOI] [PubMed] [Google Scholar]
- Zaja‐Milatovic S, Milatovic D, Schantz AM, Zhang J, Montine KS, Samii A, Deutch AY, Montine TJ (2005): Dendritic degeneration in neostriatal medium spiny neurons in Parkinson disease. Neurology 64:545–547. [DOI] [PubMed] [Google Scholar]
- Zarei M, Ibarretxe‐Bilbao N, Compta Y, Hough M, Junque C, Bargallo N, Tolosa E, Martí MJ (2013): Cortical thinning is associated with disease stages and dementia in Parkinson's disease. J Neurol Neurosurg Psychiatry 84:875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]