

Abstract
Introduction
Gray matter (GM) atrophy is common in multiple sclerosis (MS), but the relationship with white matter (WM) pathology is largely unknown. Some studies found a co‐occurrence in specific systems, but a regional analysis across the brain in different clinical phenotypes is necessary to further understand the disease mechanism underlying GM atrophy in MS. Therefore, we investigated the association between regional GM atrophy and pathology in anatomically connected WM tracts.
Methods
Conventional and diffusion tensor imaging was performed at 3T in 208 patients with long‐standing MS and 60 healthy controls. Deep and cortical GM regions were segmented and quantified, and both lesion volumes and average normal appearing WM fractional anisotropy of their associated tracts were derived using an atlas obtained by probabilistic tractography in the controls. Linear regression was then performed to quantify the amount of regional GM atrophy that can be explained by WM pathology in the connected tract.
Results
MS patients showed extensive deep and cortical GM atrophy. Cortical atrophy was particularly present in frontal and temporal regions. Pathology in connected WM tracts statistically explained both regional deep and cortical GM atrophy in relapsing‐remitting (RR) patients, but only deep GM atrophy in secondary‐progressive (SP) patients.
Conclusion
In RRMS patients, both deep and cortical GM atrophy were associated with pathology in connected WM tracts. In SPMS patients, only regional deep GM atrophy could be explained by pathology in connected WM tracts. This suggests that in SPMS patients cortical GM atrophy and WM damage are (at least partly) independent disease processes. Hum Brain Mapp, 36:1796–1807, 2015. © 2015 Wiley Periodicals, Inc.
Keywords: multiple sclerosis, magnetic resonance imaging, gray matter atrophy, volumetric magnetic resonance imaging, diffusion tensor imaging
INTRODUCTION
Multiple sclerosis (MS) is a chronic inflammatory and neurodegenerative disease of the central nervous system. Although focal white matter (WM) lesions are still the most important magnetic resonance imaging (MRI) characteristic used today in MS diagnosis [Polman et al., 2011] and clinical trials [Kappos et al., 2007; Mikol et al., 2008; Polman et al., 2006], it is now recognized that gray matter (GM) atrophy is an essential component of the disease [Daams et al., 2013; Hulst and Geurts, 2011]. GM atrophy has shown to be present early in the disease, across all different MS disease types, and to be associated with physical disability, cognitive decline, and disease duration [Calabrese et al., 2007; Chen et al., 2004; De Stefano et al., 2003; Fisniku et al., 2008; Narayana et al., 2013; Pagani et al., 2005; Roosendaal et al., 2011; Sailer et al., 2003; Sastre‐Garriga et al., 2004]. Although the exact mechanism underlying GM atrophy is unknown, several hypotheses have been postulated [Geurts and Barkhof, 2008]. They include primary damage of the GM, such as neuronal and axonal loss [Popescu et al., 2015]; but also secondary damage due to axonal transection by WM lesions [Charil et al., 2007], or gradually increasing damage due to network degeneration (i.e. gradually increasing effects of local WM and GM pathology throughout the brain).
Several MRI studies investigated the presumed relationship between GM atrophy and WM pathology. Most of these reported associations between GM loss and increased lesion load [Battaglini et al., 2009; De Stefano et al., 2003; Fisniku et al., 2008; Furby et al., 2009; Henry et al., 2008; Narayana et al., 2013; Roosendaal et al., 2011; Sailer et al., 2003; Tedeschi et al., 2005], and some additionally reported associations between atrophy and normal appearing tissue damage [Vrenken et al., 2006]. Previous work in our own group indicated that, in patients with long‐standing MS, whole‐brain and deep GM atrophy are particularly associated with WM atrophy and lesion volume, while cortical atrophy is associated with normal appearing WM integrity loss [Steenwijk et al., 2014]. To explore the spatial relationship between GM atrophy and WM pathology, some authors investigated whether regional WM pathology is related to GM atrophy in specific regions. Using voxelwise statistics, they found that regional GM atrophy can be better explained by WM pathology in colocalized areas compared to whole‐brain measures [Bodini et al., 2009; Mühlau et al., 2013; Sepulcre et al., 2009]. Only a few studies used a direct analysis of structural connectivity (using diffusion tensor imaging; DTI) to assess the association between WM pathology and GM atrophy in connected areas [Henry et al., 2009; Jehna et al., 2013]. Those studies focused mostly on specific GM regions or WM tracts in early MS patients, and found a stronger relation between GM atrophy and WM lesions in connected regions. To gain more insight in the relationship between GM atrophy and WM pathology, an analysis of these mechanisms across the entire brain and in the different disease types is necessary, but to our knowledge, this has not yet been performed.
Therefore, in this study, we aimed to assess the association between regional GM atrophy and pathology in connected WM tracts on a whole‐brain level. In our unique large cohort of long‐standing MS patients [Steenwijk et al., 2014], we quantified regional atrophy and used a custom‐made atlas of typical regional connectivity to quantify the amount of WM pathology in connected tracts. Linear regression was performed to quantify the amount of regional GM atrophy that can be explained by WM pathology in the connected tracts.
MATERIALS AND METHODS
Participants
The institutional ethics review board approved the study protocol and all subjects gave written informed consent prior to participation. All patients were diagnosed with clinically definite MS [Polman et al., 2011], and were recruited from the clinical MS database in our MS center if they had a disease duration of at least 10 years. Disease severity of the patients was measured on the day of scanning using the Expanded Disability Status Scale (EDSS) [Kurtzke, 1983]. To allow for comparison of the imaging measures in the MS patients with control values, healthy control subjects were recruited via advertisements in the hospital and from nonrelated family and friends of the patients. A more detailed description of the cohort, recruitment, and eligibility procedure can be found elsewhere [Steenwijk et al., 2014]; it should be noted that the participants and imaging data used in the current study overlapped with that previous study.
MRI
Scanning was performed on a 3T whole body scanner (GE Signa HDxt, Milwaukee, WI, USA) using an eight‐channel phased‐array head coil. The protocol included a 3D T1‐weighted fast spoiled gradient echo sequence (repetition time [TR] 7.8 ms, echo time [TE] 3 ms, inversion time [TI] 450 ms, flip angle 12°, sagittal 1.0 mm slices, 0.94 × 0.94 mm2 in‐plane resolution) for volumetric measurements and a 3D fluid attenuated inversion recovery sequence (FLAIR; TR 8,000 ms, TE 125 ms, TI 2,350 ms, sagittal 1.2 mm slices, 0.98 ×0.98 mm2 in‐plane resolution) for lesion detection. Furthermore, 2D echo‐planar DTI (TR 13,000 ms, TE 86 ms, 2.4 mm slices, 2.0 ×2.0 mm2 in‐plane resolution) was performed, including 30 volumes with noncollinear diffusion gradients (b‐value 900 s/mm2) and 5 volumes without diffusion weighting. Prior to further analysis, the 3D images were corrected for geometrical distortions due to gradient nonlinearity.
Lesion Segmentation and Whole‐Brain MRI Measures
A detailed description on the WM lesion segmentation and the whole‐brain measures in the present cohort was published previously [Steenwijk et al., 2014]. In short, WM lesions were automatically segmented using the kNN‐TTP algorithm [Steenwijk et al., 2013]. Subsequently, in each patient, the lesion volume was quantified and normalized for head size resulting in normalized lesion volume. Normalized whole brain, WM (NWMV), and GM (NGMV) were computed using the T1‐weighted image and SIENAX (part of FSL 5.0.4, http://www.fmrib.ox.ac.uk/fsl) after lesion filling [Chard et al., 2010] to minimize the impact of WM lesions on atrophy measurements.
Deep GM Structures
Deep GM structures were segmented using the T1‐weighted images and FIRST (also part of FSL). This provided segmentations of seven deep GM structures per hemisphere, including the nucleus accumbens, amygdala, caudate, hippocampus, globus pallidus, putamen, and thalamus. The volumes of all deep GM regions were measured and normalized to account for differences in head size.
Cortical Thickness
Cortical thickness was measured using the FreeSurfer 5.3 processing stream [Dale et al., 1999; Fischl et al., 1999]. In short, FreeSurfer uses the T1‐weighted images to locate the pial and WM surface of the cortex. The distance between these surfaces gives the vertexwise cortical thickness (i.e., the perpendicular thickness at each location). The cortical thickness was subsequently averaged in different “lobes” (i.e., frontal, precentral, postcentral, parietal, temporal, occipital, cingulate, and insula) for global description, and in 34 cortical regions per hemisphere for regional analysis [Desikan et al., 2006]. Vertexwise analysis (see ‘Statistical analysis’ section below) was used to compare the long‐standing MS population to previous cortical thickness studies. All cortical segmentations were manually checked and rerun after manual editing if errors occurred.
Construction of the Connectivity Atlas and Quantification of WM Pathology in Tracts
The DTI images were corrected for head movement and eddy current distortions using FMRIB's Diffusion Toolbox (also part of FSL). Subsequently the diffusion tensor was fitted, from which the fractional anisotropy (FA), mean (MD), axial (AD), and radial diffusivity (RD) were calculated.
DTI was then used to obtain the tract distributions typically associated with the previously segmented deep and cortical GM regions. Because tractography in the presence of MS pathology might lead to unreliable results, this was done by means of a probabilistic atlas based on the healthy controls. The pipeline was as follows (Fig. 1):
For each healthy control, the construction of the atlas first involved running bedpostx (also part of FSL) to estimate the voxelwise diffusion parameter distributions of a two‐fiber model. Then, for each healthy control, 82 GM region–tract pairs were obtained by running whole‐brain probabilistic tractography (probtrackx2, also part of FSL, 5,000 streamlines per voxel) for each GM region separately, using the previously obtained deep and cortical GM masks as seed regions. Except the brain mask, no stopping criteria were applied.
The probabilistic maps of the tracts were nonlinearly registered to MNI152 space, and averaged to construct a single probabilistic map for each region in standard space. The 82 probabilistic maps (left/right, 7 deep, and 34 cortical GM) formed the atlas of the tracts associated to the GM regions.
To obtain a subject‐specific segmentation, the probabilistic atlas was propagated to the individual‐space of each subject using nonlinear registration (FNIRT, also part of FSL) of the FA image while masking out the lesions from the registration cost‐function. Linear registration was used in the patients to obtain the lesion mask in FA space.
For each subject and tract distribution separately, the weighted lesion volume, as well as the weighted average FA values inside the NAWM of the tract were computed using the atlas probability values as a weighting value. Weighting was performed to emphasize lesions and FA values in the centers of the tracts associated to each GM region. To only include WM, the weighting values of voxels belonging to GM and CSF were set to zero using the previously derived SIENAX segmentation. Note that the weighted lesion volume is a relative instead of an absolute value, and cannot be compared with normal (unweighted) whole‐brain lesion volume.
Figure 1.
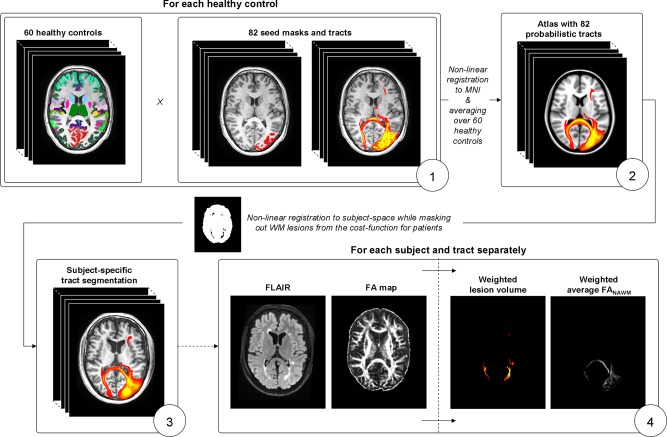
Construction of the connectivity atlas and quantification of pathology in white matter tracts. (1) For each healthy control, probabilistic tractography is run from 82 gray matter regions to identify the connected tracts; (2) the healthy controls are non‐linearly registered to standard space and averaged to obtain an atlas with 82 probabilistic tracts; (3) the atlas is nonlinearly registered to subject space to obtain an subject‐specific tract segmentation; (4) the average weighted lesion volume and weighted average FANAWM are computed for each subject and tract separately. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Statistical Analysis
Statistical analyses of the demographic, clinical and descriptive MRI measures were performed in SPSS 20 (Chicago, IL). Kolmogorov–Smirnov tests and visual inspection of the histogram were used to assess normality of the variables. When the variables were normally distributed, a multivariate general linear model (GLM) was used to assess group differences. When variables were not normally distributed, the Mann–Whitney or the Kruskal–Wallis test was used, as appropriate. Where applicable, the analyses were Bonferroni‐corrected. P‐values < 0.05 were considered as statistically significant.
To analyze local cortical thickness differences in MS patients and disease types, vertexwise GLM analysis was performed using the FreeSurfer software. Prior to vertexwise analysis, the surface maps of cortical thickness were smoothed using a Gaussian kernel with a FWHM of 10 mm. Clusterwise correction for multiple comparisons was applied using Monte Carlo Z simulation while thresholding the statistical maps at P < 0.001, using 5,000 iterations and setting the cluster‐level threshold at P < 0.05.
The relationship between atrophy in GM regions and pathology in the connected WM tracts was assessed using a two step procedure. First, linear regression was used for every region separately to remove the effects of normal ageing and sex on deep GM volume and cortical thickness. In this step, the regression coefficients were estimated based on the healthy controls and the resulting model was used to eliminate the normal ageing and sex effect from the GM atrophy measures. A second linear regression analysis was then used to assess the relationship between regional GM atrophy and pathology in the connected WM tract. Here the residuals of the previous analysis were used as dependents, and the weighted lesion volume as well as weighted average FANAWM of the tract connected to the respective GM region as independent variables. Cortical regression variables and statistics were displayed on a 3D surface to enable visual interpretation of the results. Possible differences between disease types were investigated by repeating the analysis for all disease types as well as the relapse‐onset (RO) MS patients separately. Regional regression analyses were performed in Matlab R2011a (Natick, MA).
RESULTS
Demographic, Clinical, and MRI Characteristics
A detailed discussion on the demographical, clinical, and whole‐brain MRI findings in the present cohort was published previously [Steenwijk et al., 2014]. In short, a total of 208 (67% female) patients with MS and 60 (62% female) healthy controls were included (Table 1). The MS patients had an average age of 53.70 (±9.62) years, had an average disease duration of 20.20 (±7.08) years and consisted of 130 relapsing‐remitting (RR), 53 secondary‐progressive (SP) and 25 primary‐progressive (PP) patients. Of the MS patients, 10 were using glatiramer, 40 beta‐interferon, and 9 natalizumab. Age was different between the total patient group and controls, but not between RRMS patients and controls. PPMS and SPMS patients had higher EDSS scores than RRMS patients.
Table 1.
Demographic, clinical, and MRI measuresa
| Healthy controls (n = 60) | MS patients (n = 208) | RRMS (n = 130) | SPMS (n = 53) | PPMS (n = 25) | |
|---|---|---|---|---|---|
| Age, y | 50.33 ± 7.08 | 53.70 ± 9.62** | 50.68 ± 9.53 | 57.00 ± 6.76***, ††† | 62.40 ± 7.66***, ††† |
| F/M | 37/23 | 141/67 | 97/33 | 34/19 | 10/15†† |
| Disease duration, y | — | 20.20 ± 7.08 | 19.03 ± 6.23 | 23.05 ± 8.50†† | 20.22 ± 6.44 |
| EDSSb | — | 4.0 (3.0–6.0) | 3.0 (2.5–4.0) | 6.0 (4.0–7.0)††† | 6.0 (4.5–6.5)††† |
| NBV, L | 1.49 ± 0.07 | 1.41 ± 0.09*** | 1.43 ± .0.10*** | 1.39 ± 0.08***, † | 1.41 ± 0.10*** |
| NGMV, L | 0.80 ± 0.05 | 0.75 ± 0.06*** | 0.76 ± 0.06* | 0.73 ± 0.05***, †† | 0.74 ± 0.07***, † |
| NWMV, L | 0.70 ± 0.03 | 0.66 ± 0.05*** | 0.66 ± 0.05*** | 0.66 ± 0.05*** | 0.67 ± 0.04 |
| LV, mL | — | 11.24 (3.92–20.5) | 9.70 (3.86–19.26) | 13.93 (3.76–25.56) | 12.49 (6.03–16.06) |
| NLV, mLb | — | 18.09 (9.93–29.67) | 16.80 (8.58–26.81) | 24.65 (15.92–41.50)†† | 15.24 (9.35–28.50) |
| NDGMV, mLc | 63.45 ± 4.71 | 57.27 ± 6.62*** | 58.00 ± 6.73*** | 55.21 ± 6.28***, † | 57.60 ± 6.26*** |
| Accumbens | 1.26 ± 0.20 | 1.10 ± 0.29*** | 1.16 ± 0.26* | 1.00 ± 0.28***, †† | 1.02 ± 0.36*** |
| Amygdala | 3.92 ± 0.55 | 3.64 ± 0.53*** | 3.66 ± 0.52* | 3.58 ± 0.55** | 3.65 ± 0.55 |
| Caudate | 9.30 ± +1.01 | 8.30 ± 1.12*** | 8.42 ± 1.16*** | 8.06 ± 0.93*** | 8.18 ± 1.21*** |
| Hippocampus | 10.33 ± 1.09 | 9.39 ± 1.32*** | 9.63 ± 1.20** | 9.00 ± 1.31***, †† | 8.94 ± 1.66***, † |
| Globus pallidus | 4.76 ± 0.51 | 4.36 ± 0.64*** | 4.42 ± 0.61** | 4.18 ± 0.67** | 4.43 ± 0.70 |
| Putamen | 13.01 ± 1.15 | 12.02 ± 1.60*** | 12.08 ± 1.61*** | 11.73 ± 1.63*** | 12.29 ± 1.47 |
| Thalamus | 20.83 ± 1.59 | 18.43 ± 2.40*** | 18.61 ± 2.44*** | 17.66 ± 2.36***, † | 19.09 ± 1.90** |
| Cortical thickness, mmd | 2.48 ± 0.09 | 2.39 ± 0.10*** | 2.40 ± 0.10*** | 2.35 ± 0.09***, †† | 2.38 ± 0.11*** |
| Frontal | 2.51 ± 0.09 | 2.43 ± 0.12*** | 2.45 ± 0.10** | 2.40 ± 0.10***, † | 2.39 ± 0.19*** |
| Precentral | 2.53 ± 0.11 | 2.42 ± 0.14*** | 2.45 ± 0.13*** | 2.37 ± 0.13***, †† | 2.37 ± 0.17***, † |
| Postcentral | 2.07 ± 0.11 | 1.99 ± 0.12*** | 2.01 ± 0.11* | 1.96 ± 0.11***, † | 1.97 ± 0.15* |
| Parietal | 2.35 ± 0.09 | 2.27 ± 0.12*** | 2.28 ± 0.11*** | 2.24 ± 0.11*** | 2.25 ± 0.17** |
| Temporal | 2.82 ± 0.10 | 2.70 ± 0.15*** | 2.72 ± 0.13*** | 2.65 ± 0.13***, †† | 2.67 ± 0.25*** |
| Occipital | 2.04 ± 0.09 | 1.96 ± 0.11*** | 1.97 ± 0.10*** | 1.95 ± 0.09*** | 1.95 ± 0.17** |
| Cingulate | 2.57 ± 0.13 | 2.50 ± 0.14** | 2.53 ± 0.12 | 2.48 ± 0.13** | 2.44 ± 0.19***, † |
| Insular | 3.06 ± 0.11 | 2.98 ± 0.15** | 3.00 ± 0.14** | 2.97 ± 0.09** | 2.93 ± 0.25 |
| FANAWM | 0.39 ± 0.02 | 0.37 ± 0.03*** | 0.37 ± 0.03*** | 0.36 ± 0.02***, †† | 0.37 ± 0.02** |
| MDNAWM | 0.83 ± 0.03 | 0.85 ± 0.03*** | 0.85 ± 0.03*** | 0.86 ± 0.03*** | 0.85 ± 0.03** |
| ADNAWM | 1.19 ± 0.03 | 1.20 ± 0.03 | 1.20 ± 0.03 | 1.20 ± 0.03 | 1.20 ± 0.03 |
| RDNAWM | 0.65 ± 0.03 | 0.68 ± 0.04*** | 0.68 ± 0.04*** | 0.70 ± 0.04***, † | 0.67 ± 0.04* |
Abbreviations: RRMS, relapsing‐remitting multiple sclerosis; SPMS, secondary‐progressive multiple sclerosis; PPMS, primary‐progressive multiple sclerosis; EDSS, Expanded Disability Status Scale; NBV, normalized brain volume; NGMV, normalized gray matter volume; NWMV, normalized white matter volume; NLV, normalized lesion volume; NAWM, normal‐appearing white matter, FA, fractional anisotropy; MD, mean diffusitivity; AD, axial diffusitivity; RD, radial diffusitivity.
Values listed are mean ± SD for normally distributed variables, P values were Bonferroni‐corrected where applicable.
Variables were not normally distributed and therefore median (interquartile range) is provided.
Values listed are volumes normalized for head size; left and right were averaged.
left and right were averaged.
*P < 0.05, compared with healthy controls
**P < 0.01, compared with healthy controls
***P < 0.001, compared with healthy controls
† P < 0.05, compared with RRMS
†† P < 0.01, compared with RRMS
††† P < 0.001, compared with RRMS
The total MS group displayed atrophy in all deep GM structures. Compared with the RRMS patients, SPMS patients had more severe atrophy in the accumbens, hippocampus, and thalamus, whereas in the PPMS patients only the hippocampus showed more pronounced atrophy (Table 1). The MS patients displayed reduced cortical thickness in all lobes. Compared with RRMS patients, SPMS patients showed more cortical atrophy in the frontal, precentral, postcentral, and temporal region, while in the PPMS patients more cortical atrophy was only found in the precentral and cingulate gyrus (Table 1).
Local Cortical Thickness
Supporting Information, Figure S1 shows the areas with significant cortical thinning in the MS patients as a group compared with the controls. Bilateral thinning was found in the insula, precuneus, lingual, precentral, superior frontal, superior parietal, and inferior parietal cortex. Thinning was also present in other regions, such as the left supramarginal, paracentral, and rostral middle frontal cortex, as well as in the right entorhinal, lateral occipital, post central, fusiform, and superior temporal cortex. The analyses were repeated with age and sex as covariates. Adding age as a covariate showed a slight reduction of the size, but not position of the clusters of significant cortical thinning in patients. No additional differences were seen with sex as a co‐factor (data not shown).
Comparing the individual subtypes with healthy controls revealed a more widespread pattern of cortical thinning in the SPMS patients than the RRMS patients, while the pattern of PPMS was comparable to RRMS patients (Fig. 2). A direct comparison of the SPMS to the RRMS patients revealed small regions with more thinning in the bilateral temporal lobes, and superior part of the left precentral cortex (data not shown). No significant differences were found in the direct comparison of PPMS with RRMS patients.
Figure 2.
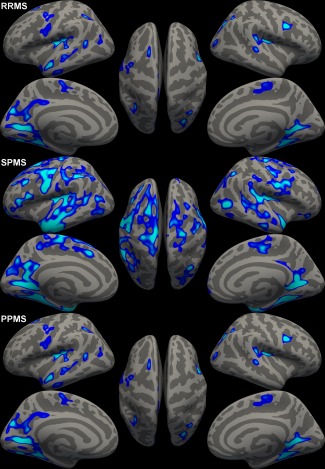
Thickness difference maps highlighting significantly thinner regions in respectively the RRMS patients, SPMS patients, and PPMS patients compared to healthy controls. Figure displays vertexwise P‐values in significant clusters. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Association Between Regional GM Atrophy and Pathology in Connected WM Tracts
Figure 3 and Table 2 display the results of the regional linear regression analyses. In the regression analyses, variables measured in the left and right hemispheres were averaged as the patterns only differed marginally. The results (i.e., the linear regression models in which regional age and sex corrected cortical thickness or deep GM volume were the dependent variables and weighted lesion volume and average FANAWM were the independent variables) are displayed in an alternative fashion in Figures 4 and 5. In general, twice as much of the variance was explained in the models for deep GM volume compared with the models for cortical thickness (in the total MS group, on average 20.0 and 10.4%, respectively). The volumes of all deep GM structures could be explained significantly, whereas only in 28 of the 34 (82%) cortical regions, atrophy could be explained on the basis of pathology in connected WM tracts. Deep GM atrophy was particularly explained by lesion volume in the connected tract (in the total MS group, on average stdβLV = −0.332 vs. stdβFA = 0.172), whereas cortical GM atrophy was particularly associated with normal appearing tract FA (in the total MS group, on average stdβFA = 0.201 vs. stdβLV = −0.116).
Figure 3.
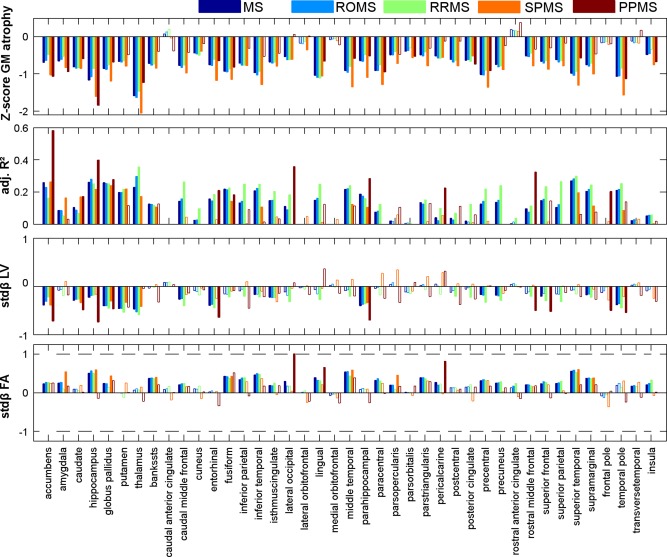
Bar charts displaying the regression results for each GM region. From top to bottom, the Z‐score of atrophy, the adjusted R 2 of the model, and associated standardized beta's are displayed for each group in different colors. Opaque bars indicate significant models and variables, whereas a transparent bar indicates non‐significant total models (in the adjusted R 2 chart) or that the variable did not contribute significantly to the model (in the standardized beta charts). [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Table 2.
Averaged regional linear regression results
| Regions | Result | MS | ROMS | RRMS | SPMS | PPMS |
|---|---|---|---|---|---|---|
| All (n = 41) | #Sign | 35 (85%) | 33 (80%) | 34 (83%) | 15 (37%) | 11 (27%) |
| Adj. R2 | 0.120 | 0.122 | 0.154 | 0.065 | 0.096 | |
| StdβLV | −0.153 | −0.144 | −0.214 | −0.063 | −0.249 | |
| StdβFA | 0.196 | 0.206 | 0.193 | 0.137 | 0.090 | |
| Subcortical (n = 7) | #Sign | 7 (100%) | 7 (100%) | 7 (100%) | 7 (100%) | 4 (57%) |
| Adj. R2 | 0.200 | 0.204 | 0.194 | 0.207 | 0.221 | |
| StdβLV | −0.332 | −0.318 | −0.349 | −0.264 | −0.438 | |
| StdβFA | 0.172 | 0.191 | 0.128 | 0.290 | 0.038 | |
| Cortical (n = 34) | #Sign | 28 (82%) | 26 (76%) | 27 (79%) | 8 (24%) | 7 (21%) |
| Adj. R2 | 0.104 | 0.105 | 0.145 | 0.036 | 0.071 | |
| StdβLV | −0.116 | −0.108 | −0.187 | −0.021 | −0.201 | |
| StdβFA | 0.201 | 0.209 | 0.207 | 0.105 | 0.101 |
Abbreviations: MS, multiple sclerosis; ROMS, relapse‐onset MS; RRMS, relapsing‐remitting MS; SPMS, secondary‐progressive MS; PPMS, primary‐progressive MS; #sign, number of regions with significant models.
Figure 4.
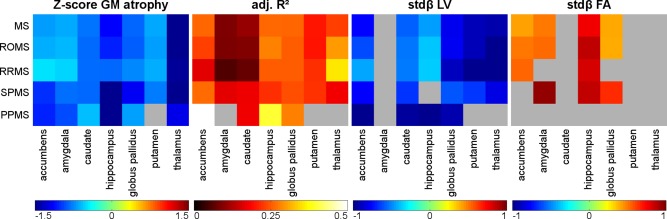
Graphical visualization of the deep GM regional regression model results. Gray areas indicate regions without significant thinning (Z‐scores), nonsignificant models (adjusted R 2) or variables nonsignificantly contributing to the model (standardized beta), whereas the colors correspond to the results of the respective regional model. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Figure 5.
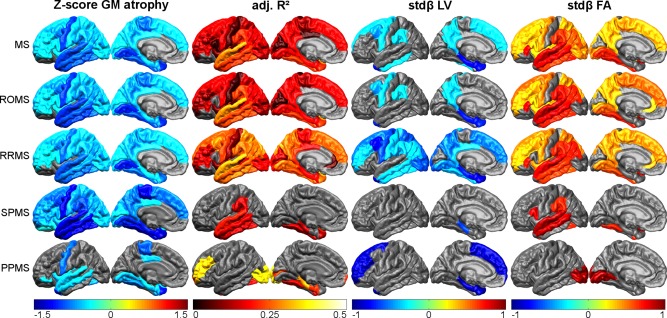
Graphical visualization of the cortical regional regression model results. Gray areas indicate regions without significant thinning (Z‐scores), nonsignificant models (adjusted R 2) or variables nonsignificantly contributing to the model (standardized beta), whereas the colors correspond to the results of the respective regional model. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Looking into the clinical subtypes (Table 2), all models for deep GM volume were significant, except the models for the amygdala, putamen, and thalamus in PPMS patients. Large differences were however observed in the models for cortical thickness: in the RRMS patients, 26 of the 34 (76%) models for regional cortical thickness were significant, whereas only 8 (24%) models were significant in the SPMS patients and 7 (21%) in the PPMS patients. To exclude the possibility that difference in group size has led to the differences between the RRMS and SPMS patients, we combined the RRMS and SPMS patients in a ROMS group. The deep GM model parameters in this ROMS group were similar to those of the separate RRMS and SPMS groups. However, in the cortical models, on average more variance was explained in the RRMS patients compared with the ROMS patients (14.5 vs. 10.5%, respectively), emphasizing the difference between RRMS and SPMS.
Typically, the models for cortical thickness in the RRMS patients explained most of the variance in the temporal, parietal, and posterior part of the frontal lobes (Fig. 5). This pattern showed a widespread reduction in the SPMS patients; only a few of the models in the temporal lobe remained (marginally) significant. In contrast to the RR and SPMS patients, the PPMS patients displayed a much more scattered pattern of explained variance, with two regions displaying very high numbers of explained variance. These regions were regions with very little atrophy, stressing that the results of the PPMS models should be interpreted with care as the PPMS group was small.
DISCUSSION
To our knowledge, this is the first study in a large sample of long‐term MS patients directly investigating the association between regional GM atrophy and pathology in connected WM tracts at the whole‐brain level. Our results demonstrate that normal‐appearing WM damage plays an important role in explaining deep and cortical GM atrophy, whereas lesion volume is particularly associated with deep GM atrophy. Large differences were observed in the association patterns of regional GM atrophy and connected WM pathology of RRMS patients and SPMS patients, indicating that GM atrophy and WM damage become (at least partly) independent after conversion to progressive disease.
Our results confirm findings from previous studies, in which MS patients displayed extensive deep and cortical GM atrophy in the bilateral temporal lobes, frontal areas, insula, precentral gyrus, and medial occipital lobes [Bergsland et al., 2012; Calabrese et al., 2007, 2013; Narayana et al., 2013; Ramasamy et al., 2009; Sailer et al., 2003; Schoonheim et al., 2012]. Compared with RRMS patients, SPMS patients showed a decreased cortical thickness, while PPMS patients did not show a significant difference. More pronounced GM atrophy in patients with SPMS has been reported before, both at a whole‐brain level and specifically in the cortical and deep GM regions [Calabrese et al., 2009; Fisher et al., 2008; Fisniku et al., 2008; Roosendaal et al., 2011]. The absence of statistically more cortical atrophy in PPMS patients compared with RRMS patients is contrary to our expectations [Pagani et al., 2005], but might be explained by a different atrophy pattern in the two groups, or by the relatively low number of PPMS patients in our study cohort.
While previous studies, including our own work on this same patient cohort, have focused on whole‐brain measures of atrophy and other pathology, this work takes the investigation of these associations to another level. By identifying specific WM tracts to anatomically defined GM regions, and quantifying the focal and diffuse damage in those WM tracts, we were able to directly assess the relationship between regional GM atrophy and pathology in connected WM tracts.
An atlas of WM connectivity based on healthy controls was used to quantify the WM pathology in the tracts connected to individual deep and cortical GM regions. To minimize the effects of atrophy and lesions on registration accuracy in the patients, nonlinear registration and masking of lesions were used. Regional linear regression models in the MS patients revealed that WM explained deep GM atrophy better than cortical GM atrophy. Whereas NAWM damage showed comparable associations with both deep and cortical GM atrophy, lesion volume showed stronger associations with deep than cortical GM atrophy. Associations were found between regional GM atrophy and pathology in connected WM tracts in the RRMS patients, but in the SPMS patients, only deep GM atrophy, and not cortical GM atrophy, could be explained by pathology in the connected WM tracts. The findings in RRMS patients are in line with previous studies reporting a spatial relationship between deep GM atrophy and WM pathology [Bodini et al., 2009; Charil et al., 2007; Henry et al., 2009; Mühlau et al., 2013]. Studies investigating regional cortical GM and whole‐brain lesion load in RRMS indicated that GM atrophy in frontotemporoparietal regions is partially dependent on increasing lesion load [Battaglini et al., 2009; Bendfeldt et al., 2009a, 2009b; Charil et al., 2007; Jehna et al., 2013]. Our results confirm these findings, with the difference that the strongest cortical associations were found in the temporal lobe.
An interesting finding is the absence of a relationship between GM atrophy and pathology in the connected WM tracts in the cortical GM of the SPMS patients. To exclude the possibility that the smaller SPMS sample size caused the absence of this relationship, we performed a post‐hoc analysis in the ROMS patients with a similar approach, but now using a median split based on EDSS (EDSS < 4 and EDSS ≥ 4, respectively) to construct two equally sized groups. This procedure resulted in a mildly impaired group of 91 patients (83 RR and 8 SPMS) and a severely impaired group of 92 patients (45 RR and 47 SPMS). The results of this post‐hoc analysis showed a similar behavior as the separate RR and SPMS analyses; indicating a stronger association between cortical GM atrophy and pathology in connected WM tracts in mildly impaired patients compared with severely impaired patients (Supporting Information, Table S1). Studies describing the relation between regional GM atrophy and WM pathology in SPMS patients are limited, but Fisher et al. [2008] described accelerated GM atrophy in these patients and Bendfeldt et al. [2012] found a clearly different GM atrophy pattern in patients with low versus high lesion load.
It should be noted that this study was performed in a cohort of patients with long‐standing disease. This might be beneficial for addressing questions related to the neurodegenerative aspects of the disease, as measures of pathologic and physical decline will be more pronounced in these patients compared with patients with a short disease duration. However, due to the same reason, some of the RRMS patients in our cohort have a rather benign (and thereby somewhat atypical) disease course.
Taken together, our data suggests that in RRMS patients GM atrophy can (at least partially) be explained by retrograde neurodegeneration, whereas GM atrophy may be much more independent of WM pathology after conversion to progressive disease. Possible explanations for this difference in SPMS may include a more widespread inflammatory disease [Kutzelnigg et al., 2005; Politis et al., 2012], or gradually increasing (indirect) remote effects of local WM and GM damage in the cortex of these patients, but future studies are required to investigate these hypotheses.
Some limitations apply to this work. Although the RRMS patients were well matched in age, the whole MS group was on average older than the healthy controls. To prevent an unwanted influence of age on the study outcomes, we removed the effects of age (and sex) based prior to performing the regional regression analyses. Although it might be that after removal of healthy ageing effects some additional age dependencies can be found in the MS data, we believe this approach is justified for various reasons: first, we selected this approach to limit the number of candidate predictors in the regional analysis and assure robust regression results; second, given that older MS patients are more likely to have more pathology, it may be that entering age (and sex) as a covariate in the regional models erroneously corrects for some of the disease‐related effects; and third, although we do currently extrapolate the healthy control data to older MS patients, brain changes due to healthy ageing are thought be fairly smooth processes and the age difference between the healthy controls and the SPMS group is fairly small (∼7 years, approximately the standard deviation of both groups). A second limitation is the relatively small size of the PPMS and SPMS groups compared with the RRMS group. Therefore, especially the results concerning PPMS should be interpreted with great care. In the SPMS patients, however, the results from the combined RRMS and SPMS (ROMS) and post‐hoc analysis (i.e., mildly impaired vs. severely impaired patients) can be used to confirm the lower amount of GM atrophy explained in SPMS patients. Indeed, the ROMS analysis displayed a lower amount of explained variance compared to the RRMS group only, whereas the post‐hoc analysis displayed less explained variance in the severely impaired patients compared with the mildly impaired patients, supporting the interpretation of a different mechanism underlying cortical atrophy in the SPMS patients. Third, although the effect of (juxta) cortical lesions on cortical atrophy is thought to be limited [Wegner et al., 2006], it cannot be ruled out that (juxta) cortical lesions have had an influence on the results as they might result in anterograde axonal degeneration [Mistry et al., 2014]. Future studies elaborating on this topic are required to overcome this limitation. Finally, the cross‐sectional design of the present study makes it impossible to make statements about causality. Longitudinal studies are required to overcome this limitation.
In conclusion, in RRMS patients, both deep and cortical GM atrophy were associated with pathology in connected WM tracts. In SPMS patients, only regional deep GM atrophy could be explained by pathology in connected WM tracts. This suggests that in SPMS patients cortical GM atrophy and WM damage are (at least partly) independent disease processes.
Supporting information
Supporting Information.
REFERENCES
- Battaglini M, Giorgio A, Stromillo ML, Bartolozzi ML, Guidi L, Federico A, De Stefano N (2009): Voxel‐wise assessment of progression of regional brain atrophy in relapsing‐remitting multiple sclerosis. J Neurol Sci 282:55–60. [DOI] [PubMed] [Google Scholar]
- Bendfeldt K, Kappos L, Radue EW, Borgwardt SJ (2009a): Progression of gray matter atrophy and its association with white matter lesions in relapsing‐remitting multiple sclerosis. J Neurol Sci 285:268–9; author reply 269. [DOI] [PubMed] [Google Scholar]
- Bendfeldt K, Kuster P, Traud S, Egger H, Winklhofer S, Mueller‐Lenke N, Naegelin Y, Gass A, Kappos L, Matthews PM, Nichols TE, Radue E‐W, Borgwardt SJ (2009b): Association of regional gray matter volume loss and progression of white matter lesions in multiple sclerosis—A longitudinal voxel‐based morphometry study. Neuroimage 45:60–67. [DOI] [PubMed] [Google Scholar]
- Bendfeldt K, Klöppel S, Nichols TE, Smieskova R, Kuster P, Traud S, Mueller‐Lenke N, Naegelin Y, Kappos L, Radue E‐W, Borgwardt SJ (2012): Multivariate pattern classification of gray matter pathology in multiple sclerosis. Neuroimage 60:400–408. [DOI] [PubMed] [Google Scholar]
- Bergsland N, Horakova D, Dwyer MG, Dolezal O, Seidl ZK, Vaneckova M, Krasensky J, Havrdova E, Zivadinov R (2012): Subcortical and cortical gray matter atrophy in a large sample of patients with clinically isolated syndrome and early relapsing‐remitting multiple sclerosis. AJNR Am J Neuroradiol 33:1573–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodini B, Khaleeli Z, Cercignani M, Miller DH, Thompson AJ, Ciccarelli O (2009): Exploring the relationship between white matter and gray matter damage in early primary progressive multiple sclerosis: An in vivo study with TBSS and VBM. Hum Brain Mapp 30:2852–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese M, Atzori M, Bernardi V, Morra A, Romualdi C, Rinaldi L, McAuliffe MJM, Barachino L, Perini P, Fischl B, Battistin L, Gallo P (2007): Cortical atrophy is relevant in multiple sclerosis at clinical onset. J Neurol 254:1212–1220. [DOI] [PubMed] [Google Scholar]
- Calabrese M, Filippi M, Rovaris M, Bernardi V, Atzori M, Mattisi I, Favaretto A, Grossi P, Barachino L, Rinaldi L, Romualdi C, Perini P, Gallo P (2009): Evidence for relative cortical sparing in benign multiple sclerosis: A longitudinal magnetic resonance imaging study. Mult Scler 15:36–41. [DOI] [PubMed] [Google Scholar]
- Calabrese M, Favaretto A, Poretto V, Romualdi C, Rinaldi F, Mattisi I, Morra A, Perini P, Gallo P (2013): Low degree of cortical pathology is associated with benign course of multiple sclerosis. Mult Scler 19:904–911. [DOI] [PubMed] [Google Scholar]
- Chard DT, Jackson JS, Miller DH, Wheeler‐Kingshott CAM (2010): Reducing the impact of white matter lesions on automated measures of brain gray and white matter volumes. J Magn Reson Imaging 32:223–228. [DOI] [PubMed] [Google Scholar]
- Charil A, Dagher A, Lerch JP, Zijdenbos AP, Worsley KJ, Evans AC (2007): Focal cortical atrophy in multiple sclerosis: Relation to lesion load and disability. Neuroimage 34:509–517. [DOI] [PubMed] [Google Scholar]
- Chen JT, Narayanan S, Collins DL, Smith SM, Matthews PM, Arnold DL (2004): Relating neocortical pathology to disability progression in multiple sclerosis using MRI. Neuroimage 23:1168–1175. [DOI] [PubMed] [Google Scholar]
- Daams M, Geurts JJG, Barkhof F (2013): Cortical imaging in multiple sclerosis. Curr Opin Neurol 26:345–352. [DOI] [PubMed] [Google Scholar]
- Dale AM, Fischl B, Sereno MI (1999): Cortical surface‐based analysis. I. Segmentation and surface reconstruction. Neuroimage 9:179–194. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Matthews PM, Filippi M, Agosta F, De Luca M, Bartolozzi ML, Guidi L, Ghezzi A, Montanari E, Cifelli A, Federico A, Smith SM (2003): Evidence of early cortical atrophy in MS: Relevance to white matter changes and disability. Neurology 60:1157–1162. [DOI] [PubMed] [Google Scholar]
- Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, Buckner RL, Dale AM, Maguire RP, Hyman BT, Albert MS, Killiany RJ (2006): An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 31:968–980. [DOI] [PubMed] [Google Scholar]
- Fischl B, Sereno MI, Dale AM (1999): Cortical surface‐based analysis. II: Inflation, flattening, and a surface‐based coordinate system. Neuroimage 9:195–207. [DOI] [PubMed] [Google Scholar]
- Fisher E, Lee J‐C, Nakamura K, Rudick R a (2008): Gray matter atrophy in multiple sclerosis: A longitudinal study. Ann Neurol 64:255–265. [DOI] [PubMed] [Google Scholar]
- Fisniku LK, Chard DT, Jackson JS, Anderson VM, Altmann DR, Miszkiel K a, Thompson AJ, Miller DH (2008): Gray matter atrophy is related to long‐term disability in multiple sclerosis. Ann Neurol 64:247–254. [DOI] [PubMed] [Google Scholar]
- Furby J, Hayton T, Altmann D, Brenner R, Chataway J, Smith KJ, Miller DH, Kapoor R (2009): Different white matter lesion characteristics correlate with distinct grey matter abnormalities on magnetic resonance imaging in secondary progressive multiple sclerosis. Mult Scler 15:687–694. [DOI] [PubMed] [Google Scholar]
- Geurts JJG, Barkhof F (2008): Grey matter pathology in multiple sclerosis. Lancet Neurol 7:841–851. [DOI] [PubMed] [Google Scholar]
- Henry RG, Shieh M, Okuda DT, Evangelista A, Gorno‐Tempini ML, Pelletier D (2008): Regional grey matter atrophy in clinically isolated syndromes at presentation. J Neurol Neurosurg Psychiatry 79:1236–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry RG, Shieh M, Amirbekian B, Chung S, Okuda DT, Pelletier D (2009): Connecting white matter injury and thalamic atrophy in clinically isolated syndromes. J Neurol Sci 282:61–66. [DOI] [PubMed] [Google Scholar]
- Hulst HE, Geurts JJG (2011): Gray matter imaging in multiple sclerosis: What have we learned? BMC Neurol 11:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jehna M, Langkammer C, Khalil M, Fuchs S, Reishofer G, Fazekas F, Ebner F, Enzinger C (2013): An exploratory study on the spatial relationship between regional cortical volume changes and white matter integrity in Multiple Sclerosis. Brain Connect:1–47. [DOI] [PubMed] [Google Scholar]
- Kappos L, Freedman MS, Polman CH, Edan G, Hartung HP, Miller DH, Montalbán X, Barkhof F, Radü E‐W, Bauer L, Dahms S, Lanius V, Pohl C, Sandbrink R (2007): Effect of early versus delayed interferon beta‐1b treatment on disability after a first clinical event suggestive of multiple sclerosis: A 3‐year follow‐up analysis of the BENEFIT study. Lancet 370:389–397. [DOI] [PubMed] [Google Scholar]
- Kurtzke JF (1983): Rating neurologic impairment in multiple sclerosis: An expanded disability status scale (EDSS). Neurology 33:1444–1452. [DOI] [PubMed] [Google Scholar]
- Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M, Schmidbauer M, Parisi JE, Lassmann H (2005): Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 128:2705–2712. [DOI] [PubMed] [Google Scholar]
- Mikol DD, Barkhof F, Chang P, Coyle PK, Jeffery DR, Schwid SR, Stubinski B, Uitdehaag BMJ (2008): Comparison of subcutaneous interferon beta‐1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): A multicentre, randomised, parallel, open‐label trial. Lancet Neurol 7:903–914. [DOI] [PubMed] [Google Scholar]
- Mistry N, Abdel‐Fahim R, Mougin O, Tench C, Gowland P, Evangelou N (2014): Cortical lesion load correlates with diffuse injury of multiple sclerosis normal appearing white matter. Mult Scler 20:227–233. [DOI] [PubMed] [Google Scholar]
- Mühlau M, Buck D, Förschler A, Boucard CC, Arsic M, Schmidt P, Gaser C, Berthele A, Hoshi M, Jochim A, Kronsbein H, Zimmer C, Hemmer B, Ilg R (2013): White‐matter lesions drive deep gray‐matter atrophy in early multiple sclerosis: Support from structural MRI. Mult Scler. [DOI] [PubMed] [Google Scholar]
- Narayana P a., Govindarajan K a., Goel P, Datta S, Lincoln J a., Cofield SS, Cutter GR, Lublin FD, Wolinsky JS (2013): Regional cortical thickness in relapsing remitting multiple sclerosis: A multi‐center study. NeuroImage Clin 2:120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani E, Rocca M a, Gallo A, Rovaris M, Martinelli V, Comi G, Filippi M (2005): Regional brain atrophy evolves differently in patients with multiple sclerosis according to clinical phenotype. AJNR Am J Neuroradiol 26:341–346. [PMC free article] [PubMed] [Google Scholar]
- Patenaude B, Smith SM, Kennedy DN, Jenkinson M (2011): A Bayesian model of shape and appearance for subcortical brain segmentation. Neuroimage 56:907–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politis M, Giannetti P, Su P, Turkheimer F, Keihaninejad S, Wu K, Waldman A, Malik O, Matthews PM, Reynolds R, Nicholas R, Piccini P (2012): Increased PK11195 PET binding in the cortex of patients with MS correlates with disability. Neurology 79:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polman CH, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, Phillips JT, Lublin FD, Giovannoni G, Wajgt A, Toal M, Lynn F, Panzara MA, Sandrock AW (2006): A randomized, placebo‐controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 354:899–910. [DOI] [PubMed] [Google Scholar]
- Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, Fujihara K, Havrdova E, Hutchinson M, Kappos L, Lublin FD, Montalban X, O'Connor P, Sandberg‐Wollheim M, Thompson AJ, Waubant E, Weinshenker B, Wolinsky JS (2011): Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 69:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu V, Klaver R, Voorn P, Galis-de Graaf Y, Knol D, Twisk J, Versteeg A, Schenk G, Van der Valk P, Barkhof F, De Vries H, Vrenken H, Geurts J. What drives MRI‐measured cortical atrophy in multiple sclerosis? Mult Scler. 2015 Jan 12. pii: 1352458514562440. [Epub ahead of print] [DOI] [PubMed]
- Ramasamy DP, Benedict RHB, Cox JL, Fritz D, Abdelrahman N, Hussein S, Minagar A, Dwyer MG, Zivadinov R (2009): Extent of cerebellum, subcortical and cortical atrophy in patients with MS: A case‐control study. J Neurol Sci 282:47–54. [DOI] [PubMed] [Google Scholar]
- Roosendaal SD, Bendfeldt K, Vrenken H, Polman CH, Borgwardt S, Radue EW, Kappos L, Pelletier D, Hauser SL, Matthews PM, Barkhof F, Geurts JJG (2011): Grey matter volume in a large cohort of MS patients: Relation to MRI parameters and disability. Mult Scler 17:1098–1106. [DOI] [PubMed] [Google Scholar]
- Sailer M, Fischl B, Salat D, Tempelmann C, Schönfeld MA, Busa E, Bodammer N, Heinze H‐J, Dale A (2003): Focal thinning of the cerebral cortex in multiple sclerosis. Brain 126:1734–1744. [DOI] [PubMed] [Google Scholar]
- Sastre‐Garriga J, Ingle GT, Chard DT, Ramió‐Torrentà L, Miller DH, Thompson AJ (2004): Grey and white matter atrophy in early clinical stages of primary progressive multiple sclerosis. Neuroimage 22:353–359. [DOI] [PubMed] [Google Scholar]
- Schoonheim MM, Popescu V, Rueda Lopes FC, Wiebenga OT, Vrenken H, Douw L, Polman CH, Geurts JJG, Barkhof F (2012): Subcortical atrophy and cognition: Sex effects in multiple sclerosis. Neurology 79:1754–1761. [DOI] [PubMed] [Google Scholar]
- Sepulcre J, Goñi J, Masdeu JC, Bejarano B, Vélez De Mendizábal N, Toledo JB, Villoslada P (2009): Contribution of white matter lesions to gray matter atrophy in multiple sclerosis: Evidence from voxel‐based analysis of T1 lesions in the visual pathway. Arch Neurol 66:173–179. [DOI] [PubMed] [Google Scholar]
- Steenwijk MD, Pouwels PJW, Daams M, Dalen JW van, Caan MWA, Richard E, Barkhof F, Vrenken H (2013): Accurate white matter lesion segmentation by k nearest neighbor classification with tissue type priors (kNN‐TTPs). NeuroImage Clin 3:462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenwijk MD, Daams M, Pouwels PJW, Balk LJ, Tewarie PK, Killestein J, Uitdehaag BMJ, Geurts JJG, Barkhof F, Vrenken H (2014): What explains gray matter atrophy in long‐standing multiple sclerosis? Radiology: 132708. [DOI] [PubMed] [Google Scholar]
- Tedeschi G, Lavorgna L, Russo P, (2005): Brain atrophy and lesion load in a large population of patients with multiple sclerosis. Neurology 65:280–285. [DOI] [PubMed] [Google Scholar]
- Vrenken H, Pouwels PJW, Geurts JJG, Knol DL, Polman CH, Barkhof F, Castelijns JA (2006): Altered diffusion tensor in multiple sclerosis normal‐appearing brain tissue: Cortical diffusion changes seem related to clinical deterioration. J Magn Reson Imaging 23:628–636. [DOI] [PubMed] [Google Scholar]
- Wegner C, Esiri MM, Chance SA, Palace J, Matthews PM (2006): Neocortical neuronal, synaptic, and glial loss in multiple sclerosis. Neurology 67:960–967. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information.