

Abstract
Blood oxygenation level‐dependent (BOLD) signal changes are often assumed to directly reflect neural activity changes. Yet the real relationship is indirect, reliant on numerous assumptions, and subject to several sources of noise. Deviations from the core assumptions of BOLD contrast functional magnetic resonance imaging (fMRI), and their implications, have been well characterized in healthy populations, but are frequently neglected in stroke populations. In addition to conspicuous local structural and vascular changes after stroke, there are many less obvious challenges in the imaging of stroke populations. Perilesional ischemic changes, remodeling in regions distant to lesion sites, and diffuse perfusion changes all complicate interpretation of BOLD signal changes in standard fMRI protocols. Most stroke patients are also older than the young populations on which assumptions of neurovascular coupling and the typical analysis pipelines are based. We present a review of the evidence to show that the basic assumption of neurovascular coupling on which BOLD‐fMRI relies does not capture the complex changes arising from stroke, both pathological and recovery related. As a result, estimating neural activity using the canonical hemodynamic response function is inappropriate in a number of contexts. We review methods designed to better estimate neural activity in stroke populations. One promising alternative to event‐related fMRI is a resting‐state‐derived functional connectivity approach. Resting‐state fMRI is well suited to stroke populations because it makes no performance demands on patients and is capable of revealing network‐based pathology beyond the lesion site. Hum Brain Mapp 36:1620–1636, 2015. © 2014 Wiley Periodicals, Inc.
Keywords: MRI, functional, perfusion, aging, neurovascular coupling, connectivity, multimodal imaging, vascular injury, brain, cerebrovascular accident
INTRODUCTION
The blood oxygenation level‐dependent (BOLD) contrast is the mainstay of functional magnetic resonance imaging (fMRI) and has great utility as a brain‐mapping tool capable of relating behavior to brain function. It is often tacitly assumed that the BOLD signal is a direct measure of neural activity. However, the BOLD signal relies on neurovascular coupling, the relationship between the hemodynamic response (HR) and the neural activity that initiates it. Neurovascular coupling is known to be variable in healthy populations [Aguirre et al., 1998; Handwerker et al., 2012; Heeger and Ress, 2002; Miezin et al., 2000]. Aging introduces further variability, complicating interpretation of BOLD signal changes as a proxy for neural activity changes in older individuals [D'Esposito et al., 1999; Grady and Garrett, 2013]. In stroke, populations' variability in neurovascular coupling remains largely uncharacterized. The HR is dependent on cerebral blood flow (CBF), blood oxygenation levels, and blood volume, all of which are likely to be compromised in stroke populations. Furthermore, stroke populations are typically older than the young populations on which assumptions of neurovascular coupling are based. This is largely unaddressed in BOLD‐fMRI studies of stroke patients. Image processing pipelines and assumptions of neurovascular coupling developed on young, healthy brains may not be appropriate for studying older, structurally atypical, and vascularly impaired brains.
Stroke populations are particularly susceptible to changes in neurovascular coupling arising from both local structural damage following focal ischemia and changes distant to the insult [de Haan et al., 2013; Pineiro et al., 2002]. As well as structural changes, alterations in baseline perfusion, neuronal reorganization, and altered vasoreactivity complicate a vascular system that is already likely to be altered with normal aging [D'Esposito et al., 2003; Feigin et al., 2003].
In this review, we present an overview of BOLD‐fMRI, including challenges to the interpretation of BOLD signal changes in task‐based fMRI in young and aging populations. We then review the current state of the evidence regarding sources of neurovascular decoupling that threaten interpretation of BOLD signal changes specific to stroke populations. We present suggestions for improving BOLD imaging in stroke populations. Finally, we examine functional imaging in the resting‐state (rsfMRI) as an alternative to task‐based studies, and introduce the burgeoning literature implementing this approach in stroke populations. While rsfMRI overcomes a number of limitations of event‐related fMRI, it is also dependent on the BOLD signal, and therefore, also subject to assumptions of neurovascular coupling which may be significantly altered by stroke.
ESTIMATING NEURAL ACTIVITY WITH BOLD‐fMRI
The core assumption of BOLD‐fMRI is that the measured blood oxygenation changes reflect the metabolic demands of neural activity. Importantly, the BOLD signal itself is downstream and delayed from the neural activity that is assumed to trigger it. In the dominant model of the BOLD signal‐measured HR, increased neural activity results in an initial dip in the signal as oxygen in the surrounding vasculature is depleted. Increased demand for oxygen and glucose, driven by the increased local metabolic needs, triggers local vascular dilation, and increased blood flow that facilitates delivery of metabolites. The increased blood flow delivers glucose in proportion to the increased metabolic needs but overcompensates in oxygen supply, resulting in a net increase in local blood oxygen levels [Buxton et al., 2004; Heeger and Ress, 2002]. These dynamic changes in the level of deoxygenated blood as the result of the vascular response to neural activity, alter local magnetic susceptibility—the basis of BOLD contrast fMRI [Ogawa et al., 1990].
The HR is often referred to as “sluggish,” because it is delayed in onset and slow to return to baseline levels relative to the speed and transience of neural activity. Nevertheless, support for the neurovascular coupling of the HR and neural activity validates the use of the BOLD signal as a reliable, if indirect, measure of neural activity in healthy populations [Logothetis, 2008; Logothetis et al., 2001; Rees et al., 2000]. For example, Logothetis et al. [2001] showed that the BOLD signal was monotonically related to stimulus evoked synaptic activity as measured intracortically in anesthetized monkeys. This has been widely interpreted as evidence of a causal relationship between neural activity, at least in terms of presynaptic activity and the BOLD signal.
Since neural activity cannot be directly measured with fMRI, it must be modeled by an impulse response function that describes the HR. Given the sluggishness of the response, when events are either adequately spaced in time to allow the HR to return to baseline, or modeled as a block of the same successive events, the exact shape of the hemodynamic response function (HRF) need not be estimated. This is also the case when spontaneous fluctuations in the signal are measured in the absence of precisely timed events, as in resting‐state paradigms. However, in the majority of task‐based fMRI studies, researchers use rapid events to more efficiently model neural activity, and to offer greater flexibility in task design than block or slow event‐related designs. The widely used canonical, double gamma HRF, modeled as a linear combination of gamma functions [Glover et al., 1999], captures the relatively slow BOLD response. As illustrated in Figure 1, the HRF peaks around 5 s poststimulus, gradually declines to below baseline in the next 4–6 s and returns to baseline over 20 s after initial stimulation. The parameters of the canonical HRF are fixed and the amplitude of the response is typically the estimated parameter of interest. For example, the double gamma HRF used in SPM (Fig. 1) is described by the formula, as expressed by Lindquist et al. [2009],
where amplitude (A) is unknown, t refers to time, β 1 and β 2 = 1, α 1 = 6, α 2 = 16, c = 1/16, and Γ is the gamma function.
Figure 1.
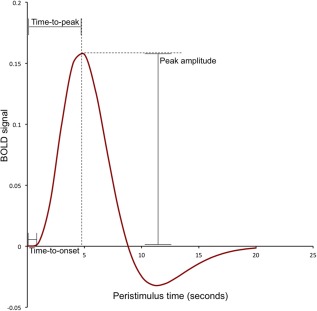
The canonical hemodynamic response (HR) function captures the slow rise, peak, and undershoot of the BOLD signal. Labels illustrate typical parameters of interest in estimation of the HR and sources of variability in the response across populations. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
The HR is assumed to sum linearly across time, allowing modeling of rapid successive events by convolution with the canonical HRF. The duration and latency of the response can also be estimated by taking into consideration the time‐to peak, time‐to‐onset, and the width of the peristimulus‐BOLD signal (Fig. 1). The derivation of these parameters will depend on the model of the HRF implemented [Lindquist et al., 2009; Miezin et al., 2000].
While the canonical HRF has been shown to be a good fit for early sensory regions, it may not adequately model the response in higher cortical regions, nor account for nonlinearity in the amplitude of response under certain conditions [Buxton et al., 2004; Heeger and Ress, 2002]. In fact the HR varies in shape, latency, and magnitude across brain regions [Handwerker et al., 2004], with task parameters [Miezin et al., 2000], across subject groups [D'Esposito et al., 1999], and even within the same subject at different time points [Aguirre et al., 1998].
VARIABILITY IN THE HRF IN YOUNG POPULATIONS
In a reaction time task in young volunteers, Aguirre et al. [1998] demonstrated significant between‐subject variability in the HRF in primary motor cortex (mean age 23 years, SD 3). Given this variability, they questioned the use of a canonical HRF as a standard in fMRI analyses. A replication and extension of this experiment by Handwerker et al. [2004] formally evaluated the impact of between‐ and within‐subject variation in the HRF and its impact on the standard general linear model statistical analyses of fMRI data. HRFs were estimated in young subjects (ages 18–38 years) in primary visual and motor cortex and frontal and supplementary eye fields in a simple sensorimotor task. The majority of subject‐derived HRFs peaked up to 2.5 s earlier than the canonical HRF (the default double gamma HRF used in the popular statistical parametric mapping (SPM) analysis package). The onset time of the empirical HRF also varied significantly across subjects and differed from the canonical HRF. There was greater between‐subject variability in the HRF within brain regions than within‐subject variability across brain regions. The HRF is, therefore, better estimated from a different region within a subject than from the same region averaged across subjects [Handwerker et al., 2004].
Variability between individuals in the HR across the brain may arise from the supporting neural vasculature [Nair, 2005]. Animal models have demonstrated variability in vascular density associated with variability in metabolic demands across the brain [Harrison, 2002]. High local metabolic demand is associated with high capillary density which increases the likelihood of detection of the BOLD signal in comparison to poorly vascularized regions [Harrison, 2002]. Variability in vascular density may, therefore, also contribute to between‐subject variability in the HRF.
Methods exist to account for the variability in the HR that assume fewer, or no, fixed parameters, including Fourier basis sets and finite impulse response (FIR) models [Aguirre et al., 1998; Henson et al., 2001; Lindquist et al., 2009]. However, the canonical HRF remains the standard in the majority of event‐related fMRI analyses. Given the variability in the HRF in young, healthy populations, it is unsurprising that further variability would be seen in aging populations in which there is more likely to be alterations as well as pathologies in the vascular system.
FURTHER VARIABILITY IN THE HRF IN AGING POPULATIONS
Caution is warranted in the interpretation of BOLD signal changes in aging populations, especially in comparison to healthy populations, where comparable neurovascular coupling cannot be assumed [D'Esposito et al., 2003; Gauthier et al., 2013; Kannurpatti et al., 2010; Samanez‐Larkin and D'Esposito, 2008]. The cerebrovascular system changes with age. Numerous studies have shown CBF to be negatively correlated with age [Kawamura et al., 1993; Krejza et al., 1999; Schultz et al., 1999]. Moreover, CBF is not uniformly reduced throughout the aging brain. Instead, perfusion appears to be reduced in a number of cortical regions, independent of regional age‐associated atrophy, but remains relatively preserved in visual and motor cortices and most subcortical regions in normal aging [Chen et al., 2011a]. Lu et al., [2011] assessed cerebral metabolic rate, blood flow, and cerebrovascular reactivity in 232 healthy individuals aged between 20 and 89 with blood flow and blood oxygen saturation sensitive MRI sequences and hypercapnia challenge. They found an increase in the rate of resting cerebral metabolic oxygen consumption (CMRO2) and a reduction in CBF (CBF) and cerebrovascular reactivity associated with aging. In line with Chen et al. [2011a], CBF was heterogeneous, not globally reduced, across the brain in aging. These complex interactions and changes in hemodynamics make the interpretation of BOLD signal changes in aging populations problematic.
In a simple reaction time task, in which task‐related neural activity is thought to be preserved in aging, the HRF (modeled with a Fourier basis set) was similar in shape and peak amplitude in younger and older subjects in motor cortex, but signal‐to‐noise ratio (SNR) was reduced in older subjects [D'Esposito et al., 1999]. This was attributed to greater noise in the BOLD signal of older subjects, a finding replicated in visual cortex during checkerboard stimulation as modeled with an empirically derived HRF [Huettel and McCarthy, 2000; Huettel et al., 2001]. Brodtmann et al. [2003] also demonstrated preserved shape, amplitude, and latency of a block task‐evoked HRF in higher visual regions into the ninth decade of life. The number of activated voxels in striate and extrastriate regions declined systematically with advancing age, again likely attributable to greater noise in the signal with aging.
Where the amplitude, shape, or latency of the task‐evoked HRF has been shown to differ between young and old populations, the direction, or extent of change has been variable [Kannurpatti et al., 2010]. Increased activation for equivalent performance has been repeatedly observed in older, as compared to younger individuals, and is hypothesized to reflect neural plasticity, compensatory recruitment to preserve performance or increased demand on resources [Cabeza, 2002; Cabeza et al., 2002, 2004; Grady, 2012; Grady et al., 2006; Heuninckx et al., 2008]. To characterize the effects of aging on cognition and neural activity, it is critical to separate age‐related neural activity changes from age‐related vascular or hemodynamic changes.
Scaling the BOLD signal with the signal obtained in breath holding [Kannurpatti et al., 2010] and hypercapnia challenge [Gauthier et al., 2013; Thomason et al., 2007] removes some of the variability associated with vascular differences between subjects so that BOLD signal differences across groups can be more confidently attributed to neural activity differences. When the BOLD signal is calibrated or vascular variability is accounted for in this way, BOLD signal is attenuated in older subjects, and this can now be more directly attributable to neural activity changes with aging [Kannurpatti et al., 2010, 2011].
Pathological changes to the vascular system are more likely to be seen in the aging brain, including clinically silent infarcts, cerebral atherosclerosis, and small‐vessel cerebrovascular disease [Brown and Thore, 2011; Fazekas et al., 1993; Wardlaw et al., 2013]. White matter lesions visible on MR images, known as white matter hyperintensities (WMH) of presumed vascular origin [Wardlaw et al., 2013], are frequently seen in normal aging [Brown and Thore, 2011; Christiansen et al., 1994]. WMH have been detected in over 90% of large cohorts of subjects in their sixties [Söderlund et al., 2003; Wen and Sachdev, 2004]. WMH increase the risk of stroke, even after adjustment for known associated vascular risk factors such as hypertension and smoking [Debette and Markus, 2010], suggesting the presence of WMH are associated with a pathological vascular state. In support of this, people with WMH have been demonstrated to have reduced CBF and cerebrovascular reactivity [Hatazawa et al., 1997; Kawamura et al., 1993; Kobari et al., 1990; Marstrand et al., 2002].
SOURCES OF VARIABILITY IN THE HRF IN STROKE POPULATIONS
Many of the pathologies associated with aging are also seen in stroke populations. This is because these pathologies create a predisposition to stroke, and because stroke populations are generally older. The incidence of stroke increases with each decade [Feigin et al., 2003] compounding the changes to the vascular system seen in normal aging with the changes that result from damage to the brain following stroke.
Vascular Pathologies
Stroke populations are heterogeneous in the location and extent of their lesions and in the presence of associated vascular pathologies. Patients should, therefore, be carefully assessed before making assumptions about the state of neurovascular coupling underlying the BOLD response. The risk factors that lead to stroke are implicated in both large and small vessel disease in the heart and the brain. Optimal regulation of CBF is known to be affected by vascular risk factors such as normal aging, hypertension, diabetes, and smoking [de la Torre, 2012; Muller et al., 2012; Toda, 2012]. Subsequently, many stroke patients have a combination of large and small vessel disease. The resulting neurovascular dysfunction is associated with suppression of the physiological CBF increases that should occur during neural activity [Østergaard et al., 2013]. There is also evidence that strokes are associated with abnormal autonomic regulation, meaning that vascular changes that ensure normal blood pressure, and hence brain perfusion, are also affected [Xiong et al., 2014]. This appears to be irrespective of stroke etiology [Xiong et al., 2014]. In support of this, Lin et al. [2011] found impaired neurovascular coupling in ischemic stroke patients with either large or small vessel disease [Lin et al., 2011].
The presence of cerebrovascular occlusive disease is a clear source of abnormal HR [Carusone et al., 2002; Hamzei et al., 2003; Roc et al., 2006; Rossini et al., 2004], because of the chronic cerebral perfusion pressure reductions that accompany occlusion. Patients with severe vascular disease can exhibit a low perfusion state known as misery perfusion [Yamauchi et al., 1999]. Misery perfusion is defined as reduced CBF and increased oxygen extraction fraction (as assessed with PET) and is a strong predictor of recurrent stroke [Yamauchi et al., 2012]. This state of chronic hemodynamic compromise is specific to the vascular territory of occluded arteries, resulting in further within‐subject variability in the HR in this patient population. Misery perfusion represents a further case of compromised perfusion in seemingly healthy tissue. Conversely, luxury perfusion, a state of hyperperfusion, may not show the expected HR to neural activity because additional blood flow would not be necessary or triggered by neural activity in this state. Interestingly, cerebrovascular occlusive disease presents a sufficient but not necessary condition for altered HR in stroke patients [Carusone et al., 2002; Hamzei et al., 2003; Roc et al., 2006], with evidence of altered HR in patients in the absence of cerebrovascular occlusion [Krainik et al., 2005; Promjunyakul et al., 2013]. Altered HR in the absence of cerebrovascular occlusive disease indicates further sources of neurovascular decoupling beyond perfusion changes.
Stroke Etiology
There are many different classifications of stroke, but the Oxford Community Stroke Project (OCSP, Bamford or Oxford) and the Trial of ORG 10172 in acute stroke treatment (TOAST) criteria remain the most popular [Adams et al., 1993; Bamford et al., 1991]. Significant differences in natural history have been noted when infarcts are divided into anterior (carotid) circulation infarcts, posterior (vertebrobasilar) circulation infarcts (POCI), and lacunar infarct syndromes [Bamford et al., 1991]. TOAST is a series of 11 definitions to classify patients with ischemic stroke into five major etiologic/pathophysiological subtypes, including atherosclerotic, cardioembolic, lacunar, and other determined or undetermined etiology [Adams et al., 1993; Goldstein et al., 2001; Kolominsky‐Rabas et al., 2001].
These classifications are clinically useful as stroke site determines likely etiology. For example, the majority of POCIs are caused by cardioemboli propagating up the vertebral arteries and lodging in a posterior circulation branch [Caplan et al., 2005]. More rarely, thrombi can form on intracranial vessels, causing POCIs. Anterior circulation events are also likely to be embolic, but can be either due to large artery embolism from a downstream carotid lesion or via cardioembolism, especially in patients with atrial fibrillation.
A cardioembolic stroke usually occurs in the context of previously adequate brain perfusion, causing infarction to occluded branch territories, and therefore, making perfusion changes beyond the stroke site less likely. In contrast, patients with large artery embolic or occlusive disease, especially internal carotid artery stenosis, may have years of preceding waning perfusion into their ipsilateral anterior circulation territory (see Focal damage, global effects section). The resulting effects are complex, but this relative hypoperfusion may in fact be protective, reducing focal ischemic damage via pre‐existing vascular remodeling with recruitment and proliferation of collateral vessels [Kim et al., 2008].This process is termed ischemic preconditioning. There has been no systematic investigation on the effects of ischemic preconditioning on the BOLD response, and while it has been demonstrated unequivocally in animal models, clinical translational evidence has been harder to achieve [Zhao, 2013].
Focal Damage, Global Effects
It is often assumed that disruptions to cerebral hemodynamics will be localized in focal stroke, with these disruptions only affecting the cortex globally in the case of global ischemia or hemorrhagic stroke [Girouard and Iadecola, 2006]. However, as has been outlined, there is accumulating evidence that suggests diffuse altered hemodynamics beyond focal ischemic sites [de Haan et al., 2013] and related to stroke etiology and comorbid vascular pathologies. Richardson et al., [2011] examined cerebral perfusion in patients with left hemisphere cortical strokes using arterial spin labeling (ASL). Cerebral perfusion was reduced across the affected hemisphere, especially in peri‐infarct tissue. Larger infarcts were associated with greater decreases in perfusion. Importantly, these perfusion changes were evident months and years (up to 20) after the initial ischemic stroke, demonstrating that perfusion changes also persist beyond the acute stages of stroke. The relationship between lesion size and perfusion further complicates matters, since lesion volume cannot be easily associated with behavioral or neural activation changes, without acknowledging the added reductions in cerebral perfusion that accompany larger infarcts [Richardson et al., 2011].
Pineiro et al. [2002] examined the evoked BOLD response in sensorimotor cortex in response to a block designed hand and finger‐tapping task in patients with a subcortical lacunar stroke compared to controls. The maximum signal magnitude and rate of rise in both the ipsilesional and contralesional cortical hemispheres was markedly reduced in patients in comparison to age‐matched controls even when behavioral performance was matched between groups. The reduced BOLD signal in cortical hemispheres remote from the subcortical lesion locations may reflect diffuse neurovascular decoupling. This may be either the result of compromised baseline perfusion, which may have predisposed patients to stroke, or vascular changes as the result of stroke. Alternatively, this may reflect compensatory activity, reorganization as a result of stroke, or deafferentation from subcortical regions. Patients with lacunar infarction invariably have pre‐existing, often widespread, small vessel disease. Pineiro et al's [2002] findings may, therefore, reflect vascular changes as the result of widespread small vessel disease. More recently, Makedonov et al. [2013] found that cardiac sampling BOLD metrics were altered in white matter in both normal aging and in the presence of small vessel disease: evidence for how pervasive these microvascular changes are and their impact on BOLD signal.
Recovery and Reorganization
Regardless of stroke location, neuronal reorganization following stroke will alter task evoked cerebral hemodynamics. This is likely to complicate interpretation of neural activity associated with recovery or rehabilitation. Time poststroke is an important factor. Cerebral autoregulation, as indexed by blood pressure and CBF velocity has been shown to be preserved in patients in the acute phase but disrupted in the subacute phase 5–12 days poststroke [Altamura et al., 2009; Reinhard et al., 2005]. The physiological cause of this preserved, and then disrupted, cerebral autoregulation is unclear. The result is a delayed time to peak in the HR, modeled with FIR functions, that is not evident in the acute phase [Altamura et al., 2009]. A number of studies, not specifically looking at recovery or rehabilitation, have demonstrated altered hemodynamics in the chronic stroke phase [Blicher et al., 2012; Richardson et al., 2011] and even after full recovery [Krainik et al., 2005]. This has significant implications for longitudinal studies, or those seeking to evaluate rehabilitative interventions as BOLD signal changes can be misinterpreted as functional recovery or plasticity but may simply reflect hemodynamic changes. To add to these complications, recovery, and plasticity are likely mediated by γ‐aminobutyric acid (GABA) [Blicher et al., 2009, 2014; Swayne et al., 2008], whose activity is associated with brain energy metabolism [Buzsáki et al., 2007; Donahue et al., 2010; Logothetis et al., 2001].
GABA levels, measured with MR spectroscopy (MRS), are inversely related to BOLD signal changes in healthy individuals [Donahue et al., 2010; Muthukumaraswamy et al., 2009]. This is thought to reflect individual differences in the balance between cortical excitation and inhibition, with increases in inhibitory activity reflected in decreased BOLD signal [Donahue et al., 2010; Muthukumaraswamy et al., 2009]. This relationship may explain coupling between energy consumption in the brain and CBF [Logothetis and Wandell, 2004]; with energetically expensive neurotransmitter signaling driving the HR. Individual variability in baseline GABA levels is likely to be exacerbated in stroke patients.
Recently, Blicher et al. [2014] demonstrated reduced GABA levels, as measured with GABA‐edited MRS, in patients 3–12 months after stroke compared to healthy controls. To further complicate matters, GABA levels are not stable after stroke but vary with time poststroke [Swayne et al., 2008] and between healthy and lesioned cortical hemispheres [Liepert, 2006]. Blicher et al. [2014] found a correlation between reduced GABA levels pre to postrehabilitative training and motor improvements. Modulation of GABA levels may facilitate recovery [Blicher et al., 2009, 2013; Swayne et al., 2008]. The dynamic changes in GABA levels complicate interpretation of BOLD signal given its inverse relationship with it. At a group level, if baseline GABA levels are reduced in stroke patients, even at chronic stages [Blicher et al., 2014], compared to healthy controls, BOLD signal changes may not reflect changes in neural activity but alterations in neurotransmitter driven neurovascular coupling [D'Esposito et al., 2003]. Similarly, interpretation of BOLD signal across patient groups or within patients longitudinally may reflect dynamic changes in GABA levels associated with reorganization or recovery as opposed to neural activity changes.
Vascular Reactivity Changes
A number of studies have examined vascular reactivity in patients with cerebrovascular occlusive disease and stroke as a source of neurovascular decoupling [Carusone et al., 2002; Krainik et al., 2005; Rossini et al., 2004]. Vascular reactivity collectively refers to CBF and cerebral blood volume (CBV) and its autoregulation in response to neural activity [Iannetti and Wise, 2007; Jackman and Iadecola, 2014]. Alterations in either direction, that is reduced vascular reactivity or increased compensatory reactivity, may result in false‐negative or false‐positive detection of neural activation, respectively.
Krainik et al. [2005] assessed vascular reactivity using hyperventilation and task‐related BOLD signal change in motor cortex in patients with frontal lobe strokes. Hyperventilation is a simple method of inducing hypocapnia, a state of reduced carbon dioxide levels, leading to cerebral vasoconstriction, reduced CBF and attenuated MR signal. Block task‐related BOLD response in ipsilesional eloquent motor cortex was attenuated in stroke patients and predicted by impaired vascular reactivity. Additionally, vascular reactivity was shown to be heterogeneous across brain regions in both patient and control populations. Importantly, the patient group showed an additional source of variability in reactivity, attenuation of BOLD signal in the ipsilesional hemisphere, beyond the lesion itself and in spite of preserved performance and anatomical integrity of motor regions. These findings are supported by those in patients with cerebral occlusive disease in which event‐related BOLD signal was found to be reduced in both magnitude and latency in the hemisphere ipsilateral to stenosis, and associated with reduced cerebrovascular reactivity as measured with transcranial Doppler (TCD) ultrasonography [Carusone et al., 2002]. Rossini et al. [2004] comprehensively investigated the uncoupling of HR and neural activity in patients with cerebral occlusive disease and a history of stroke. Despite clear magnetoencephalographic signals in response to median nerve stimulation in patients and controls, BOLD response to the same stimulation was absent in some patients in both the diseased and healthy hemisphere. The absence of detectable fMRI activation was not associated with lesion site, WMH load, medication or risk factors but was strongly correlated with cerebrovascular reactivity as assessed by TCD during CO2 inhalation.
Low SNR
Low SNR [Bonakdarpour et al., 2007; Krainik et al., 2005; Murata et al., 2006] and progressive attenuation of the magnitude of BOLD response [Carvalho et al., 2008; Mazzetto‐Betti et al., 2010] may lead to underestimation of neural activity in patients after stroke. As previously discussed, low SNR is already a problem in the detection of neural activity in aging populations [Brodtmann et al., 2003; D'Esposito et al., 1999; Huettel et al., 2001], and this may be compounded in older stroke patients. Bonakdarpour et al. [2007] had previously observed an absence of activation in aphasic patients, despite preserved performance in language tasks. This motivated an analysis of the BOLD time to peak and temporal SNR in patients, in a long‐event related design, to evaluate whether the observed lack of activation reflected changes in neural activation levels or poor detection of the BOLD signal related to changes in the HR. They confirmed reduced SNR and delayed time to peak in the damaged cortex of aphasic stroke patients. Mazzetto‐Betti et al. [2010] examined the stability of the BOLD response in patients with unilateral middle cerebral artery territory stroke performing a motor task across successive blocks. The amplitude of the BOLD signal decreased across successive blocks of the tasks in patients, resulting in an underestimation of BOLD activation. Correcting for this progressive attenuation in modeling the HR resulted in reliable detection of activation [Mazzetto‐Betti et al., 2010].
A multimodal imaging approach used by Blicher et al. [2012], to localize premotor cortex activation in chronic stroke patients, showed BOLD to be highly variable between patients and absent at a group level. They compared CBV‐ and CBF‐weighted imaging with BOLD‐fMRI and showed significant functional activation for all three imaging modalities in primary motor cortex in controls. In patients, BOLD signal responses were absent but CBF and CBV associated activation was comparable to controls (Fig. 2). BOLD‐fMRI, therefore, performed worse in terms of detection of neural activation and there was clear neurovascular decoupling in stroke patients.
Figure 2.
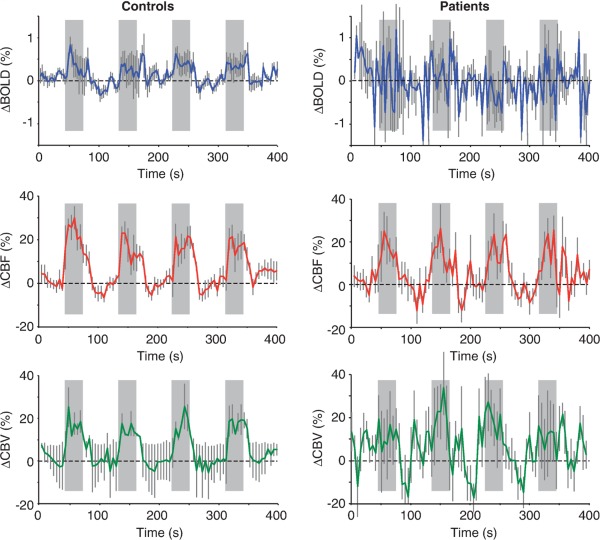
Average blood oxygen level‐dependent (BOLD), cerebral blood flow‐weighted (CBF), and CBV‐weighted (CBV), signal time course in M1 associated with cued hand movements (gray bars). Error bars represent standard error in each group. Reproduced with permission from Blicher et al., J Cereb Blood Flow Metab, 2012, Vol. 32, 2044–2054, © Macmillan Publishers.
BOLD signal may also be underestimated in stroke patients if errors are introduced in the normalization of lesioned brains into a common reference space. The brains of stroke patients are particularly susceptible to poor intersubject registration because of the atypical anatomy that results from structural damage [Crinion et al., 2007]. This can cause misclassification of tissue types in segmentation and normalization procedures developed for use with structurally intact brains [Brett et al., 2001; Rorden et al., 2012]. This presents a further source of variability in HR unrelated to neural activity, one that is potentially greater in stroke patients than in controls.
The risk of stroke increases with age [Feigin et al., 2003] and the stroke populations in imaging studies are typically in their sixties [Rorden et al., 2012]. Despite this, the brains of stroke patients are routinely warped into variations of the montreal neurological institute's, (MNI), brain templates, all of which are based on relatively young brains. For example, MNI 305 is based on the brains of 305 people whose average age is just 23 years (SD 4). Similarly, the newer MNI 152 template, distributed with many popular imaging analysis software packages, is based on brains with an average age of 25 years (SD 5) [Rorden et al., 2012]. The brain changes with age, showing regional changes in volume [Park and Reuter‐Lorenz, 2009; Raz et al., 2005] and cortical thinning [Salat et al., 2004], and therefore, the young template brains are not representative of the brains of average stroke patients [Salat et al., 2004]. Errors in spatial normalization, whether due to atypical structure or warping to inappropriate template brains, can result in misalignment of fMRI activations and consequently an underestimation of fMRI activation at a group level.
IMPROVING BOLD IMAGING OF STROKE POPULATIONS
Reliance on BOLD signal changes as a proxy for neural activity changes is challenged whenever the vascular response deviates from the predicted response to neural activity. The predicted response, captured by the canonical HRF, is based on a healthy vascular system. Yet even within a healthy system, there is significant within and between subject variability [Handwerker et al., 2004; Miezin et al., 2000]. Healthy aging and age‐related vascular pathology add several more sources of variability to the vascular response [D'Esposito et al., 2003; Grady et al., 2006]. The local and global effects of stroke compromise and alter the vascular system compounding the variability already seen in aging. These sources of variability and their effects have been outlined in this review and are summarized in Table 1. Next, we suggest methods to improve BOLD imaging in stroke populations (summarized in Table 1).
Table 1.
Source of variability in the HR and suggestions for improving BOLD imaging
| Sources of variability in the HR | Suggestions to improve BOLD imaging | References |
|---|---|---|
| Variability in all populations | ||
| Individual differences; variability in HR across brain regions or by task parameters | Individualized, empirically derived HRFs | Bonakdarpour et al. [2007]; Handwerker et al. [2004]; Promjunyakul et al. [2013] |
| HR modeling options, e.g., flexible basis sets, multivariate modeling | Aguirre et al. [1998]; Henson et al. [2001]; Lindquist et al. [2009] | |
| Additional age‐related variability | ||
| Signal‐to‐noise ratio (SNR) | Multimodal imaging; Assessment and correction for low SNR; Improved intersubject registration, BOLD scaling by hypercapnia challenge or breath‐holding | Blicher et al. [2012]; Crinion et al. [2007]; Gauthier et al. [2013]; Kannurpatti et al. [2010]; Mazzetto‐Betti et al. [2010]; Thomason et al. [2007] |
| Vascular pathologies | Assessment of vascular status and comorbidities | Carusone et al. [2002] |
| Variability in stroke populations | ||
| Baseline perfusion changes | Assessment of baseline perfusion: ASL or BOLD response to vascular challenge; normalization with event‐related or rsfMRI; CBV weighted MRI, e.g., Vascular space occupancy imaging | Bangen et al. [2009]; Blicher et al. [2012]; Buxton et al. [2004]; Iannetti and Wise [2007]; Liu [2013]; Richardson et al. [2011] |
| Cerebrovascular reactivity | TCD, BOLD response to vascular challenge | Carusone et al. [2002]; Liu [2013]; Rossini et al. [2004] |
| Stroke location and extent | Assessment of baseline perfusion and vascular status | Richardson et al. [2011] |
| Intersubject registration errors | Lesion masking; age‐specific normalization templates | Andersen et al. [2010]; Brett et al. [2001]; Rorden et al. [2012] |
| Diaschisis and functional reorganization | Resting‐state derived functional connectivity analysis, Baseline GABA measurement | Blicher et al. [2014]; Carter et al. [2010]; Chen and Schlaug [2013]; Grefkes and Fink [2011]; Park et al. [2011]; Rehme and Grefkes [2013] |
As well as the variability in the HR seen across all populations, stroke patients have additional sources of variability associated with normal aging, vascular pathologies, and variability directly resulting from damage due to stroke. References refer to articles demonstrating improved functional imaging with suggested methods either explicitly, or as a consequence of the results obtained and conclusions drawn.
Within‐Subject Characterization of the HRF
To account for the variability of the BOLD signal and improve within‐subject estimation of the HRF to neural activity, individualized HRFs can be estimated from an additional, independent functional scan and applied to the statistical modeling of the experimental paradigm [Aguirre et al., 1998]. Handwerker et al., [2004] evaluated this method and found improved model fitting and detection of neural activation with individualized HR estimates in healthy subjects. The benefit of individualized HRFs was somewhat limited by within‐subject variation in the HR across brain regions. Nevertheless, between‐subject variation in HR was larger than within‐subject variation, validating the superiority of individualized HRFs over the canonical HRF, especially in the context of group level statistics. Misestimating the onset time of the HRF by as little as 1 s affected statistical sensitivity, resulting in false negatives in their idealized, simulated data. Factoring in head motion and other sources of noise in real data, HRF misestimates are likely to lead to even greater underestimates of activation. Since fMRI studies in stroke populations often seek to compare neural activations against controls on a group level, this has direct applicability to the interpretation of studies in which the canonical HRF is applied across populations with diverse vascular pathologies.
Within‐subject characterization of the HRF has the potential to more accurately model neural activity in stroke patients when the canonical HRF may not reflect the neurovascular coupling assumed in healthy brains and where the spatiotemporal profile of the HR may vary within stroke patient populations, as discussed above [Bonakdarpour et al., 2007; Handwerker et al., 2004; Mazzetto‐Betti et al., 2010; Newton et al., 2002]. Modeling with the canonical HRF, Bonakdarpour et al. [2007] failed to detect significant activation in one patient, but using the patient's own HRF, they increased sensitivity to activation. An additional 6.5 min of scan time has the potential to increase detection of activation and better model neural activity in stroke patients [Handwerker et al., 2004]. Of course, the additional functional scans needed to estimate individualized HRFs may not be practical in some populations, where scanning time needs to be limited to what patients can comfortably tolerate.
Promjunyakul et al., [2013] evaluated the relative benefit of estimating individualized HRFs in patients with cortical and subcortical strokes and chronic poststroke hemiparesis. They used an event‐related design to estimate individualized HRFs from sensorimotor cortex in a lower limb movement task and compared results obtained in an independent block‐design task when these individualized HRFs were used compared to when a canonical HRF was used in the analysis. While the use of individualized HRFs as opposed to the canonical HRF did not improve detection of task‐related BOLD activity in their stroke patients, there were differences in the spatiotemporal profile of the HR depending on stroke location. Notably, the patients in Promjunyakul et al.'s [2013] study showed no evidence of cerebrovascular occlusive disease, a significant contributor to abnormal HR seen in stroke patients [Bonakdarpour et al., 2007; Carusone et al., 2002; Rossini et al., 2004]. The limited variability in the HR in their patient population precluded them from providing a rule of thumb as to the threshold at which the HR is significantly abnormal to be more effectively modeled with individualized HRFs. The authors advised examining the spatiotemporal profile of the HR obtained in stroke populations because of variability within populations and likely alterations in neurovascular coupling. They also caution the application of the canonical HRF to stroke populations with significant cerebrovascular disease.
An assessment of the likely vascular status of the population will help to determine whether the additional scanning time will significantly improve estimation of neural activation. Use of an empirical HRF, derived from a separate 6.5‐min scan, improves estimation more than an additional 6.5 min spent collecting experimental data and fitting it to the canonical HRF [Handwerker et al., 2004]. If stroke populations are heterogeneous, or if the strokes are accompanied by known cerebrovascular occlusive disease, this would be highly recommended.
Recognizing and Correcting for Altered Baseline Perfusion or Cerebrovascular Reactivity
Hypoperfusion is a cardinal feature of acute stroke [Chalela et al., 2000]. It has also been demonstrated more recently in perilesional and healthy cortex in chronic stroke patients, even months after their infarct [de Haan et al., 2013; Richardson et al., 2011]. Characterization of baseline perfusion can aid interpretation of BOLD signal changes in stroke patients. Baseline perfusion can be directly assessed with ASL; an MR sequence sensitive to the magnetization of water in arterial blood from which blood flow can be estimated. ASL is noninvasive, unlike traditional perfusion measures, which rely on the injection of contrast agents. This is a major advantage for patient populations in which invasive procedures are difficult to implement, either for practical reasons or due to contraindications to gadolinium‐based contrast agents, such as renal impairment [Hellman, 2011]. ASL is, therefore, also suitable for longitudinal scanning protocols that require repeated scan sessions, ideal for stroke recovery studies. ASL requires an additional 5–10 min of scanning time, which may not be feasible in all protocols, but in some contexts may be mandatory for interpretation of BOLD signal changes, or even preferable to BOLD‐weighted fMRI entirely. Blicher et al. [2012] found CBF, as measured with ASL, to be a better index of motor cortex activity than BOLD signal in chronic stroke patients (Fig. 2). The authors recommend multimodal imaging because individual differences in the hemodynamic status of their stroke patients meant no single imaging technique was sensitive to neural activity in all patients.
Bangen et al. [2009] found reduced CBF levels in the resting state in healthy older adults. ASL revealed an increased CBF response to a picture‐encoding task, for equivalent performance, in older as compared to younger adults. This was not reflected in the BOLD signal, showing that vital additional information could be gained from ASL in situations of altered neurovascular coupling. Without this information, the neural activity, estimated from BOLD signal changes, would have been assumed to be preserved in older populations. The quantification of baseline perfusion can be used to assess regional hemodynamic compromise as well as provide a covariate in group level analyses where differences in baseline perfusion levels exist within or between groups [Buxton et al., 2004; Iannetti and Wise, 2007].
ASL is not without limitations. Early methods suffered a low SNR. This has been improved by continuous ASL, whose long radiofrequency labeling pulse sequences improved signal detection, but required specialized hardware. Recently, pseudocontinuous ASL has provided improved signal‐to‐noise with the benefit of being easily implemented on standard MRI scanners [Chen et al., 2011b; Wu et al., 2007]. Early issues surrounding signal loss in brain regions far from the reference labeling site have been overcome with technical advances to ASL [Donahue et al., 2012]. Donahue et al., [2012] provide a comprehensive review of the use of ASL in cerebrovascular disease, including advances that have seen new ASL sequences providing CBF estimates on par with the gold standard positron emission tomography (PET) and single‐photon emission computed tomography (SPECT) imaging.
In healthy subjects, modulations of CBF through hypocapnia and hypercapnia have been shown to influence the magnitude and temporal characteristics of the BOLD response in primary visual cortex [Cohen et al., 2002], a region in which the canonical HRF is thought to be representative of the true HR [Miezin et al., 2000]. The BOLD response to these vascular challenges can be used to quantify regional and global baseline cerebrovascular reactivity. The BOLD response to vascular challenge can also be used to normalize the event‐related BOLD response [Liu, 2013]. The advantage of this technique is its ease of implementation, especially in cases where multimodal imaging is not feasible.
Processing Pipelines Specific for Stroke Patients
Analysis pipelines and templates designed to allow normalization of individual brains to standard stereotaxic space are not always optimized for the aging and structurally damaged brains of stroke patients. Crinion et al. [2007] found improved task‐related activation, modeled with a canonical HRF, associated with improved intersubject registration. Creating a mask of the lesion can improve results by aiding normalization algorithms and segmentation and classification of cerebral tissue types. A significant hurdle to masking the lesion before normalization is the time it takes to create a lesion mask for each patient. A precise mask can take around 8 h to trace, adding a significant burden to the analysis process, especially for large cohort studies. However, even roughly drawn masks, taking only 30 min to trace, can significantly improve normalization [Andersen et al., 2010; Brett et al., 2001]. Not masking lesions can result in underestimation of the lesion volume and misclassification of perilesional tissue, particularly so for large infarcts. The underestimation of lesion volume can be clearly seen in Figure 3 in four representative brains normalized with and without lesion masking [Andersen et al., 2010].
Figure 3.
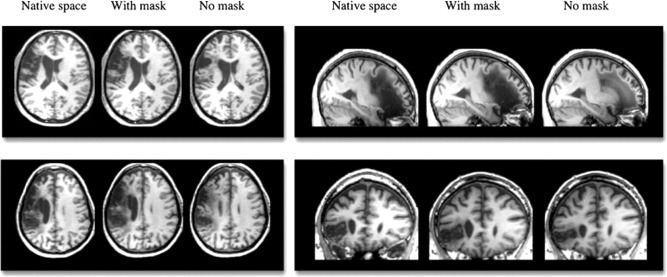
Four representative brains, all with left hemisphere lesions, shown in native space compared to after normalization with and without cost function masking. Reproduced with permission from Andersen et al., Neuroimage, 2010, Vol. 53, 78–84, © Elsevier.
If manual masking is not feasible, automated methods are available for lesion identification within normalization routines, such as automatic lesion identification or diffeomorphic anatomical registration using exponentiated lie algebra [Ashburner, 2007; Seghier et al., 2008]. Ripollés et al., [2012] carried out a comprehensive review of 10 normalization routines, with varying degrees of manual and automatic lesion delineation, and found performance varied significantly across routines, with no single method outperforming on all aspects tested. Importantly, while some automated methods performed very well, they were liable to underestimate lesion volume. If lesion volume is central to the hypotheses of the study, the costs of underestimating lesion volume are significant. Cost‐function masking with unified segmentation provided a good trade‐off, with superior performance in stroke patients and in lesion volume estimation [Ripollés et al., 2012]. The initial investment in masking the lesion is worthwhile, particularly since rough masking is sufficient, given the potential for improved detection and localization of activation.
Normalization to a common reference space can also be improved by using age appropriate templates. Many imaging analysis software suites, including SPM8 and FSL, include functions for creating templates from subjects or patients specific to your study. Rorden et al. [2012] created stroke age‐appropriate computerized axial tomography and MRI templates and found improved normalization performance with these templates. The use of study‐specific templates does not allow reporting of localization of activation in terms of stereotaxic space. However, the advantages of using a common reference space are invalidated if the brains of the patient group have been erroneously warped, tissue has been misclassified or the template is not representative of the patient group.
Analytical Methods Accounting for Variable HR
Brain imaging software packages now routinely come with analysis options that make it easier to move beyond modeling with the canonical HRF. Reducing the number of fixed parameters in estimating the HR allows for more flexibility in the model to account for variability within and across subjects. For example, SPM allows the inclusion of temporal and dispersion derivatives that allow some flexibility in the time to peak and width of the canonical HRF. However, the 1 s of variability around the canonical HRF peak that the temporal derivative allows may not be sufficient to account for the changes in the HR seen in some stroke patients. For example, Bonakdarpour et al. [2007] reported a significantly delayed time‐to‐peak of 16.5 s in a subset of their patients.
Further flexibility in estimating the HR can be obtained by specifying basis functions, such as a set of sinusoidal functions [Bullmore et al., 1996], with assumptions or constraints on the shape of the response [Harms and Melcher, 2003]. FIR [Goutte et al., 2000] offer even greater flexibility by making no assumptions about the shape of HR, estimating the parameters of the HRF at every time point. These options are readily available in most brain imaging software packages. A compromise between the increased power of BOLD signal detection from using non‐canonical basis functions and the flexibility but lack of specificity of FIR models may be in the use of Bayesian techniques implementing anatomically or data driven constraints on the shape of the HR [Gössl et al., 2001; Marrelec et al., 2003; Woolrich, 2012].
Model free analyses, including independent components analysis, partial least squares, and other multivariate approaches such as multivoxel pattern analysis and representational similarity analysis, are less dependent of the specific shape of the HR [Kriegeskorte et al., 2006; McIntosh and Lobaugh, 2004; Mourão‐Miranda et al., 2006; Mur et al., 2009]. The parameter of interest is no longer the amplitude of the response, as is typically the case in univariate analyses, but rather the spatiotemporal pattern of activation across voxels. A detailed analysis of the pros and cons of these methods is beyond the scope of this review and the choice of method will depend on the questions the researcher is seeking to answer.
RESTING‐STATE BOLD IMAGING: EXPLORING FUNCTIONAL CONNECTIVITY
rsfMRI presents a special case of BOLD‐fMRI that is not reliant on evoked BOLD signals, nor modeled with the canonical HRF. Remarkably stable and reliable functional networks emerge when subjects are scanned in the absence of task demands, in the “resting‐state” [Biswal et al., 2010; Shehzad et al., 2009]. These networks are often defined by temporal correlations in spontaneous low‐frequency BOLD signal fluctuations between brain regions, and they are highly reproducible and consistent [Damoiseaux et al., 2006; Zuo et al., 2010]. This consistency has been demonstrated in large group studies, including across over 1,414 subjects and 35 centers [Biswal et al., 2010]. They have also been shown to closely correspond to task‐based functional networks [Smith et al., 2009].
One of the key challenges in scanning stroke patients is in designing tasks that patients are capable of accomplishing [Price and Friston, 2002; Price et al., 2006]. The functions of a focally damaged region cannot be directly assessed and researchers need to account for alternative performance strategies or neural processing pathways. The selection of patients to task‐based studies can also be relatively arbitrary. Assigning patients based on symptoms, such as motor or visual deficits, is complicated by the heterogeneous collections of symptoms seen in stroke cohorts. Resting‐state derived connectivity analysis is, therefore, particularly useful in the context of fMRI in stroke patients, as it overcomes the need to equalize task performance across populations. There are no performance demands on patients and multiple functional networks can be examined simultaneously [Greicius et al., 2012].
Connectivity analyses have revealed encouraging and replicable evidence of network disruptions in clinical disorders including Alzheimer's disease [Greicius et al., 2009; Wang et al., 2007; Zhang and Raichle, 2010], depression [Seminowicz et al., 2004], and schizophrenia [Lynall et al., 2010]. Diagnostic utility has also been demonstrated in Alzheimer's disease [Li et al., 2002]. This, combined with the wealth of evidence that the effects of stroke are not confined to the site of injury, suggests connectivity analysis may be a promising avenue for research into the effects of, and recovery from, stroke [Carter et al., 2010; Chen and Schlaug, 2013; Grefkes and Fink, 2011, 2014; Park et al., 2011; Rehme and Grefkes, 2013].
He et al. [2007] reported disruptions to functional networks associated with poststroke neglect, in both the lesion‐disrupted ventral attentional network and the lesion‐free dorsal attentional network. Similarly, Carter et al. [2010] demonstrated that disruption to interhemispheric functional connectivity in attentional and somatosensory networks was associated with reduced behavioral performance in tasks specific to each network that were completed outside the scanner. This emphasizes the behavioral significance of disrupted network connectivity without the need for task‐related fMRI [Carter et al., 2010].
Functional connectivity analyses rely on the temporal correlation of BOLD signal in spatially independent regions in the brain. The tight relationship between BOLD and CBF [Tak et al., 2014; Viviani et al., 2011] and BOLD and CMRO2 [Fukunaga et al., 2008; Wu et al., 2009], suggest these correlated spontaneous signal fluctuations reflect neuronal activity and not just hemodynamic oscillations or vascular drainage [Fukunaga et al., 2008; Wu et al., 2009]. Although this lends weight to the use of functional connectivity methods for assessing network‐based neural activity, it also means these methods are not immune to issues associated with abnormal neurovascular coupling in stroke patients. As with task‐related activity, the concern is that BOLD signal changes will be taken to reflect changes in neural activity, in this case, in functional networks, but may simply reflect alterations in neurovascular coupling.
Research into the effect of neurovascular decoupling on resting‐state connectivity analyses is in its infancy. Initial results suggest characterization of baseline perfusion levels, for example, through hypercapnia, can aid interpretation of connectivity changes [Liu, 2013]. As with event‐related fMRI, care should be taken in the analysis of rsfMRI in stroke populations. Spurious correlations in low frequency signal fluctuations result from excessive movement, which may be greater in stroke populations [Power et al., 2012], and from physiological noise as the result of cardiovascular processes [Chang and Glover, 2009]. Low SNR, as is evident in event‐related fMRI in aging populations, may lead to false negatives. Liu [2013] recently reviewed approaches to assessing and correcting alterations to neurovascular coupling which threaten interpretation of rsfMRI in clinical populations. As with event‐related fMRI, multimodal imaging can improve interpretation of rsfMRI in clinical populations. Where this is not feasible, careful characterization of the hemodynamic status of the clinical population is critical, at least until we have a better understanding of the mechanisms underlying rsfMRI. Nevertheless, because multiple networks can be examined simultaneously in resting‐state imaging, and patients are not selected based on specific functional deficits or performance criteria, rsfMRI can be useful in exploring network disruption and recovery after stroke.
CONCLUSIONS
The proliferation of BOLD‐fMRI as a research tool over the last two decades belies the fact that there remains uncertainty about the precise mechanisms that relate the BOLD signal to neural activity. Inferring neural activation changes based on stimulus evoked BOLD signal changes is particularly problematic given several sources of neurovascular decoupling in the aging, structurally atypical and vascularly compromised brains of stroke patients. Several methods exist to improve the characterization of the HR in stroke patients including estimation of baseline perfusion, assessment of vascular reactivity, use of individualized HRFs and multimodal imaging protocols. At the very least, cognizance of the network wide impact of stroke on the vascular system will aid interpretation of the BOLD signal in stroke populations.
REFERENCES
- Adams HP, Bendixen BH, Kappelle LJ, Biller J, Love BB, Gordon DL, Marsh EE (1993): Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 24:35–41. [DOI] [PubMed] [Google Scholar]
- Aguirre GK, Zarahn E, D'Esposito M (1998): The variability of human, BOLD hemodynamic responses. Neuroimage 8:360–369. [DOI] [PubMed] [Google Scholar]
- Altamura C, Reinhard M, Vry M‐S, Kaller CP, Hamzei F, Vernieri F, Rossini PM, Hetzel A, Weiller C, Saur D (2009): The longitudinal changes of BOLD response and cerebral hemodynamics from acute to subacute stroke. A fMRI and TCD study. BMC Neurosci 10:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen SM, Rapcsak SZ, Beeson PM (2010): Cost function masking during normalization of brains with focal lesions: Still a necessity? Neuroimage 53:78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner J (2007): A fast diffeomorphic image registration algorithm. Neuroimage 38:95–113. [DOI] [PubMed] [Google Scholar]
- Bamford J, Sandercock P, Dennis M, Warlow C, Burn J (1991): Classification and natural history of clinically identifiable subtypes of cerebral infarction. Lancet 337:1521–1526. [DOI] [PubMed] [Google Scholar]
- Bangen KJ, Restom K, Liu TT, Jak AJ, Wierenga CE, Salmon DP, Bondi MW (2009): Differential age effects on cerebral blood flow and BOLD response to encoding: Associations with cognition and stroke risk. Neurobiol Aging 30:1276–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswal BB, Mennes M, Zuo X‐N, Gohel S, Kelly C, Smith SM, Beckmann CF, Adelstein JS, Buckner RL, Colcombe S, Dogonowski A‐M, Ernst M, Fair D, Hampson M, Hoptman MJ, Hyde JS, Kiviniemi VJ, Kötter R, Li S‐J, Lin C‐P, Lowe MJ, Mackay C, Madden DJ, Madsen KH, Margulies DS, Mayberg HS, McMahon K, Monk CS, Mostofsky SH, Nagel BJ, Pekar JJ, Peltier SJ, Petersen SE, Riedl V, Rombouts SARB, Rypma B, Schlaggar BL, Schmidt S, Seidler RD, Siegle GJ, Sorg C, Teng G‐J, Veijola J, Villringer A, Walter M, Wang L, Weng X‐C, Whitfield‐Gabrieli S, Williamson P, Windischberger C, Zang Y‐F, Zhang H‐Y, Castellanos FX, Milham MP (2010): Toward discovery science of human brain function. Proc Natl Acad Sci USA 107:4734–4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonakdarpour B, Parrish TB, Thompson CK (2007): Hemodynamic response function in patients with stroke‐induced aphasia: Implications for fMRI data analysis. NeuroImage. 36:322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blicher JU, Jakobsen J, Andersen G, Nielsen JF (2009): Cortical excitability in chronic stroke and modulation by training: A TMS study. Neurorehabil Neural Repair 23:486–493. [DOI] [PubMed] [Google Scholar]
- Blicher JU, Stagg CJ, O'Shea J, Østergaard L, MacIntosh BJ, Johansen‐Berg H, Jezzard P, Donahue MJ (2012): Visualization of altered neurovascular coupling in chronic stroke patients using multimodal functional MRI. J Cereb Blood Flow Metab 32:2044–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blicher JU, Near J, Næss‐Schmidt E, Østergaard L, Johansen‐Berg H, Ho Y‐CL (2013): Intracortical GABA is decreased after stroke and further suppressed with successful rehabilitation. J Neurol Sci 333:e561–e562. [Google Scholar]
- Blicher JU, Near J, Næss‐Schmidt E, Stagg CJ, Johansen‐Berg H, Nielsen JF, Ostergaard L, Ho Y‐CL (2014): GABA levels are decreased after stroke and GABA changes during rehabilitation correlate with motor improvement. Neurorehabil Neural Repair. doi: 10.1177/1545968314543652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett M, Leff AP, Rorden C, Ashburner J (2001): Spatial normalization of brain images with focal lesions using cost function masking. Neuroimage 14:486–500. [DOI] [PubMed] [Google Scholar]
- Brodtmann A, Puce A, Syngeniotis A, Darby D, Donnan G (2003): The functional magnetic resonance imaging hemodynamic response to faces remains stable until the ninth decade. Neuroimage 20:520–528. [DOI] [PubMed] [Google Scholar]
- Brown WR, Thore CR (2011): Review: Cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol Appl Neurobiol 37:56–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullmore E, Brammer M, Williams SC, Rabe‐Hesketh S, Janot N, David A, Mellers J, Howard R, Sham P (1996): Statistical methods of estimation and inference for functional MR image analysis. Magn Reson Med 35:261–277. [DOI] [PubMed] [Google Scholar]
- Buxton RB, Uludağ K, Dubowitz DJ, Liu TT (2004): Modeling the hemodynamic response to brain activation. Neuroimage 23 Suppl 1:S220–S233. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Kaila K, Raichle M (2007): Inhibition and brain work. Neuron 56:771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabeza R (2002): Hemispheric asymmetry reduction in older adults: The HAROLD model. Psychol Aging 17:85–100. [DOI] [PubMed] [Google Scholar]
- Cabeza R, Anderson ND, Locantore JK, McIntosh AR (2002): Aging gracefully: Compensatory brain activity in high‐performing older adults. Neuroimage 17:1394–1402. [DOI] [PubMed] [Google Scholar]
- Cabeza R, Daselaar SM, Dolcos F, Prince SE, Budde M, Nyberg L (2004): Task‐independent and Task‐specific age effects on brain activity during working memory, visual attention and episodic retrieval. Cereb Cortex 14:364–375. [DOI] [PubMed] [Google Scholar]
- Caplan L, Chung C‐S, Wityk R, Glass T, Tapia J, Pazdera L, Chang H‐M, Dashe J, Chaves C, Vemmos K, Leary M, Dewitt L, Pessin M (2005): New England medical center posterior circulation stroke registry: I. Methods, data base, distribution of brain lesions, stroke mechanisms, and outcomes. J Clin Neurol 1:14–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AR, Astafiev SV, Lang CE, Connor LT, Rengachary J, Strube MJ, Pope DLW, Shulman GL, Corbetta M (2010): Resting interhemispheric functional magnetic resonance imaging connectivity predicts performance after stroke. Ann Neurol 67:365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carusone LM, Srinivasan J, Gitelman DR, Mesulam MM, Parrish TB (2002): Hemodynamic response changes in cerebrovascular disease: implications for functional MR imaging. AJNR Am J Neuroradiol 23:1222–1228. [PMC free article] [PubMed] [Google Scholar]
- Carvalho SMF, Pontes‐Neto OM, Fabio SRC, Leite JP, Santos AC, de Araujo DB (2008): Rapid BOLD fMRI signal loss in the primary motor cortex of a stroke patient. Arq Neuropsiquiatr 66:885–887. [DOI] [PubMed] [Google Scholar]
- Chalela JA, Alsop DC, Gonzalez‐Atavales JB, Maldjian JA, Kasner SE, Detre JA (2000): Magnetic resonance perfusion imaging in acute ischemic stroke using continuous arterial spin labeling. Stroke 31:680–687. [DOI] [PubMed] [Google Scholar]
- Chang C, Glover GH (2009): Relationship between respiration, end‐tidal CO2, and BOLD signals in resting‐state fMRI. Neuroimage 47:1381–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JL, Schlaug G (2013): Resting state interhemispheric motor connectivity and white matter integrity correlate with motor impairment in chronic stroke. Front Neurol 4:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, Rosas HD, Salat DH (2011a): Age‐associated reductions in cerebral blood flow are independent from regional atrophy. Neuroimage 55:468–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wang DJJ, Detre JA (2011b): Test‐retest reliability of arterial spin labeling with common labeling strategies. J Magn Reson Imaging 33:940–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen P, Larsson HB, Thomsen C, Wieslander SB, Henriksen O (1994): Age dependent white matter lesions and brain volume changes in healthy volunteers. Acta Radiol 35:117–122. [PubMed] [Google Scholar]
- Cohen ER, Ugurbil K, Kim S‐G (2002): Effect of basal conditions on the magnitude and dynamics of the blood oxygenation level‐dependent fMRI response. J Cereb Blood Flow Metab 22:1042–1053. [DOI] [PubMed] [Google Scholar]
- Crinion J, Ashburner J, Leff A, Brett M, Price C, Friston K (2007): Spatial normalization of lesioned brains: Performance evaluation and impact on fMRI analyses. Neuroimage 37:866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Esposito M, Zarahn E, Aguirre GK, Rypma B (1999): The effect of normal aging on the coupling of neural activity to the bold hemodynamic response. Neuroimage 10:6–14. [DOI] [PubMed] [Google Scholar]
- D'Esposito M, Deouell LY, Gazzaley A (2003): Alterations in the BOLD fMRI signal with ageing and disease: A challenge for neuroimaging. Nat Rev Neurosci 4:863–872. [DOI] [PubMed] [Google Scholar]
- Damoiseaux JS, Rombouts SARB, Barkhof F, Scheltens P, Stam CJ, Smith SM, Beckmann CF (2006): Consistent resting‐state networks across healthy subjects. Proc Natl Acad Sci USA 103:13848–13853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debette S, Markus HS (2010): The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: Systematic review and meta‐analysis. BMJ 341:c3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue MJ, Near J, Blicher JU, Jezzard P (2010): Baseline GABA concentration and fMRI response. Neuroimage 53:392–398. [DOI] [PubMed] [Google Scholar]
- Donahue MJ, Strother MK, Hendrikse J (2012): Novel MRI approaches for assessing cerebral hemodynamics in ischemic cerebrovascular disease. Stroke 43:903–915. [DOI] [PubMed] [Google Scholar]
- Fazekas F, Kleinert R, Offenbacher H, Schmidt R, Kleinert G, Payer F, Radner H, Lechner H (1993): Pathologic correlates of incidental MRI white matter signal hyperintensities. Neurology 43:1683–1683. [DOI] [PubMed] [Google Scholar]
- Feigin VL, Lawes CM, Bennett DA, Anderson CS (2003): Stroke epidemiology: A review of population‐based studies of incidence, prevalence, and case‐fatality in the late 20th century. Lancet Neurol 2:43–53. [DOI] [PubMed] [Google Scholar]
- Fukunaga M, Horovitz SG, de Zwart JA, van Gelderen P, Balkin TJ, Braun AR, Duyn JH (2008): Metabolic origin of BOLD signal fluctuations in the absence of stimuli. J Cereb Blood Flow Metab 28:1377–1387. [DOI] [PubMed] [Google Scholar]
- Gauthier CJ, Madjar C, Desjardins‐Crépeau L, Bellec P, Bherer L, Hoge RD (2013): Age dependence of hemodynamic response characteristics in human functional magnetic resonance imaging. Neurobiol Aging 34:1469–1485. [DOI] [PubMed] [Google Scholar]
- Girouard H, Iadecola C (2006): Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol 100:328–335. [DOI] [PubMed] [Google Scholar]
- Glover GH (1999): Deconvolution of impulse response in event‐related BOLD fMRI1. NeuroImage 9:416–429. doi: 10.1006/nimg.1998.0419. [DOI] [PubMed] [Google Scholar]
- Goldstein LB, Jones MR, Matchar DB, Edwards LJ, Hoff J, Chilukuri V, Armstrong SB, Horner RD (2001): Improving the reliability of stroke subgroup classification using the Trial of ORG 10172 in Acute Stroke Treatment (TOAST) criteria. Stroke 32:1091–1098. [DOI] [PubMed] [Google Scholar]
- Gössl C, Fahrmeir L, Auer DP (2001): Bayesian modeling of the hemodynamic response function in BOLD fMRI. Neuroimage 14:140–148. [DOI] [PubMed] [Google Scholar]
- Goutte C, Nielsen FA, Hansen LK (2000): Modeling the haemodynamic response in fMRI using smooth FIR filters. IEEE Trans Med Imaging 19:1188–1201. [DOI] [PubMed] [Google Scholar]
- Grady C (2012): The cognitive neuroscience of ageing. Nat Rev Neurosci 13:491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady CL, Garrett DD (2013): Understanding variability in the BOLD signal and why it matters for aging. Brain Imaging Behav 8:274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady CL, Springer M V., Hongwanishkul D, McIntosh AR, Winocur G (2006): Age‐related changes in brain activity across the adult lifespan. J Cogn Neurosci 18:227–241. [DOI] [PubMed] [Google Scholar]
- Grefkes C, Fink GR (2011): Reorganization of cerebral networks after stroke: New insights from neuroimaging with connectivity approaches. Brain 134:1264–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grefkes C, Fink GR (2014): Connectivity‐based approaches in stroke and recovery of function. Lancet Neurol 13:206–216. [DOI] [PubMed] [Google Scholar]
- Greicius MD, Supekar K, Menon V, Dougherty RF (2009): Resting‐state functional connectivity reflects structural connectivity in the default mode network. Cereb Cortex 19:72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greicius M, Seeley W, Carter AR, Shulman GL, Corbetta M (2012): Why use a connectivity‐based approach to study stroke and recovery of function? Neuroimage 62:2271–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan B, Rorden C, Karnath H‐O (2013): Abnormal perilesional BOLD signal is not correlated with stroke patients' behavior. Front Hum Neurosci 7:669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamzei F, Knab R, Weiller C, Röther J (2003): The influence of extra‐ and intracranial artery disease on the BOLD signal in FMRI. Neuroimage 20:1393–1399. [DOI] [PubMed] [Google Scholar]
- Handwerker DA, Ollinger JM, D'Esposito M (2004): Variation of BOLD hemodynamic responses across subjects and brain regions and their effects on statistical analyses. Neuroimage 21:1639–1651. [DOI] [PubMed] [Google Scholar]
- Handwerker DA, Gonzalez‐Castillo J, D'Esposito M, Bandettini PA (2012): The continuing challenge of understanding and modeling hemodynamic variation in fMRI. Neuroimage 62:1017–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms MP, Melcher JR (2003): Detection and quantification of a wide range of fMRI temporal responses using a physiologically‐motivated basis set. Hum Brain Mapp 20:168–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison R V. (2002): Blood capillary distribution correlates with hemodynamic‐based functional imaging in cerebral cortex. Cereb Cortex 12:225–233. [DOI] [PubMed] [Google Scholar]
- Hatazawa J, Shimosegawa E, Satoh T, Toyoshima H, Okudera T (1997): Subcortical hypoperfusion associated with asymptomatic white matter lesions on magnetic resonance imaging. Stroke 28:1944–1947. [DOI] [PubMed] [Google Scholar]
- He BJ, Snyder AZ, Vincent JL, Epstein A, Shulman GL, Corbetta M (2007): Breakdown of functional connectivity in frontoparietal networks underlies behavioral deficits in spatial neglect. Neuron 53:905–918. [DOI] [PubMed] [Google Scholar]
- Heeger DJ, Ress D (2002): What does fMRI tell us about neuronal activity? Nat Rev Neurosci 3:142–151. [DOI] [PubMed] [Google Scholar]
- Hellman RN (2011): Gadolinium‐induced nephrogenic systemic fibrosis. Semin Nephrol 31:310–316. [DOI] [PubMed] [Google Scholar]
- Henson R, Rugg MD, Friston KJ (2001): The choice of basis functions in event‐related fMRI. Neuroimage 13:149. [Google Scholar]
- Heuninckx S, Wenderoth N, Swinnen SP (2008): Systems neuroplasticity in the aging brain: Recruiting additional neural resources for successful motor performance in elderly persons. J Neurosci 28:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettel SA, McCarthy G (2000): Evidence for a refractory period in the hemodynamic response to visual stimuli as measured by MRI. Neuroimage 11:547–553. [DOI] [PubMed] [Google Scholar]
- Huettel SA, Singerman JD, McCarthy G (2001): The effects of aging upon the hemodynamic response measured by functional MRI. Neuroimage 13:161–175. [DOI] [PubMed] [Google Scholar]
- Iannetti GD, Wise RG (2007): BOLD functional MRI in disease and pharmacological studies: Room for improvement? Magn Reson Imaging 25:978–988. [DOI] [PubMed] [Google Scholar]
- Jackman K, Iadecola C (2014): Neurovascular regulation in the ischemic brain. Antioxid Redox Signal (in press). doi: 10.1089/ars.2013.5669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannurpatti SS, Motes MA, Rypma B, Biswal BB (2010): Neural and vascular variability and the fMRI‐BOLD response in normal aging. Magn Reson Imaging 28:466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannurpatti SS, Motes MA, Rypma B, Biswal BB (2011): Increasing measurement accuracy of age‐related BOLD signal change: Minimizing vascular contributions by resting‐state‐fluctuation‐of‐amplitude scaling. Hum Brain Mapp 32:1125–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura J, Terayama Y, Takashima S, Obara K, Pavol MA, Meyer JS, Mortel KF, Weathers S (1993): Leuko‐araiosis and cerebral perfusion in normal aging. Exp Aging Res 19:225–240. [DOI] [PubMed] [Google Scholar]
- Kim SH, Kim EH, Lee BI, Heo JH (2008): Chronic cerebral hypoperfusion protects against acute focal ischemia, improves motor function, and results in vascular remodeling. Curr Neurovasc Res 5:28–36. [DOI] [PubMed] [Google Scholar]
- Kobari M, Meyer JS, Ichijo M (1990): Leuko‐araiosis, cerebral atrophy, and cerebral perfusion in normal aging. Arch Neurol 47:161–165. [DOI] [PubMed] [Google Scholar]
- Kolominsky‐Rabas PL, Weber M, Gefeller O, Neundoerfer B, Heuschmann PU (2001): Epidemiology of ischemic stroke subtypes according to TOAST criteria: Incidence, recurrence, and long‐term survival in ischemic stroke subtypes: A population‐based study. Stroke 32:2735–2740. [DOI] [PubMed] [Google Scholar]
- Krainik A, Hund‐Georgiadis M, Zysset S, von Cramon DY (2005): Regional impairment of cerebrovascular reactivity and BOLD signal in adults after stroke. Stroke 36:1146–1152. [DOI] [PubMed] [Google Scholar]
- Krejza J, Mariak Z, Walecki J, Szydlik P, Lewko J, Ustymowicz A (1999): Transcranial color Doppler sonography of basal cerebral arteries in 182 healthy subjects: Age and sex variability and normal reference values for blood flow parameters. AJR Am J Roentgenol 172:213–218. [DOI] [PubMed] [Google Scholar]
- Kriegeskorte N, Goebel R, Bandettini P (2006): Information‐based functional brain mapping. Proc Natl Acad Sci USA 103:3863–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre JC (2012): Cerebral hemodynamics and vascular risk factors: Setting the stage for Alzheimer's disease. J Alzheimers Dis 32:553–567. [DOI] [PubMed] [Google Scholar]
- Li S‐J, Li Z, Wu G, Zhang M‐J, Franczak M, Antuono PG (2002): Alzheimer disease: Evaluation of a functional MR imaging index as a marker. Radiology 225:253–259. [DOI] [PubMed] [Google Scholar]
- Liepert J (2006): Motor cortex excitability in stroke before and after constraint‐induced movement therapy. Cogn Behav Neurol 19:41–47. [DOI] [PubMed] [Google Scholar]
- Lin WH, Hao Q, Rosengarten B, Leung WH, Wong KS (2011): Impaired neurovascular coupling in ischaemic stroke patients with large or small vessel disease. Eur J Neurol 18:731–736. [DOI] [PubMed] [Google Scholar]
- Lindquist MA, Meng Loh J, Atlas LY, Wager TD (2009): Modeling the hemodynamic response function in fMRI: Efficiency, bias and mis‐modeling. Neuroimage 45:S187–S198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TT (2013): Neurovascular factors in resting‐state functional MRI. Neuroimage 80:339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis NK (2008): What we can do and what we cannot do with fMRI. Nature 453:869–878. [DOI] [PubMed] [Google Scholar]
- Logothetis NK, Wandell BA (2004): Interpreting the BOLD signal. Annu Rev Physiol 66:735–769. [DOI] [PubMed] [Google Scholar]
- Logothetis NK, Pauls J, Augath M, Trinath T, Oeltermann A (2001): Neurophysiological investigation of the basis of the fMRI signal. Nature 412:150–157. [DOI] [PubMed] [Google Scholar]
- Lu H, Xu F, Rodrigue KM, Kennedy KM, Cheng Y, Flicker B, Hebrank AC, Uh J, Park DC (2011): Alterations in cerebral metabolic rate and blood supply across the adult lifespan. Cereb Cortex 21:1426–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynall M‐E, Bassett DS, Kerwin R, McKenna PJ, Kitzbichler M, Muller U, Bullmore E (2010): Functional connectivity and brain networks in schizophrenia. J Neurosci 30:9477–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makedonov I, Black SE, Macintosh BJ (2013): BOLD fMRI in the white matter as a marker of aging and small vessel disease. PloS One 8:e67652. doi: 10.1371/journal.pone.0067652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrelec G, Benali H, Ciuciu P, Pélégrini‐Issac M, Poline J‐B (2003): Robust Bayesian estimation of the hemodynamic response function in event‐related BOLD fMRI using basic physiological information. Hum Brain Mapp 19:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marstrand JR, Garde E, Rostrup E, Ring P, Rosenbaum S, Mortensen EL, Larsson HBW (2002): Cerebral perfusion and cerebrovascular reactivity are reduced in white matter hyperintensities. Stroke 33:972–976. [DOI] [PubMed] [Google Scholar]
- Mazzetto‐Betti KC, Leoni RF, Pontes‐Neto OM, Santos AC, Leite JP, Silva AC, de Araujo DB (2010): The stability of the blood oxygenation level‐dependent functional MRI response to motor tasks is altered in patients with chronic ischemic stroke. Stroke 41:1921–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh AR, Lobaugh NJ (2004): Partial least squares analysis of neuroimaging data: Applications and advances. Neuroimage 23 Suppl 1:S250–S263. [DOI] [PubMed] [Google Scholar]
- Miezin FM, Maccotta L, Ollinger JM, Petersen SE, Buckner RL (2000): Characterizing the hemodynamic response: Effects of presentation rate, sampling procedure, and the possibility of ordering brain activity based on relative timing. Neuroimage 11:735–759. [DOI] [PubMed] [Google Scholar]
- Mourão‐Miranda J, Reynaud E, McGlone F, Calvert G, Brammer M (2006): The impact of temporal compression and space selection on SVM analysis of single‐subject and multi‐subject fMRI data. Neuroimage 33:1055–1065. [DOI] [PubMed] [Google Scholar]
- Muller M, van der Graaf Y, Visseren FL, Mali WPTM, Geerlings MI (2012): Hypertension and longitudinal changes in cerebral blood flow: The SMART‐MR study. Ann Neurol 71:825–833. [DOI] [PubMed] [Google Scholar]
- Mur M, Bandettini P a, Kriegeskorte N (2009): Revealing representational content with pattern‐information fMRI–an introductory guide. Soc Cogn Affect Neurosci 4:101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y, Sakatani K, Hoshino T, Fujiwara N, Kano T, Nakamura S, Katayama Y (2006): Effects of cerebral ischemia on evoked cerebral blood oxygenation responses and BOLD contrast functional MRI in stroke patients. Stroke 37:2514–2520. [DOI] [PubMed] [Google Scholar]
- Muthukumaraswamy SD, Edden RAE, Jones DK, Swettenham JB, Singh KD (2009): Resting GABA concentration predicts peak gamma frequency and fMRI amplitude in response to visual stimulation in humans. Proc Natl Acad Sci USA 106:8356–8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair DG (2005): About being BOLD. Brain Res Rev 50:229–243. [DOI] [PubMed] [Google Scholar]
- Newton J, Sunderland A, Butterworth SE, Peters AM, Peck KK, Gowland PA (2002): A pilot study of event‐related functional magnetic resonance imaging of monitored wrist movements in patients with partial recovery. Stroke 33:2881–2887. [DOI] [PubMed] [Google Scholar]
- Ogawa S, Lee TM, Kay AR, Tank DW (1990): Brain magnetic resonance imaging with contrast dependent on blood oxygenation. Proc Natl Acad Sci USA 87:9868–9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Østergaard L, Jespersen SN, Mouridsen K, Mikkelsen IK, Jonsdottír KÝ, Tietze A, Blicher JU, Aamand R, Hjort N, Iversen NK, Cai C, Hougaard KD, Simonsen CZ, Von Weitzel‐Mudersbach P, Modrau B, Nagenthiraja K, Riisgaard Ribe L, Hansen MB, Bekke SL, Dahlman MG, Puig J, Pedraza S, Serena J, Cho T‐H, Siemonsen S, Thomalla G, Fiehler J, Nighoghossian N, Andersen G (2013): The role of the cerebral capillaries in acute ischemic stroke: The extended penumbra model. J Cereb Blood Flow Metab 33:635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DC, Reuter‐Lorenz P (2009): The adaptive brain: Aging and neurocognitive scaffolding. Annu Rev Psychol 60:173–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, Chang WH, Ohn SH, Kim ST, Bang OY, Pascual‐Leone A, Kim Y‐H (2011): Longitudinal changes of resting‐state functional connectivity during motor recovery after stroke. Stroke 42:1357–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineiro R, Pendlebury S, Johansen‐Berg H, Matthews PM (2002): Altered hemodynamic responses in patients after subcortical stroke measured by functional MRI. Stroke 33 :103–109. [DOI] [PubMed] [Google Scholar]
- Power JD, Barnes KA, Snyder AZ, Schlaggar BL, Petersen SE (2012): Spurious but systematic correlations in functional connectivity MRI networks arise from subject motion. Neuroimage 59:2142–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price CJ, Friston KJ (2002): Functional imaging studies of neuropsychological patients: Applications and limitations. Neurocase 8:345–354. [DOI] [PubMed] [Google Scholar]
- Price CJ, Crinion J, Friston KJ (2006): Design and analysis of fMRI studies with neurologically impaired patients. J Magn Reson Imaging 23:816–826. [DOI] [PubMed] [Google Scholar]
- Promjunyakul N, Schmit BD, Schindler‐Ivens S (2013): Changes in hemodynamic responses in chronic stroke survivors do not affect fMRI signal detection in a block experimental design. Magn Reson Imaging 31:1119–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Lindenberger U, Rodrigue KM, Kennedy KM, Head D, Williamson A, Dahle C, Gerstorf D, Acker JD (2005): Regional brain changes in aging healthy adults: General trends, individual differences and modifiers. Cereb Cortex 15:1676–1689. [DOI] [PubMed] [Google Scholar]
- Rees G, Friston K, Koch C (2000): A direct quantitative relationship between the functional properties of human and macaque V5. Nat Neurosci 3:716–723. [DOI] [PubMed] [Google Scholar]
- Rehme AK, Grefkes C (2013): Cerebral network disorders after stroke: Evidence from imaging‐based connectivity analyses of active and resting brain states in humans. J Physiol 591:17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard M, Roth M, Guschlbauer B, Harloff A, Timmer J, Czosnyka M, Hetzel A (2005): Dynamic cerebral autoregulation in acute ischemic stroke assessed from spontaneous blood pressure fluctuations. Stroke 36:1684–1689. [DOI] [PubMed] [Google Scholar]
- Richardson JD, Baker JM, Morgan PS, Rorden C, Bonilha L, Fridriksson J (2011): Cerebral perfusion in chronic stroke: Implications for lesion‐symptom mapping and functional MRI. Behav Neurol 24:117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripollés P, Marco‐Pallarés J, de Diego‐Balaguer R, Miró J, Falip M, Juncadella M, Rubio F, Rodriguez‐Fornells A (2012): Analysis of automated methods for spatial normalization of lesioned brains. Neuroimage 60:1296–1306. [DOI] [PubMed] [Google Scholar]
- Roc AC, Wang J, Ances BM, Liebeskind DS, Kasner SE, Detre JA (2006): Altered hemodynamics and regional cerebral blood flow in patients with hemodynamically significant stenoses. Stroke 37:382–387. [DOI] [PubMed] [Google Scholar]
- Rorden C, Bonilha L, Fridriksson J, Bender B, Karnath H‐O (2012): Age‐specific CT and MRI templates for spatial normalization. Neuroimage 61:957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossini PM, Altamura C, Ferretti A, Vernieri F, Zappasodi F, Caulo M, Pizzella V, Del Gratta C, Romani G‐L, Tecchio F (2004): Does cerebrovascular disease affect the coupling between neuronal activity and local haemodynamics? Brain 127:99–110. [DOI] [PubMed] [Google Scholar]
- Salat DH, Buckner RL, Snyder AZ, Greve DN, Desikan RSR, Busa E, Morris JC, Dale AM, Fischl B (2004): Thinning of the cerebral cortex in aging. Cereb Cortex 14:721–730. [DOI] [PubMed] [Google Scholar]
- Samanez‐Larkin GR, D'Esposito M (2008): Group comparisons: Imaging the aging brain. Soc Cogn Affect Neurosci 3:290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz SK, O'Leary DS, Boles Ponto LL, Watkins GL, Hichwa RD, Andreasen NC (1999): Age‐related changes in regional cerebral blood flow among young to mid‐life adults. Neuroreport 10:2493–2496. [DOI] [PubMed] [Google Scholar]
- Seghier ML, Ramlackhansingh A, Crinion J, Leff AP, Price CJ (2008): Lesion identification using unified segmentation‐normalisation models and fuzzy clustering. Neuroimage 41:1253–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seminowicz D., Mayberg H., McIntosh A., Goldapple K, Kennedy S, Segal Z, Rafi‐Tari S (2004): Limbic–frontal circuitry in major depression: A path modeling metanalysis. Neuroimage 22:409–418. [DOI] [PubMed] [Google Scholar]
- Shehzad Z, Kelly AMC, Reiss PT, Gee DG, Gotimer K, Uddin LQ, Lee SH, Margulies DS, Roy AK, Biswal BB, Petkova E, Castellanos FX, Milham MP (2009): The resting brain: Unconstrained yet reliable. Cereb Cortex 19:2209–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Fox PT, Miller KL, Glahn DC, Fox PM, Mackay CE, Filippini N, Watkins KE, Toro R, Laird AR, Beckmann CF (2009): Correspondence of the brain's functional architecture during activation and rest. Proc Natl Acad Sci USA 106:13040–13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderlund H, Nyberg L, Adolfsson R, Nilsson L‐G, Launer LJ (2003): High prevalence of white matter hyperintensities in normal aging: Relation to blood pressure and cognition. Cortex 39:1093–1105. [DOI] [PubMed] [Google Scholar]
- Swayne OBC, Rothwell JC, Ward NS, Greenwood RJ (2008): Stages of motor output reorganization after hemispheric stroke suggested by longitudinal studies of cortical physiology. Cereb Cortex 18:1909–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak S, Polimeni JR, Wang DJJ, Yan L, Chen J (2014): Associations of resting‐state fMRI functional connectivity with flow‐BOLD coupling and regional vasculature. Brain Connect. doi: 10.1089/brain.2014.0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason ME, Foland LC, Glover GH (2007): Calibration of BOLD fMRI using breath holding reduces group variance during a cognitive task. Hum Brain Mapp 28:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda N (2012): Age‐related changes in endothelial function and blood flow regulation. Pharmacol Ther 133:159–176. [DOI] [PubMed] [Google Scholar]
- Viviani R, Messina I, Walter M (2011): Resting state functional connectivity in perfusion imaging: Correlation maps with BOLD connectivity and resting state perfusion. PLoS One 6:e27050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Liang M, Wang L, Tian L, Zhang X, Li K, Jiang T (2007): Altered functional connectivity in early Alzheimer's disease: A resting‐state fMRI study. Hum Brain Mapp 28:967–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, Lindley RI, O'Brien JT, Barkhof F, Benavente OR, Black SE, Brayne C, Breteler M, Chabriat H, Decarli C, de Leeuw F‐E, Doubal F, Duering M, Fox NC, Greenberg S, Hachinski V, Kilimann I, Mok V, Oostenbrugge R , van Pantoni L , Speck O, Stephan BCM, Teipel S, Viswanathan A, Werring D, Chen C, Smith C, van Buchem M, Norrving B, Gorelick PB, Dichgans M (2013): Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12:822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen W, Sachdev P (2004): The topography of white matter hyperintensities on brain MRI in healthy 60‐ to 64‐year‐old individuals. Neuroimage 22:144–154. [DOI] [PubMed] [Google Scholar]
- Woolrich MW (2012): Bayesian inference in FMRI. Neuroimage 62:801–810. [DOI] [PubMed] [Google Scholar]
- Wu W‐C, Fernández‐Seara M, Detre JA, Wehrli FW, Wang J (2007): A theoretical and experimental investigation of the tagging efficiency of pseudocontinuous arterial spin labeling. Magn Reson Med 58:1020–1027. [DOI] [PubMed] [Google Scholar]
- Wu CW, Gu H, Lu H, Stein EA, Chen J‐H, Yang Y (2009): Mapping functional connectivity based on synchronized CMRO2 fluctuations during the resting state. Neuroimage 45:694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong L, Leung HW, Chen XY, Leung WH, Soo OY, Wong KS (2014): Autonomic dysfunction in different subtypes of post‐acute ischemic stroke. J Neurol Sci 337:141–146. [DOI] [PubMed] [Google Scholar]
- Yamauchi H, Fukuyama H, Nagahama Y, Nabatame H, Ueno M, Nishizawa S, Konishi J, Shio H (1999): Significance of increased oxygen extraction fraction in five‐year prognosis of major cerebral arterial occlusive diseases. J Nucl Med 40:1992–1998. [PubMed] [Google Scholar]
- Yamauchi H, Higashi T, Kagawa S, Nishii R, Kudo T, Sugimoto K, Okazawa H, Fukuyama H (2012): Is misery perfusion still a predictor of stroke in symptomatic major cerebral artery disease? Brain 135:2515–2526. [DOI] [PubMed] [Google Scholar]
- Zhang D, Raichle ME (2010): Disease and the brain's dark energy. Nat Rev Neurol 6:15–28. [DOI] [PubMed] [Google Scholar]
- Zhao H (2013): Hurdles to clear before clinical translation of ischemic postconditioning against stroke. Transl Stroke Res 4:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo X‐N, Di Martino A, Kelly C, Shehzad ZE, Gee DG, Klein DF, Castellanos FX, Biswal BB, Milham MP (2010): The oscillating brain: complex and reliable. Neuroimage 49:1432–1445.M. [DOI] [PMC free article] [PubMed] [Google Scholar]