

Abstract
Response inhibition is a basic mechanism in cognitive control and dysfunctional in major psychiatric disorders. The neuronal mechanisms are in part driven by dopamine in the striatum. Animal data suggest a regulatory role of glutamate on the level of the striatum. We used a trimodal imaging procedure of the human striatum including F18‐DOPA positron emission tomography, proton magnetic resonance spectroscopy, and functional magnetic resonance imaging of a stop signal task. We investigated dopamine synthesis capacity and glutamate concentration in vivo and their relation to functional properties of response inhibition. A mediation analysis revealed a significant positive association between dopamine synthesis capacity and inhibition‐related neural activity in the caudate nucleus. This relationship was significantly mediated by striatal glutamate concentration. Furthermore, stop signal reaction time was inversely related to striatal activity during inhibition. The data show, for the first time in humans, an interaction between dopamine, glutamate, and the neural signature of response inhibition in the striatum. This finding stresses the importance of the dopamine–glutamate interaction for behavior and may facilitate the understanding of psychiatric disorders characterized by impaired response inhibition. Hum Brain Mapp 36:4031–4040, 2015. © 2015 Wiley Periodicals, Inc.
Keywords: multimodal imaging, dopamine, glutamate, striatum, functional magnetic resonance imaging, magnetic resonance spectroscopy, positron emission tomography, response inhibition
INTRODUCTION
Response inhibition is a core aspect of human nature as it forms a basic mechanism to control one's own behavior [Bari and Robbins, 2013] by suppression of inappropriate or irrelevant actions. This represents a key component of flexible behavior in changing environments [Verbruggen and Logan, 2009]. Insufficient inhibitory control or dysfunctional inhibitory processes can have a strong impact on everyday life, leading for instance to overly impulsive behavior [Bari and Robbins, 2013]. Furthermore, psychiatric disorders such as attention deficit/hyperactivity disorder and drug addiction are characterized by deficiencies in inhibitory control [Bari and Robbins, 2013].
The neuronal basis of response inhibition consists of a complex fronto‐cortico‐basal‐ganglia circuitry. Indeed, within this circuitry, the neuromodulator dopamine has been shown to play an important role as reduced dopamine D2‐receptor expression is related to poor behavioral control in different species [Cropley et al., 2006; Dalley et al., 2007; Hamidovic et al., 2009; Lawrence et al., 1998]. In humans, there is evidence that striatal dopaminergic neurotransmission is related to impulsive personality characteristics [Buckholtz et al., 2010; de Wit et al., 2002] as well as to psychiatric disorders [Koob and Volkow, 2010; Swanson et al., 2007; Verbruggen and Logan, 2008]. In addition, the synaptic plasticity underlying reinforcement learning has been associated with neurochemical interactions of different neurotransmitters at the level of the striatum, with dopamine shown to play a key role [Cools and D'Esposito, 2011; Nair et al., 2014].
However, there is a lack of understanding with regard to regulation of basic dopamine mechanisms at the level of the striatum. Although preliminary evidence indicates an association between in vivo dopamine and striatal blood oxygenation level‐dependent (BOLD) activation [Ghahremani et al., 2012; Schott et al., 2008], the role of the excitatory neurotransmitter glutamate in the striatum has rarely been studied in humans. In animal studies, it has been shown that glutamate and dopamine interact on approximately 95% of striatal neurons [Nair et al., 2014]. In particular, the convergence of cortical glutamate projections onto striatal dopamine neurons may be of critical importance for dopaminergic neurotransmission [Carlsson et al., 1999]. In addition, different types of glutamatergic inputs from brain stem areas, such as the substantia nigra pars compacta, appear to regulate dopaminergic activity in the striatum [Carlsson et al., 1999; Nair et al., 2014]. Studies in animals have shown that administration of glutamate in the ventral striatum reduces the reuptake of dopamine resulting in higher dopamine availability in the synaptic cleft [Morales et al., 2012; Segovia and Mora, 2001]. Interacting dopamine/glutamate strongly modulate molecular and cellular properties of striatal neurons and the strength of corticostriatal synapses [Nair et al., 2014].
Considering pharmacological studies in humans, dopamine receptor imaging studies involving an administration of glutamatergic N‐Methyl‐D‐aspartic acid (NMDA) receptor antagonists have shown heterogeneous results. While some studies found a relationship between the pharmacological intervention and dopaminergic neurotransmission [Breier et al., 1998; Smith et al., 1998; Vernaleken et al., 2013; Vollenweider et al., 2000], some studies showed no effect [Aalto et al., 2002; Kegeles et al., 2002]. Thus, the specific role of glutamate in the striatum remains unclear.
Within the basal ganglia, the striatum represents a key structure for response inhibition driven by dopaminergic modulation [Ghahremani et al., 2012; Vink et al., 2005]. While Ghahremani et al. [2012] investigated postsynaptic dopaminergic neurotransmission, here we assume that presynaptic dopamine neurotransmission may also be related to the neuronal signature of response inhibition. In particular, BOLD activity may reflect neural activity including local synaptic activity [Logothetis et al., 2001]. Thus, it seems to be plausible that presynaptic levels of dopamine predict the actual stimulus‐associated BOLD activity. Furthermore, animal studies indicated a crucial role for glutamate in striatal dopamine activity [Morales et al., 2012; Segovia and Mora, 2001]. In accordance, Carlsson et al. [1999] have supposed that cortical glutamate and striatal dopamine are in homoeostasis in a healthy population and, finally, some pharmacological studies in humans also suggest a glutamate–dopamine interaction in the striatum [e.g. Breier et al., 1998]. Therefore, we hypothesize that the interaction between dopaminergic neurotransmission and the neuronal signature of response inhibition is mediated by glutamatergic neurotransmission on the level of the striatum. In this study, we aim to investigate presynaptic dopamine function via dopamine synthesis capacity from positron emission tomography (PET) scans with the radiotracer 6‐[18F]fluoro‐l‐DOPA (FDOPA) in healthy participants. To assess striatal neuronal signature during response inhibition in the same participants, we used a stop signal paradigm during functional magnetic resonance imaging (fMRI). Finally, we applied single voxel proton magnetic resonance spectroscopy (1H‐MRS) to measure glutamate concentrations in the striatum.
MATERIALS AND METHODS
Participants
We recruited 44 healthy adults via online advertisements and local newspapers. Before participants were recruited, a telephone interview was conducted. During this interview, we asked standardized questions regarding history of psychological and medical disease. In addition, the Structured Clinical Interview for Axis I Disorders (SCID‐I) was administered by an experienced clinician. A short demographic questionnaire and a magnetic resonance imaging (MRI) safety assessment were also completed. Left‐handed participants, participants with prior neurological disease or history of brain surgery, current psychological problems, lifetime psychopharmacological treatment, and MRI contraindications (e.g., nonremovable ferromagnetic material) were not included in the study. The study was approved by the local ethics committee of the Charité – Universitätsmedizin Berlin. All participants gave written informed consent and participants received financial compensation for participation.
The final sample consisted of 38 participants (mean age = 47.3 years, standard deviation (SD) = 19.3 years; age range: 20–80 years; 19 females). Six participants were excluded, either due to excessive head movements during fMRI scanning (two participants exceeded the motion cut‐off of 3° rotation or 3 mm translation in fMRI session) or inadequate performance in the stop signal task (four participants had more than 60% successful withheld responses in stop trials indicating that they intentionally responded slower in order to avoid unsuccessful responses). We further assessed years of education (mean years of education = 16.1 years, SD = 3.6 years), smoking status (29 nonsmoker), alcohol consumption per week (mean = 58.8 g, SD = 71.1 g), and memory ability (assessed by Digit Symbol Test [Tewes, 1991], correct items after 90 s: mean = 56, SD = 15).
Stop Signal Paradigm
Participants completed an adaptive stop signal paradigm [Logan and Cowan, 1984] during the fMRI scanning session. The paradigm was programmed using Presentation software (Version 14.9, Neurobehavioral Systems). We used a slightly modified version of the task assessed by Ghahremani et al. [2012] including a condition with an execution of a motor command (go trials) and another condition, where an already initiated motor command had to be withheld (stop trials). Participants were instructed to respond as fast as possible to a white arrow pointing either to right or left direction by pressing right or left button (using right and left thumb, respectively). For stop trials (25% of trials), participants were instructed to stop their response when the white arrow changed color to red after a particular delay (stop signal delay, SSD). The adaptive character of the task was carried out by a continuous adaption of the SSD using two independent staircases (for detailed explanation, see below) through the experiment, in order to reach a performance level of approximately 50% successfully inhibited responses in each participant.
All trials started with a cue period, which lasted for 500 ms. During this time, a white circular ring was presented in the center of the screen. Subsequently an arrow appeared either in the right (>) or left (<) direction in the center of the ring. The direction of the arrow was balanced across the experiment in a pseudorandomized order. A go trial (144 of 192 trials) ended in the moment of a button press, but lasted a maximum of 1,000 ms. In the stop trials (48 of 192 trials), the arrow and the ring changed the color to red after the time of the adaptive SSD indicating to withhold the response. Stop trials also lasted a maximum of 1,000 ms and ended, if the participant was not able to withhold the response. At the end of a trial, a blank black screen was presented and the next trial started after a variable delay ranging between 0.5 and 4 s (mean = 1 s) that was characterized by an exponentially decreasing function. Additionally, 24 nullevents were pseudorandomly uniformly interspersed over the 192 experimental trials, in order to improve the modeling of the BOLD response.
Before the fMRI scanning session, participants were familiarized with the task in a standardized way and performed a behavioral pretest session containing 96 trials (72 go and 24 stop trials). This pretest was used to estimate the individual SSD using two independent staircases. The staircases started at 150 and 200 ms, respectively. A staircase was incremented in case of a successful stop trial (no button press) or decremented in case of an unsuccessful stop trial (executed button press) by 50 ms. Staircases were updated independently in a pseudorandomized balanced fashion. Mean SSD of the pretest were used as starting values for the two staircases during the experiment. The SSD was also updated continuously during the experiment in the same way as described for the pretest.
Logan and Cowan [1984] supposed that the go and the stop processes are two independent processes, which compete against each other typically described as a race. The finishing time of these processes determines a successful response inhibition. The time of the go process could easily be measured by the reaction time of the go trials, whereas the time of the stop process cannot be directly measured and has to be estimated. Therefore, the so‐called stop signal reaction time (SSRT) is calculated by subtracting the mean SSD from the median of the go reaction time (GoRT) [Verbruggen and Logan, 2009]. This well‐established approach supposes a similar distribution of the stop and go processes and about 50% successful withheld responses in stop trials. The resulting SSRT is an estimation of the length of inhibition process, and therefore, reflects inhibition performance.
fMRI
Imaging acquisition
Imaging data was acquired at the Berlin Center for Advanced Neuroimaging on the Campus Charité Mitte. A 3‐Tesla Siemens TIM Trio Scanner was used that was equipped with a 12‐channel head coil. The stop signal task was presented via a video projector on a mirror system on top of the head coil. Functional images were collected using axially aligned gradient echo planar imaging (EPI) with the following parameters: 33 slices, descending slice order, time to repeat (TR) = 2 s, time to echo (TE) = 30 ms, field of view (FoV) = 192 × 192, flip angle = 78°, matrix size = 64 × 64, voxel size = 3 × 3 × 3 mm3. Additionally, three‐dimensional (3D) anatomical images of the whole brain were obtained by T1‐weighted magnetization prepared gradient‐echo sequence for anatomical reference (MPRAGE; 192 sagittal slices, TR = 1,900 ms, TE = 2.52 ms, flip angle = 9°, FoV = 256 × 256, matrix size = 256 × 256, voxel size = 1 × 1 × 1 mm3).
Image processing
Functional imaging data was analyzed using Statistical Parametric Mapping software package (SPM8, Wellcome Department of Imaging Neuroscience). EPIs were corrected for slice timing and head motion and transformed into the stereotactic normalized standard space of the Montreal Neuroimaging Institute using the unified segmentation algorithm. Finally, EPIs were resampled (voxel size = 3 × 3 × 3 mm3) and spatially smoothed with a 3D Gaussian kernel of 7‐mm full width at half maximum.
Statistical Analysis with Classical Event‐Related Approach
A two‐stage mixed‐effects general linear model (GLM) was applied. On the single subject level, event‐related separate regressors were included for go trials, successful stop trials, and unsuccessful stop trials. Additionally, a regressor of no interest was included that modeled invalid trials, in which a participant did not respond during go trials or made discrimination errors (e.g., pressed right button when left‐directed arrow was presented). Finally, the six rigid body movement parameters were also included in the single subject GLM. Differential t‐contrasts for successful stop trials versus go trials were calculated and taken to group level analysis. On the second level, these differential t‐contrast images were entered into a one sample t‐test. Whole brain effects were corrected for multiple comparisons with an error probability of P < 0.05 using a whole brain familywise error (FWE)‐correction as implemented in SPM8.
MRS
Acquisition and data analysis
MRS data was collected at the same 3‐Tesla scanner as fMRI. Metabolites in the magnetic resonance spectrum were quantified immediately after acquisition of a high‐resolution T1 structural image using 3‐Tesla 1H‐MRS (point resolved spectroscopy, 128/8 averages, 90° flip angle, TE = 80 ms, TR = 3 s, automatic shimming, 128/8 spectra with and without water‐suppression, respectively). The linear combination of model spectra commercial spectral‐fitting package [LCmodel; Provencher, 1993; Göttingen, Germany] was used to analyze MRS data. For estimation of glutamate concentration within the striatal voxel, we used a threshold of ≤30% for Cramér‐Rao lower bounds due to lower signal to noise ratio in subcortical structures (mean fit deviation = 22.5%, SD = 4.36; range: 15–30%).
Localization of the striatum voxel
A 20 × 20 × 20 mm3 voxel was placed individually in the left medial dorsal striatum (see Supporting Information Fig. S1). First, the voxel was placed individually in the striatum including the medial dorsal striatum in the center of the voxel. Therefore, the voxel was shifted dorsally/ventrally and/or tilted to maximize inclusion of striatal tissue and minimize insula gray matter (GM) as well as white matter (WM) and cerebrospinal fluid (CSF). Finally, the voxel was shifted and tilted on the transversal and sagittal planes.
Individual adjustment for GM and WM
To correct for individual differences in GM, WM, and CSF fractions within the MRS voxel, absolute glutamate concentrations were transformed via the formula: Glutamate adjusted = Glutamate absolute × (GM + WM)−1 [Gleich et al., 2014]. Adjusted glutamate concentrations were expressed in mmol l−1. For quantification of GM, WM, and CSF, we used the unified segmentation approach of SPM8 and extracted voxel‐specific fractions of GM, WM, and CSF using python scripts (Python Software Foundation, Beaverton, OR).
PET
Data acquisition and preprocessing
To investigate presynaptic dopamine‐related neurotransmission, the radioisotope [18F]fluoro‐3,4‐dihydroxyphenyl‐l‐alanine (F‐DOPA) was used, which reflects the activity of endogenous l‐DOPA in the brain. In particular, following uptake via the blood–brain barrier, F‐DOPA propagates across the brain, is metabolized to 6‐[18F]fluoro‐dopamine and trapped in vesicles in nerve terminals. These terminals are predominantly located in the striatum. Striatal F‐DOPA PET can thus be used to investigate the dopamine synthesis capacity in the brain, mainly reflecting tonic presynaptic dopamine function [Howes et al., 2012; Kumakura and Cumming, 2009; Schlagenhauf et al., 2013]. PET imaging data was acquired using a time‐of‐flight PET/computed tomography (CT) scanner (Philips Gemini TF16) in 3D acquisition mode. Together with the start of intravenous administration of 120–200 MBq F‐18‐FDOPA as a prolonged bolus (about 10 s), a 60 min “list‐mode” PET emission scan was initiated. Emission data were sorted into 20 frames (3 × 20 s, 3 × 1 min, 3 × 2 min, 3 × 3 min, 7 × 5 min, 1 × 6 min) and reconstructed into transaxial images using the 3D iterative reconstruction algorithm of the system software with default parameter settings. Further data preprocessing was performed with SPM8 software. SPM8's realignment procedure was used to correct for head motion during the PET emission scan. Maximum head motion was 3.5° rotation or 3 mm translation, thus we included all participants. Then MRI T1 images were coregistered to the mean of PET frames 13–20. The T1 image was then spatially normalized and the computed normalization parameters were applied to all PET emission recording frames.
Statistical PET analysis
To estimate dopamine synthesis capacity voxelwise, FDOPA Ki (min−1) values were computed using Gjedde‐Patlak graphical analysis with the cerebellum (excluding vermis) as defined in the automated anatomical labeling brain atlas [Tzourio‐Mazoyer et al., 2002] as reference region. The linear fit was restricted to the time interval 20–60 min after recording start. The resulting individual FDOPA Ki maps were finally smoothed with a 3D isotropic Gaussian kernel of 5‐mm full width at half maximum.
Multimodal imaging analysis
For multimodal imaging analysis, we focused on the left striatum. For glutamate concentration, the aforementioned GM‐corrected glutamate values derived from the MRS measurement were used. Further, data from fMRI and PET were in a voxel‐by‐voxel format. We concentrated on the part of the striatum that was functionally and behaviorally involved in response inhibition. To this end, we used the cluster in the left caudate nucleus that was revealed by fMRI analysis (successful stop vs. go, see results). For fMRI data, individual mean parameter estimates was extracted from this cluster in the caudate nucleus. PET analysis was also to this cluster by calculating individual mean Ki values extracted from FDOPA Ki maps of this fMRI‐driven cluster.
Further, we analyzed the multimodal imaging data set in two steps. First, the measurements of the three imaging modalities were correlated using Pearson correlation coefficients. Due to inadequate fit deviation during MRS, data from only 31 participants were available for glutamate concentration. Using the outlier labeling rule [Hoaglin et al., 1986] two further participants were identified as outlier resulting in a final MRS sample of 29 participants. Thus, correlations with glutamate were calculated with this smaller sample. Second, we tested the relationship between all three measurements in a pathway model using Preacher and Hayes SPSS (Statistical Package for the Social Sciences, Chicago, IL) Macro for multiple mediation [Preacher and Hayes, 2008] with the smaller sample.
RESULTS
The striatum was targeted by three different imaging methodologies: (1) fMRI BOLD activity of the left caudate nucleus, (2) FDOPA‐PET Ki of the left caudate nucleus, a measure of dopamine synthesis capacity, and (3) 1H‐MRS, measuring absolute glutamate concentrations in a voxel covering the left striatum including the caudate nucleus. Before conducting correlation analyses, dependent variables were all checked for normal distribution using Kolmogorov–Smirnov tests. These analyses revealed that all variables were normally distributed (all P's > 0.345).
Stop Signal Task: Behavior and Neuronal Signature
An adaptive stop signal paradigm was conducted [Ghahremani et al., 2012; Logan and Cowan, 1984]. Behaviorally, participants successfully inhibited their response to the stop signal in 52% (SD = 4%). Based on a horse race model in response inhibition [Logan and Cowan, 1984], we estimated the SSRT by subtracting the mean SSD time (interstimulus interval between go signal and stop signal) from the median of the GoRT [Ghahremani et al., 2012]. Participants had on average a median GoRT of 462.6 ms (SD = 66.8 ms) and a mean SSRT of 228.1 ms (SD = 42.8 ms). Neuroimaging data showed elevated BOLD activity in a well‐established frontostriatal network including bilateral caudate nucleus during successful stop trials contrasted against go trials (see Supporting Information including Supporting Information Table S1 and Figs. S2 and S3, for details).
Due to our a priori hypothesis, a correlational analysis between striatal activation and behavioral inhibition performance (SSRT) was conducted. Individual mean BOLD activity of the contrast successful stop versus go was extracted from the significant cluster covering 11 voxels in the left caudate nucleus. This striatal BOLD activity correlated inversely with the SSRT (r(38) = 0.352; P = 0.03) indicating better inhibition performance in individuals with stronger striatal neuronal activity during stopping (see Supporting Information Fig. S4). As control analysis, we further conducted a correlation analysis between GoRT and BOLD activity of the same significant cluster resulting in a nonsignificant result (r(38) = 0.125; P = 0.455).
Additionally, the relationship between the behavioral aspects of response inhibition (GoRT and SSRT) and the measurements of the neurotransmitter systems (striatal glutamate concentration and dopamine synthesis capacity) was investigated. No significant associations were observed (all P's > 0.564).
Trimodal Imaging of the Left Striatum
The analysis focused on the interaction of the three imaging measures within the striatum. We restricted the extraction of the mean parameter estimates to the significant effect of response inhibition (successful stop vs. go) in the fMRI analysis. In order to parallel this with the PET measurement, the mean Ki values were extracted from this cluster. We observed significant positive relationships between (1) BOLD activity and dopamine synthesis capacity (r(38) = 0.326; P = 0.046), (2) absolute glutamate concentrations and BOLD activity (r(29) = 0.685; P < 0.001), as well as (3) dopamine synthesis capacity and absolute glutamate concentrations (r(29) = 0.43; P = 0.02; Fig. 1).
Figure 1.
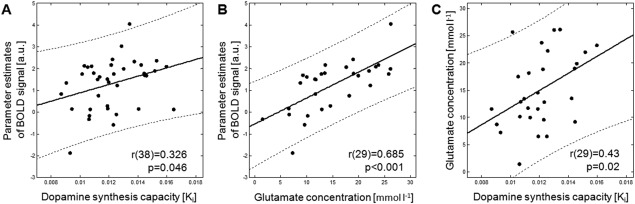
Correlations of the trimodal imaging of the left striatum. (A) Association between dopamine synthesis capacity (assessed with PET) and parameter estimates of BOLD signal during stop signal task (assessed with fMRI, n = 38) and (B) association between glutamate concentration (assessed with MRS) and parameter estimates of the BOLD signal (n = 29). (C) Association between dopamine synthesis capacity and glutamate concentration (n = 29). MRS = Magnetic resonance spectroscopy; PET = Positron emission tomography; BOLD = Blood oxygen level dependent; fMRI = functional magnetic resonance imaging.
Pathmodel of Multimodal Imaging Measurements
We used mediation analysis [Preacher and Hayes, 2008] to test the hypothesis that higher levels of dopamine synthesis capacity were associated with enhanced functional striatal BOLD activity, mediated by striatal glutamate concentrations (Fig. 2). This analysis revealed a significant positive relationship between dopamine synthesis capacity and striatal BOLD activity (C path: ß = 292.2; standard error (SE) = 108.9; t(26) = 2.68; P = 0.012). This association was statistically significant when glutamate was included as a mediator but not without glutamate: C′ path: ß = 128.4; SE = 97.5; t(26) = 1.32; P = 0.199. Furthermore, the mediator variable (glutamate concentration) was positively associated with both dopamine synthesis capacity (A path: ß = 1,572.5; SE = 636.7; t(26) = 2.47; P = 0.02) and BOLD activity (B path: ß = 0.104; SE = 0.026; t(26) = 3.9.9; P = 0.006). We tested the significance of the mediation effect using bootstrapping procedures, and therefore, computed 10.000 bootstrapped samples. The bias corrected 95% confidence interval of the bootstrapped indirect mediation effect ranged from 38.6 to 363 indicating a significant mediation effect. Overall, the model fit was significant (R 2 = 0.503, F (2,26) = 13.16; P = 0.001). In conclusion, this pathmodel analysis revealed glutamate as crucial mediator in the relationship between dopamine synthesis capacity and BOLD activity in the striatum.
Figure 2.
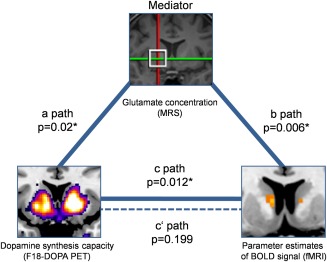
Pathmodel of multimodal imaging measurements. Mediation analysis (n = 29) revealed that the influence of dopamine synthesis capacity on BOLD parameter estimates (c path, solid arrow) was mediated by glutamate concentration within the striatum. When excluding mediator influence, the dopamine‐BOLD relationship did not remain significant (c′ path, dashed line). Furthermore, mediator glutamate concentration was positively associated with dopamine synthesis capacity (a path) and BOLD parameter estimates (b path). MRS = Magnetic resonance spectroscopy; PET = Positron emission tomography; BOLD = Blood oxygen level dependent; fMRI = functional magnetic resonance imaging. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Furthermore, we calculated two control analyses. First, we conducted a partial correlation analysis between dopamine synthesis capacity and inhibition‐related neuronal activity controlling for glutamate concentration. As expected, this partial correlation was not significant (r(26) = 0.262, P = 0.177). Second, a moderator analysis was calculated with glutamate concentration as moderator for the dopamine–BOLD relationship (see Supporting Information for details). The results of these additional analyses were in line with the result of the mediation analysis indicating that when taking glutamate concentration into account for the dopamine–BOLD relationship, this relationship remained no longer significant.
Effect of Age
In this study, a wide age range was used in order to increase the variability in the imaging parameters. No correlation was observed between age and striatal glutamate concentration (r(29) = 0.273; P = 0.152), dopamine synthesis capacity (r(29) = 0.172; P = 0.302), and BOLD activity in caudate nucleus cluster that was active during response inhibition (r(38) = 0.188; P = 0.259), respectively (see also Supporting Information Table S2). When using age as covariate in the mediation analysis, no significant influence of age was shown (ß = 0.01; SE = 0.009; t(25) = 1.22; P = 0.234) indicating that age does not explain the observed mediation.
DISCUSSION
The data shows for the first time a direct interaction of dopamine, glutamate, and BOLD activity during response inhibition within the striatum in humans. A positive association between dopamine synthesis capacity and inhibition‐related neuronal activity was observed. Moreover, glutamate seems to play a crucial role in this dopamine–BOLD relationship, because it was associated with both measurements. Trimodal imaging is a capable tool to investigate the interaction of major neurotransmitters as a basic mechanism of striatal processing in vivo.
The role of dopamine in response inhibition is a focus of current research. A recent study by Ghahremani et al. [2012] highlighted the positive relationship in caudate nucleus between inhibition‐related BOLD activity and dopamine D2/D3 receptor availability, which is mainly associated with postsynaptic dopaminergic function [Ito et al., 2011]. Dopamine synthesis capacity, which was measured in this study, is associated with endogenous presynaptic dopaminergic neurotransmission, reflecting a background level of dopamine receptor stimulation [Ito et al., 2011; Schlagenhauf et al., 2013]. Thus, not only post synaptic but also presynaptic dopaminergic status seems to be associated with functional properties of response inhibition. Still, a recent study by Ito et al. [2011] showed that these properties are negatively related across the striatum, as a whole, but not in the caudate nucleus. This finding is highly plausible, as presynaptic dopamine release very likely leads to increases in postsynaptic receptor occupancy [Kandel et al., 2000]. Taken together, we were able to add evidence regarding presynaptic dopaminergic neurotransmission to the complex neurochemical interactions with response inhibition in the caudate nucleus.
Furthermore, in this study, we showed that better individual inhibition performance (i.e., faster SSRTs) was accompanied by stronger inhibition‐related BOLD activity in caudate nucleus that is in line with previous research [Ghahremani et al., 2012; Ray Li et al., 2008]. While one study reported an association between higher D2/D3 receptor availability and better response inhibition performance [Ghahremani et al., 2012], another study showed a positive association between impulsivity personality trait and striatal dopamine release also assessed by D2/D3 receptor availability [Buckholtz et al., 2010]. In our data, we were unable to demonstrate a significant relationship between presynaptic dopamine function and inhibition performance. This discrepancy indicates that rather postsynaptic than presynaptic dopaminergic neurotransmission may directly be related to response inhibition behavior. Future research might investigate the presynaptic and postsynaptic contributions to response inhibition systematically.
Response inhibition may be considered as an important characteristic for executive function in general. Interestingly, previous studies investigating executive functions such as working memory reported a positive association between dopamine synthesis capacity of caudate nucleus and working memory performance [Cools et al., 2008; Landau et al., 2009]. Indeed, striatal dopamine is thought to play a major role during the updating of current goal representations in general executive functions (stopping can be conceptualized as an updating process of a current goal) in terms of a gating mechanism. Therefore, striatal dopamine‐related neurotransmission may be linked to higher cognitive function in the prefrontal cortex [Cools and D'Esposito, 2011].
Of note, in this study, the striatal glutamate concentration was positively associated with striatal inhibition‐related BOLD activity. To our knowledge, this is the first study showing a relationship of striatal glutamate concentration to a neuronal correlate of response inhibition. Studies quantifying in vivo glutamate concentrations in the striatum are very scarce. However, in a recent MRS study a positive relationship of striatal glutamate concentration with performance in tests of executive functions was observed [Zahr et al., 2008]. This observation may argue for the role of glutamate within the frontostriatal network, with relevance for executive functions and motor commands [Chambers et al., 2009; Zahr et al., 2008].
According to converging evidence from animal investigations for a close interaction of glutamate and dopamine, a positive relationship between striatal dopamine synthesis capacity and glutamate concentration was observed for the first time in a human sample. A higher presynaptic dopamine level was associated with higher concentrations of glutamate in the striatum. This observation is in line with animal research showing that higher glutamatergic activity (e.g., through local infusions of the glutamate agonist) within the striatum was accompanied by increased extracellular dopamine concentration [Mora et al., 2008; Segovia and Mora, 2001]. However, apart from this local striatal interaction, glutamate acts from distant brain areas onto the striatum, particularly via prefrontal efferents [Carlsson et al., 1999; Chambers et al., 2009; Schwartz et al., 2012; Zahr et al., 2008]. Furthermore, prefrontal glutamatergic connections modulate striatal dopamine release via inhibitory GABAergic interneurons [Carlsson et al., 1999]. In this circuit, reduced prefrontal glutamate activity was suggested to be associated with an increased striatal dopamine function and represents a core mechanism of the pathobiological model of schizophrenia [Carlsson et al., 1999]. Interestingly, in schizophrenic patients abnormal dopamine function and glutamate concentrations have been reported in the striatum as well as in the prefrontal cortex [Bloemen et al., 2011; de la Fuente‐Sandoval et al., 2011; de la Fuente‐Sandoval et al., 2013; Hietala et al., 1995; Howes and Kapur, 2009; Howes et al., 2012;Meyer‐Lindenberg et al., 2002; Nozaki et al., 2009] together with deficits in response inhibition [Enticott et al., 2008; Thoma et al., 2007].
Regarding to age effects, we did not find a relationship between dopamine synthesis capacity and age. Such an age‐related decline was reported previously, but not consistently [Braskie et al., 2008; Kumakura et al., 2010]. Furthermore, age did not contribute to the findings of mediation analysis indicating that the striatal dopamine–glutamate interaction might be independent from age effects. Animal studies support this finding by showing a dorsal–ventral gradient of age effects in the glutamate–dopamine interaction, that is, the dorsal parts of the striatum are less affected by aging than the more ventral parts [Mora et al., 2008].
There are some limitations that need to be addressed. First, in this study, we focused on the left striatum in order to limit the measuring time. Recent studies showed a bilateral striatal neural response during inhibition processes [Vink et al., 2005], which was also bilaterally related to dopaminergic neurotransmission [Ghahremani et al., 2012]. Thus, future studies should investigate bilateral contributions of glutamate more closely. Second, the striatum receives glutamatergic input not only from different brain areas in the prefrontal cortex, but also from subcortical structures, for example, amygdala and hippocampus [Mora et al., 2008]. Future studies might focus also on brain structures that are linked to the striatum with glutamatergic connections. Third, the measurement of glutamate concentration with MRS in the striatum represents all existing glutamate including extracellular and intracellular glutamate of neurons and glia cells in this voxel [Rothman et al., 1999]. It is likely that the glutamate concentration reflects the neurotransmission of glutamatergic afferents from the prefrontal inhibition‐related brain areas. However, also other aspects may contribute to the measured glutamate concentration in the striatum. More research is needed for a better understanding of the functional role of the striatal glutamate concentration as assessed by MRS. Fourth, single voxel MRS is restricted to a cuboid‐shaped volume. We tried to optimize the coverage of the caudate nucleus by an individual voxel placement, however, the MRS voxel of the striatum does not cover the whole caudate nucleus and also parts of the putamen were covered by the voxel. Therefore, the MRS results reflect only an approximation of the glutamate concentration of the caudate nucleus. Fifth, this study has a cross‐sectional design and correlational data analyses techniques were applied for integration of the different imaging modalities. Therefore, it is not allowed drawing any causal conclusions. However, the relationships that were observed in this study might be used to propose new hypothesis in future longitudinal studies with, for example, pharmacological treatments like ketamine (for glutamate intervention) or l‐DOPA (for a dopamine intervention) administration.
Considering all results together, we found a coupling between presynaptic striatal dopaminergic status and inhibition‐related neuronal activity mediated by glutamate. We believe that the finding of glutamate as mediator variable in the dopamine–BOLD coupling adds a significant piece of evidence about frontostriatal neurochemical interactions to the field of human neuroscience. Glutamatergic regulation seems to be critical for inhibition behavior and its neuronal correlate in caudate nucleus. Future studies should investigate the neurochemical basis of response inhibition in psychiatric disorders like schizophrenia and drug addiction since these disorders are characterized by an imbalanced glutamate–dopamine interaction as well as deficits in response inhibition.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The authors thank J. Frenzel, L. Adam, S. Luo, and S. Schweinzer for assistance during fMRI/MRS data collection as well as S. Lücke for organization and assistance during FDOPA PET. The authors declare no conflict of interest.
REFERENCES
- Aalto S, Hirvonen J, Kajander J, Scheinin H, Någren K, Vilkman H, Gustafsson L, Syvälahti E, Hietala J (2002): Ketamine does not decrease striatal dopamine D2 receptor binding in man. Psychopharmacology (Berl) 164:401–406. [DOI] [PubMed] [Google Scholar]
- Bari A, Robbins TW (2013): Inhibition and impulsivity: Behavioral and neural basis of response control. Prog Neurobiol 108:44–79. [DOI] [PubMed] [Google Scholar]
- Bloemen OJ, Gleich T, de Koning MB, da Silva Alvis F, de Haan L, Linszen DH, Booij J, van Amelsvoort TA (2011): Hippocampal glutamate levels and striatal dopamine D(2/3) receptor occupancy in subjects at ultra high risk of psychosis. Biol Psychiatry 70:e1–2. [DOI] [PubMed] [Google Scholar]
- Braskie MN, Wilcox CE, Landau SM, O'Neil JP, Baker SL, Madison CM, Kluth JT, Jagust WJ (2008): Relationship of striatal dopamine synthesis capacity to age and cognition. J Neurosci 28:14320–14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breier A, Adler CM, Weisenfeld N, Su TP, Elman I, Picken L, Malhotra AK, Pickar D (1998): Effects of NMDA antagonism on striatal dopamine release in healthy subjects: application of a novel PET approach. Synapse 29:142–147. [DOI] [PubMed] [Google Scholar]
- Buckholtz JW, Treadway MT, Cowan RL, Woodward ND, Li R, Ansari MS, Baldwin RM, Schwartzman AN, Shelby ES, Smith CE, Kessler RM, Zald DH (2010): Dopaminergic network differences in human impulsivity. Science 329:532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A, Waters N, Carlsson ML (1999): Neurotransmitter interactions in schizophrenia–therapeutic implications. Biol Psychiatry 46:1388–1395. [DOI] [PubMed] [Google Scholar]
- Chambers CD, Garavan H, Bellgrove MA (2009): Insights into the neural basis of response inhibition from cognitive and clinical neuroscience. Neurosci Biobehav Rev 33:631–646. [DOI] [PubMed] [Google Scholar]
- Cools R, D'Esposito M (2011): Inverted‐U‐shaped dopamine actions on human working memory and cognitive control. Biol Psychiatry 69:e113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cools R, Gibbs SE, Miyakawa A, Jagust W, D'Esposito M (2008): Working memory capacity predicts dopamine synthesis capacity in the human striatum. J Neurosci 28:1208–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cropley VL, Fujita M, Innis RB, Nathan PJ (2006): Molecular imaging of the dopaminergic system and its association with human cognitive function. Biol Psychiatry 59:898–907. [DOI] [PubMed] [Google Scholar]
- Dalley JW, Fryer TD, Brichard L, Robinson ESJ, Theobald DEH, Laane K, Pena Y, Murphy ER, Shah Y, Probst K, Abakumova I, Aigbirhio FI, Richards HK, Hong Y, Baron JC, Everitt BJ, Robbins TW (2007): Nucleus accumbens D2/3 receptors predict trait impulsivity and cocaine reinforcement. Science 315:1267–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Fuente‐Sandoval C, León‐Ortiz P, Favila R, Stephano S, Mamo D, Ramírez‐Bermúdez J, Graff‐Guerrero A (2011): Higher levels of glutamate in the associative‐striatum of subjects with prodromal symptoms of schizophrenia and patients with first‐episode psychosis. Neuropsychopharmacology 36:1781–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Fuente‐Sandoval C, León‐Ortiz P, Azcárraga M, Favila R, Stephano S, Graff‐Guerrero A (2013): Striatal glutamate and the conversion to psychosis: a prospective 1H‐MRS imaging study. Int J Neuropsychopharmacol 16:471–475. [DOI] [PubMed] [Google Scholar]
- De Wit H, Enggasser JL, Richards JB (2002): Acute administration of d‐amphetamine decreases impulsivity in healthy volunteers. Neuropsychopharmacology 27:813–825. [DOI] [PubMed] [Google Scholar]
- Enticott PG, Ogloff JR, Bradshaw JL (2008): Response inhibition and impulsivity in schizophrenia. Psychiatry Res 157:251–254. [DOI] [PubMed] [Google Scholar]
- Ghahremani DG, Lee B, Robertson CL, Tabibnia G, Morgan AT, De Shetler N, Brown AK, Monterosso JR, Aron AR, Mandelkern MA, Poldrack RA, London ED (2012): Striatal dopamine D2/D3 receptors mediate response inhibition and related activity in frontostriatal neural circuitry in humans. J Neurosci off J Soc Neurosci 32:7316–7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleich T, Lorenz RC, Pöhland L, Raufelder D, Deserno L, Beck A, Heinz A, Kühn S, Gallinat J (2014): Frontal glutamate and reward processing in adolescence and adulthood. Brain Struct Funct. Advance online publication. [DOI] [PubMed] [Google Scholar]
- Hamidovic A, Dlugos A, Skol A, Palmer AA, de Wit H (2009): Evaluation of genetic variability in the dopamine receptor D2 in relation to behavioral inhibition and impulsivity/sensation seeking: an exploratory study with d‐amphetamine in healthy participants. Exp Clin Psychopharmacol 17:374–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietala J, Syvälahti E, Vuorio K, Räkköläinen V, Bergman J, Haaparanta M, Solin O, Kuoppamäki M, Kirvelä O, Ruotsalainen U (1995): Presynaptic dopamine function in striatum of neuroleptic‐naive schizophrenic patients. Lancet 346:1130–1131. [DOI] [PubMed] [Google Scholar]
- Hoaglin DC, Iglewicz B, Tukey JW (1986): Performance of some resistant rules for outlier labeling. J Am Stat Assoc 81:991–999. [Google Scholar]
- Howes OD, Kapur S (2009): The dopamine hypothesis of schizophrenia: version III–The final common pathway. Schizophr Bull 35:549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes OD, Kambeitz J, Kim E, Stahl D, Slifstein M, Abi‐Dargham A, Kapur S (2012): The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch Gen Psychiatry 69:776–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Kodaka F, Takahashi H, Takano H, Arakawa R, Shimada H, Suhara T (2011): Relation between presynaptic and postsynaptic dopaminergic functions measured by positron emission tomography: Implication of dopaminergic tone. J Neurosci 31:7886–7890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER, Schwartz JH, Jessell TM (2000): Principles of Neural Science Auflage, 4th Revised ed. New York: Mcgraw‐Hill Professional. [Google Scholar]
- Kegeles LS, Martinez D, Kochan LD, Hwang D‐R, Huang Y, Mawlawi O, Suckow RF, Van Heertum RL, Laruelle M (2002): NMDA antagonist effects on striatal dopamine release: positron emission tomography studies in humans. Synapse 43:19–29. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND (2010): Neurocircuitry of addiction. Neuropsychopharmacology 35:217–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumakura Y, Cumming P (2009): PET Studies of Cerebral Levodopa Metabolism: A Review of Clinical Findings and Modeling Approaches. Neuroscientist. 15:635–650. [DOI] [PubMed] [Google Scholar]
- Kumakura Y, Vernaleken I, Buchholz H‐G, Borghammer P, Danielsen E, Gründer G, Heinz A, Bartenstein P, Cumming P (2010): Age‐dependent decline of steady state dopamine storage capacity of human brain: An FDOPA PET study. Neurobiol Aging 31:447–463. [DOI] [PubMed] [Google Scholar]
- Landau SM, Lal R, O'Neil JP, Baker S, Jagust WJ (2009): Striatal dopamine and working memory. Cereb Cortex 19:445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence AD, Weeks RA, Brooks DJ, Andrews TC, Watkins LH, Harding AE, Robbins TW, Sahakian BJ (1998): The relationship between striatal dopamine receptor binding and cognitive performance in Huntington's disease. Brain J Neurol 121:1343–1355. [DOI] [PubMed] [Google Scholar]
- Logan GD, Cowan WB (1984): On the ability to inhibit thought and action: A theory of an act of control. Psychol Rev 91:295–327. [DOI] [PubMed] [Google Scholar]
- Logothetis NK, Pauls J, Augath M, Trinath T, Oeltermann A (2001): Neurophysiological investigation of the basis of the fMRI signal. Nature 412:150–157. [DOI] [PubMed] [Google Scholar]
- Meyer‐Lindenberg A, Miletich RS, Kohn PD, Esposito G, Carson RE, Quarantelli M, Weinberger DR, Berman KF (2002): Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat Neurosci 5:267–271. [DOI] [PubMed] [Google Scholar]
- Mora F, Segovia G, del Arco A (2008): Glutamate–dopamine–GABA interactions in the aging basal ganglia. Brain Res Rev 58:340–353. [DOI] [PubMed] [Google Scholar]
- Morales I, Fuentes A, Ballaz S, Obeso JA, Rodriguez M (2012): Striatal interaction among dopamine, glutamate and ascorbate. Neuropharmacology 63:1308–1314. [DOI] [PubMed] [Google Scholar]
- Nair AG, Gutierrez‐Arenas O, Eriksson O, Jauhiainen A, Blackwell KT, Kotaleski JH (2014): Modeling intracellular signaling underlying striatal function in health and disease. Prog Mol Biol Transl Sci 123:277–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki S, Kato M, Takano H, Ito H, Takahashi H, Arakawa R, Okumura M, Fujimura Y, Matsumoto R, Ota M, Takano A, Otsuka A, Yasuno F, Okubo Y, Kashima H, Suhara T (2009): Regional dopamine synthesis in patients with schizophrenia using L‐[β‐11C]DOPA PET. Schizophr Res 108:78–84. [DOI] [PubMed] [Google Scholar]
- Preacher KJ, Hayes AF (2008): Asymptotic and resampling strategies for assessing and comparing indirect effects in multiple mediator models. Behav Res Methods 40:879–891. [DOI] [PubMed] [Google Scholar]
- Provencher SW (1993): Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med 30:672–679. [DOI] [PubMed] [Google Scholar]
- Ray Li C‐S, Yan P, Sinha R, Lee T‐W (2008): Subcortical processes of motor response inhibition during a stop signal task. NeuroImage 41:1352–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman DL, Sibson NR, Hyder F, Shen J, Behar KL, Shulman RG (1999): In vivo nuclear magnetic resonance spectroscopy studies of the relationship between the glutamate–glutamine neurotransmitter cycle and functional neuroenergetics. Philos Trans R Soc Lond B Biol Sci 354:1165–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlagenhauf F, Rapp MA, Huys QJ, Beck A, Wüstenberg T, Deserno L, Buchholz H‐G, Kalbitzer J, Buchert R, Bauer M, Kienast T, Cumming P, Plotkin M, Kumakura Y, Grace AA, Dolan RJ, Heinz A (2013): Ventral striatal prediction error signaling is associated with dopamine synthesis capacity and fluid intelligence. Hum Brain Mapp 34:1490–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott BH, Minuzzi L, Krebs RM, Elmenhorst D, Lang M, Winz OH, Seidenbecher CI, Coenen HH, Heinze H‐J, Zilles K, Düzel E, Bauer A (2008): Mesolimbic functional magnetic resonance imaging activations during reward anticipation correlate with reward‐related ventral striatal dopamine release. J Neurosci off J Soc Neurosci 28:14311–14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz TL, Sachdeva S, Stahl SM (2012): Glutamate Neurocircuitry: Theoretical Underpinnings in Schizophrenia. Front Pharmacol 3:195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segovia G, Mora F (2001): Involvement of NMDA and AMPA/kainate receptors in the effects of endogenous glutamate on extracellular concentrations of dopamine and GABA in the nucleus accumbens of the awake rat. Brain Res Bull 54:153–157. [DOI] [PubMed] [Google Scholar]
- Smith GS, Schloesser R, Brodie JD, Dewey SL, Logan J, Vitkun SA, Simkowitz P, Hurley A, Cooper T, Volkow ND, Cancro R (1998): Glutamate modulation of dopamine measured in vivo with positron emission tomography (PET) and 11C‐raclopride in normal human subjects. Neuropsychopharmacology 18:18–25. [DOI] [PubMed] [Google Scholar]
- Swanson JM, Kinsbourne M, Nigg J, Lanphear B, Stefanatos GA, Volkow N, Taylor E, Casey BJ, Castellanos FX, Wadhwa PD (2007): Etiologic subtypes of attention‐deficit/hyperactivity disorder: brain imaging, molecular genetic and environmental factors and the dopamine hypothesis. Neuropsychol Rev 17:39–59. [DOI] [PubMed] [Google Scholar]
- Tewes U (1991): Hamburg‐Wechsler Intelligenztest für Erwachsene – Revision 1991 (HAWIE‐R). Bern: Huber. [Google Scholar]
- Thoma P, Wiebel B, Daum I (2007): Response inhibition and cognitive flexibility in schizophrenia with and without comorbid substance use disorder. Schizophr Res 92:168–180. [DOI] [PubMed] [Google Scholar]
- Tzourio‐Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, Mazoyer B, Joliot M (2002): Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single‐subject brain. Neuroimage 15:273–289. [DOI] [PubMed] [Google Scholar]
- Verbruggen F, Logan GD (2008): Response inhibition in the stop‐signal paradigm. Trends Cogn Sci 12:418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbruggen F, Logan GD (2009): Models of response inhibition in the stop‐signal and stop‐change paradigms. Neurosci Biobehav Rev 33:647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernaleken I, Klomp M, Moeller O, Raptis M, Nagels A, Rösch F, Schaefer WM, Cumming P, Gründer G (2013): Vulnerability to psychotogenic effects of ketamine is associated with elevated D2/3‐receptor availability. Int J Neuropsychopharmacol 16:745–754. [DOI] [PubMed] [Google Scholar]
- Vink M, Kahn RS, Raemaekers M, van den Heuvel M, Boersma M, Ramsey NF (2005): Function of striatum beyond inhibition and execution of motor responses. Hum Brain Mapp 25:336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollenweider FX, Vontobel P, Oye I, Hell D, Leenders KL (2000): Effects of (S)‐ketamine on striatal dopamine: a [11C]raclopride PET study of a model psychosis in humans. J Psychiatr Res 34:35–43. [DOI] [PubMed] [Google Scholar]
- Zahr NM, Mayer D, Pfefferbaum A, Sullivan EV (2008): Low striatal glutamate levels underlie cognitive decline in the elderly: evidence from in vivo molecular spectroscopy. Cereb Cortex 18:2241–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information