

Abstract
Objectives
To investigate both cross‐sectional and time‐related changes of striatal and whole‐brain microstructural properties in different stages of Huntington's disease (HD) using diffusion tensor imaging.
Experimental design
From the TRACK‐HD study, premanifest gene carriers (preHD), early manifest HD and controls were scanned at baseline and 2‐year follow‐up. Stratification of the preHD group into a far (preHD‐A) and near (preHD‐B) to predicted disease onset was performed. Age‐corrected histograms of whole‐brain white matter (WM), gray matter (GM) and striatal diffusion measures were computed and normalised by the number of voxels in each subject's data set.
Principle observations
Higher cross‐sectional mean, axial and radial diffusivities were found in both WM (P ≤ 0.001) and GM (P ≤ 0.001) of the manifest HD compared to the preHD and control groups. In preHD, only WM axial diffusivity (AD) was higher than in controls (P ≤ 0.01). This finding remained valid only in preHD‐B (P ≤ 0.001). AD was also higher in the striatum of preHD‐B compared to controls and preHD‐A (P ≤ 0.01). Fractional anisotropy (FA) lacked sensitivity in differentiating between the groups. Histogram peak heights were generally lower in manifest HD compared to the preHD and control groups. No longitudinal differences were found in the degree of diffusivity change between the groups in the two year follow‐up. There was a significant relationship between diffusivity and neurocognitive measures.
Conclusions
Alterations in cross‐sectional diffusion profiles between manifest HD subjects and controls were evident, both in whole‐brain and striatum. In the preHD stage, only AD alterations were found, a finding suggesting that this metric is a sensitive marker for early change in HD prior to disease manifestation. The individual diffusivities were superior to FA in revealing pathologic microstructural brain alterations. Diffusion measures were well related to clinical functioning and disease stage. Hum Brain Mapp 36:2061–2074, 2015. © 2015 Wiley Periodicals, Inc.
Keywords: Huntington's disease, premanifest, diffusion tensor imaging, longitudinal biomarker, white matter, gray matter, striatum, cognitive, TRACK‐HD, histogram analysis
INTRODUCTION
Huntington's disease (HD) is a neurodegenerative autosomal dominant disorder. It is caused by an increased CAG (Cytosine–Adenine–Guanine) repeat within the huntingtin gene on the short arm of chromosome 4 [The Huntington's Disease Collaborative Research Group, 1993]. The mutant huntingtin protein triggers a pathogenic cascade responsible for neuropathology in the brain [Ross and Tabrizi, 2011; Tobin and Signer, 2000]. This results in cognitive, motor, and psychiatric symptoms. The brain as a whole is impacted, though preferential striatal volume loss has been extensively documented by postmortem histopathological as well as in vivo magnetic resonance imaging (MRI) studies [Aylward et al., 2012; Hadzi et al., 2012; Stoffers et al., 2010; Tabrizi et al., 2009; Van den Bogaard et al., 2011; Vonsattel et al., 2011].
Even though no medication is currently available to cure or slow‐down the disease, it remains crucial to have a clear understanding of the typical evolution of brain changes in the disease to determine when microstructural changes start and how fast degeneration occurs. This is necessary to define optimal intervention starting points as well as possibly providing an objective tool to determine the impact of candidate therapies, especially in the premanifest (preHD) phase where clinical measures are lacking.
Diffusion tensor imaging (DTI) is an MRI technique that can quantify water diffusion within tissue [Basser et al., 1994; Jones and Leemans, 2011; Pierpaoli et al., 1996; Tournier et al., 2011]. The diffusion tensor in every voxel can be described by its three eigenvectors and eigenvalues (λ1, λ2, λ3). These eigenvalues quantify the diffusion in three orthogonal orientations and are typically synthesized to axial (=λ1) and radial [= (λ2 + λ3)/2] diffusivities.
Another popular diffusion measure is fractional anisotropy (FA), which is a function of the eigenvalues, and ranges from 0 (completely isotropic diffusion) to 1 (completely anisotropic diffusion), with higher values generally corresponding to a higher directional coherence of tissue organization. High FA occurs for example in healthy white matter (WM) which typically has a parallel‐oriented microarchitecture. Another commonly reported diffusivity measure is the mean diffusivity (MD), which is the average of the three eigenvalues. In this study, we evaluate and report these measures as well as the separate underlying eigenvalues, as these may provide complementary information about the nature of microstructural change [Alexander et al., 2007; Concha, 2014]. It is possible that certain metrics are more selectively affected and, therefore, might be more sensitive to longitudinal change. For example, when changes in axial diffusivity (AD) are proportional to radial diffusivity (RD), the FA value may not be very informative [Acosta‐Cabronero et al., 2010].
In a previous study, we evaluated cross‐sectional group differences in FA and MD between controls, preHD and manifest HD subjects using a region‐of‐interest and fiber tractography analysis approach [Dumas et al., 2012]. In that study, MD proved to be more sensitive in differentiating between the groups compared to FA. Findings from previous longitudinal reports remain inconsistent [Vandenberghe et al., 2009; Weaver et al., 2009; Sritharan et al., 2010]. With inherent limitations such as inter‐user variability to nonautomated methods such as hand drawn regions‐of‐interest, we chose an automated histogram analysis method in this work to assess cross‐sectional as well as time‐related changes of diffusivity measures occurring within 2 years. We hypothesized that lower FA and higher MD, AD, and RD values would be found in subjects with manifest HD when compared to preHD subjects and controls, reflective of higher microstructural disorganization in the manifest group. In addition, we hypothesized that MD would be elevated in preHD subjects when compared to controls based on results from our previous work [Dumas et al., 2012]. Gray matter (GM) diffusivity was assessed separately to assess potential higher sensitivity toward alteration compared to WM, fully bearing in mind the limitations of the tensor model in GM. Associations between neurocognitive measures and diffusivity findings were assessed for potential usage as surrogate markers or predictors for these findings. Also, associations between diffusivity and the expected time to disease onset were assessed to test the hypothesis that sensitivity of diffusivity measures in detecting disturbances in preHD subjects increases with shorter proximity to expected disease onset.
As a subanalysis, diffusion in the left and right hemispheres was assessed individually. This was done to explore the hypothesis of preferential degeneration of the dominant versus the nondominant hemisphere. Plausibly increased lifetime excitotoxic exposure due to higher activation could lead to such a finding in HD. We hypothesized that diffusion parameters indicative of greater neuronal damage were represented more readily in the dominant hemisphere, as findings from previous studies have suggested [Lambrecq et al., 2013; Muhlau et al., 2007; Rosas et al., 2002; Thieben et al., 2002]. To the best of our knowledge, this is the first study exploring this hypothesis and the first to apply histogram analysis to (longitudinal) DTI data in HD as well as to separately assess microstructural properties of both whole‐brain GM and WM.
MATERIALS AND METHODS
Participants
As part of the TRACK‐HD study, 90 participants were included at baseline at the Leiden University Medical Center (LUMC) study site (for details see Tabrizi et al., 2009]. DTI was added to the standard MRI protocol. At baseline, DTI was not performed in 10 participants because of claustrophobia, and another nine were excluded from analysis due to excessive movement artefacts. Of the remaining 71 subjects, 62 subjects completed DTI scans at both visits. Of these 62, a further six subjects were excluded from analysis due to excessive movement artefacts at the second visit. The longitudinal cohort included in this work was thus comprised of 56 subjects: 24 healthy controls, 22 preHD and 10 early manifest HD (Table 1).
Table 1.
Group characteristics and clinical scores
| Healthy controls | preHD (A and B) | preHD‐A | preHD‐B | Manifest HD | ||
|---|---|---|---|---|---|---|
| N | 24 | 22‡ | 11 | 11 | 10 | |
| Gender male/female | 11/13 | 9/13 | 4/7 | 5/6 | 4/6 | |
| Age in years (at V1), mean (SD) | 49.0 (8.2) | 43.6 (8.7) | 44.2 (5.7) | 43.0 (11.2) | 50.2 (9.3) | |
| Handedness R/L | 20/4 | 18/4 | 9/2 | 9/2 | 9/1 | |
| Level of education (ISCED), median (range) | 4 (3) | 4 (3) | 4 (3) | 4 (3) | 4 (3) | |
| DART‐IQ, mean (SD) | 105.0 (9.4) | 100.5 (11.2) | 101.3 (9.7) | 99.6 (13.0) | 101.8 (13.5) | |
| CAG repeat length, mean (SD) | n/a | 42.6 (2.7) | 41.3 (1.4) | 43.9 (3.1)^ | 42.5 (1.2) | |
| Estimated years to onset, mean (SD) | n/a | 11.8 (4.7) | 14.9 (4.7) | 8.6 (1.8)^ | n/a | |
| Total functional capacity, mean (SD) | ||||||
| V1 | 13.0 (0.2) | 12.8 (0.5) | 12.7 (0.7) | 12.8 (0.4) | 11.0 (1.5)Φ | |
| V2 | 12.9 (0.5) | 12.6 (0.9) | 12.7 (0.6) | 12.5 (1.0) | 10.3 (2.2)Φ | |
| UHDRS‐TMS, mean (SD) | ||||||
| V1 | 2.6 (2.5) | 2.6 (1.5) | 2.0 (1.5) | 3.1 (1.2) | 14.6 (7.7)Φ | |
| V2 | 2.1 (1.6) | 5.7 (5.1) ¥ | 3.5 (2.2) | 8.3 (6.1)*^ | 23.0 (12.1)Φ | |
| SDMT, mean (SD) | ||||||
| V1 | 49.4 (8.9) | 50.1 (11.0) | 53.5 (9.3) | 46.7 (11.9) | 41.2 (9.2)Φ | |
| V2 | 50.9 (9.3) | 50.6 (10.0) | 54.7 (10.0) | 46.6 (8.5)^ | 39.2 (10.6)Φ | |
| SWR, mean (SD) | ||||||
| V1 | 100.1 (13.2) | 91.9 (14.2)* | 95.6 (9.6) | 88.3 (17.3)* | 87.7 (14.7)* | |
| V2 | 102.0 (15.6) | 87.9 (15.7)* | 91.4 (9.4) | 84.4 (20.0)* | 86.4 (18.6)* | |
| BDI‐II, mean (SD) | ||||||
| V1 | 4.1 (4.4) | 6.4 (6.4) | 4.9 (6.0) | 7.9 (6.8) | 10.2 (8.2)* | |
| V2 | 3.9 (4.1) | 5.1 (5.6) | 3.2 (4.9) | 6.9 (5.9) | 8.2 (8.4) | |
| Between‐scan interval in months, mean (SD) | 23.0 (0.8) | 23.0 (0.7) | 23.2 (0.6) | 22.7 (0.7) | 23.5 (0.7) |
N = number of participants, SD = Standard deviation, n/a = not applicable, ISCED = International Standard Classification of Education, DART‐IQ = Dutch Adult Reading Test Intelligence Quotient, CAG = Cytosine‐Adenine‐Guanine, UHDRS‐TMS = Unified Huntington's Disease Rating Scale‐Total Motor Score, SDMT = Symbol Digit Modalities Test, SWR = Stroop Word Reading task, BDI‐II = Beck Depression Inventory‐II, V1 = visit 1, V2 = visit 2.
Significance at P ≤ 0.05 level: * significantly different from controls, Φ significantly different from controls and preHD, ¥ significantly different from controls and HD, ^ significantly different from preHD‐A.
Including five subjects progressing to the early manifest stage during the two year follow‐up period.
Inclusion criteria for the preHD group were a CAG repeat ≥ 40 with a total motor score on the Unified Huntington's Disease Rating Scale (UHDRS‐TMS) ≤ five. Inclusion criteria for the early manifest HD group were a CAG repeat ≥ 40, with a UHDRS‐TMS ≥ five and a Total Functional Capacity score (TFC) ≥ seven. A further inclusion criterion for both the preHD and early manifest HD group consisted of a burden of pathology score greater than 250 [(CAG repeat length − 35.5) × age) [Penney et al., 1997; Tabrizi et al., 2009]. Healthy gene negative family members or partners were recruited as control subjects. None of the participants suffered from a concomitant neurological disorder, a major psychiatric diagnosis or had a history of severe head injury.
Hemispheric dominance was defined using a standardised neuropsychological questionnaire [Oldfield, 1971]. For preHD subjects, the predicted years to disease onset was calculated using the CAG repeat length and age‐based survival analysis of Langbehn et al. [2004].
As previously applied by Tabrizi et al. [2009], to assess the effect of expected proximity to disease onset on diffusion parameters, the preHD group was divided at baseline according to the median (10.9 years) for the predicted years to disease onset into preHD‐A (≥ 10.9 years) and preHD‐B (<10.9). This resulted in two groups each consisting of eleven subjects (Table 1).
The study was approved by the Medical Ethics Committee of the LUMC and written informed consent was obtained from all participants. For full details of study parameters, see Tabrizi et al. [2009].
Clinical Measures
To monitor disease state, the following clinical measures were evaluated longitudinally for all groups: UHDRS‐TMS, TFC, Symbol Digit Modalities Test (SDMT), Stroop Word Reading (SWR) and Beck Depression Inventory‐II (BDI‐II) scores.
The UHDRS‐TMS is the traditional measure which defines manifest disease state in HD. The SDMT and SWR in particular have been shown to be sensitive longitudinal neurocognitive measures in HD, independent of disease related motor effects [Tabrizi et al., 2011].
Magnetic Resonance Imaging Acquisition
MRI acquisition was performed with a 3‐Tesla whole‐body scanner (Philips Achieva, Healthcare, Best, The Netherlands) with an eight channel SENSE head coil. T1‐weighted image volumes were acquired using a 3D MPRAGE acquisition sequence with the following imaging parameters: TR = 7.7 ms, TE = 3.5 ms, FOV = 24 × 24 cm2, matrix size 224 × 224, number of slices = 164, slice thickness = 1.00 mm, and no slice gap. A single‐shot echo‐planar diffusion tensor imaging sequence was applied with 32 measurement directions and the following scan parameters [Jones and Leemans, 2011]: TR = 10,004 ms, TE = 56 ms, FOV = 220 × 220 mm2 with an acquisition matrix of 112 × 110, 2.00 mm slice thickness, transversal slice orientation, no slice gap, flip angle = 90°, reconstruction voxel dimensions of 1.96 × 1.96 × 2.00 mm3, number of slices = 64, b‐value = 1,000 s/mm2, halfscan factor = 0.61. Parallel imaging (SENSE) was used with a reduction factor of two, NSA = one, and fat suppression was applied. DTI acquisition time was 6.55 min.
Image Processing
The DTI data was processed as described in Deprez et al. [2013]. In summary, this consisted of the following steps: (1) Correction for subject motion and eddy current induced distortions [Leemans and Jones, 2009]; (2) Correction for echo planar images based deformations due to magnetic field inhomogeneities by registration to the T1‐weighted images [Irfanoglu et al., 2012]; and (3) Tensor estimation using the iteratively reweighted linear least squares approach after outlier detection and removal by REKINDLE (κ = 6) [Tax et al., 2014; Veraart et al., 2013].
The brain regions were segmented into WM and GM regions (Fig. 1) using SPM 8 with default settings (revision 4667, 27‐Feb‐2012) [Ashburner and Friston, 2005]. Brain regions were left/right divided with the method described by Kuijf et al. [2013].
Figure 1.
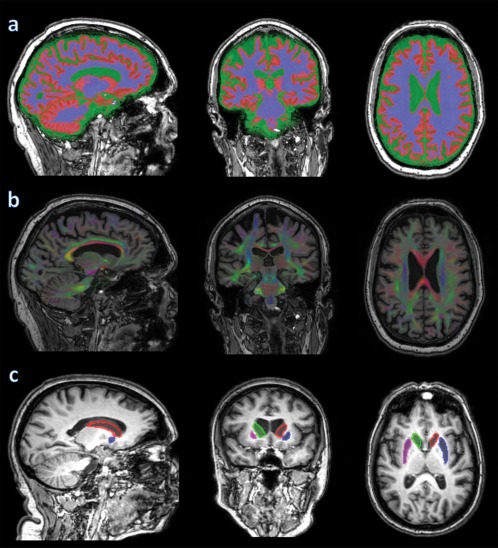
From left to right: sagittal, coronal and axial images: (a) brain segmentation into WM (blue), GM (red) and CSF (green); (b) directionally colour encoded fractional anisotropy map; (c) striatal mask: red = left caudate nucleus and blue = left putamen; green = right caudate nucleus and pink = right putamen. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Histogram Analysis
A spherical erosion filter (radius 2 mm) was applied to the brain masks (WM/GM; left/right) to minimize the inclusion of partial‐volume affected voxels [Cercignani, 2010; Van den Boomgaard and van Balen, 1992]. The histograms of the diffusion measures were computed from these segmented brain regions. Subsequently, histograms were normalised by the number of voxels in each subject's data set to create the group mean histograms [Roine et al., 2013].
With histogram analysis, frequency distributions of selected DTI measures of designated voxels can be obtained. While not providing any region‐specific information, this type of analysis is highly sensitive in detecting differences as the entire brain is included. Moreover, it provides a straightforward, fully automated and objective approach for interrogating imaging data. The resulting summarizing whole‐brain measures are suitable for comparing diffusion between groups [Deprez et al., 2013] and its value has been previously demonstrated in multiple sclerosis [Cercignani et al., 2001; Vrenken et al., 2006] and CADASIL [Holtmannspotter et al., 2005]. This type of analysis can also be applied to any given selection of voxels of interest. Given the importance of the striatum in the histopathological profile of HD, diffusion values for this structure were additionally evaluated in this study. The following diffusion features for whole‐brain WM were investigated: FA, MD, AD and RD. In addition, for the whole‐brain GM (including striatum) the MD, AD and RD were studied. The outcome measures were the mean and distribution peak heights of the histograms. Because two outcome measures were tested against two tissue types, P‐values for omnibus F‐tests were Bonferroni corrected to adjust for the increased risk of type one error and considered to be statistically significant at P ≤ 0.05/4 = 0.0125.
Obtaining Striatal Masks
Striatal masks were obtained as described previously [Van den Bogaard et al., 2012]. In summary, T1‐weighted images were segmented with the FAST [Zhang et al., 2001] and FIRST [Patenaude et al., 2011] tools from the fMRI of the Brain Software Library (http://www.fmrib.ox.ac.uk/fsl/) [Smith et al., 2004]. This provided individual brain masks for the following structures: the caudate nucleus and the putamen, both of these forming the striatum. Figure 1 shows such a segmentation result superimposed on a T1‐weighted image. To correct for potential partial volume effects, an eroded mask of these segmentations was created by removing one voxel in‐plane for all the aforementioned voxels of interest.
Statistical Analysis
We used linear mixed models (in R version 3.0.0, R Foundation for Statistical Computing, Vienna, Austria) to model the various outcome variables with patient as a random factor to accommodate the within‐person repeated nature of the data and to assess the effect of group, corrected for age at time of scanning as a co‐variable. Correlations between neurocognitive measures and DTI findings were tested in the model.
Statistical analyses of group demographics were performed with SPSS (version 20, IBM, USA). Distributions and assumptions were checked. Either Analysis of Variance (ANOVA) or Chi‐squared tests were applied where this was appropriate. Potential longitudinal change in clinical measures between the groups was also investigated. Difference values were computed and an ANOVA was performed on these delta‐scores to evaluate potential group differences. In case of a significant omnibus F‐test, exploratory post hoc analysis using Fisher's least significant difference was performed to assess which means were significantly different from each other. Differences in group demographics between preHD‐A and preHD‐B were compared using either independent‐samples t‐tests or Chi‐squared tests, where appropriate.
Paired‐samples t‐tests were performed to assess cross‐sectional interhemispheric differences in DTI measures within the groups after excluding lefthanders. Lefthanders consisted of four control, four preHD and one manifest HD subjects. The longitudinal evolution of the interhemispheric diffusion measures was assessed with the aforementioned linear mixed model.
RESULTS
Group Characteristics and Clinical Scores
The groups did not differ significantly in terms of gender, handedness, level of education, intelligence quotient, or body mass index. A trend toward a difference in age between the groups was found (P = 0.06), with premanifest subjects being generally younger compared to both controls and subjects with manifest HD. No statistical difference was found in CAG repeat count between preHD and manifest HD subjects. The between‐scan interval was not significantly different between the groups.
At baseline, significantly lower scores for subjects with manifest HD were found in TFC, SDMT and SWR when compared to both controls and preHD subjects. Higher scores for subjects with manifest HD were found for UHDRS‐TMS and BDI‐II when compared to both controls and preHD subjects. For the preHD group, a significantly lower baseline score compared to controls was found for SWR (Table 1).
Repeated assessment after 2‐year follow‐up revealed similar score differences between the groups. Progression of five of the 22 preHD subjects to the early manifest stage during the follow‐up period gave rise to a significantly higher UHDRS‐TMS when compared to controls. The only significant difference in longitudinal change of clinical scores was found in higher UHDRS‐TMS, both when considering the preHD group (including those progressing to the early manifest stage) and the manifest HD group. Other scores showed no significant longitudinal differences in this cohort (Supporting Information Table 1).
Comparing the preHD‐A and preHD‐B groups, no significant cross‐sectional score differences were found during the first visit. At the second visit, the preHD‐B group showed a significantly higher UHDRS‐TMS and lower SDMT score compared to preHD‐A. Significant longitudinal change was found only in the UHDRS‐TMS, where the difference was higher in preHD‐B relative to preHD‐A (Table 1; longitudinal change data not shown).
Diffusion Tensor Imaging Histogram Measures
Diffusivity values of whole‐brain white matter
At baseline, all whole‐brain WM diffusivity measures in the manifest HD group differed significantly from both controls and preHD subjects (Table 2): FA values were reduced and MD, AD and RD were increased. Upon applying Bonferroni correction for multiple testing, all these differences remained statistically significant except for the difference in FA (see Supporting Information Figs. 1 and 2 for group and visit histogram plots of WM FA, including separate plots for the left and right hemisphere). Elevations in MD, AD and RD were all highly significant (P ≤ 0.001) (see Fig. 2 for histogram plots of WM MD).
Table 2.
Mean whole‐brain DTI parameters. MD, AD and RD are shown x103 for readability
| Healthy controls | preHD (A and B) | preHD‐A | preHD‐B | Manifest HD | |
|---|---|---|---|---|---|
| N | 24 | 22 | 11 | 11 | 10 |
| FA‐WM | 0.434 (0.008) | 0.435 (0.012) | 0.435 (0.014) | 0.435 (0.014) | 0.421 (0.014)Φ* |
| MD‐WM | 0.754 (0.010) | 0.764 (0.016) | 0.758 (0.017) | 0.767 (0.017) | 0.783 (0.018)Φ*** |
| MD‐GM | 0.767 (0.004) | 0.777 (0.010) | 0.768 (0.024) | 0.778 (0.024) | 0.805 (0.012)Φ*** |
| AD‐WM | 1.123 (0.005) | 1.140 (0.011)¥** | 1.131 (0.012) | 1.149 (0.012)϶*** | 1.172 (0.013)Φ*** |
| AD‐GM | 0.924 (0.013) | 0.934 (0.019) | 0.923 (0.025) | 0.938 (0.025)¤ | 0.965 (0.021)Φ*** |
| RD‐WM | 0.560 (0.011) | 0.566 (0.017) | 0.562 (0.019) | 0.568 (0.019) | 0.589 (0.019)Φ*** |
| RD‐GM | 0.702 (0.004) | 0.711 (0.010) | 0.706 (0.012) | 0.716 (0.012)^ | 0.736 (0.012)Φ*** |
Data is shown as mixed model‐based estimates of the group means corrected for age (S.E.).
Φ significantly different from controls and preHD, ¥ significantly different from controls and HD, ϶ significantly different from controls, preHD‐A and HD, *P ≤ 0.05 **P ≤ 0.01 ***P ≤ 0.001, bold values indicate sustained significant difference following Bonferroni correction (P ≤ 0.0125), ¤ P = 0.08, ^ P = 0.07.
FA = fractional anisotropy; MD = mean diffusivity; AD = axial diffusivity; RD = radial diffusivity; WM = white matter; GM = gray matter.
Figure 2.
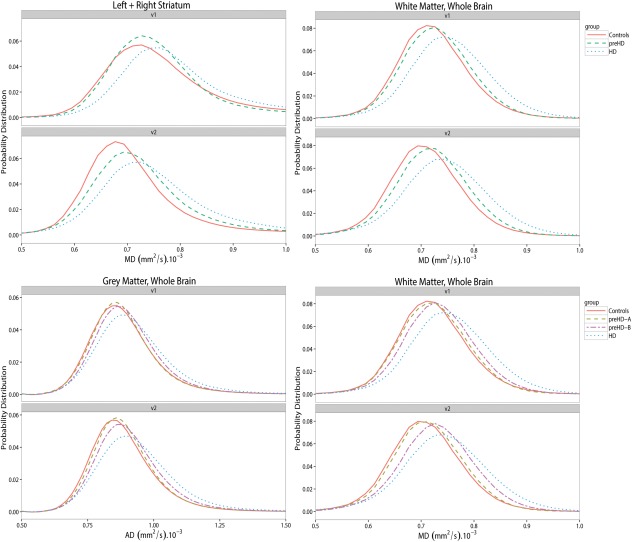
Histogram plots of MD (= mean diffusivity) and AD (= axial diffusivity) in whole brain white, gray matter and the striatum. Group diffusivities are plotted against the visits. v1 = visit 1, v2 = visit 2. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Only AD in the preHD group differed significantly from both controls and subjects with manifest HD and was lower for the controls and higher for subjects with manifest HD, even after applying Bonferroni correction (P ≤ 0.01). No statistically significant differences in FA (P = 0.83), MD (P = 0.10) or RD (P = 0.33) were found between controls and preHD subjects.
Dividing the preHD group in preHD‐A and preHD‐B revealed higher AD values only in the preHD‐B group compared to both preHD‐A and controls, even after Bonferroni correction (P ≤ 0.001). No significant differences were observed in any of the diffusivity measures between controls and preHD‐A (Table 2). No significant longitudinal differences were found in the degree of whole‐brain WM diffusivity change in any of the measures between the groups (without correction for multiple testing). Results of histogram peak height comparison of whole‐brain WM are provided in Supporting Information.
Diffusivity values of whole‐brain gray matter and striatum
At baseline, MD, AD and RD values of whole‐brain GM were significantly higher for the manifest HD group compared to both controls and preHD subjects (P ≤ 0.001; Table 2). This remained the case after Bonferroni correction for multiple testing. Figure 2 shows histogram plots for whole‐brain GM AD.
No significant differences in whole‐brain GM diffusivity measures were found between preHD subjects and controls. Upon dividing the preHD group in preHD‐A and preHD‐B, a trend was found in the preHD‐B group toward higher values of AD and RD compared to controls (P = 0.08 and P = 0.07, respectively; Table 2).
Baseline MD, AD and RD values in the striatum of subjects with manifest HD were significantly higher compared to both controls and preHD subjects (Table 3). Upon applying Bonferroni correction for multiple testing, these differences remained statistically significant except for RD. See Figure 2 for group histogram plots of striatal MD. Separate plots for MD of the left and right striatum are shown in Supporting Information Figure 3.
Table 3.
Mean striatal DTI parameters. Values are of left and right striatum together. MD, AD and RD are shown ×103 for readability
| Healthy controls | preHD (A and B) | preHD‐A | preHD‐B | Manifest HD | |
|---|---|---|---|---|---|
| N | 24 | 22 | 11 | 11 | 10 |
| MD | 0.686 (0.075) | 0.695 (0.037) | 0.648 (0.044) | 0.758 (0.043) | 0.816 (0.045)Φ** |
| AD | 1.130 (0.093) | 1.177 (0.027)¤ | 1.127 (0.032) | 1.227 (0.031)϶** | 1.235 (0.034)Φ** |
| RD | 0.658 (0.108) | 0.641 (0.039) | 0.595 (0.048) | 0.684 (0.047) | 0.764 (0.049)Φ* |
Data is shown as mixed model‐based estimates of the group means corrected for age (S.E.)
Φ significantly different from controls and preHD, ϶ significantly different from controls and preHD‐A, ¤ P = 0.08 (compared to controls), *P ≤ 0.05 **P ≤ 0.01, bold values indicate sustained significant difference following Bonferroni correction (P ≤ 0.0125).
MD = mean diffusivity; AD = axial diffusivity; RD = radial diffusivity.
Figure 3.
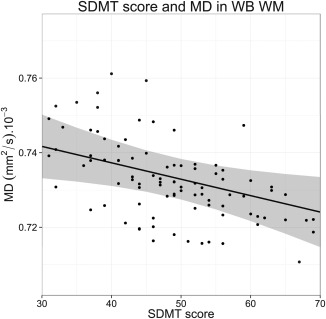
Relationship plot of Symbol Digit Modalities Test (SDMT) score and whole brain (WB) white matter (WM) mean diffusivity (MD). Data points shown are mixed model‐based estimates.
No significant baseline differences in striatal diffusivity measures were found between preHD subjects and controls, only a trend toward a higher AD in the preHD group (P = 0.08). Upon dividing the preHD group in preHD‐A and preHD‐B, a significantly higher Bonferroni corrected striatal AD value was found in preHD‐B only, compared to both controls and preHD‐A (P ≤ 0.01; Table 3). Exploratory analysis to assess whether this effect was more prominent when assessing striatal substructures separately, revealed a trend toward AD elevation in the caudate and a significantly higher AD in the putamen in preHD‐B (caudate: P = 0.06; putamen: P = 0.02) compared to both controls and preHD‐A. This result was therefore less sensitive than the combined assessment of both substructures (P ≤ 0.01), and would not have survived Bonferroni correction. No significant longitudinal differences were found in the degree of whole‐brain GM or in striatal diffusivity change in any of the measures between the groups (without correction for multiple testing). Results of histogram peak height comparison of whole‐brain GM and striatum are provided in Supporting Information.
Neurocognitive and diffusivity measures
In Table 4, significant correlations between neurocognitive measures and baseline whole‐brain diffusivity measures are shown (correlations with peak heights are not shown). As no specific group effects were found on correlations between diffusion parameters and neurocognitive measures, the following applied to all participants included in the study with a CAG repeat expansion irrespective of their group. The SDMT score was found to predict WM FA (P ≤ 0.01): the higher the SDMT score, the higher the FA (Supporting Information Fig. 4). The SDMT score was also found to predict WM MD (P ≤ 0.01): the higher the SDMT score, the lower the MD (Fig. 3).
Table 4.
Mean whole‐brain DTI parameters and neurocognitive measures correlations (corrected for age)
| Diffusion parameter | SDMT score | P | Diffusion parameter | SWR score | P | |
|---|---|---|---|---|---|---|
| FA‐WM | ↑1.2% | ↑10 points | ≤ 0.01 | / | / | / |
| MD‐WM | ↓0.7% | ↑10 points | ≤ 0.01 | / | / | / |
| MD‐GM | / | / | / | ↓0.4% | ↑10 points | ≤ 0.05 |
| AD‐WM | / | / | / | / | / | / |
| AD‐GM | ↓0.5% | ↑10 points | ≤0.05 | ↓0.4% | ↑10 points | ≤0.05 |
| RD‐WM | ↓1.0% | ↑10 points | ≤0.01 | / | / | / |
| RD‐GM | / | / | / | / | / | / |
This table is valid for all participants with a CAG repeat expansion included in the study, as no specific group effects were found on correlations between diffusion parameters and neurocognitive measures. ↑ = increase, ↓ = decrease, / = no significant correlation.
FA = fractional anisotropy; MD = mean diffusivity; AD = axial diffusivity; RD = radial diffusivity; WM = white matter; GM = gray matter.
The SWR score was found to predict GM MD (P ≤ 0.05): the higher the SWR score, the lower the MD (Supporting Information Fig. 5). The SDMT score was found to predict peak height in GM MD (P ≤ 0.05): the higher the SDMT score, the higher the peak height. The SDMT score was also found to predict peak height of WM AD (P ≤ 0.01): the higher the SDMT score, the lower the peak height.
Both SDMT and SWR scores were found to predict GM AD (P ≤ 0.05): the higher the score, the lower the AD. The SDMT score was found to predict peak height of GM AD (P ≤ 0.05): the higher the SDMT score, the higher the peak height.
The SDMT score was found to predict WM RD (P ≤ 0.01): the higher the SDMT score, the lower the RD. In the striatum, the SDMT score alone was found to predict AD (P ≤ 0.05): the higher the SDMT score, the lower the AD (data not shown).
Interhemispheric differences in diffusivity measures
In Supporting Information Table 3, baseline differences in diffusivity measures of the left minus right hemisphere are shown, both for WM and GM. Only right handed subjects were included for this analysis. Many small, though significant interhemispheric differences were found. The magnitude and direction of these differences were similar in all groups (controls, preHD and manifest HD) with no statistical significance in these differences between the groups.
No significant interhemispheric longitudinal differences between the groups were found in the degree of change of any of the diffusion measures of the WM, GM and the striatum, neither in the means nor histogram peak heights (without correction for multiple testing).
DISCUSSION
The major findings from this study were significantly higher MD, AD, and RD values in both WM and GM in subjects with manifest HD compared to preHD and control subjects. In preHD subjects, only WM AD proved to be a sensitive measure to differentiate between the study groups. This finding remained valid only in preHD‐B upon dividing the preHD group according to the median predicted years to onset. Another significantly different finding in preHD subjects was observed again only in preHD‐B in a higher AD of the striatum compared to both controls and preHD‐A. No significant longitudinal differences were found in any of the diffusivity measures between any of the groups, neither in the means nor peak heights. Finally, significant relationships between neurocognitive and diffusivity measures were demonstrated.
Findings of increased MD, AD and RD values in subjects with manifest HD are in line with results from previous reports [Bohanna et al., 2011; Della et al., 2010; Hobbs et al., 2012]. Although a reduction in WM FA in manifest HD was found, this finding did not maintain significance after correction for multiple testing, rendering it a far less sensitive marker for disease state in HD. This finding of individual diffusivities providing more sensitive measures for revealing pathologic microstructural brain alterations compared to FA, was in line with findings from a previous study in HD [Hobbs et al., 2012] and Alzheimer's disease [Acosta‐Cabronero et al., 2010]. The results presented here are also in agreement with previous findings by our group, where MD was reported to be a more sensitive measure than FA in distinguishing HD subjects from controls [Dumas et al., 2012]. Just as in the Alzheimer's disease study of Acosta‐Cabronero et al. [2010], changes found in this study were more prominent in AD than in RD, yet not enough to substantially influence FA. This provides a possible explanation for the seemingly discrepant findings of FA alterations in HD research, as the proportions of eigenvalues could be more specifically altered in studies of distinct WM regions giving rise to a modified FA.
The presence of an increased AD in whole‐brain WM and in the striatum of preHD‐B, provides evidence for ongoing neurodegeneration prior to disease manifestation, a finding that is echoed by results from previous MRI volumetric investigations in preHD [Aylward et al., 2012; Paulsen et al., 2010; Stoffers et al., 2010; Tabrizi et al., 2009; Van den Bogaard et al., 2011]. Higher AD in preHD has been previously reported by Stoffers et al. [2010], although in that study this finding was highly localized and accompanied by more pronounced and widespread increases in RD, a finding which was not replicated here. Furthermore, in the study of Stoffers et al., RD seemed to correlate with the predicted years to disease onset, while AD lacked such correlation. This stands in contrast to our findings of lack of significant increases in RD irrespective of preHD group stratification and higher AD being found primarily in preHD individuals who are closest to predicted years to disease onset. The discrepancy in these findings could very well be attributed to the differing methodologies applied in analysing the data and possibly due to the difference in scanner field strength used. In GM, no significantly different diffusivities were present between preHD subjects and controls, except for the aforementioned higher AD in the striatum of preHD‐B, which is a deep GM structure. The differences found in peak heights were only present in subjects with manifest HD, not in the preHD group, alluding to a less sensitive measure in detecting differences between manifest HD, preHD, and controls.
Exploration of the longitudinal evolution of diffusivity measures, without correction for multiple comparisons, provided no significant group differences. Results from previous longitudinal DTI studies in HD are heterogeneous. In the study of Weaver et al. [2009], significant longitudinal decreases in WM FA and AD were reported over a one year period. That study consisted of seven controls, four preHD and three manifest HD subjects, where the seven (pre)manifest subjects were compared to the controls. In another study by Sritharan et al. [2010] with 17 controls and 18 manifest HD subjects, no longitudinal change in the MD of the caudate, putamen, thalamus and corpus callosum could be demonstrated over a one year period, while baseline MD was significantly higher in the caudate and putamen of subjects with manifest HD compared to controls. A similar finding in MD was reported by Vandenberghe et al. [2009] in eight manifest HD subjects over a 2‐year period. Results from the present study are in agreement with findings from the latter two studies, with significant cross‐sectional differences found in combination with a lack of significant longitudinal differences in the evolution of these measures within the 2‐year study‐period. The lack of longitudinal differences in the diffusion profile between the groups in this study could be due to a low sensitivity of this approach in detecting small changes over time or due to a true absence of observable significant alterations of this profile using DTI in the 2‐year time‐frame.
Relationships between neurocognitive and diffusivity measures were demonstrated in our study. The SDMT and SWR scores were associated with some diffusivity measures, where the SDMT seemed to be more readily associated with WM diffusivity measures, while SWR showed associations only with GM AD and MD. The only exception to this pattern in the whole‐brain analysis, was the inverse relationship found between SDMT scores and GM AD values. These findings are important in light of selecting the most suitable cognitive measures to assess, depending on the prime target of a treatment intervention. The SDMT, considered to be a measure for information‐processing speed and working memory, has also been found to be more associated with white than gray matter lesions in multiple sclerosis [Papadopoulou et al., 2013]. In the current study, the SDMT provided for the best predictive value for baseline diffusivity measures, as reflected by both the magnitude as well as the statistical significance of these associations. As was the case in the recent study by Poudel et al. [2014], we found a significant inverse relationship between SDMT and WM RD in HD. Our results did not, however, reproduce their finding for the same inverse relationship with SWR. In the striatum, an inverse relationship was found only between the SDMT score and AD. This finding is reinforced by the recent morphometric analysis report in preHD by Harrington et al. [2014], where the SDMT score was found to be positively associated with putaminal volume.
Additional findings from our interhemispheric subanalysis of diffusion parameters revealed very small, though highly significant interhemispheric differences in diffusivity measures within the groups. There were, however, no indications for a preferential degeneration to the dominant hemisphere in (pre)HD subjects, as no significant group differences were found in interhemispherical diffusion parameters. To the best of our knowledge this is the first study exploring this hypothesis using DTI in (pre)HD subjects. Interhemispheric variations in diffusivity measures in the healthy human brain have been previously reported [Park et al., 2004; Yoshiura et al., 2010].
It should be stressed that inferral of underlying alterations to biological substance through changes in eigenvalues is not trivial, especially in GM [Beaulieu, 2002; Jones et al., 2013]. As such, it is quite challenging to draw solid conclusions about underlying neuropathology based on diffusion parameters. The progressive histopathological features of HD are numerous. Disturbed membrane systems of neurons, with derangement of all membranes that form the cell were found in a histological study by Tellez‐Nagel et al. [1974]. Loss of small spiny neurons in the caudate and putamen with subsequent astrocytosis [Vonsattel et al., 1985], and decreased neuronal densities with increased oligodendroglial densities [Myers et al., 1991], the latter found already in preHD [Gomez‐Tortosa et al., 2001], have been described. The primary role of the oligodendrocyte is providing myelin to neuronal axons. In HD mouse models, inhibition of the peroxisome‐proliferator‐activated receptor gamma coactivator 1 α in oligododrocytes by mutant huntingtin was found to be responsible for abnormal myelination [Xiang et al., 2011]. WM atrophy due to myelin breakdown is supported by histological and imaging examinations in HD subjects [Bartzokis et al., 2007]. Significantly reduced total brain, GM and WM volumes through atrophy have been demonstrated through a post mortem study in seven HD brains [Halliday et al., 1998]. These various, diverse changes could result in a competing influence on the diffusion tensor model based on the individual contributions and timing of each change. In a DTI‐histological study of the quinolinic acid rat model of HD, Van Camp et al. [2012] demonstrated that DTI was more sensitive in detecting subtle changes in the affected structures compared to histology. In that study, increases in MD, AD, and RD were detected six weeks after neurotoxin infusion as compared to the sham injected control group, with histological findings of necrotic cells involvement with shrunken cytoplasm and spongiosis.
In this study, the pattern found in the manifest HD group of higher MD, AD, and RD values without substantial changes to FA, likely reflects an increase in tissue permeability, extracellular space fluid and interaxonal spacing due to neural tissue loss [Sen and Basser, 2005; Sotak, 2004], allowing the three eigenvalues to grow proportionally due to faster diffusion of water, hereby effecting only the size of the tensor without influencing its shape [Acosta‐Cabronero et al., 2010]. This pattern of diffusivity changes, which has been associated with chronic WM degeneration [Burzynska et al., 2010; Concha et al., 2006], has previously been reported in HD [Hobbs et al., 2012] and other neurodegenerative disorders, such as amyotrophic lateral sclerosis [Metwalli et al., 2010] and hereditary spastic paraplegia [Oguz et al., 2013]. Findings from the histologically verified DTI study of the quinolinic acid rat model of HD, suggest that this pattern could point to cytoplasmic alterations and spongiosis [Van et al., 2012]. In our complete preHD cohort, only WM AD showed a significantly raised value compared to controls. Increased AD may indicate WM axonal atrophy and was suggested to be useful in identifying early changes in persons with a high risk at developing Alzheimer's disease, prior to cognitive decline [Gold et al., 2012]. Taken together, these findings suggest that both axonal degeneration as well as demyelination play an important role in WM pathophysiology of HD and are present throughout the entire brain. Given that the earliest detected abnormality is represented in the WM AD in preHD subjects, this could indicate that axonal degeneration precedes myelin abnormalities in WM at this stage of the neurodegenerative process, reinforcing findings by Hobbs et al. [2012] and further supporting this hypothesis. The GM diffusivity findings presented here suggest that tissue boundaries become less well defined in the cortical ribbon and the striatum in HD [Beaulieu, 2002].
Strengths of this study include the longitudinal design which has the advantage of evaluating the evolution of diffusivity measures in a well‐defined study group with a similar between‐scan interval. All scans were acquired on the same scanner using the same protocol, which keeps test–retest variation in DTI to a minimum [Takao et al., 2012]. Exploration of the full tensor behaviour is a further strength, as demonstrated by the better sensitivity in revealing differences between the groups in this study relative to FA characteristics. For the whole‐brain analyses we applied an automated histogram analysis, which reduces user error and provides a more suitable standardized analysis method in multicentre study settings. The limitation presented with whole‐brain analysis is the loss of topographic information. Also, proper interpretation of the underlying biological causes to alterations found in the diffusion profile remains restricted, as many different fiber orientations are found in diffusion images of the brain [Jeurissen et al., 2013]. That does not, however, preclude the ability of assessing the value of this type of analysis for identifying biomarker potential and tracking disease‐related modifications to the diffusion profile in time. This limitation was nonetheless addressed by applying this analysis specifically to the striatum. A further limitation was the relatively low number of manifest participants. This was mainly driven by disease progression in the cohort, where longitudinal scans or the ability to comply with study protocol deemed impossible, leaving the outcome measures presented here to more likely be an underestimation of the true extent of diffusion disturbances in the HD brain.
To conclude, alterations in cross‐sectional diffusion profiles between manifest HD subjects and controls were evident both in whole‐brain and striatum. In preHD, only AD alterations were found, a finding that applied only to preHD‐B upon group stratification. This suggests that AD may be a sensitive marker for early change in HD gene carriers prior to disease manifestation. The individual diffusivities proved to be more sensitive in distinguishing pathologic microstructural alterations to the HD brain than FA characteristics. This study showed no longitudinal differences in any of the diffusivity measures between the groups. Larger study samples could provide additional information on the longitudinal biomarker potential of DTI measures. However, based on the results presented here, this potential is expected to be limited.
Supporting information
Supplementary Information Figure 1.
Supplementary Information Figure 2.
Supplementary Information Figure 3.
Supplementary Information Figure 4.
Supplementary Information Figure 5.
Supplementary Information Tables.
ACKNOWLEDGMENTS
TRACK‐HD is supported by CHDI/High Q Foundation Inc., a not for profit organization dedicated to finding treatments for Huntington's disease. The authors wish to thank Sarah Tabrizi, University College London, who is the global PI for TRACK‐HD and clinical site PI for London. The authors also wish to extend their gratitude to the TRACK‐HD investigators responsible for collecting the data and to the study participants and their families. The authors declare that there are no conflicts of interest.
REFERENCES
- Acosta‐Cabronero J, Williams GB, Pengas G, Nestor PJ (2010): Absolute diffusivities define the landscape of white matter degeneration in Alzheimer's disease. Brain 133:529–539. [DOI] [PubMed] [Google Scholar]
- Alexander AL, Lee JE, Lazar M, Field AS (2007): Diffusion tensor imaging of the brain. Neurotherapeutics 4:316–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner J, Friston KJ. Unified segmentation (2005): Neuroimage; 26:839–851. [DOI] [PubMed] [Google Scholar]
- Aylward EH, Liu D, Nopoulos PC, Ross CA, Pierson RK, Mills JA, Long JD, Paulsen JS (2012): Striatal volume contributes to the prediction of onset of Huntington disease in incident cases. Biol Psychiatry 71:822–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartzokis G, Lu PH, Tishler TA, Fong SM, Oluwadara B, Finn JP, Huang D, Bordelon Y, Mintz J, Perlman S (2007): Myelin breakdown and iron changes in Huntington's disease: pathogenesis and treatment implications. Neurochem Res 32:1655–1664. [DOI] [PubMed] [Google Scholar]
- Basser PJ, Mattiello J, LeBihan D (1994): MR diffusion tensor spectroscopy and imaging. Biophys J 66:259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu C (2002): The basis of anisotropic water diffusion in the nervous system ‐ a technical review. NMR Biomed 15:435–455. [DOI] [PubMed] [Google Scholar]
- Bohanna I, Georgiou‐Karistianis N, Sritharan A, Asadi H, Johnston L, Churchyard A, Egan G (2011): Diffusion tensor imaging in Huntington's disease reveals distinct patterns of white matter degeneration associated with motor and cognitive deficits. Brain Imaging Behav 5:171–180. [DOI] [PubMed] [Google Scholar]
- Burzynska AZ, Preuschhof C, Backman L, Nyberg L, Li SC, Lindenberger U, Heekeren HR (2010): Age‐related differences in white matter microstructure: region‐specific patterns of diffusivity. Neuroimage 49:2104–2112. [DOI] [PubMed] [Google Scholar]
- Cercignani M (2010): Strategies for patient‐control comparison of diffusion MR Data In: Jones DK, editor. Diffusion MRI: Theory, Methods, and Applications. Oxford, UK: Oxford University Press; pp 485–499. [Google Scholar]
- Cercignani M, Inglese M, Pagani E, Comi G, Filippi M (2001): Mean diffusivity and fractional anisotropy histograms of patients with multiple sclerosis. AJNR Am J Neuroradiol 22:952–958. [PMC free article] [PubMed] [Google Scholar]
- Concha L (2014): A macroscopic view of microstructure: Using diffusion‐weighted images to infer damage, repair, and plasticity of white matter. Neuroscience 276:14–28. [DOI] [PubMed] [Google Scholar]
- Concha L, Gross DW, Wheatley BM, Beaulieu C (2006): Diffusion tensor imaging of time‐dependent axonal and myelin degradation after corpus callosotomy in epilepsy patients. Neuroimage 32:1090–1099. [DOI] [PubMed] [Google Scholar]
- Della NR, Ginestroni A, Tessa C, Giannelli M, Piacentini S, Filippi M, Mascalchi M (2010): Regional distribution and clinical correlates of white matter structural damage in Huntington disease: A tract‐based spatial statistics study. AJNR Am J Neuroradiol 31:1675–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deprez S, Billiet T, Sunaert S, Leemans A (2013): Diffusion tensor MRI of chemotherapy‐induced cognitive impairment in non‐CNS cancer patients: A review. Brain Imaging Behav 7:409–435. [DOI] [PubMed] [Google Scholar]
- Dumas EM, van den Bogaard SJ, Ruber ME, Reilman RR, Stout JC, Craufurd D, Hicks SL, Kennard C, Tabrizi SJ, van Buchem MA, van der Grond J, Roos RA (2012): Early changes in white matter pathways of the sensorimotor cortex in premanifest Huntington's disease. Hum Brain Mapp 33:203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold BT, Johnson NF, Powell DK, Smith CD (2012): White matter integrity and vulnerability to Alzheimer's disease: preliminary findings and future directions. Biochim Biophys Acta 1822:416–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Tortosa E, MacDonald ME, Friend JC, Taylor SA, Weiler LJ, Cupples LA, Srinidhi J, Gusella JF, Bird ED, Vonsattel JP, Myers RH (2001): Quantitative neuropathological changes in presymptomatic Huntington's disease. Ann Neurol 49:29–34. [PubMed] [Google Scholar]
- Hadzi TC, Hendricks AE, Latourelle JC, Lunetta KL, Cupples LA, Gillis T, Mysore JS, Gusella JF, MacDonald ME, Myers RH, Vonsattel JP (2012): Assessment of cortical and striatal involvement in 523 Huntington disease brains. Neurology 79:1708–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday GM, McRitchie DA, Macdonald V, Double KL, Trent RJ, McCusker E (1998): Regional specificity of brain atrophy in Huntington's disease. Exp Neurol 154:663–672. [DOI] [PubMed] [Google Scholar]
- Harrington DL, Liu D, Smith MM, Mills JA, Long JD, Aylward EH, Paulsen JS (2014): Neuroanatomical correlates of cognitive functioning in prodromal Huntington disease. Brain Behav 4:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs NZ, Cole JH, Farmer RE, Rees EM, Crawford HE, Malone IB, Roos RA, Sprengelmeyer R, Durr A, Landwehrmeyer B, Scahill RI, Tabrizi SJ, Frost C (2012): Evaluation of multi‐modal, multi‐site neuroimaging measures in Huntington's disease: Baseline results from the PADDINGTON study. Neuroimage Clin 2:204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmannspotter M, Peters N, Opherk C, Martin D, Herzog J, Bruckmann H, Samann P, Gschwendtner A, Dichgans M (2005): Diffusion magnetic resonance histograms as a surrogate marker and predictor of disease progression in CADASIL: A two‐year follow‐up study. Stroke 36:2559–2565. [DOI] [PubMed] [Google Scholar]
- Irfanoglu MO, Walker L, Sarlls J, Marenco S, Pierpaoli C (2012): Effects of image distortions originating from susceptibility variations and concomitant fields on diffusion MRI tractography results. Neuroimage 61:275–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeurissen B, Leemans A, Tournier JD, Jones DK, Sijbers J (2013): Investigating the prevalence of complex fiber configurations in white matter tissue with diffusion magnetic resonance imaging. Hum Brain Mapp 34:2747–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DK, Knosche TR, Turner R (2013): White matter integrity, fiber count, and other fallacies: the do's and don'ts of diffusion MRI. Neuroimage 73:239–254. [DOI] [PubMed] [Google Scholar]
- Jones DK, Leemans A (2011): Diffusion tensor imaging. Methods Mol Biol 711:127–144. [DOI] [PubMed] [Google Scholar]
- Kuijf HJ, van Veluw SJ, Geerlings MI, Viergever MA, Biessels GJ, Vincken KL (2013): Automatic Extraction of the Midsagittal Surface from Brain MR Images using the Kullback‐Leibler Measure. Neuroinformatics 12:395–403. [DOI] [PubMed] [Google Scholar]
- Lambrecq V, Langbour N, Guehl D, Bioulac B, Burbaud P, Rotge JY (2013): Evolution of brain gray matter loss in Huntington's disease: A meta‐analysis. Eur J Neurol 20:315–321. [DOI] [PubMed] [Google Scholar]
- Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR (2004): A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clin Genet 65:267–277. [DOI] [PubMed] [Google Scholar]
- Leemans A, Jones DK (2009): The B‐matrix must be rotated when correcting for subject motion in DTI data. Magn Reson Med 61:1336–1349. [DOI] [PubMed] [Google Scholar]
- Metwalli NS, Benatar M, Nair G, Usher S, Hu X, Carew JD (2010): Utility of axial and radial diffusivity from diffusion tensor MRI as markers of neurodegeneration in amyotrophic lateral sclerosis. Brain Res 1348:156–164. [DOI] [PubMed] [Google Scholar]
- Muhlau M, Gaser C, Wohlschlager AM, Weindl A, Stadtler M, Valet M, Zimmer C, Kassubek J, Peinemann A (2007): Striatal gray matter loss in Huntington's disease is leftward biased. Mov Disord 22:1169–1173. [DOI] [PubMed] [Google Scholar]
- Myers RH, Vonsattel JP, Paskevich PA, Kiely DK, Stevens TJ, Cupples LA, Richardson EP, Jr. , Bird ED (1991): Decreased neuronal and increased oligodendroglial densities in Huntington's disease caudate nucleus. J Neuropathol Exp Neurol 50:729–742. [DOI] [PubMed] [Google Scholar]
- Oguz KK, Sanverdi E, Has A, Temucin C, Turk S, Doerschner K (2013): Tract‐based spatial statistics of diffusion tensor imaging in hereditary spastic paraplegia with thin corpus callosum reveals widespread white matter changes. Diagn Interv Radiol 19:181–186. [DOI] [PubMed] [Google Scholar]
- Oldfield RC (1971): The assessment and analysis of handedness: The Edinburgh inventory. Neuropsychologia 9:97–113. [DOI] [PubMed] [Google Scholar]
- Papadopoulou A, Muller‐Lenke N, Naegelin Y, Kalt G, Bendfeldt K, Kuster P, Stoecklin M, Gass A, Sprenger T, Radue EW, Kappos L, Penner IK (2013): Contribution of cortical and white matter lesions to cognitive impairment in multiple sclerosis. Mult Scler 19:1290–1296. [DOI] [PubMed] [Google Scholar]
- Park HJ, Westin CF, Kubicki M, Maier SE, Niznikiewicz M, Baer A, Frumin M, Kikinis R, Jolesz FA, McCarley RW, Shenton ME (2004): White matter hemisphere asymmetries in healthy subjects and in schizophrenia: a diffusion tensor MRI study. Neuroimage 23:213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patenaude B, Smith SM, Kennedy DN, Jenkinson M (2011): A Bayesian model of shape and appearance for subcortical brain segmentation. Neuroimage 56:907–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen JS, Nopoulos PC, Aylward E, Ross CA, Johnson H, Magnotta VA, Juhl A, Pierson RK, Mills J, Langbehn D, Nance M (2010): Striatal and white matter predictors of estimated diagnosis for Huntington disease. Brain Res Bull 82:201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penney JB, Jr. , Vonsattel JP, MacDonald ME, Gusella JF, Myers RH (1997): CAG repeat number governs the development rate of pathology in Huntington's disease. Ann Neurol 41:689–692. [DOI] [PubMed] [Google Scholar]
- Pierpaoli C, Jezzard P, Basser PJ, Barnett A, Di CG (1996): Diffusion tensor MR imaging of the human brain. Radiology 201:637–648. [DOI] [PubMed] [Google Scholar]
- Poudel G, Stout JC, Dominguez DJ, Salmon L, Churchyard A, Chua P, Georgiou‐Karistianis N, Egan GF (2014): White matter connectivity reflects clinical and cognitive status in Huntington's disease. Neurobiol Dis 65:180–187. [DOI] [PubMed] [Google Scholar]
- Roine U, Roine T, Salmi J, Nieminen‐von WT, Leppamaki S, Rintahaka P, Tani P, Leemans A, Sams M (2013): Increased coherence of white matter fiber tract organization in adults with Asperger syndrome: a diffusion tensor imaging study. Autism Res 6:642–650. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Liu AK, Hersch S, Glessner M, Ferrante RJ, Salat DH, van der Kouwe A, Jenkins BG, Dale AM, Fischl B (2002): Regional and progressive thinning of the cortical ribbon in Huntington's disease. Neurology 58:695–701. [DOI] [PubMed] [Google Scholar]
- Ross CA, Tabrizi SJ (2011): Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10:83–98. [DOI] [PubMed] [Google Scholar]
- Sen PN, Basser PJ (2005): A model for diffusion in white matter in the brain. Biophys J 89:2927–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TE, Johansen‐Berg H, Bannister PR, De LM, Drobnjak I, Flitney DE, Niazy RK, Saunders J, Vickers J, Zhang Y, De SN, Brady JM, Matthews PM (2004): Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 23 Suppl 1:S208–S219. [DOI] [PubMed] [Google Scholar]
- Sotak CH (2004): Nuclear magnetic resonance (NMR) measurement of the apparent diffusion coefficient (ADC) of tissue water and its relationship to cell volume changes in pathological states. Neurochem Int 45:569–582. [DOI] [PubMed] [Google Scholar]
- Sritharan A, Egan GF, Johnston L, Horne M, Bradshaw JL, Bohanna I, Asadi H, Cunnington R, Churchyard AJ, Chua P, Farrow M, Georgiou‐Karistianis N (2010): A longitudinal diffusion tensor imaging study in symptomatic Huntington's disease. J Neurol Neurosurg Psychiatry 81:257–262. [DOI] [PubMed] [Google Scholar]
- Stoffers D, Sheldon S, Kuperman JM, Goldstein J, Corey‐Bloom J, Aron AR (2010): Contrasting gray and white matter changes in preclinical Huntington disease: an MRI study. Neurology 74:1208–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Langbehn DR, Leavitt BR, Roos RA, Durr A, Craufurd D, Kennard C, Hicks SL, Fox NC, Scahill RI, Borowsky B, Tobin AJ, Rosas HD, Johnson H, Reilmann R, Landwehrmeyer B, Stout JC (2009): Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK‐HD study: cross‐sectional analysis of baseline data. Lancet Neurol 8:791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Scahill RI, Durr A, Roos RA, Leavitt BR, Jones R, Landwehrmeyer GB, Fox NC, Johnson H, Hicks SL, Kennard C, Craufurd D, Frost C, Langbehn DR, Reilmann R, Stout JC (2011): Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK‐HD study: The 12‐month longitudinal analysis. Lancet Neurol 10:31–42. [DOI] [PubMed] [Google Scholar]
- Takao H, Hayashi N, Kabasawa H, Ohtomo K (2012): Effect of scanner in longitudinal diffusion tensor imaging studies. Hum Brain Mapp 33:466–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tax CM, Otte WM, Viergever MA, Dijkhuizen RM, Leemans A (2014): REKINDLE: Robust Extraction of Kurtosis INDices with Linear Estimation. Magn Reson Med. [DOI] [PubMed] [Google Scholar]
- Tellez‐Nagel I, Johnson AB, Terry RD (1974): Studies on brain biopsies of patients with Huntington's chorea. J Neuropathol Exp Neurol 33:308–332. [DOI] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group (1993): A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72:971–983. [DOI] [PubMed] [Google Scholar]
- Thieben MJ, Duggins AJ, Good CD, Gomes L, Mahant N, Richards F, McCusker E, Frackowiak RS (2002): The distribution of structural neuropathology in pre‐clinical Huntington's disease. Brain 125:1815–1828. [DOI] [PubMed] [Google Scholar]
- Tobin AJ, Signer ER(2000): Huntington's disease: the challenge for cell biologists. Trends Cell Biol 10:531–536. [DOI] [PubMed] [Google Scholar]
- Tournier JD, Mori S, Leemans A (2011): Diffusion tensor imaging and beyond. Magn Reson Med 65:1532–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bogaard SJ, Dumas EM, Ferrarini L, Milles J, van Buchem MA, van der Grond J, Roos RA (2011): Shape analysis of subcortical nuclei in Huntington's disease, global versus local atrophy–A results from the TRACK‐HD study. J Neurol Sci 307:60–68. [DOI] [PubMed] [Google Scholar]
- Van den Bogaard SJ, Dumas EM, Milles J, Reilmann R, Stout JC, Craufurd D, van Buchem MA, van der Grond J, Roos RA (2012): Magnetization transfer imaging in premanifest and manifest Huntington disease. AJNR Am J Neuroradiol 33:884–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Boomgaard R, van Balen R (1992): Methods for fast morphological image transforms using bitmapped binary images. Graphical Models Image Processing 54:252–258. [Google Scholar]
- Van CN, Blockx I, Camon L, de VN, Verhoye M, Veraart J, Van HW, Martinez E, Soria G, Sijbers J, Planas AM, Van der Linden A (2012): A complementary diffusion tensor imaging (DTI)‐histological study in a model of Huntington's disease. Neurobiol Aging 33:945–959. [DOI] [PubMed] [Google Scholar]
- Vandenberghe W, Demaerel P, Dom R, Maes F (2009): Diffusion‐weighted versus volumetric imaging of the striatum in early symptomatic Huntington disease. J Neurol 256:109–114. [DOI] [PubMed] [Google Scholar]
- Veraart J, Sijbers J, Sunaert S, Leemans A, Jeurissen B (2013): Weighted linear least squares estimation of diffusion MRI parameters: strengths, limitations, and pitfalls. Neuroimage 81:335–346. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Keller C, Cortes Ramirez EP (2011): Huntington's disease–Neuropathology. Handb Clin Neurol 100:83–100. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP, Jr. (1985): Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44:559–577. [DOI] [PubMed] [Google Scholar]
- Vrenken H, Pouwels PJ, Geurts JJ, Knol DL, Polman CH, Barkhof F, Castelijns JA (2006): Altered diffusion tensor in multiple sclerosis normal‐appearing brain tissue: cortical diffusion changes seem related to clinical deterioration. J Magn Reson Imaging 23:628–636. [DOI] [PubMed] [Google Scholar]
- Weaver KE, Richards TL, Liang O, Laurino MY, Samii A, Aylward EH (2009): Longitudinal diffusion tensor imaging in Huntington's Disease. Exp Neurol 216:525–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Valenza M, Cui L, Leoni V, Jeong HK, Brilli E, Zhang J, Peng Q, Duan W, Reeves SA, Cattaneo E, Krainc D (2011): Peroxisome‐proliferator‐activated receptor gamma coactivator 1 alpha contributes to dysmyelination in experimental models of Huntington's disease. J Neurosci 31:9544–9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiura T, Noguchi T, Hiwatashi A, Togao O, Yamashita K, Nagao E, Kamano H, Honda H (2010): Intra‐ and interhemispheric variations of diffusivity in subcortical white matter in normal human brain. Eur Radiol 20:227–233. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Brady M, Smith S (2001): Segmentation of brain MR images through a hidden Markov random field model and the expectation‐maximization algorithm. IEEE Trans Med Imaging 20:45–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information Figure 1.
Supplementary Information Figure 2.
Supplementary Information Figure 3.
Supplementary Information Figure 4.
Supplementary Information Figure 5.
Supplementary Information Tables.