

Abstract
Gait disturbances, including freezing of gait, are frequent and disabling symptoms of Parkinson's disease. They often respond poorly to dopaminergic treatments. Although recent studies have shed some light on their neural correlates, their modulation by dopaminergic treatment remains quite unknown. Specifically, the influence of levodopa on the networks involved in motor imagery (MI) of parkinsonian gait has not been directly studied, comparing the off and on medication states in the same patients. We therefore conducted an [H2 150] Positron emission tomography study in eight advanced parkinsonian patients (mean disease duration: 12.3 ± 3.8 years) presenting with levodopa‐responsive gait disorders and FoG, and eight age‐matched healthy subjects. All participants performed three tasks (MI of gait, visual imagery and a control task). Patients were tested off, after an overnight withdrawal of all antiparkinsonian treatment, and on medication, during consecutive mornings. The order of conditions was counterbalanced between subjects and sessions. Results showed that imagined gait elicited activations within motor and frontal associative areas, thalamus, basal ganglia and cerebellum in controls. Off medication, patients mainly activated premotor‐parietal and pontomesencephalic regions. Levodopa increased activation in motor regions, putamen, thalamus, and cerebellum, and reduced premotor‐parietal and brainstem involvement. Areas activated when patients are off medication may represent compensatory mechanisms. The recruitment of these accessory circuits has also been reported for upper‐limb movements in Parkinson's disease, suggesting a partly overlapping pathophysiology between imagined levodopa‐responsive gait disorders and appendicular signs. Our results also highlight a possible cerebellar contribution in the pathophysiology of parkinsonian gait disorders through kinesthetic imagery. Hum Brain Mapp 36:959–980, 2015. © 2014 Wiley Periodicals, Inc.
Keywords: Parkinson's disease, gait disorders, freezing of gait, kinesthetic motor imagery, PET imaging, levodopa
Abbreviations
- AW
actual walking
- BA
Brodmann's areas
- C
control task
- DBS
deep brain stimulation
- FoG
freezing of gait
- FWE
family wise error
- MDS‐UPDRS
movement disorders society unified parkinson disease rating scale
- FWHM
full width at half maximum
- MI
motor imagery
- off
off dopaminergic medication
- on
on dopaminergic medication
- PD
Parkinson's disease
- PET
positron emission tomography
- rCBF
regional cerebral blood flow
- VI
visual imagery
INTRODUCTION
Gait disorders—ranging from hypokinetic gait to a sudden inability to produce steps at initiation or during walking, called freezing of gait (FoG) [Giladi and Nieuwboer, 2008]‐are very debilitating and poorly understood features of Parkinson's disease [Nutt et al., 2011a, 2011b]. They can be responsible for falls, and greatly affect patients' mobility, autonomy, and quality of life [Bloem et al., 2004; Nutt et al., 2011a, 2011b]. In advanced Parkinson's disease, their response to pharmacological and surgical therapies is often unsatisfactory, despite initial improvement at onset of treatment [Ferraye et al., 2008]. The diversity of gait difficulties and their complex and inconstant response to dopaminergic medication [Espay et al., 2012; Fox, 2013; Vercruysse et al., 2012] suggest the involvement of nondopaminergic structures beyond the nigrostriatal disruption [Bonnet et al., 1987; Devos et al., 2010]. Structural and/or functional alterations of fronto‐parieto‐occipital areas, cerebellum and brainstem have been evoked [Bartels and Leenders, 2008; Bartels et al., 2006; Crémers et al., 2012; Fling et al., 2013; Kostic et al., 2012; Peterson et al., 2014a, 2014b; Shine et al., 2013a, 2013b; Snijders et al., 2011; Tessitore et al., 2012a, 2012b]. Yet, a better understanding of the mechanisms underlying parkinsonian gait disorders and their modulation by treatments is needed.
As current neuroimaging techniques are not compatible with the study of actual gait, motor imagery (MI), that is, the mental simulation of a movement without overt execution [Jeannerod, 1994], appears well suited to address this issue. Over the last decades, numerous studies have used MI to explore the neural correlates of normal and pathological movement, highlighting significant overlap between the substrates of actual and imagined actions, including in patients with Parkinson's disease [Guillot et al., 2009, 2012; Munzert and Zentgraf, 2009; Samuel et al., 2001; Solodkin et al., 2004; Stephan et al., 1995; Thobois et al., 2000, 2002]. However, the use of MI to study the neural basis of motor control calls for methodological caution. First, the subjects' ability to adequately imagine actions needs to be ascertained [Maillet et al., 2012a, 2013], as this ability varies among individuals [Mulder et al., 2007; van der Meulen et al., 2014] and can be further affected by age [Saimpont et al., 2012, 2013; Zapparoli et al., 2013] and disease [Cohen et al., 2011; Helmich et al., 2007]. Furthermore, kinesthetic MI, that is, evoking the sensations triggered by the movements, is particularly relevant to study motor control [Guillot et al., 2009; Stinear et al., 2006; Voisin et al., 2011]. Specific tools enable assessment of MI ability, such as the kinesthetic and visual imagery (VI) questionnaire [KVIQ, Malouin et al., 2007], recently validated in Parkinson's disease [Heremans et al., 2011; Pickett et al., 2013; Randhawa et al., 2010]. Acquisition of reliable neuroimaging data also depends on the subjects actually performing the targeted task during scanning. This can be controlled using Fitts' law [Fitts, 1954], which states that the time needed to perform an action increases with its difficulty. Regarding gait, previous studies have confirmed that the time to physically or mentally walk along a path increases with increasing length and decreasing width, in controls and in parkinsonian patients with or without medication [Bakker et al., 2007, 2008; Maillet et al., 2013; Snijders et al., 2011; Stevens, 2005].
In recent years, MI has been increasingly used to investigate the neural control of normal and parkinsonian gait [Allali et al., 2014; Bakker et al., 2008; Crémers et al., 2011, 2012; Karachi et al., 2010, 2012; La Fougère et al., 2010; Malouin et al., 2003; Peterson et al., 2014a, 2014b; Snijders et al., 2011; Wai et al., 2012; see review by Maillet et al., 2012a]. Overall, these studies have confirmed the involvement of frontoparietal areas, basal ganglia (BG), cerebellum, and brainstem during MI of natural gait in controls. Regarding parkinsonian patients with gait disorders, frontoparietal and cingulate hypoactivations have been highlighted, both on [Crémers et al., 2012] and off [Peterson et al., 2014a, 2014b; Snijders et al., 2011] medication. In addition, a pontomesencephalic overactivation was also reported during imagined gait off medication in patients with, compared to without, FoG [Snijders et al., 2011]. Conversely, pontomesencephalic hypoactivation was reported in patients under dopaminergic treatment relative to controls [Crémers et al., 2012]. Yet, to date, the influence of levodopa on the networks involved in MI of parkinsonian gait has not been directly studied, comparing the off and on medication states in the same patients.
We therefore adapted for [H2 150]‐positron emission tomography (PET) a previously validated protocol of MI of gait [Bakker et al., 2007, 2008; Snijders et al., 2011] to explore within subjects the modulation by levodopa of the gait‐related cerebral correlates in eight patients with levodopa‐responsive gait disorders and FoG. As this study was part of a larger project including patients with levodopa‐resistant FoG treated by pedunculopontine nucleus stimulation (not included in the present study), we used PET instead of fMRI that is contra‐indicated for patients with deep brain stimulation (DBS).
MATERIAL AND METHODS
Participants
Eight patients (four men; mean age = 63.3 ± 6.3 years, range [52–73]) with advanced Parkinson's disease (mean Hoehn and Yahr stage = 3.4 ± 0.5; mean disease duration = 12.3 ± 3.8 years, range [8–19]), gait disorders and FoG, and eight age and gender‐matched healthy controls (four men; mean age = 62.9 ± 6.7 years; range [49–70]) were recruited in the Grenoble, Lyon, and Clermont‐Ferrand University Hospitals (Table 1). All subjects were right‐handed [Edinburgh Handedness Inventory score > 70/100—Oldfield, 1971]. Exclusion criteria included cognitive impairment [mini mental state examination score < 27/30—Folstein et al., 1975; frontal assessment battery score < 14/18—Dubois et al., 2000; and for patients, Mattis dementia rating scale score < 130/144—Schmidt et al., 1994], orthopedic or psychiatric disorders, marked resting tremor, and neurosurgery. The patients fulfilled the UK Parkinson's disease Brain Bank Criteria [Gibb and Lees, 1988] for the diagnosis of idiopathic Parkinson's disease and received chronic dopaminergic antiparkinsonian treatment. They were included if their gait disorders were improved by at least 1 point on compared to off medication with on scores < 2/4 for both the gait and FoG items of the Movement Disorders Society Unified Parkinson Disease Rating Scale motor section [MDS‐UPDRS III—Goetz et al., 2008] during objective assessment. The study was approved by the Grenoble ethics committee and conducted in accordance with the Declaration of Helsinki. All participants provided written consent.
Table 1.
Main clinical characteristics of the patients
| Patients/most affected side | Hoehn & Yahr staging (/5) | MDS‐UPDRS total motor score (/132) | MDS‐UPDRS Gait score (/4) | MDS‐UPDRS FoG score (/4) | GABS Score (/93) | FoG‐Q score (/24) | MATTIS score (/144) | Levodopa Equivalent Daily Dose (mg) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| State | OFF | ON | OFF | ON | OFF | ON | OFF | ON | OFF | ON | on | ON | – |
| 1/L | 3 | 2 | 33 | 18 | 2 | 0 | 2 | 0 | 25 | 9 | 17 | 144 | 800 |
| 2/R | 4 | 2 | 39 | 18 | 2 | 1 | 3 | 1 | 41 | 12 | 20 | 140 | 1150 |
| 3/R | 3 | 2 | 32 | 14 | 1 | 0 | 1 | 0 | 31 | 6 | 20 | 140 | 500 |
| 4/R | 3 | 2 | 46 | 26 | 2 | 1 | 4 | 1 | 35 | 14 | 14 | 138 | 950 |
| 5/R | 4 | 3 | 32 | 9 | 2 | 0 | 2 | 1 | 45 | 16 | 20 | 140 | 1100 |
| 6/L | 3 | 2 | 36 | 14 | 1 | 0 | 2 | 0 | 32 | 7 | 19 | 141 | 1000 |
| 7/L | 3 | 2 | 29 | 9 | 1 | 0 | 2 | 0 | 31 | 9 | 11 | 143 | 875 |
| 8/L | 4 | 3 | 55 | 11 | 3 | 0 | 4 | 0 | 38 | 17 | 23 | 138 | 950 |
| Mean ± SD | 3.4 ± 0.5 | 2.3 ± 0.5 | 37.8 ± 8.7 | 14.9 ± 5.7 | 1.8 ± 0.7 | 0.3 ± 0.5 | 2.5 ± 1.1 | 0.4 ± 0.5 | 34.8 ± 6.4 | 11.3 ± 4.1 | 18 ± 3,9 | 140.5 ± 2.1 | 916 ± 203 |
| Percentages of improvement (between off & on states) | 32.4 % | 60.6 % | 83.4 % | 84 % | 67.5 % | – | – | – | |||||
Abbreviations: FoG‐Q = Freezing of Gait Questionnaire—Giladi et al., 2000; GABS = Gait and Balance Scale; L = left; LEDD = Levodopa Equivalent Daily Dose, calculated according to Tomlinson et al., 2010; MATTIS = Mattis Dementia Rating scale; mg = milligrams; on = under chronic medication; R = right; SD = standard deviation.
Assessment of MI abilities
Participants' ability to perform MI was assessed using the KVIQ, which includes visual (KVIQ‐v) and kinesthetic (KVIQ‐k) subscales, administered as prescribed by Malouin et al. [2007]. This 100‐points scale requests physical execution of 10 movements (involving the head, shoulders, trunk, upper, and lower limbs), followed by their mental representation, for each of the two modalities. Participants are asked to rate the clarity of the visual image or the intensity of the sensations associated with each imagined action on a 5‐points scale (from 0 = “no image/sensation” to 5 = “very clear image/intense sensation”), a higher score reflecting a greater MI ability. Patients were assessed under usual treatment. Subjects were then further trained to kinesthetically imaging segmental and whole body movements. Briefly, this training first consisted in inducing a relaxation state. Subjects were then asked to actually perform a given motor task, the eyes closed, while specifically focusing on the sensations associated with the action. After a few repetitions, they had to mentally evoke the sensations of the action without actually making it. They were also asked to rate the intensity of the sensations perceived during the mental representation, using the 5‐points scale of the KVIQ. The training involved actual and mental performance of seven different movements, including gait tasks. They were included if their KVIQ‐k score was ≥30/50 after training.
Experimental Design
PET acquisitions were preceded by a behavioral session designed to familiarize the subjects with the experimental tasks. A maximum of 4 weeks separated the behavioral and the scanning sessions. For the patients, each session included two assessments on two consecutive mornings, without “off” and with “on” dopaminergic medication. The off state evaluation was performed after an overnight (12 h) withdrawal of all antiparkinsonian treatment [Langston et al., 1992]. The on assessment was performed after administration of 120% of the usual morning levodopa‐equivalent dose. Clinical assessments also included the motor section of the MDS‐UPDRS and the Gait and balance scale (GABS; Thomas et al., 2004]. The order of conditions was counterbalanced between subjects and sessions. Controls were tested only once. The patients' clinical features are detailed in Table 1.
Behavioral Session
Experimental tasks
During the behavioral session, the subjects performed three tasks, that is, actual walking (AW), MI of gait, and VI [Bakker et al., 2007, 2008].
AW was performed on black linoleum paths of two lengths (6 and 10 m) and widths (27 and 9 cm) placed in a corridor, and was videotaped. Subjects had to walk along the paths at a comfortable pace. The wide paths allowed for natural walking, while the narrow paths required close attention to feet placement. The starting and end points were marked by a green and a yellow square, respectively. Duration of AW and FoG episodes was recorded with a stopwatch. AW was performed after MI to minimize the effect of tacit knowledge of the time needed to actually walk along the paths.
The imagery tasks were supported by photographs of the paths presented on a computer screen using the Presentation® software (Neurobehavioral Systems, Albany). During MI, subjects had to imagine walking on the paths from the green square to the yellow square (Fig. 1a,b), using a kinesthetic perspective without producing any actual movements. Patients were instructed to imagine walking in their actual state, that is, with the motor difficulties experienced under each medication condition. Before performing MI, participants were allowed to walk few meters along each path to get acquainted with the bandwidth constraints. The VI task consisted in imagining a blue puck moving along the paths until it reached the yellow square (Fig. 1c,d). Prior to VI, the subjects watched a video showing the puck moving in the corridor. Subjects were requested to press a button to indicate when they started, and again when they, or the puck, reached the end mark. These tasks were performed sitting with the eyes closed, as the patients could not sustain prolonged standing under the off treatment condition. Subjects were asked to report the ease with which they had evoked the sensations or images and, for the patients, whether they had imagined FoG. Three trials on each path were performed for each task, for a total of 12 randomized trials. The three tasks were performed in successive sessions of about 30 min each, separated by a break. Imagery task order was counterbalanced across subjects, groups, and conditions. The patients also performed the KVIQ‐k before each assessment, in the off and on conditions.
Figure 1.

Examples of photographs used for MI and VI trials during the behavioral session. Pictures showing black paths of different lengths (6 or 10 m) and widths (9 or 27 cm) on which subjects were asked to imagine walking or seeing a puck moving. The same paths were used for actual gait. A green square marked the starting point and a yellow square the end point (at 6 or 10 meters from the starting point) for the MI trials. For the VI trials, the participants were asked to imagine seeing a blue puck moving along the different paths until it reached the yellow square (adapted from Bakker et al., 2007). (a) and (b): MI [(a) = 6 m*27 cm path; (b) = 10 m*9 cm path]; c and d: VI [(c) = 10 m*27 cm path; (d) = 6 m*9 cm path]. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Behavioral analysis
Because of the limited sample size, non‐parametric statistics were used, Wilcoxon tests for within‐group, and Mann–Whitney tests for between‐group, comparisons. To ascertain the effect of levodopa on the patients' motor signs, the off and on clinical scores were compared. The averaged KVIQ‐k scores before (k1) and after (k2) training, and with (k‐on) or without (k‐off) treatment in patients, were compared within groups (KVIQ‐k1 vs. KVIQ‐k2 for both groups; KVIQ‐k‐off vs. KVIQ‐k‐on; KVIQ‐k‐off vs. KVIQ‐k1; KVIQ‐k‐off vs. KVIQ‐k2; KVIQ‐k‐on vs. KVIQ‐k1; KVIQ‐k‐on vs. KVIQ‐k2 for patients) and between groups (KVIQ‐k1‐controls vs. KVIQ‐k1‐patients; KVIQ‐k2‐controls vs. KVIQ‐k2‐patients; KVIQ‐k1‐controls vs. KVIQ‐k‐off; KVIQ‐k1‐controls vs. KVIQ‐k‐on; KVIQ‐k2‐controls vs. KVIQ‐k‐off; KVIQ‐k2‐controls vs. KVIQ‐k‐on). Comparisons between the KVIQ‐k and KVIQ‐v scores were also performed both within (KVIQ‐k1 vs. KVIQ‐v; KVIQ‐k2 vs. KVIQ‐v for all groups; KVIQ‐k‐off vs. KVIQ‐v; KVIQ‐k‐on vs. KVIQ‐v for patients) and between (KVIQ‐v‐controls vs. KVIQ‐v‐patients) groups.
We also examined the influence of path length and width on the time needed to perform AW, MI, and VI. Task duration was defined as the time elapsed between the two button presses. Mean durations (±SD, in seconds) were compared to examine the effects of task difficulty (6 m vs. 10 m; 27 cm vs. 9 cm), condition (off vs. on medication), and group (controls vs. patients). Statistics were performed under Statistica® (STATISTICA 8, Statsoft, Tulsa). The significance level was fixed at P < 0.05 with Bonferroni corrections for multiple comparisons.
PET Session
PET images were acquired while subjects performed two imagery tasks and a control task. Because of the limitation in the number of radioactive injections that subjects can receive, data were collected for the wider paths only (6 and 10 m). Before scanning, the subjects were allowed a few training trials. Task performance was assessed off‐line, testing the effects of path length (6 m vs. 10 m), condition (off vs. on levodopa), and group (controls vs. patients) on the MI and VI times recorded during PET‐scans.
Experimental procedure
MI of gait
The trial started with a 7‐s presentation of a picture of one of the two paths, showing the starting and end points. When the picture disappeared, subjects were asked to close their eyes, imagine standing on the left of the starting point, then step on the path and walk to the end mark. They were requested to press a button when they imagined stepping on the path and again when reaching the end point. An auditory signal indicated they could reopen their eyes. A white cross was displayed on the screen until the next picture (Fig. 2A). This sequence was repeated three times for each path, for a total of six randomized trials, in a 2‐min block‐design. Each block was triplicated. Electromyographic (EMG) activity of the right gastrocnemius and tibialis anterior muscles was recorded to control for the absence of overt leg movements (Noraxon® Telemyo 2400, Scottsdale, USA).
Figure 2.
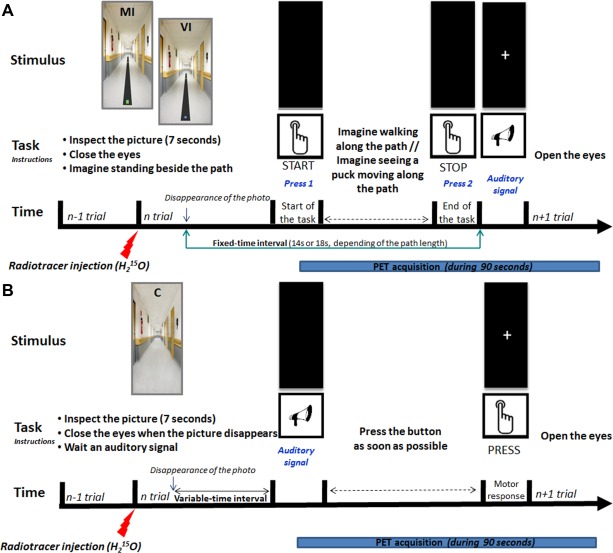
Setup and time‐course of the tasks during the PET session. (A) Time course of the MI and VI trials—For each task, after inspecting (7 s) a picture representing one of the two paths (6 m*27 cm or 10 m*27 cm), subjects were asked to close their eyes and imagine standing on the left of the green square (MI trials) or blue puck (VI trials). During MI trials they were asked to imagine walking along the path from the green square (starting point) to the yellow square (end point) using a kinesthetic perspective, and to press a button when they started and finished their mental gait. Patients were instructed to imagine walking in their current motor state. During the VI trials, subjects had to imagine seeing the blue puck moving along the paths, and press the button when they imagined the puck starting to move and reaching the yellow square. The time elapsed between the two button presses was taken as the time needed to perform the task. An auditory signal indicated that the subjects could reopen their eyes. The auditory signal was issued 14 s (for the 6 m*27 cm path) or 18 s (for the 10 m*27 cm path) after the beginning of the trial (i.e., the disappearance of the picture). A white cross on the screen announced the next trial. This sequence was repeated six times during a block (3 randomly ordered trials for each path) [adapted from Bakker et al., 2008 ; Snijders et al., 2011]. (B) Time course of the control task—Subjects were asked to inspect a neutral image, that is, a picture of an empty corridor, close their eyes when the picture disappeared and press the button as soon as possible when they heard an auditory signal presented after a variable time‐interval. They then reopened their eyes and waited for a new auditory signal. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Visual imagery
Participants first watched three repetitions of a 9‐s movie showing the puck sliding in an empty corridor. Pictures showing the 6 m*27 cm or 10 m*27 cm paths with the blue puck at the start and the yellow end mark were then displayed. When the picture disappeared, subjects were instructed to imagine the blue puck moving along the path. They were requested to press the button when the puck started to move and when it reached the target (Fig. 2A). They performed three trials for each path, leading to three 2‐min blocks of six randomized trials.
Control task
For the control task (C), participants saw a photograph of an empty corridor during 7 s, and closed their eyes when it disappeared. An auditory signal occurred after a variable‐time interval, on which they were to press the button as fast as possible. They then reopened their eyes and watched a cross displayed on the screen until the next trial (Fig. 2B). Three 2‐min blocks of six trials each were collected. This task was introduced to remove activations related to visual inspection of the pictures, hearing of the auditory signal, and manual pressure on the button from the MI and VI brain profiles during data processing. A reaction‐time task was chosen as control to ensure that the subjects did not perform MI or VI during acquisitions.
Scanning procedure and data collection
Subjects were positioned supine on the scanner bed, head maintained. A camera allowed control of the head's position before and after each acquisition. PET‐scans were acquired on a PET/CT tomograph (Siemens Biograph mCT/S 64—with a spatial transverse resolution of 4.4 mm) at the CERMEP cyclotron centre (Bron, France) in a 3D mode during 90 min. In total, nine scans were obtained for the controls and 18 scans (9 in each condition) for the patients. For each scan, the rCBF was measured by recording the radioactivity distribution after a bolus injection of 7 mCi (270 MBq) of [H2 150] over 10 s through an intravenous catheter. Emission data were collected for 90 s, starting 30 s after injection. The tasks started a few seconds before scanning. A 10‐min interval between scans allowed for adequate radioactivity decay. Before acquisitions, a low‐dose CT‐scan (<0.3 mSV) was carried out. After scanning, the participants reported the ease with which they had performed the tasks (intensity of perceived sensations for MI, and clarity of images for VI). The patients also reported the occurrence of imagined FoG.
PET images processing
After scanning, PET data were reconstructed using the CT‐scan to correct for tissue attenuation. Only the first 60 s of each scan—corresponding to the maximum of the radioactivity—were retained. Images displayed a transaxial resolution of 5 mm FWHM in a 200 × 200 pixel‐matrix, resulting in 109 slices generating approximately voxel size of 2 × 2 × 2 mm. To perform a parametric analysis of the images [Friston et al., 1994], data were then preprocessed as follows using the SPM8 software (Statistical Parametric Mapping, Wellcome Department for Cognitive Neuroscience, London, UK) implemented in Matlab® 7.1 (Math Works, Natick, MA): (1) for each subject, all images were spatially realigned to the first volume acquired to correct for interscan head movements and a mean image was then generated; (2) the mean image was first normalized onto the standard PET Montreal Neurological Institute (MNI) space template, and the normalization parameters were then applied to the individual images; and (3) the normalized images were thereafter smoothed using an isotropic Gaussian Kernel filter (10 mm FWHM) to reduce the variance due to interindividual anatomical variability and to improve the signal/noise ratio in individual data. Global flow variations across subjects and scans were removed by proportionally scaling each image to have an arbitrary level of 50 mL/100 mL/min.
PET analysis
To examine the effects of Parkinson's disease and levodopa on each imagery task, a multisubjects x conditions and covariates model was used. More precisely, a flexible factorial design based on repeated measures ANOVAs was used for both within and between‐group statistical comparisons.
Within‐group analysis
For each group, we assessed the global increase of rCBF associated with MI and VI, compared with C, using the contrasts [MI‐C] and [VI‐C]. We also studied the increase and decrease of rCBF related to MI compared to VI with the [(MI‐C)‐(VI‐C)] and [(VI‐C)‐(MI‐C)] contrasts, respectively.
Influence of levodopa
To reveal the effect of levodopa on the patients' activation profiles, we examined the increase and decrease of rCBF associated with MI and VI, compared with C, using the contrasts [(MI‐C)patients‐on‐(MI‐C)patients‐off], [(VI‐C)patients‐on‐(VI‐C)patients‐off], [(MI‐C)patients‐off‐(MI‐C)patients‐on], and [(VI‐C)patients‐off‐(VI‐C)patients‐on].
Between‐group comparisons
To assess the impact of Parkinson's disease on brain profiles, we examined the increase and the decrease of rCBF associated with MI and VI in patients off compared to controls, using the contrasts [(MI‐C)patients‐off‐(MI‐C)controls], [(VI‐C)patients‐off‐(VI‐ C)controls], and [(MI‐C)controls‐(MI‐C)patients‐off], [(VI‐C)controls‐(VI‐C)patients‐off].
Statistical analysis
Global changes in rCBF were covaried out for all voxels. Because of a priori hypotheses regarding the involvement of cortical (mainly in frontal, premotor, and motor areas), subcortical (thalamus and BG), and brainstem regions, an uncorrected threshold (P < 0.001) for multiple comparisons across the whole brain (Z‐scores > 3.10) was applied at the voxel level using flexible factorial design with the linear contrasts defined above. This level of significance has been used in previous neuroimaging studies having used MI method with a similar, or higher, number of subjects [e.g., Guillot et al., 2009; Jahn et al., 2004; Karachi et al., 2010, 2012; Malouin et al., 2003; Samuel et al., 2001; Thobois et al., 2000, 2002; van der Meulen et al., 2014]. Only voxels exceeding a threshold of a P‐value < 0.001 uncorrected and clusters of a minimum of 20 contiguous voxels were considered significant. Such combination has been recently described to be consistent for studying neural changes with weak amplitudes [Lieberman and Cunningham, 2009], as expected for mental imagery tasks [van der Meulen et al., 2014]. To strengthen data, small volume corrections with a radius of 5 mm for the volume of interest were also applied for brain regions with a strong a priori hypothesis (i.e., frontal, motor, premotor and parietal areas, BG, thalamus, cerebellum, and brainstem), irrespective of the voxel uncorrected P‐value, and a voxel‐based FWE‐corrected P‐value < 0.05 was considered as significant. Reported coordinates conform to the MNI space. Anatomical labels of activated clusters were determined using the SPM Anatomical Automatic Labeling toolbox [Tzourio‐Mazoyer et al., 2002] and a probabilistic atlas [Gousias et al., 2008; Hammers et al., 2003].
RESULTS
Clinical and Behavioral Data
Effect of levodopa on parkinsonian symptoms
All motor scores (MDS‐UPDRS III total score, Gait and FoG items of the MDS‐UPDRS III, GABS) were significantly improved under levodopa (60.6, 83.4, 84, and 67.5, respectively; zwilcoxon = 2.52; P < 0.02 for all scores; Table 1).
MI abilities
KVIQ‐k scores were significantly improved after training in controls [KVIQ‐k1 = 38.6 ± 5.6; KVIQ‐k2 = 42.1 ± 4.6; z Wilcoxon = 2.36; P < 0.02] and patients [KVIQ‐k1 = 37.1 ± 4.9; KVIQ‐k2 = 41.5 ± 5.3; z Wilcoxon = 2.52; P < 0.02], but not differ between groups, whether before (z Mann–Whitney = 0.94; P = 0.38) or after (z Mann–Whitney = 0.37; P = 0.72) training. Compared with KVIQ‐v scores, the KVIQ‐k1 scores did not statistically differ in both groups [KVIQ‐v‐controls = 43.6 ± 5.8; z Wilcoxon = 1.05; P = 0.29; KVIQ‐v‐patients = 37.6 ± 5.3; z Wilcoxon = 0.42; P = 0.67], whereas the KVIQ‐k2 scores were significantly different in patients [z Wilcoxon = 2.20; P < 0.03], but not in controls [z Wilcoxon = 0.42; P = 0.67]. The between‐group comparison of the KVIQ‐v scores did not reveal any difference [z Mann–Whitney = 1.79; P = 0.08].
In patients, the off and on KVIQ‐k scores did not significantly differ [KVIQ‐k‐off = 39.6 ± 5.3; KVIQ‐k‐on = 39.5 ± 4.9; z Wilcoxon = 0.365; P = 0.72]. These scores were not statistically different from their KVIQ‐v scores [KVIQ‐k‐off vs. KVIQ‐v‐patients: z Wilcoxon = 0.98; P = 0.33; KVIQ‐k‐on vs. KVIQ v‐patients: z Wilcoxon = 1.26; P = 0.21], and both from the KVIQ‐k1 scores [KVIQ‐k‐off vs. KVIQ‐k1‐controls: z Mann–Whitney = −0.10; P = 0.96; KVIQ‐k‐on vs. KVIQ‐k1‐controls: z Mann–Whitney = 0.10; P = 0.96] and the KVIQ‐k2 scores [KVIQ‐k‐off vs. KVIQ‐k2‐controls: z Mann–Whitney = 0.89; P = 0.38; KVIQ‐on vs. KVIQ‐k2‐controls: z Mann–Whitney = 1.05; P = 0.33] obtained in controls. However, these scores were significantly different from the KVIQ‐k1 and KVIQ‐k2 scores acquired under chronic medication, for three of the four comparisons performed [KVIQ‐k‐off vs. KVIQ‐k1‐patients: z Wilcoxon = 1.94; P = 0.052; KVIQ‐k‐off vs. KVIQ‐k2‐patients: z Wilcoxon = 2.02; P < 0.05; KVIQ‐k‐on vs. KVIQ‐k1‐patients: z Wilcoxon = 2.37; P < 0.02; KVIQ‐k‐on vs. KVIQ‐k2‐patients: z Wilcoxon = 2.20; P < 0.03].
Actual and imagined performance
Figure 3 summarizes the behavioral data for all groups, conditions and tasks. Detailed statistical values are provided in Table 2.
Figure 3.
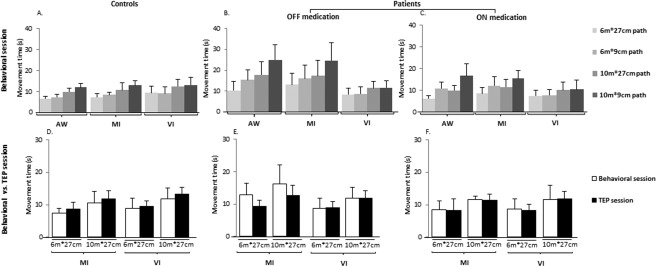
(A–C) Behavioral data. Movement times (in seconds, mean ± SD) recorded in all groups and conditions (controls, patients off and on medication) during the behavioral session for AW, MI and VI tasks (for the 6 m*27 cm, 6 m*9 cm, 10 m*27 cm, and 10 m*9 cm paths); (D–F) Comparison of the MI and VI times (in seconds, mean ± SD) obtained during the behavioral and PET sessions for all groups and conditions (controls, patients off and on medication), for the 6 m*27 cm and 10 m*27 cm paths.
Table 2.
Detailed statistical values obtained regarding the behavioral data collected during the behavioral and PET sessions
| Session | Effect | Task | Path | Controls | Patients off | Patients on | |
|---|---|---|---|---|---|---|---|
| Within‐group comparisons | Behavioral session | Length (6 m vs. 10 m) | AW/MI/VI | 27 cm/9 cm | z = 2.52; P < 0.02 | z = 2.52; P < 0.02 | z = 2.52; P < 0.02 |
| Width (27 cm vs. 9 cm) | AW | 6 m/10 m | z = 2.52; P < 0.02 | z = 2.52; P < 0.02 | z = 2.52; P < 0.02 | ||
| MI | 6 m | z = 2.38; P < 0.02 | z = 1.68; P = 0.093 | z = 2.52; P < 0.02 | |||
| 10 m | z = 2.24; P < 0.03 | z = 2.52; P < 0.02 | z = 2.52; P < 0.02 | ||||
| VI | 6 m | z = 1.26; P = 0.207 | z = 1.68; P = 0.093 | z = 1.54; P = 0.123 | |||
| 10 m | z = 1.82; P = 0.069 | z = 0.70; P = 0.483 | z = 0.7; P = 0.483 | ||||
| PET session | Length (6 m vs. 10 m) | MI | 27 cm | z = 2.52; P < 0.02 | z = 2.36; P < 0.02 | z = 2.52; P < 0.02 | |
| VI | 27 cm | z = 2.52; P < 0.02 | z = 2.52; P < 0.02 | z = 2.52; P < 0.02 |
| Session | Task | Path | Patients off vs. on | Controls vs. Patients off | Controls vs. Patients on | |
|---|---|---|---|---|---|---|
| Between‐medication states & Between‐group comparisons | Behavioral session | AW | 6 m * 27 cm | z = 2.52; P < 0.02 | z = −2.31; P < 0.03 | z = 0.63; P = 0.58 |
| 6 m * 9 cm | z = 2.38; P < 0.02 | z = −2.94; P < 0.002 | z = −1.26; P = 0.24 | |||
| 10 m * 27 cm | z = 2.52; P < 0.02 | z = −2.73; P < 0.005 | z = −0.10; P = 0.96 | |||
| 10 m * 9 cm | z = 2.52; P < 0.02 | z = −3.15; P < 0.001 | z = −1.36; P = 0.20 | |||
| MI | 6 m * 27 cm | z = 2.52; P < 0.02 | z = −3.25; P < 0.001 | z = −0.53; P = 0.65 | ||
| 6 m * 9 cm | z = 2.52; P < 0.02 | z = −2.94; P < 0.002 | z = −2.31; P < 0.03 | |||
| 10 m * 27 cm | z = 2.52; P < 0.02 | z = −2.10; P < 0.04 | z = −0.84; P = 0.45 | |||
| 10 m * 9 cm | z = 2.52; P < 0.02 | z = −2.94; P < 0.002 | z = −1.05; P = 0.33 | |||
| VI | 6 m * 27 cm | z = 0.14; P = 0.88 | z = −0.31; P = 0.80 | z = −0.10; P = 0.96 | ||
| 6 m * 9 cm | z = 0.28; P = 0.77 | z = −0.52; P = 0.65 | z = −0.52; P = 0.65 | |||
| 10 m * 27 cm | z = 0.14; P = 0.88 | z = 0.10; P = 0.96 | z = 0.31; P = 0.80 | |||
| 10 m * 9 cm | z = 0.28; P = 0.77 | z = 0.10; P = 0.96 | z = 0.31; P = 0.80 | |||
| PET session | MI | 6 m * 27 cm | z = 2.36; P < 0.02 | z = −0.57; P = 0.62 | z = 0.84; P = 0.44 | |
| 10 m * 27 cm | z = 2.20; P < 0.03 | z = −0.57; P = 0.62 | z = 0.42; P = 0.72 | |||
| VI | 6 m * 27 cm | z = 1.96; P < 0.05 | z = 0.21; P = 0.88 | z = 1.15; P = 0.28 | ||
| 10 m * 27 cm | z = 2.10; P < 0.04 | z = 0.63; P = 0.58 | z = 1.05; P = 0.33 |
Non‐parametric Wilcoxon tests were used for within‐group comparisons, and Mann‐Whitney tests for between‐group comparisons. All significant statistical differences are highlighted in bold.
Analyses of the behavioral data revealed a significant effect of path length for all tasks, whatever the group and condition. The effect of path width was significant on AW times in all groups and conditions, and on MI times, except for untreated patients on the 6 m distance. In contrast, path width did not influence VI times. In patients, AW and MI times, but not VI times, significantly decreased on levodopa compared to off. Between‐groups comparisons revealed that AW and MI durations differed between controls and patients off, but not between controls and patients on, except for MI on the 6 m by 9 cm path, where patients, although they imagined walking faster than when off treatment, remained slower than controls. VI times did not differ between groups or conditions. Thus, overall, the behavioral data confirmed that AW and MI times increased with path length and decreased with path width, whatever the group and condition. In contrast, as expected, VI times were only influenced by path length.
Regarding the imagery duration data acquired during the PET session, analyses revealed a significant effect of path length on MI and VI times for all groups and conditions. In patients, the effect of condition was also significant on both MI and VI times. There was no difference of either MI or VI times between controls and patients, whether off or on.
PET Data
EMG recordings confirmed that subjects did not produce any overt leg movements during PET‐scans.
Within‐group analysis
In controls, gait imagery, compared with VI, revealed greater recruitment of motor and somatosensory regions (i.e., left rostral supplementary motor area (pre‐SMA), right caudal SMA, right motor (M1), right somatosensory (S1) cortices), associative areas [i.e., bilateral dorsolateral (DLPFC) and ventrolateral (VLPFC) prefrontal cortices, right orbitofrontal (OFC) cortex], left dorsal anterior (dACC) and ventral posterior (vPCC) cingulate cortices, right insula, temporal areas, as well as left thalamus, putamen, and pallidum. In patients off, compared to VI, MI of gait elicited greater activations in the left caudal SMA, lateral premotor cortex (PMC), and right S1, DLPFC, VLPFC, OFC, ventral ACC (vACC), dACC, superior parietal lobule (SPL), and pontomesencephalic area. Relative to MI, VI mainly engaged parieto‐occipital and cerebellar regions in all groups (Table 3).
Table 3.
MNI coordinates for the main brain areas displaying increased and decreased rCBF during MI of gait in controls and in patients off medication
| Location of the main cerebral areas | Coordinates | ||||
|---|---|---|---|---|---|
| Functional label | Brodmann's areas (BA) | Laterality | x, y, z | Z−score | k |
| Controls ∼ [(MI−C)−(VI−C)] | |||||
| Supplementary Motor Area (pre−SMA & SMA)* | BA 6 | L | −6, 10, 50 | 4.939 | 130 |
| R | 8, −20, 51 | 3.721 | 39 | ||
| Primary motor cortex (M1)* | BA 4 | R | 8, −26, 60 | 3.734 | 72 |
| Somatosensory cortex (S1)* | BA 1/2/3 | R | 6, −28, 56 | 3.968 | 117 |
| Dorsolateral prefrontal cortex (DLPFC)* | BA 9/10/46 | L | −24, 52, 36 | 3,534 | 89 |
| R | 30, 48, 30 | 3.562 | 76 | ||
| Orbitofrontal cortex (OFC)* | BA 11/12 | R | 6, 62, −22 | 3.556 | 22 |
| Premotor/associative cortex* | BA 8 | L | −6, 40, 49 | 3.947 | 50 |
| Ventrolateral prefrontal cortex (VLPFC)* | BA 47 | L | −46, 37, −2 | 4.050 | 110 |
| R | 46, 36, −13 | 3.432 | 23 | ||
| Dorsal part of anterior cingulate cortex (dACC) | BA 32 | L | −6, 30, 34 | 4.939 | 230 |
| Insula | ND | R | 46, 14, −10 | 4.424 | 216 |
| Temporal lobe | BA 38 | L | −30, 10, −20 | 3.617 | 94 |
| Ventral part of posterior cingulate cortex (PCC) | BA 23 | L | −4, −26, 38 | 3.872 | 117 |
| Superior temporal cortex | BA 22 | L | −50, 14, −4 | 3.439 | 41 |
| Anterior part of entorhinal cortex | BA 34 | L | −28, 6, −16 | 3.907 | 94 |
| R | 16, 0, −16 | 3.565 | 60 | ||
| Thalamus* | ND | L | −14, −18, 16 | 3.217 | 31 |
| Putamen* | ND | L | −22, 12, 2 | 3.382 | 32 |
| Pallidum* | ND | L | −12, 2, −4 | 3.511 | 79 |
| Controls ∼ [(VI−C)−(MI−C)] | |||||
| Lateral premotor cortex (lateral PMC)* | BA 6 | R | 48, 0, 54 | 3.618 | 57 |
| Superior parietal lobule (SPL)/Precuneus | BA 7 | L | −22, −60, 58 | 3.629 | 40 |
| R | 22, −76, 48 | 4.762 | 1309 | ||
| Inferior parietal lobule (IPL)/Supramarginal gyrus* | BA 40 | L | −38, −56, 54 | 4.036 | 69 |
| Inferior parietal lobule (IPL)/Angular gyrus* | BA 39 | R | 36, −76, 26 | 4.596 | 276 |
| Fusiform gyrus | BA 37 | L | −34, −60, −14 | 4.331 | 494 |
| R | 62, −50, −24 | 3.475 | 20 | ||
| Primary visual cortex | BA 17 | L | −12, −96, 6 | 3.679 | 281 |
| R | 8, −84, 8 | 3.546 | 1111 | ||
| Secondary visual cortex | BA 18 | L | −12, −82, −2 | 5.817 | 5237 |
| R | 6, −72, 0 | 6.128 | 5237 | ||
| Associative visual cortex | BA 19 | L | −36, −80, 22 | 5.565 | 5237 |
| R | 36, −78, 24 | 5.795 | 13.9 | ||
| Cerebellum (hemisphere)/Paravermis* | ND | L | −10, −68, −16 | 3.410 | 26 |
| Patients off medication ∼ [(MI−C)−(VI−C)] | |||||
| Supplementary Motor Area (caudal SMA)* | BA 6 | L | −10, −14, 62 | 3.540 | 30 |
| Lateral premotor cortex (lateral PMC)* | BA 6 | L | −46, 8, 46 | 3.543 | 226 |
| Somatosensory cortex (S1)* | BA 1/2/3 | R | 34, −20, 44 | 3.287 | 20 |
| Dorsolateral prefrontal cortex (DLPFC)* | BA 9/10/46 | R | 19, 49, 24 | 3.501 | 31 |
| Orbitofrontal cortex (OFC)* | BA 11/12 | R | 22, 54, −18 | 3.378 | 27 |
| Ventrolateral prefrontal cortex (VLPFC)* | BA 47 | R | 48, 32, −6 | 3.214 | 20 |
| Ventral part of anterior cingulate cortex (vACC) | BA 24 | R | 4, 35, 12 | 3.343 | 21 |
| Dorsal part of anterior cingulate cortex (dACC) | BA 32 | R | 10, 46, 22 | 4.180 | 71 |
| Superior parietal lobule (SPL)/Paracentral lobule* | BA 5 | R | 18, −45, 72 | 3.360 | 125 |
| Temporal pole | BA 38 | L | −36, 14, −22 | 3.425 | 20 |
| R | 34, 8, −15 | 3.606 | 23 | ||
| Superior temporal cortex | BA 22 | L | −50, −10, −9 | 4.472 | 226 |
| Anterior part of entorhinal cortex | BA 34 | R | 27, −1, −20 | 3.392 | 48 |
| Ponto−mesencephalic tegmentum* | ND | R | 12, −10, −16 | 4.510 | 244 |
| R | 6, −26, −26 | 3.845 | 28 | ||
| Patients off medication ∼ [(VI−C)−(MI−C)] | |||||
| Superior parietal lobule (SPL)/Precuneus* | BA 7 | L | −6, −70, 60 | 3.750 | 30 |
| Primary visual cortex | BA 17 | L | −10, −92, −4 | 4.108 | 342 |
| R | 12, −92, 6 | 5.826 | 1824 | ||
| Secondary visual cortex | BA 18 | L | −16, −84, 0 | 3.835 | 342 |
| R | 28, −90, −12 | 4.806 | 1824 | ||
| Associative visual cortex | BA 19 | R | 28, −72, −6 | 3.813 | 56 |
| Cerebellum (hemisphere)/Paravermis* | ND | L | −20, −40, −50 | 3.983 | 57 |
| R | 16, −82, −36 | 3.685 | 80 | ||
Abbreviations: k = cluster size (number of voxels); L = left; ND = no defined as Brodmann's Areas; R = right.
The stereotaxic coordinates, reported in the MNI space, represent the peak voxel within a cluster that was present above the statistical threshold in the whole−brain analysis. Z−scores indicate the statistical value of the most significant voxel in the associated cluster at a P−value < 0.001 uncorrected and k > 20 contiguous voxels. The regions which survived at a FWE−corrected P−value in the whole−brain analysis are indicated in italics. The asterisk points out the brain areas which survived at a FWE−corrected P−value after small volume correction.
Effect of levodopa
During MI of gait compared to C, levodopa intake led to increased rCBF within the left pre‐SMA, DLPFC and thalamus, and in the right M1, lateral PMC, PCC, putamen and cerebellum, as well as decreased rCBF within the caudal SMA, left lateral PMC, in the right DLPFC, dACC, SPL and pontomesencephalic area. Under medication, VI‐related activations were stronger within the left SPL, inferior parietal, superior temporal cortices, in the right cerebellum, and in occipital regions. No areas were more involved in the off than in the on condition (Table 4 and Fig. 4).
Table 4.
MNI coordinates for the main brain areas more activated during MI of gait, or VI, in comparison with the C task (1) in patients without treatment than in patients with treatment, and (2) in patients with treatment than in patients without treatment
| [MI−C] | (1) Patients off − on med. | (2) Patients on − off med. | ||||||
|---|---|---|---|---|---|---|---|---|
| Localization of the main cerebral areas | Laterality | Coordinates | Z−score | k | Coordinates | Z−score | k | |
| Functional label | Brodmann's areas (BA) | x, y, z | x, y, z | |||||
| Supplementary Motor Area (pre−SMA & SMA)* | BA 6 | L | −6, −16, 64 | 3.843 | 62 | −6, 4, 66 | 4.612 | 33 |
| R | 10, −6, 54 | 3.273 | 24 | − | − | − | ||
| Lateral premotor cortex (PMC)* | BA 6 | L | −32, 8, 45 | 3.214 | 28 | − | − | − |
| R | − | − | − | 40, −4, 48 | 3.278 | 25 | ||
| Primary motor cortex (M1)* | BA 4 | R | − | − | − | 18, −30, 62 | 3.528 | 38 |
| Dorsolateral prefrontal cortex (DLPFC)* | BA 9/10/46 | L | − | − | − | −35, 54, 8 | 3.546 | 23 |
| R | 24, 38, 32 | 3.306 | 20 | − | − | − | ||
| Dorsal part of anterior cingulate cortex (dACC) | BA 32 | R | 18, 42, 12 | 4.270 | 58 | − | − | − |
| Superior parietal cortex (SPL) / Paracentral lobule* | BA 5 | R | 8, −50, 57 | 3.765 | 91 | − | − | − |
| Ventral part of posterior cingulate cortex (PCC) | BA 23 | R | − | − | − | 8, −14, 40 | 3.186 | 23 |
| Thalamus* | ND | L | − | − | − | −10, −16, 4 | 3.486 | 30 |
| Putamen* | ND | R | − | − | − | 29, 16, 0 | 3.607 | 21 |
| Cerebellum (hemisphere) / Paravermis* | ND | R | − | − | − | 40, −68, −25 | 3.251 | 24 |
| Ponto−mesencephalic tegmentum* | ND | R | 12, −14, −20 | 3.544 | 123 | − | − | − |
| [VI−C] | (1) Patients off − on med. | (2) Patients on − off med. | ||||||
|---|---|---|---|---|---|---|---|---|
| Location of the main cerebral areas | Laterality | Coordinates | Z−score | k | Coordinates | Z−score | k | |
| Functional label | Brodmann's areas (BA) | x, y, z | x, y, z | |||||
| Superior parietal cortex (SPL)/ Precuneus* | BA 7 | L | No suprathreshold clusters | −24, −56, 50 | 3.674 | 129 | ||
| Inferior parietal lobule (IPL) / Supramarginal gyrus* | BA 40 | L | −36, −56, 62 | 3.543 | 125 | |||
| Superior temporal gyrus | BA 22 | L | −52, −12, −8 | 3.613 | 31 | |||
| Primary visual cortex | BA 17 | L | −16, −102, 4 | 3.649 | 72 | |||
| Secondary visual cortex | BA 18 | L | −6, −88, 12 | 3.869 | 84 | |||
| R | 22, −74, 32 | 3.935 | 49 | |||||
| Associative visual cortex | BA 19 | L | −28, −70, −4 | 3.494 | 22 | |||
| Cerebellum (hemisphere) / Paravermis* | ND | R | 26, −28, −28 | 4.063 | 62 | |||
Abbreviations: k = cluster size (number of voxels); L = left; ND = no defined as Brodmann's Areas; R = right.
The stereotaxic coordinates, reported in the MNI space, represent the peak voxel within a cluster that was present above the statistical threshold in the whole‐brain analysis. Z‐scores indicate the statistical value of the most significant voxel in the associated cluster at a P‐value < 0.001 uncorrected and k > 20 contiguous voxels. The asterisk points out the brain areas which survived at a FWE‐corrected P‐value after small volume correction.
Figure 4.
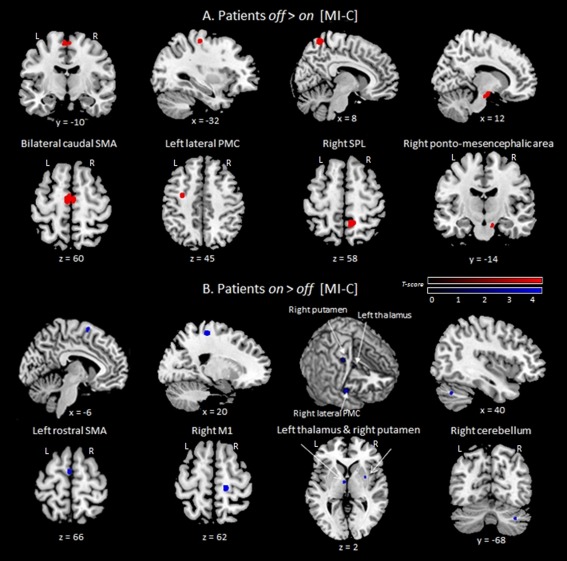
Brain areas with decreased (A) and increased (B) rCBF induced by MI of gait‐related activity in patients on medication relative to off (Statistical t‐maps were thresholded to a P‐value < 0.001 uncorrected, with a minimal cluster size k = 20). Caudal SMA = caudal part of supplementary motor area; lateral PMC = lateral premotor cortex; L = left; M1 = primary motor cortex; R = right; SPL = superior parietal lobule; x = medio‐lateral, y = rostro‐caudal, z = dorso‐ventral coordinates according to the MNI space. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Between‐group comparisons
Relative to controls, gait imagery in patients off led to greater activation in the left caudal SMA, lateral PMC, and right dACC, SPL, and pontomesencephalic area. Activation was reduced in the pre‐SMA, DLPFC, the left dACC and the right M1, S1, lateral PMC, insula, thalamus, putamen, cerebellum, and red nucleus. In patients, VI‐related activity was stronger in the left frontal cortex, and reduced in the right lateral PMC, VLPFC, SPL and cerebellum (Table 5 and Fig. 5).
Table 5.
MNI coordinates for the main brain areas more activated during MI of gait, or VI, in comparison with the C task (1) in controls than in patients without treatment, and (2) in patients without treatment than in controls
| [MI−C] | (1) Controls − Patients off med. | (2) Patients off med.−Controls | ||||||
|---|---|---|---|---|---|---|---|---|
| Location of the main cerebral areas | Laterality | Coordinates | Z‐score | k | Coordinates | Z−score | k | |
| Functional label | Brodmann's areas (BA) | x, y, z | x, y, z | |||||
| Supplementary Motor Area (pre‐SMA & SMA)* | BA 6 | L | −10, 14, 70 | 3.185 | 40 | −6, −12, 66 | 3.845 | 75 |
| R | 6,4,66 | 3.181 | 32 | − | − | − | ||
| Lateral premotor cortex (lateral PMC)* | BA 6 | L | − | − | − | −30, −16, 50 | 4.835 | 36 |
| R | 52, −3, 47 | 4.564 | 146 | − | − | − | ||
| Primary motor cortex (M1)* | BA 4 | R | 14, −26, 64 | 3.964 | 49 | − | − | − |
| Somatosensory cortex (S1)* | BA 1/2/3 | R | 40, −16, 38 | 3.839 | 146 | − | − | − |
| Dorsolateral prefrontal cortex (DLPFC)* | BA 9/10/46 | L | −29, 43, 32 | 3.888 | 21 | − | − | − |
| R | 24, 50, 40 | 3.703 | 32 | − | − | − | ||
| Dorsal part of anterior cingulate cortex (dACC) | BA 32 | L | −10, 16, 40 | 3.476 | 65 | − | − | − |
| R | − | − | − | 16, 42, 6 | 3.182 | 26 | ||
| Superior parietal cortex (SPL)/Paracentral lobule* | BA 5 | R | − | − | − | 16, −52, 55 | 3.215 | 105 |
| Hippocampus | ND | L | − | − | − | −16, −5, −11 | 3.256 | 28 |
| Insula | ND | R | 46, 12, −2 | 3.427 | 45 | − | − | − |
| Thalamus* | ND | R | 16, −20, 4 | 3.126 | 31 | − | − | − |
| Putamen* | ND | R | 24, 6, 4 | 3.112 | 28 | − | − | − |
| Cerebellum (hemisphere) /Paravermis* | ND | R | 40, −60, −36 | 3.946 | 112 | − | − | − |
| Red nucleus* | ND | R | 6, −18, −6 | 3.547 | 37 | − | − | − |
| Ponto‐mesencephalic tegmentum* | ND | R | − | − | − | 6, −28, −28 | 3.847 | 53 |
| [VI−C] | (1) Controls − Patients off med. | (2) Patients off med. − Controls | ||||||
|---|---|---|---|---|---|---|---|---|
| Location of the main cerebral areas | Laterality | Coordinates | Z−score | k | Coordinates | Z−score | k | |
| Functional label | Brodmann's areas (BA) | x, y, z | x, y, z | |||||
| Premotor cortex (PMC)* | BA 6 | R | 52, −4, 52 | 3.702 | 56 | − | − | − |
| Ventrolateral prefrontal cortex (VLPFC)* | BA 47 | R | 46, 32, −4 | 3.994 | 62 | − | − | − |
| Premotor/associative cortex* | BA 8 | L | − | − | − | −31, 16, 50 | 4.835 | 82 |
| Superior parietal cortex (SPL)/Precuneus* | BA 7 | R | 12, −50, 8 | 3.519 | 32 | − | − | − |
| Fusiform gyrus | BA 37 | R | 24, −32, −16 | 3.805 | 26 | − | − | − |
| Cerebellum (hemisphere)/Paravermis* | ND | R | 26, −66, −40 | 3.341 | 20 | − | − | − |
Abbreviations: k = cluster size (number of voxels); L = left; ND = no defined as Brodmann's Areas; R = right.
The stereotaxic coordinates, reported in the MNI space, represent the peak voxel within a cluster that was present above the statistical threshold in the whole‐brain analysis. Z‐scores indicate the statistical value of the most significant voxel in the associated cluster at a P‐value < 0.001 uncorrected and k > 20 contiguous voxels. The asterisk points out the brain areas which survived at a FWE‐corrected P‐value after small volume correction.
Figure 5.
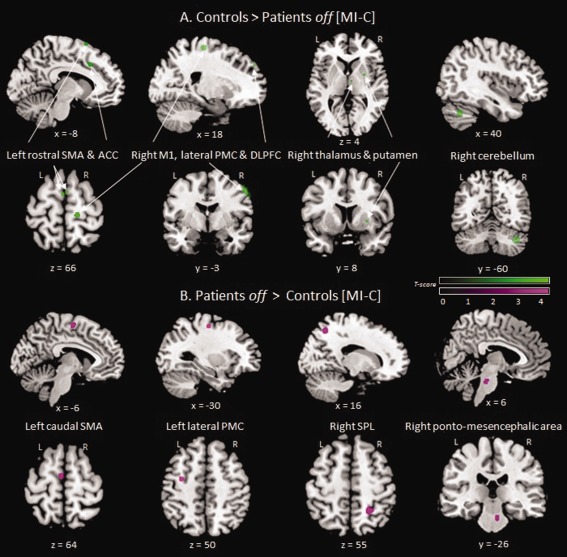
Brain areas with decreased (A) and increased (B) rCBF induced by MI of gait‐related activity in patients off medication relative to controls (statistical t‐maps were thresholded to a P‐value < 0.001 uncorrected, with a minimal cluster size k = 20). ACC = anterior cingulate cortex; Caudal SMA = caudal part of supplementary motor area; DLPFC = dorsolateral prefrontal cortex; lateral PMC = lateral premotor cortex; L = left; M1 = primary motor cortex; pre‐SMA = rostral part of supplementary area; R = right; SPL = superior parietal lobule; x = medio‐lateral; y = rostro‐caudal; z = dorso‐ventral coordinates according to the MNI space. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
DISCUSSION
The primary purpose of this study was to examine the influence of levodopa on the networks involved in MI of gait in advanced Parkinson's disease. Our results confirmed prior works, showing that imagined parkinsonian gait disturbances are related to decreased brain activations in motor and frontal associative areas, BG, thalamus and cerebellum, and increased activations within premotor‐parietal cortices and pontomesencephalic tegmentum, relative to controls. Levodopa mostly restored activations in the primary motor cortex, BG, thalamus and cerebellum. Before discussing our results, it must be kept in mind that, despite careful training of the subjects to MI, and as much as possible indirect measures of MI quality within the scanner, the present study still represents an indirect approach to actual PD gait impairments. We will examine methodological issues before discussing the modulation by levodopa of frontostriatal networks, premotor‐parietal and mesencephalic regions, and the significance of reduced activation of the cerebellum in brain activity related to levodopa‐responsive parkinsonian gait disorders.
Validation of the Research Design
Behavioral data confirmed that patients with advanced Parkinson's disease had preserved MI abilities, and were able to adequately perform MI from a kinesthetic perspective. Comparison of the KVIQ scores showed similar visual and kinesthetic aptitudes in our patients, independent of medication status, and age‐matched healthy individuals, as previously reported [Heremans et al., 2011; Maillet et al., 2013; Peterson et al., 2012; Pickett et al., 2013]. In both groups, the kinesthetic ability further improved with training, suggesting that it is possible to increase the reliability of neuroimaging using MI by providing adequate training. Our data also confirmed that parkinsonian patients can imagine moving in their actual motor state, which is important for study designs based on MI. Finally, the lack of difference in kinesthetic ability between controls and patients enables comparison of gait‐related brain activities between the groups without confounds due to MI performance itself.
Regarding the experimental protocol, analysis of task duration confirmed that both actual and imagined gait were similarly affected by the characteristics of the paths in all groups and conditions, as expected from previous works [Maillet et al., 2013; Snijders et al., 2011]. Furthermore, VI times were only modulated by path length, and not by path width. In other words, only MI shared the properties of actual movement. This difference confirms that participants deployed different mental activities during the two tasks. Such control was important as the methodological constraints of PET did not allow the use of the narrow paths during brain acquisitions.
MI and VI activated different networks, confirming that VI is an adequate behavioral control during brain acquisitions. The VI task was included to control for brain activations associated to nonspecific aspects of the mental processes. Although MI revealed activity within the M1 and S1 cortices, VI mostly involved the parieto‐occipital areas. The M1 paramedian location corresponds to the lower limbs' area of Penfield's homunculus (1954). EMG recordings confirmed that the motor activations seen during MI did not result from actual movements. Such motor activations during gait imagery have already been reported [Karachi et al., 2012; Malouin et al., 2003; Miyai et al., 2001; Sacco et al., 2006; van der Meulen et al., 2014], and could thus be specifically related to the use of kinesthetic MI.
Neural Substrates of Parkinsonian Gait Disorders
In controls, MI of gait recruited a widespread supraspinal network, including motor and frontal associative areas, cingulate and insular cortices, the BG, thalamus, cerebellum, and red nucleus, consistent with the well‐documented cortico‐subcortical circuitry previously reported for healthy natural gait [e.g., Allali et al., 2014; Bakker et al., 2008; Crémers et al., 2011, 2012; Karachi et al., 2010, 2012; La Fougère et al., 2010; Malouin et al., 2003; Snijders et al., 2011; van der Meulen et al., 2014; see Maillet et al., 2012a]. Relative to controls, patients off medication mostly exhibited a hypoactivation of a right frontal‐motor network, putamen, thalamus, and cerebellum, together with increased recruitment of premotor, parietal and mesencephalic regions when performing gait imagery. Thus, the cerebral activity related to parkinsonian levodopa‐responsive gait disorders, including FoG, is linked with disrupted functioning of several areas at the cortical, subcortical and brainstem levels of the locomotor system.
Decreased gait‐related cerebral activity in advanced Parkinson's disease
The frontostriatal hypoactivation observed in patients off, already described for actual and imagined upper‐limbs movements in Parkinson's disease [Buhmann et al., 2003; Jahanshahi et al., 2010; Sabatini et al., 2000; Wu and Hallet, 2005; Wu et al., 2010] is usually interpreted as a direct consequence of the degeneration of the dopaminergic neurons in both the substantia nigra pars compacta and the ventral tegmental area, disturbing the motor and associative cortico‐subcortical pathways. In keeping with these findings, previous studies have shown reduced activity, or metabolism, in several structures of this network (i.e., the pre‐SMA, sensorimotor and OFC cortices, putamen, caudate nucleus, globus pallidus) in patients with gait disorders, at rest and during actual and imagined gait, or in a virtual reality gait proxy task [Bartels et al., 2006; Hanakawa et al., 1999a, 1999b; Imamura et al., 2012; Matsui et al., 2005; Mito et al., 2006; Ouchi et al., 2001; Peterson et al., 2014a, 2014b; Snijders et al., 2011; Shine et al., 2013a, 2013b]. Other works have also shown atrophy and reduced connectivity in the frontostriatal network of patients with gait difficulties, compared to those without, or compared to controls [Fling et al., 2013; Herman et al., 2014; Kostic et al., 2012; Rosenberg‐Katz et al., 2013; Shine et al., 2013b; Tessitore et al., 2012a, 2012b].
Thus, the reduced activation observed within the BG during imagined gait in patients off medication is consistent with a disconnection, or at least an abnormal communication, between subcortical and cortical areas, as one possible key mechanisms of parkinsonian gait disorders [Bohnen and Jahn, 2013; Lewis and Barker, 2009; Shine et al., 2013a, 2013b, 2013c]. Indeed, as the BG are involved in self‐generated step initiation, automaticity of gait processes, and integrative control of locomotion, disruption of these regions in Parkinson's disease would impair gait [Bohnen and Jahn, 2013; Shine et al., 2013a, 2013b, 2013c; Takakusaki et al., 2004, 2008]. The thalamic deactivation observed in patients off is also in line with such subcortico‐thalamic‐cortical failure. Previous reports have highlighted the role of the thalamus in postural control and maintenance of an erect posture [Jahn et al., 2004, 2008; Muller et al., 2013], while others have suggested that a thalamic hypoactivation, or atrophy, could contribute to gait disorders, FoG, and to an increased risk of falling in Parkinson's disease [Bohnen et al., 2009, 2012; Fling et al., 2013; Sunwoo et al., 2013].
At the cortical level, the activation deficits observed within M1, pre‐SMA, DLPFC, and ACC in patients off also points to a disruption of the motor and nonmotor cortico‐subcortical pathways in imagined parkinsonian gait disorders. Alteration of the motor circuit could be responsible for deficits in sensorimotor integration, voluntary action, or motor planning [Benecke et al., 1987; Cunnington et al., 1996; Knobl et al., 2011] inducing impaired symmetry, rhythmicity, and bilateral coordination during parkinsonian gait [Iansek et al., 2006; Jacobs et al., 2009]. The decreased step length has been linked to pre‐SMA hypoactivation, a hypothesized substrate of hypokinesia [Nachev et al., 2008; Sabatini et al., 2000], in relation with a defective maintenance by the BG [Chee et al., 2009; Snijders et al., 2011]. In addition, it has been shown that adequate executive, and therefore frontal lobe, functioning is required for efficient gait [Amboni et al., 2008; Collette et al., 2006; Yogev‐Seligmann et al., 2008]. Although our patients were screened for preserved executive function, suggesting that the mental representation of their gait difficulties did not mainly depend on cognitive processing alterations, one cannot exclude that a disruption of the associative and limbic frontostriatal loops might contribute to difficulties in updating action plans or adjusting the time‐course of movements [Dirnberger and Jahanshahi, 2013; Helmich et al., 2009; Snijders et al., 2011]. The ACC hypoactivation could explain the reduced capacity of patients to switch from one action to another, which may be important during gait [Naismith et al., 2010; Shine et al., 2013b, 2013c]. Levodopa reversed the M1, pre‐SMA, thalamic and putaminal hypoactivations observed during MI of gait disorders, restoring a better connectivity between motor cortical and subcortical areas [Shine et al., 2013b], together with improved actual gait performance.
The reduced activation in the DLPFC, normally involved in response selection [Frith et al., 2000; Jahanshahi et al., 2000], could also be a hallmark of brain activity related to parkinsonian gait impairments, although the precise role of the DLPFC in these gait disorders is not clear. Shine et al. [2013a] reported its bilateral overactivation in patients with FoG and suggested a compensatory mechanism involving the recruitment of high‐level cognitive control. Hence, frontal involvement could reflect an attempt at counterbalancing the loss of automaticity through goal‐directed behavior [Lewis and Barker, 2009; Shine et al. 2013a, 2013c]. We, however, observed hypoactivation of the DLPFC. The discrepancy between our and Shine et al. [2013a, 2013c] results could be related to differences in methodology, patients' characteristics (i.e., disease duration and levodopa‐responsiveness of gait symptoms), and also to the fact that Shine et al. [2013a] specifically examined brain activations during FoG episodes, while we aimed at better understanding the brain networks disrupted in levodopa responsive gait disorders.
Finally, our study also supports the notion that cerebral activity related to parkinsonian gait disorders could be associated with a predominant functional deficit within the right hemisphere. This is of interest as several previous studies have indeed reported a structural and functional impairment of this hemisphere in patients suffering from gait disorders and freezing, both in cortical (that is, in fronto‐parieto‐occipital areas) and subcortical (BG, cerebellum, brainstem) structures [Bartels and Leenders, 2008; Bartels et al., 2006; Crémers et al., 2012; Fling et al., 2013; Peterson et al., 2014b; Snijders et al., 2011; Tessitore et al., 2012a]. Such result is also in line with the right hemispheric predominance in visuo‐spatial and attentional processes [Crémers et al., 2011; Foxe et al., 2003].
Increased gait‐related cerebral activity in advanced Parkinson's disease
Our results provide evidence of an increased recruitment of premotor‐parietal and pontomesencephalic areas during MI of gait in patients off medication, relative to controls. These results are in keeping with those of previous neuroimaging studies relying on actual gait or virtual reality gait proxy [Hanakawa et al., 1999a; Shine et al., 2013a]. In contrast, Snijders et al. [2011] reported a parietal deactivation during imagined gait in patients off medication. Once again, comparing data is difficult as previous authors did not distinguish between levodopa‐responsive and levodopa‐resistant FoG, which is likely to have a significant bearing on gait‐related brain activation profiles.
The premotor‐parietal overactivation, already reported for upper‐limb movements [Frackowiak et al., 2003; Haslinger et al., 2001; Jahanshahi et al., 2010; Maillet et al., 2012b; Sabatini et al., 2000; Samuel et al., 1997; Wu and Hallet, 2005; Wu et al., 2010; Yu et al., 2007], could reflect an attempt at compensating for frontostriatal disruption. Indeed, the lateral PMC is known to be involved in action programming, visuo‐motor coordination and proprioceptive integration [Rizzolatti et al., 1988] through its connections with the parietal cortex, which is involved in visuo‐spatial processing [Fletcher et al., 1995]. Particularly, the possible compensatory role of the left lateral PMC is supported here by its overactivation in patients off medication compared to controls, and the reduction of its activation under levodopa, concomitantly with imagined gait improvement. Thus, we could discuss an imbalance between the right and left hemisphere, and a specific compensation by the left lateral PMC, as previously suggested in another study on paradoxical gait in PD [Hanakawa et al., 1999a]. Moreover, the caudal SMA also receives inputs from the parietal cortex [Luppino et al., 1993]. Its hyperactivity in patients off could therefore contribute to overcoming the M1 and pre‐SMA deficits, supporting the compensatory hypothesis [Sabatini et al., 2000].
According to this hypothesis, the functional switch from the deficient nigro‐striatal loop, involved in self‐generated actions, to a better preserved and functional circuit controlling execution of visually guided movements, would enable execution of a formerly automatic action under conscious control. This idea is supported by the beneficial effect of visual cues on actual parkinsonian gait [Azulay et al., 2006; Lee et al., 2012; Nieuwboer, 2008; Park et al., 2014], mediated by the recruitment of this lateral parietal‐premotor circuit [Azulay et al., 1999; Hanakawa et al. 1999a]. Finally, the reduced activation of the frontoparietal network under levodopa suggests that this recruitment is no longer useful when the frontostriatal loop functioning is partly restored. The same effect has already been reported for real and imagined upper‐limb movements in Parkinson's disease [Cools et al., 2002; Haslinger et al., 2001; Jenkins et al., 1992; Kraft et al., 2009; Maillet et al., 2012b]. Ours is however, the first study to demonstrate it for MI of gait in advanced Parkinson's disease. Cortical or transcranial magnetic stimulation studies will be needed to confirm the hypothetical compensatory role of these areas in parkinsonian gait disorders.
The pontomesencephalic overactivation observed in patients without treatment was suppressed by levodopa, which could suggest another possible subcortical compensatory mechanism to counterbalance the frontostriatal deficit responsible for parkinsonian gait disorders. This result is consistent with those of Snijders et al. [2011] and Crémers et al. [2012], but not with those of Shine et al. [2013a] and Peterson et al. [2014b]. Again, such inconsistent findings may relate to the different activation tasks and neuroimaging techniques used, as well as to the clinical characteristics of the patients. Actually, it is not possible to ascertain whether the pontomesencephalic area plays a compensatory or pathogenic role here. Indeed, within this specific region, the area overactivated in the “patients off medication versus controls” contrast differs in terms of topography from the one observed in the “patients off medication versus patients on medication” contrast. It is therefore difficult to conclude that dopaminergic treatment improves gait disorders by normalizing a pathological pontomesencephalic overactivation. In addition, given the high individual anatomical variability in the brainstem, even more pronounced in PD patients—as this region in known to be atrophied in subjects suffering from gait disorders [Chastan et al., 2009; Snijders et al., 2011]—, the unavailability of recent and specific atlas for this structure, as well as the difficulty to study this region using PET technique (due to the limited spatial resolution and the impact of data preprocessing), we should remain extremely cautious regarding the pathophysiological interpretation of these activation changes within the brainstem. Further studies will be therefore, needed to better understand the role of brainstem structures in the pathophysiology of parkinsonian gait disorders.
A cerebellar involvement in parkinsonian gait disorders?
Another important finding of our study is the contribution of the cerebello‐thalamo‐cortical loop to the brain activity related to the pathophysiology of parkinsonian gait. Indeed, we observed a right cerebellar hemisphere activation during gait imagery in controls and in patients on, but not in patients off. A cerebellar hypoactivation has been previously reported during actual [Hanakawa et al., 1999b], and imagined [Crémers et al., 2012; Peterson et al., 2014b] gait, while the cerebellum has also been shown to have reduced connectivity with other areas involved in motor control [Fling et al., 2013; Schweder et al., 2010] in patients with gait disorders. The BG and cerebellum are known to modulate both motor and nonmotor aspects of behavior through two anatomically distinct but interconnected loops [Bostan et al., 2010; Middleton and Strick, 2000; Wu and Hallett, 2013]. The cerebellum is particularly involved in balance control [Gilman et al., 2010; Morton and Bastian, 2004], real‐time motor adjustments based on sensorimotor and visual informations [Blakemore and Sirigu, 2003; Purzner et al., 2007; Takakusaki, 2013], and regulation of on‐going movement velocity and rhythm [Nadkarni et al., [Link]; Takakusaki, 2013]. Its increased activity under medication might thus be related to enhanced limb co‐ordination during gait imagery in patients, in link with the partial restoration of the BG circuit. The improvement of motor function and FoG in patients with levodopa‐resistant signs, related to an increased activity or connectivity of the SMA, thalamus, and cerebellum under pedunculopontine nucleus stimulation [Ballanger et al., 2009; Schweder et al., 2010; Strafella et al., 2008; Thevathasan et al., 2012a, 2012b] also supports the hypothesis of a combined BG and cerebellar dysfunction in link with advanced parkinsonian gait disorders.
CONCLUSION
A better understanding of the neural correlates of parkinsonian gait disturbances is needed for the development of more efficient therapeutic strategies. The results of our study based on kinesthetic imagery approach show that brain disruption related to levodopa‐responsive parkinsonian gait disorders might be precipitated by dysfunction across coordinated motor and cognitive neural networks, resulting in both frontostriatal disruption and abnormal recruitment of premotor‐parietal and pontomesencephalic regions. Levodopa restores a more physiological brain activation profile, and suppresses the recruitment of the accessory circuits. Similar results have been reported for actual and imagined upper‐limb movements in Parkinson's disease, suggesting common pathophysiological mechanisms. In the future, it would be of great interest to explore the anatomo‐functional basis of levodopa‐resistant parkinsonian gait disorders.
ACKNOWLEDGMENTS
The authors gratefully thank the CERMEP PET staff, that is, Fabienne Vey, Véronique Berthier and Christine Vighi, for their helpful support in performing this study and patients' management, as well as all the subjects who kindly agreed to be part of this research.
REFERENCES
- Allali G, van der Meulen M, Beauchet O, Rieger SW, Vuilleumier P, Assal F (2014): The neural basis of age‐related changes in motor imagery of gait: An fMRI study. J Gerontol A Biol Sci Med Sci 69:1389–1398. [DOI] [PubMed] [Google Scholar]
- Amboni M, Cozzolino A, Longo K, Picillo M, Barone P (2008): Freezing of gait and executive functions in patients with Parkinson's disease. Mov Disord 23:395–400. [DOI] [PubMed] [Google Scholar]
- Azulay JP, Mesure S, Blin O (2006): Influence of visual cues on gait in Parkinson's disease: Contribution to attention or sensory dependence? J Neurol Sci 248:192–195. [DOI] [PubMed] [Google Scholar]
- Azulay JP, Mesure S, Amblard B, Blin O, Sangla I, Pouget J (1999): Visual control of locomotion in Parkinson's disease. Brain 122:111–120. [DOI] [PubMed] [Google Scholar]
- Bakker M, de Lange FP, Stevens JA, Toni I, Bloem BR (2007): Motor imagery of gait: A quantitative approach. Exp Brain Res 179:497–504. [DOI] [PubMed] [Google Scholar]
- Bakker M, de Lange FP, Helmich RC, Scheeringa R, Bloem BR, Toni I (2008): Cerebral correlates of motor imagery of normal and precision gait. Neuroimage 41:998–1010. [DOI] [PubMed] [Google Scholar]
- Ballanger B, Lozano AM, Moro E, van Eimeren T, Hamani C, Chen R, Cilia R, Houle S, Poon YY, Lang AE, Strafella AP (2009): Cerebral blood flow changes induced by pedunculopontine nucleus stimulation in patients with advanced Parkinson's disease: A [(15)O] H2O PET study. Hum Brain Mapp 30:3901–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels AL, Leenders KL (2008): Brain imaging in patients with freezing of gait. [Review]. Mov Disord 23:1639–1640. [DOI] [PubMed] [Google Scholar]
- Bartels AL, de Jong BM, Giladi N, Schaafsma JD, Maguire RP, Veenma L, Pruim J, Balash Y, Youdim MB, Leenders KL (2006): Striatal dopa and glucose metabolism in PD patients with freezing of gait. Mov Disord 21:1326–1332. [DOI] [PubMed] [Google Scholar]
- Benecke R, Rothwell JC, Dick JP, Day BL, Marsden CD (1987): Disturbance of sequential movements in patients with Parkinson's disease. Brain 110:361–379. [DOI] [PubMed] [Google Scholar]
- Blakemore SJ, Sirigu A (2003): Action prediction in the cerebellum and in the parietal lobe. Exp Brain Res 153:239–245. [DOI] [PubMed] [Google Scholar]
- Bloem BR, Hausdorff JM, Visser JE, Giladi N (2004): Falls and freezing of gait in Parkinson's disease: A review of two interconnected, episodic phenomena. Mov Disord 19:871–884. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, Jahn K (2013): Imaging: What can it tell us about parkinsonian gait? [Review]. Mov Disord 28:1492–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen NI, Muller ML, Koeppe RA, Studenski SA, Kilbourn MR, Frey KA, Albin RL (2009): History of falls in Parkinson disease is associated with reduced cholinergic activity. Neurology 73:1670–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen NI, Muller ML, Kotagal V, Koeppe RA, Kilbourn MR, Gilman S, Albin RL, Frey KA (2012): Heterogeneity of cholinergic denervation in Parkinson's disease without dementia. J Cereb Blood Flow Metab 32:1609–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet AM, Loria Y, Saint‐Hilaire MH, Lhermitte F, Agid Y (1987): Does long‐term aggravation of Parkinson's disease result from non‐dopaminergic lesions? Neurology 37:1539–1542. [DOI] [PubMed] [Google Scholar]
- Bostan AC, Dum RP, Strick PL (2010): The basal ganglia communicate with the cerebellum. PNAS 107:8452–8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhmann C, Glauche V, Stürenburg HJ, Oechsner M, Weiller C, Büchel C (2003): Pharmacologically modulated fMRI‐‐cortical responsiveness to levodopa in drug‐naive hemiparkinsonian patients. Brain 126:451–461. [DOI] [PubMed] [Google Scholar]
- Chastan N, Do MC, Bonneville F, Torny F, Bloch F, Westby GW, Dormont D, Agid Y, Welter ML (2009): Gait and balance disorders in Parkinson's disease: Impaired active braking of the fall of centre of gravity. Mov Disord 24:188–195. [DOI] [PubMed] [Google Scholar]
- Chee R, Murphy A, Danoudis M, Georgiou‐Karistianis N, Iansek R (2009): Gait freezing in Parkinson's disease and the stride length sequence effect interaction. Brain 132:2151–2160. [DOI] [PubMed] [Google Scholar]
- Cohen RG, Chao A, Nutt JG, Horak FB (2011): Freezing of gait is associated with a mismatch between motor imagery and motor execution in narrow doorways, not with failure to judge doorway passability. Neuropsychol 49:3981–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collette F, Hogge M, Salmon E, Van der Linden M (2006): Exploration of the neural substrates of executive functioning by functional neuroimaging. Neuroscience 139:209–221. [DOI] [PubMed] [Google Scholar]
- Cools R, Stefanova E, Barker RA, Robbins TW, Owen AM (2002): Dopaminergic modulation of high‐level cognition in Parkinson's disease: The role of the prefrontal cortex revealed by PET. Brain 125:584–594. [DOI] [PubMed] [Google Scholar]
- Crémers J, Dessoullières A, Garraux G (2011): Hemispheric specialization during mental motor imagery of brisk walking. Hum Brain Mapp 33:873–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crémers J, D'Ostilio K, Stamatakis J, Delvaux V, Garraux G (2012): Brain activation pattern related to gait disturbances in Parkinson's disease. Mov Disord 27:1498–1505. [DOI] [PubMed] [Google Scholar]
- Cunnington R, Iansek R, Thickbroom GW, Laing BA, Mastaglia FL, Bradshaw JL, Phillips JG (1996): Effects of magnetic stimulation over supplementary motor area on movement in Parkinson's disease. Brain 119:815–822. [DOI] [PubMed] [Google Scholar]
- Devos D, Defebvre L, Bordet R (2010): Dopaminergic and non‐dopaminergic pharmacological hypotheses for gait disorders in Parkinson's disease. [Review]. Fundam Clin Pharmacol 24:407–421. [DOI] [PubMed] [Google Scholar]
- Dirnberger G, Jahanshahi M (2013): Executive dysfunction in Parkinson's disease: A review. [Review]. J Neuropsychol 7:193–224. [DOI] [PubMed] [Google Scholar]
- Dubois B, Slachevsky A, Litvan I, Pillon B (2000): The FAB: A frontal assessment battery at bedside. Neurology 55:1621–1626. [DOI] [PubMed] [Google Scholar]
- Espay AJ, Fasano A, van Nuenen BFL, Payne MM, Snijders AH, Bloem BR (2012): “On” state freezing of gait in Parkinson disease. A paradoxical levodopa‐induced complication. Neurology 78:454–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraye MU, Debû B, Fraix V, Xie‐Brustolin J, Chabardès S, Krack P, Benabid AL, Pollak P (2008): Effects of subthalamic nucleus stimulation and levodopa on freezing of gait in Parkinson's disease. Neurology 70 (16 Pt 2):1431–1437. [DOI] [PubMed] [Google Scholar]
- Fitts PM (1954): The informed capacity of the human motor system in controlling the amplitude of movements. J Exp Psychol 121:262–269. [DOI] [PubMed] [Google Scholar]
- Fletcher PC, Frith CD, Grasby PM, Shallice T, Frackowiak RSJ, Dolan RJ (1995): The mind's eye precuneus activation in memory related imagery. Neuroimage 2:195–200. [DOI] [PubMed] [Google Scholar]
- Fling BW, Cohen RG, Mancini M, Nutt JG, Fair DA, Horak FB (2013): Asymmetric pedunculopontine network connectivity in parkinsonian patients with freezing of gait. Brain 136:2405–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR (1975): “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12:189–198. [DOI] [PubMed] [Google Scholar]
- Fox SH (2013): Non‐dopaminergic treatments for motor control in Parkinson's disease. [Review]. Drugs 73:1405–1415. [DOI] [PubMed] [Google Scholar]
- Foxe JJ, Mc Court ME, Javitt DC (2003): Right hemisphere control of visuospatial attention: Line‐bisection judgments evaluated with high‐density electrical mapping and source analysis. NeuroImage 19:710–726. [DOI] [PubMed] [Google Scholar]
- Frackowiak RS, Dowsey‐Limousin P, Jahanshahi M, Hariz M (2003): Brain imaging and mobility in Parkinson's disease. [Article in French]. Bull Acad Natl Med 187:295–304. [PubMed] [Google Scholar]
- Friston KJ, Holmes AP, Worsley KJ, Poline JP, Frith CD (1994): Statistical parametric maps in functional imaging: A general linear approach. Hum Brain Mapp 2:189–210. [Google Scholar]
- Frith CD, Blakemore SJ, Wolpert DM (2000): Abnormalities in the awareness and control of action. Phil Trans R Soc London 355:1771–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb WR, Lees AJ (1988): The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. JNNP 51:745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giladi N, Nieuwboer A (2008): Understanding and treating freezing of gait in parkinsonism, proposed working definition, and setting the stage. [Review]. Mov Disord 23:425–425. [DOI] [PubMed] [Google Scholar]
- Giladi N, Shabtai H, Simon ES, Biran S, Tal J, Korczyn AD (2000): Construction of freezing of gait questionnaire for patients with parkinsonism. Parkinsonism Relat Disord 6:165–170.x. [DOI] [PubMed] [Google Scholar]
- Gilman S, Koeppe RA, Nan B, Wang CN, Wang X, Junck L, Chervin RD, Consens F, Bhaumik A (2010): Cerebral cortical and subcortical cholinergic deficits in parkinsonian syndromes. Neurology 74:1416–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz CG, Poewe W, Rascol O, Sampaio C, Stebbins GT, Counsell C, Giladi N, Holloway RG, Moore CG, Wenning GK, Yahr MD, Seidl L; Movement Disorders Society Task Force report on Rating scales for Parkinson's disease (2004): Movement Disorders Society task force report on the Hoehn and Yahr staging scale; status and recommendations. Mov Disord 19:1020–1028. [DOI] [PubMed] [Google Scholar]
- Goetz CG, Tilly BC, Shaftman SR, Stebbins GT, Fahn S (2008): Movement Disorders Society ‐ sponsored revision of the Unified Parkinson's Disease Rating Scale (MDS‐UPDRS): Scale presentation and clinimetric testing results. Mov Disord 15, 2129–2170. [DOI] [PubMed] [Google Scholar]
- Gousias IS, Rueckert D, Heckemann RA, Dyet LE, Boardman JP (2008): Automatic segmentation of brain MRIs of 2‐years‐olds into 83 regions of interest. Neuroimage 40 672–684. [DOI] [PubMed] [Google Scholar]
- Guillot A, Collet C, Nguyen VA, Malouin F, Richards C, Doyon J (2009): Brain activity during visual versus kinesthetic imagery: An fMRI study. Hum Brain Mapp 30:2157–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot A, Di Rienzo F, Collet C (2012): The neurofunctional architecture of motor imagery. [Review] In: Papageorgiou TD, Christopoulos G, Smirnakis S, editor. Functional Magnetic Resonance Imaging/Book 1, (InTech). [Google Scholar]
- Hammers A, Allom R, Koepp MJ, Free SL, Myers R (2003): Three‐dimensional maximum probability atlas of the human brain, with particular reference to the temporal lobe. Hum Brain Mapp 19 224–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanakawa T, Fukuyama H, Katsumi Y, Honda M, Shibasaki H (1999a): Enhanced lateral premotor activity during paradoxical gait in Parkinson's disease. Annu Neurol 45:329–336. [DOI] [PubMed] [Google Scholar]
- Hanakawa T, Katsumi Y, Fukuyama H, Honda M, Hayashi T, Kimura J, Shibasaki H (1999b): Mechanisms underlying gait disturbance in Parkinson's disease: A single photon emission computed tomography study. Brain 122, 1271–1282. [DOI] [PubMed] [Google Scholar]
- Haslinger B, Erhard P, Kampfe N, Boecker H, Rummeny E, Schwaiger M, Conrad B, Ceballos‐Baumann O (2001): Event‐related functional magnetic resonance imaging in Parkinson's disease before and after levodopa. Brain 124:558–570. [DOI] [PubMed] [Google Scholar]
- Helmich RC, de Lange FP, Bloem BR, Toni I (2007): Cerebral compensation during motor imagery in Parkinson's disease. Neuropsychol 45:2201–2215. [DOI] [PubMed] [Google Scholar]
- Helmich RC, Aarts E, de Lange FP, Bloem BR, Toni I (2009): Increased dependence of action selection on recent motor history in Parkinson's disease. J Neurosci 29:6105–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heremans E, Feys P, Nieuwboer A, Vercruysse S, Vandenberghe W, Sharma N, Helsen W (2011): Motor imagery ability in patients with early‐ and mid‐stage Parkinson disease. Neurorehabil Neural Repair 25:168–177. [DOI] [PubMed] [Google Scholar]
- Herman T, Rosenberg‐Katz K, Jacob Y, Giladi N, Hausdorff JM (2014): Gray matter atrophy and freezing of gait in Parkinson's disease: Is the evidence black on white? Mov Disord 29:134–139. Doi: 10.1002/mds.25697. [DOI] [PubMed] [Google Scholar]
- Hoehn MM, Yahr MD (1967): Parkinsonism: Onset, progression, and mortality. Neurology 17:427–442. [DOI] [PubMed] [Google Scholar]
- Iansek R, Huxman F, Mc Ginley J (2006): The sequence effect and gait festination in Parkinson disease: Contributors to freezing of gait? Mov Disord 21:1419–1424. [DOI] [PubMed] [Google Scholar]
- Imamura K, Okayasu N, Nagatsu T (2012): Cerabral blood flow and freezing of gait in Parkinson's disease. Act Neurol Scand 126:210–218. [DOI] [PubMed] [Google Scholar]
- Jacobs JV, Lou JS, Kraakevik JA, Horak FB (2009): The supplementary motor area contributes to the timing of the anticipatory postural adjustment during step initiation in participants with or without Parkinson's disease. Neuroscience 164:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahanshahi M, Dimberger G, Fuller R, Frith CD (2000): The role of the dorsolateral prefrontal cortex in random number generation: A study with positron emission tomography. NeuroImage 12:713–725. [DOI] [PubMed] [Google Scholar]
- Jahanshahi M, Jones CR, Zijlmans J, Katzenschlager R, Lee L, Quinn N, Frith CD, Lees AJ (2010): Dopaminergic modulation of striato‐frontal connectivity during motor timing in Parkinson's disease. Brain 133:727–745. [DOI] [PubMed] [Google Scholar]
- Jahn K, Deutschländer A, Stephan T, Strupp M, Wiesmann M, Brandt T (2004): Brain activation patterns during imagined stance and locomotion in functional magnetic resonance imaging. NeuroImage 22:1722–1731. [DOI] [PubMed] [Google Scholar]
- Jahn K, Deutschländer A, Stephan T, Kalla R, Hufner K, Wagner J, Strupp M, Brandt T (2008): Supraspinal locomotor control in quadrupeds and humans. [Review]. Prog Brain Res 171:353–362. [DOI] [PubMed] [Google Scholar]
- Jeannerod M (1994): The representing brain. Neural correlates of motor intention and imagery. [Review]. Behav Brain Sci 17:187–245. [Google Scholar]
- Jenkins IH, Fernandez W, Playford ED, Lees AJ, Frackowiak RS (1992): Impaired activation of the supplementary motor area in Parkinson's disease is reversed when akinesia is treated with apomorphine. Annu Neurol 32:749–757. [DOI] [PubMed] [Google Scholar]
- Karachi C, Grabli D, Bernard FA, Tandé D, Wattiez N, Belaid H, Bardinet E, Prigent A, Nothacker HP, Hunot S, Hartmann A, Lehéricy S, Hirsch EC, François C (2010): Cholinergic mesencephalic neurons are involved in gait and postural disorders in Parkinson disease. J Clin Invest 120:2745–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karachi C, André A, Bertasi E, Bardinet E, Lehéricy S, Bernard FA (2012): Functional parcellation of the lateral mesencephalus. J Neurosci 32:9396–9401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobl P, Kielstra L, Almeida Q (2011): The relationship between motor planning and freezing of gait in Parkinson's disease. JNNP 83:98–101. [DOI] [PubMed] [Google Scholar]
- Kostic VS, Agosta F, Pievani M, Stefanova E, Jecmenica‐Lukic M, Scarale A, Spica V, Filippi M (2012): Pattern of brain tissue loss associated with freezing of gait in Parkinson disease. Neurology 78:409–416. [DOI] [PubMed] [Google Scholar]
- Kraft E, Loichinger W, Diepers M, Lule D, Schwarz J, Ludolph AC, Storch A (2009): Levodopa‐induced striatal activation in Parkinson's disease: A functional MRI study. Parkinsonism Relat Disord 15:558–563. [DOI] [PubMed] [Google Scholar]
- La Fougère C, Zwergal A, Rominger A, Forster S, Fesl G, Dieterich M, Brandt T, Strupp M, Bartenstein P, Jahn K (2010): Real versus imagined locomotion: A [18F]‐FDG PET‐fMRI comparison. Neuroimage 50:1589–1598. [DOI] [PubMed] [Google Scholar]
- Langston JW, Widner H, Goetz CG, Brooks D, Fahn S, Freeman T, Watts R (1992): Core assessment program for intracerebral transplantations (CAPIT). Mov Disord 7:2–13. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Yoo JY, Ryu JS, Park HK, Chung SJ (2012): The effects of visual and auditory cues on freezing of gait in patients with Parkinson disease. Am Phys Med Rehabil 91:2–11. [DOI] [PubMed] [Google Scholar]
- Lewis SJ, Barker RA (2009): A pathophysiological model of freezing of gait in Parkinson's disease. Parkinsonism Relate Disord 15:333–338. [DOI] [PubMed] [Google Scholar]
- Lieberman MD, Cunningham WA (2009): Type I and type II error concerns in fMRI research: Re‐balancing the scale. SCAN 4:423–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luppino G, Matelli M, Camarda R, Rizzolatti G (1993): Corticocortical connections of area F3 (SMA proper) and area F6 (pre‐SMA) in the macaque monkey. J Comput Neurol 338:114–140. [DOI] [PubMed] [Google Scholar]
- Maillet A, Pollak P, Debû B (2012a): Imaging gait disorders in Parkinsonism: A review. [Review]. JNNP 83:986–993. [DOI] [PubMed] [Google Scholar]
- Maillet A, Krainik A, Debû B, Troprès I, Lagrange C, Thobois S, Pollak P, Pinto S (2012b): Levodopa effects on hand and speech movements in patients with Parkinson's disease: A fMRI study. PLoS One 7:e46541 Doi: 10.1371/journal.pone.0046541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillet A, Fraix V, Thobois S, Derost P, Bloem BR, Pollak P, Debû B (2013): Kinesthetic imagery of gait in advanced Parkinson's disease. Mov Sport Sci 82:115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malouin F, Richards CL, Jackson PL, Dumas F, Doyon J (2003): Brain activations during motor imagery of locomotor‐related tasks: A PET study. Hum Brain Mapp 19:47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malouin F, Richards CL, Jackson PL, Lafleur MF, Durand A, Doyon J (2007): The Kinesthetic and Visual Imagery Questionnaire (KVIQ) for assessing motor imagery in persons with physical disabilities: A reliability and construct validity study. J Neurol Phys Ther 31:20–29. [DOI] [PubMed] [Google Scholar]
- Matsui H, Udaka F, Miyoshi T (2005): Three dimensional stereotaxic surface projection study of freezing of gait and brain perfusion image in Parkinson's disease. Mov Disord 20:1272–1277. [DOI] [PubMed] [Google Scholar]
- Middleton FA, Strick PL (2000): Basal ganglia and cerebellar loops: Motor and cognitive circuits. [Review]. Brain Res Rev 31:236–250. [DOI] [PubMed] [Google Scholar]
- Mito Y, Yoshida K, Yabe I, Makino K, Tashiro K, Kikuchi S, Sasaki H (2006): Brain SPECT analysis by 3D‐SSP and phenotype in Parkinson's disease. J Neurol Sci 241:67–72. [DOI] [PubMed] [Google Scholar]
- Miyai I, Tanabe HC, Sase I, Eda H, Oda I, Konishi I, Tsunazawa Y, Suzuki T, Yanagida T, Kubota K (2001): Cortical mapping of gait in humans: An near infrared spectroscopic topography study. NeuroImage 14:1186–1192. [DOI] [PubMed] [Google Scholar]
- Morton SM, Bastian AJ (2004): Cerebellar control of balance and locomotion. [Review]. Neuroscientist 10:247–259. [DOI] [PubMed] [Google Scholar]
- Mulder T, Hochstenbach JB, van Heuvelen MJ, den Otter AR (2007): Motor imagery: The relation between age and imagery capacity. Hum Mov Sci 26:203–211. [DOI] [PubMed] [Google Scholar]
- Muller ML, Albin RL, Kotagal V, Koeppe RA, Scott PJ, Frey KA, Bohnen NI (2013): Thalamic cholinergic innervation and postural sensory integration function in Parkinson's disease. Brain 136:3282–3289. Doi: 10.1093/brain/awt247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzert J, Zentgraf K (2009): Motor imagery and its implications for understanding the motor system. [Review]. Prog Brain Res 174:219–229. [DOI] [PubMed] [Google Scholar]
- Nachev P, Kennard C, Husain M (2008): Functional role of the supplementary and pre‐supplementary areas. [Review]. Nat Rev Neurosci 9:856–869. [DOI] [PubMed] [Google Scholar]
- Nadkarni NK, Nunley KA, Aizenstein H, Harris TB, Yaffe K, Satterfield S, Newman AB, Rosano C; for the Health ABC Study (2014): Association between cerebellar gray matter volumes, gait speed, and information‐processing ability in older adults enrolled in the health ABC study. J Gerontol A Biol Sci Med Sci 69:996–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naismith SL, Shine JM, Lewis SJ (2010): The specific contributions of set‐shifting to freezing of gait in Parkinson's disease. [Review]. Mov Disord 25:1000–1004. [DOI] [PubMed] [Google Scholar]
- Nieuwboer A (2008): Cueing for freezing of gait in patients with Parkinson's disease: A rehabilitation perspective. Mov Disord 23:475–481. [DOI] [PubMed] [Google Scholar]
- Nutt JG, Horak FB, Bloem BR (2011a): Milestones in gait, balance, and falling. [Review]. Mov Disord 26:1166–1674. [DOI] [PubMed] [Google Scholar]
- Nutt JG, Bloem BR, Giladi N, Hallett M, Horak FB, Nieuwboer A (2011b): Freezing of gait: Moving forward on a mysterious clinical phenomenon. [Review]. Lancet Neurol 10:734–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield RC (1971): The assessment and analysis of handedness: The Edinburgh Inventory. Neuropsychol 9:97–114. [DOI] [PubMed] [Google Scholar]
- Ouchi Y, Kanno T, Okada H, Yoshikawa E, Tutasubashi M, Nobezawa S, Torizuka T, Tanaka K (2001): Changes in dopamine availability in the nigrostriatal and mesocortical dopaminergic systems by gait in Parkinson's disease. Brain 124:784–792. [DOI] [PubMed] [Google Scholar]
- Park HK, Yoo JY, Kwon M, Lee JH, Lee SJ, Kim SR, Kim MJ, Lee MC, Lee SM, Chung SJ (2014): Gait freezing and speech disturbance in Parkinson's disease. Neurol Sci 35:357–363. [DOI] [PubMed] [Google Scholar]
- Peterson DS, Pickett KA, Earhart GM (2012): Effects of levodopa on vividness of motor imagery in Parkinson's disease. J Parkinsons Dis 2:127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson DS, Pickett KA, Duncan RP, Perlmutter JS, Earhart GM (2014a): Brain activity during complex imagined tasks in Parkinson disease. Clin Neurophysiol 125:995–1005. Doi: S1388‐2457(13)01129‐2. 10.1016/j.clinph.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson DS, Pickett KA, Duncan RP, Perlmutter JS, Earhart GM (2014b): Gait‐related Brain activity in people with Parkinson disease with freezing of gait. PloS One 9:e90634 Doi: 10.1371/journal.pone.0090634. eCollection 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickett KA, Peterson DS, Earhart GM (2013): Motor imagery of gait tasks in individuals with Parkinson's disease. J Parkinsons Dis 2:19–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purzner J, Paradiso GO, Cunic D, Saint‐Cyr JA, Hoque T, Lozano AM, Lang AE, Moro E, Hodaie M, Mazzella F, Che R (2007): Involvement of the basal ganglia and cerebellar motor pathways in the preparation of self‐initiated and externally triggered movements in humans. J Neurosci 27:6029–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randhawa B, Harris S, Boyd LA (2010): The kinesthetic and visual imagery questionnaire is a reliable tool for individuals with Parkinson's disease. J Neurol Phys Therapy 34:161–167. [DOI] [PubMed] [Google Scholar]
- Rizzolatti G, Camarda R, Fogassi L, Gentulicci M, Luppino G, Matelli M (1988): Functional organization of inferior area 6 in the macaque monkey II. Area F5 and the control of distal movement. Exp Brain Res 7:491–507. [DOI] [PubMed] [Google Scholar]
- Rosenberg‐Katz K, Herman T, Jacob Y, Giladi N, Hendler T, Hausdorff JM (2013): Gray matter atrophy distinguishes between Parkinson disease motor subtypes. Neurology 80:1476–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini U, Boulanouar K, Fabre N, Martin F, Carel C, Colonnese C, Bozzao L, Berry I, Montastruc JL, Chollet F, Rascol O (2000): Cortical motor reorganization in akinetic patients with Parkinson's disease: A functional MRI study. Brain 123:394–403. [DOI] [PubMed] [Google Scholar]
- Sacco K, Cauda F, Cerliani L, Mate D, Duca S, Geminiani GC (2006): Motor imagery of walking following training in locomotor attention: The effects of ‘the tango lesson’. NeuroImage 32:1441–1449. [DOI] [PubMed] [Google Scholar]
- Saimpont A, Malouin F, Tousignant B, Jackson PL (2012): The influence of body configuration on motor imagery of walking in younger and older adults. Neuroscience 222:49–57. [DOI] [PubMed] [Google Scholar]
- Saimpont A, Malouin F, Tousignant B, Jackson PL (2013): Motor imagery and ageing. J Motor Behav 45:21–28. [DOI] [PubMed] [Google Scholar]
- Samuel M, Ceballos‐Baumann AO, Blin J, Uema T, Boecker H, Passingham RE, Brooks DJ (1997): Evidence for lateral premotor and parietal overactivity in Parkinson's disease during sequential and bimanual movements. A PET study. Brain 120:963–976. [DOI] [PubMed] [Google Scholar]
- Samuel M, Ceballos‐Baumann AO, Boecker H, Brooks DJ (2001): Motor imagery in normal subjects and Parkinson's disease patients: An H2015 PET study. Neuroreport 12:821–828. [DOI] [PubMed] [Google Scholar]
- Schmidt R, Freidl W, Fazekas F, Reinhart B, Grieshofer P, Koch M, Eber B, Schumacher M, Polmin K, Lechner H (1994): The Mattis dementia rating scale: Normative data from 1,001 healthy subjects volunteers. Neurology 44:964–966. [DOI] [PubMed] [Google Scholar]
- Schweder PM, Hansen PC, Green AL, Quaghebeur G, Stein J, Aziz TZ (2010): Connectivity of the pedunculopontine nucleus in parkinsonian freezing of gait. Neuroreport 21:914–916. [DOI] [PubMed] [Google Scholar]
- Shine JM, Matar E, Ward PB, Bolitho SJ, Gilat M, Pearson M, Naismith SL, Lewis SJ (2013a): Exploring the cortical and subcortical functional magnetic resonance imaging changes associated with freezing in Parkinson's disease. Brain 136:1204–1215. [DOI] [PubMed] [Google Scholar]
- Shine JM, Matar E, Ward PB, Frank MJ, Moustafa AA, Pearson M, Naismith SL, Lewis SJ (2013b): Freezing of gait in Parkinson's disease is associated with functional decoupling between the cognitive control network and the basal ganglia. Brain 136(Pt 12):3671–3681. [DOI] [PubMed] [Google Scholar]
- Shine JM, Moustafa AA, Matar E, Frank MJ, Lewis SJ (2013c): The role of frontostriatal impairment in freezing of gait in Parkinson's disease. [Review]. Front Syst Neurosci 7:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijders AH, Leunissen I, Bakker M, Overeem, S , Helmich RC, Bloem BR, Toni I (2011): Gait related cerebral alterations in patients with Parkinson's disease with freezing of gait. Brain 134:59–72. [DOI] [PubMed] [Google Scholar]
- Solodkin A, Hlustik P, Chen EE, Small SL (2004): Fine modulation in network activation during motor execution and motor imagery. Cereb Cortex 14:1246–1255. [DOI] [PubMed] [Google Scholar]
- Stephan KM, Fink GR, Passingham RE, Silbersweig D, Ceballos‐Baumann AO, Frith CD, Frackowiak RS (1995): Functional anatomy of the mental representation of upper extremity movements in healthy subjects. J Neurophysiol 73:373–386. [DOI] [PubMed] [Google Scholar]
- Stevens JA (2005): Interference effects demonstrate distinct roles for visual and motor imagery during the mental representation of human action. Cognition 95:329–350. [DOI] [PubMed] [Google Scholar]
- Stinear CM, Byblow WD, Steyvers M, Levin O, Swinnen SP (2006): Kinesthetic, but not visual, motor imagery modulates corticomotor excitability. Exp Brain Res 168:157–164. [DOI] [PubMed] [Google Scholar]
- Strafella AP, Lozano AM, Ballanger B, Poon YY, Lang AE, Moro E (2008): RCBF changes associated with PPN stimulation in a patient with Parkinson's disease: A PET study. Mov Disord 23:1051–1054. [DOI] [PubMed] [Google Scholar]
- Sunwoo MK, Choo KH, Hong JY, Lee JE, Sohn YH, Lee PH (2013): Thalamic volume and related visual recognition are associated with freezing of gait in non‐demented patients with Parkinson's disease. Parkinsonism Relat Disord. 19:1106–1109. Doi: 10.1016/j.parkreldis.2013.07.023. [DOI] [PubMed] [Google Scholar]
- Takakusaki K (2013): Neurophysiology of gait: From the spinal cord to the frontal lobe. [Review]. Mov Disord 28:1483–1491. [DOI] [PubMed] [Google Scholar]
- Takakusaki K, Saitoh K, Harada H, Kashiwayanagi M (2004): Role of basal ganglia‐brainstem pathways in the control of motor behaviors. [Review]. Neurosci Res 50:137–151. [DOI] [PubMed] [Google Scholar]
- Takakusaki K, Tomita N, Yano M (2008): Substrates for normal and pathophysiology of gait disturbances with respect to the basal ganglia dysfunction. [Review]. J Neurol 4:19–29. [DOI] [PubMed] [Google Scholar]
- Tessitore A, Amboni M, Esposito F, Russo A, Picillo M, Marcuccio L, Pellecchia MT, Vitale C, Cirillo M, Tedeschi G, Barone P (2012a): Resting‐state brain connectivity with Parkinson's disease and freezing of gait. Parkinsonism Relat Disord 18:781–787. [DOI] [PubMed] [Google Scholar]
- Tessitore A, Amboni M, Cirillo G, Corbo D, Picillo M, Russo A, Vitale C, Santangelo G, Erro R, Cirillo M, Esposito F, Barone P, Tedeschi G (2012b): Regional gray matter atrophy in patients with Parkinson's disease and freezing of gait. Am J Neuroradiol 33:1804–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevathasan W, Pogosyan A, Hyam JA, Jenkinson N, Foltynie T, Limousin P, Bogdanovic M, Zringo L, Green AL, Aziz TZ, Brown P (2012a): Alpha oscillations in the pedunculopontine nucleus correlate with gait performance in parkinsonism. Brain 135:148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevathasan W, Cole MH, Graepel CL, Hyam JA, Jenkinson N, Brittain JS, Coyne TJ, Silburn PA, Aziz TZ, Kerr G, Brown P (2012b): A spatiotemporal analysis of gait freezing and the impact of pedunculopontine nucleus stimulation. Brain 135:1446–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thobois S, Dominey PF, Decety J, Pollak P, Gregoire MC, Le Bars D, Broussolle E (2000): Motor imagery in normal subjects and in asymmetrical Parkinson's disease: A PET study. Neurology 55:996–1002. [DOI] [PubMed] [Google Scholar]
- Thobois S, Dominey P, Fraix V, Mertens P, Guenot M, Zimmer L, Pollak P, Benabid AL, Broussolle E (2002): Effects of subthalamic nucleus stimulation on actual and imagined movement in Parkinson's disease: A PET study. J Neurol 249:1689–1698. [DOI] [PubMed] [Google Scholar]
- Thomas M, Jankovic J, Suteerawattananon M, Wankadia S, Caroline KS, Vuong, KD , Protas E (2004): Clinical gait and balance scale (GABS): Validation and utilization. J Neurol Sci 217:89–99. [DOI] [PubMed] [Google Scholar]
- Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE (2010): Systematic review of levodopa dose equivalency reporting in Parkinson's disease. [Review]. Mov Disord 15; 25:2649–2653. [DOI] [PubMed] [Google Scholar]
- Tzourio‐Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Étard O, Delcroix N, Mazoyer B, Joliot M (2002): Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single‐subject brain. NeuroImage 15:273–289. [DOI] [PubMed] [Google Scholar]
- van der Meulen M, Allali G, Rieger SW, Assal F, Vuilleumier P (2014): The influence of individual motor imagery ability on cerebral recruitment during motor gait imagery. Hum Brain Mapp 35:455–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercruysse S, Devos H, Spildooren J, Vandenbossche J, Vanderberghe W, Nieuwboer A, Heremans E (2012): Explaining freezing of gait in Parkinson's disease: Motor and cognitive determinants. [Review]. Mov Disord 27:1644–1651. [DOI] [PubMed] [Google Scholar]
- Voisin JI, Mercier C, Jackson PL, Richards CL, Malouin F (2011): Is somatosensory excitability more affected by the perspective or modality content of motor imagery? Neurosci Lett 493:33–37. [DOI] [PubMed] [Google Scholar]
- Wai YY, Wang JJ, Weng YH, Lin WY, Ma HK, Ng SH, Wan YL, Wang CH (2012): Cortical involvement in a gait‐related imagery task: Comparison between Parkinson's disease and normal aging. Parkinsonism Relat Disord 18:537–542. [DOI] [PubMed] [Google Scholar]
- Wu T, Hallet M (2005): A functional MRI study of automatic movements in patients with Parkinson's disease. Brain 128:2250–2259. [DOI] [PubMed] [Google Scholar]
- Wu T, Hallett M (2013): The cerebellum in Parkinson's disease. [Review]. Brain 136:696–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Wang L, Hallett M, Li K, Chan P (2010): Neural correlates of bimanual anti‐phase and in‐phase movements in Parkinson's disease. Brain 133:2394–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogev‐Seligmann G, Hausdorff JM, Giladi N (2008): The role of executive function and attention in gait. [Review]. Mov Disord 23:329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Sternad D, Corcos DM, Vaillancourt DE (2007): Role of hyperactive cerebellum and motor cortex in Parkinson disease. NeuroImage 35:222–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapparoli L, Invernizzi P, Gandola M, Verardi M, Berlingeri M, Sberna M, De Santis A, Zerbi A, Banfi G, Bottini G, Paulesu E (2013): Mental images across the adult lifespan: A behavioural and fMRI investigation of motor execution and motor imagery. Exp Brain Res 224:519–540. [DOI] [PubMed] [Google Scholar]