

Abstract
Huntington's disease is an incurable neurodegenerative disease caused by inheritance of an expanded cytosine‐adenine‐guanine (CAG) trinucleotide repeat within the Huntingtin gene. Extensive volume loss and altered diffusion metrics in the basal ganglia, cortex and white matter are seen when patients with Huntington's disease (HD) undergo structural imaging, suggesting that changes in basal ganglia‐cortical structural connectivity occur. The aims of this study were to characterise altered patterns of basal ganglia‐cortical structural connectivity with high anatomical precision in premanifest and early manifest HD, and to identify associations between structural connectivity and genetic or clinical markers of HD. 3‐Tesla diffusion tensor magnetic resonance images were acquired from 14 early manifest HD subjects, 17 premanifest HD subjects and 18 controls. Voxel‐based analyses of probabilistic tractography were used to quantify basal ganglia‐cortical structural connections. Canonical variate analysis was used to demonstrate disease‐associated patterns of altered connectivity and to test for associations between connectivity and genetic and clinical markers of HD; this is the first study in which such analyses have been used. Widespread changes were seen in basal ganglia‐cortical structural connectivity in early manifest HD subjects; this has relevance for development of therapies targeting the striatum. Premanifest HD subjects had a pattern of connectivity more similar to that of controls, suggesting progressive change in connections over time. Associations between structural connectivity patterns and motor and cognitive markers of disease severity were present in early manifest subjects. Our data suggest the clinical phenotype in manifest HD may be at least partly a result of altered connectivity. Hum Brain Mapp 36:1728–1740, 2015. © 2015 Wiley Periodicals, Inc.
Keywords: Huntington's disease, basal ganglia, cortical, connectivity, tractography, diffusion MRI
INTRODUCTION
Huntington's disease is a devastating and incurable neurodegenerative disease caused by autosomal dominant inheritance of an expanded CAG trinucleotide repeat within the Huntingtin (HTT) gene, and primarily characterised pathologically by loss of the striatal medium spiny neurons [Vonsattel et al., 1985]. Striatal volume loss is the earliest and most characteristic structural abnormality seen using brain imaging in Huntington's disease [Aylward et al., 1994; Tabrizi et al., 2009], but, as the disease progresses, histological and structural brain changes become widespread. Clinical disease onset is typically in late‐middle age and comprises motor, cognitive and neuropsychiatric symptoms.
Structural imaging can be used to show extensive volume loss [Rosas et al., 2003; Tabrizi et al., 2009, 2011, 2012] and altered water diffusion metrics [Bohanna et al., 2011a, 2011b; Della Nave et al., 2010; Delmaire et al., 2013; Di Paola et al., 2012; Douaud et al., 2009; Dumas et al., 2012; Georgiou‐Karistianis et al., 2013; Kloppel et al., 2008; Mandelli et al., 2010; Mascalchi et al., 2004; Reading et al., 2005; Rosas et al., 2006, 2010; Seppi et al., 2006; Sritharan et al., 2010; Stoffers et al., 2010; Vandenberghe et al., 2009; Weaver et al., 2009] in the basal ganglia, cortex and white matter in Huntington's disease. As diffusion metrics can be thought of as markers of microstructural cohesion and volumetric measurements indicate macroscopic structure, the two sets of values are strongly linked. These structural changes suggest a loss of structural connectivity in Huntington's disease; this can be assessed in vivo using tractography based on diffusion‐weighted magnetic resonance imaging data. To our knowledge, reductions in subcortical–cortical structural connections have been shown in Huntington's disease subjects in comparison with healthy controls in four studies to date [Bohanna et al., 2011a,b; Kloppel et al., 2008; Marrakchi‐Kacem et al., 2010; Marrakchi‐Kacem et al., 2013]. Connections to associative [Marrakchi‐Kacem et al., 2013] and sensorimotor [Bohanna et al., 2011a, 2011b; Marrakchi‐Kacem et al., 2013] cortex appear particularly affected. Altered diffusion metrics within tractography‐generated white matter pathways have also been shown [Douaud et al., 2009; Dumas et al., 2012].
To date, only three studies have reported associations between tractography findings and clinical and genetic markers of Huntington's disease progression. These associations were between striatum‐sensorimotor cortex connections and Unified Huntington's Disease Rating Scale motor score [Bohanna et al., 2011a,b], between frontal cortex‐caudate connections and (a) estimated time to manifest disease onset and (b) saccade latencies in premanifest subjects [Kloppel et al., 2008], between prefrontal cortex‐putamen connections and tests of executive function [Poudel et al 2014], and between frontoparietal connections and both tests of executive function and Unified Huntington's Disease Rating Scale motor score [Poudel et al 2014]. There is more extensive evidence of associations between diffusion metrics and genetic [Magnotta et al., 2009; Dumas et al., 2012] and clinical [Bohanna et al., 2011a,b; Della Nave et al., 2010; Dumas et al., 2012; Georgiou‐Karistianis et al., 2013; Kloppel et al., 2008; Rosas et al., 2006; Rosas et al., 2010; Sritharan et al., 2010] markers of Huntington's disease, however, suggesting that altered connectivity is associated with clinical disease development beyond the findings reported to date.
The objectives of this study were: (a) to define differential patterns of change with high anatomical precision in basal ganglia‐cortical structural connectivity in premanifest and early manifest Huntington's disease and (b) to relate altered connectivity patterns to markers of Huntington's disease genetic load, and to the clinical—motor, cognitive, and behavioural—features of Huntington's disease. To do this, we used a novel voxel‐based structural connectivity analysis technique developed by Draganski and colleagues [Draganski et al., 2008] to define patterns of basal ganglia‐cortical connections in premanifest and early manifest Huntington's disease gene carriers, and to generate topographical maps of the basal ganglia connectivity in these subjects with unprecedented anatomical detail. This method allows multiple target regions to be reached from a single seed voxel (as opposed to the simpler and more commonly used “winner takes all” method, which has been used in a previous study of structural connectivity in Huntington's disease, for example [Bohanna et al., 2011a,b]). We hypothesised that Huntington's disease‐associated changes in structural connectivity would be extensive and interdependent, so, instead of examining multiple white matter pathways separately, we used standard multivariate analyses to characterise disease‐associated patterns of altered connectivity across all structural connections between the basal ganglia and cortex. Because Huntington's disease is both pathophysiologically and clinically complex, we predicted that the relationship between connections and clinical features would depend on multiple inter‐relating factors. Our multivariate analyses therefore tested for high‐dimensional associations between the patterns of connectivity and genetic and clinical markers. This is the first study of which we are aware in which all potential basal ganglia‐cortical connections have been tested collectively in this multivariate fashion.
MATERIALS AND METHODS
Subjects and Genetics
Fourteen Huntington's disease gene carriers with early symptoms [10 female; mean age (SD) = 51.0 (10.8) years], 17 premanifest Huntington's disease subjects [10 female; mean age (SD) = 41.3 (8.7) years] and 18 sex‐matched controls [10 female; mean age (SD) = 44.6 (10.1) years] were tested. The onset of manifest Huntington's disease is defined as the point at which characteristic motor signs are seen [Huntington Study Group, 1996]: the subjects in this study were deemed manifest if they had scores of >5 in the motor section of the Unified Huntington's Disease Rating Scale [Huntington Study Group, 1996]. The manifest subjects were significantly older than the premanifest subjects: this is common in Huntington's disease studies because Huntington's disease is a progressive disease so older subjects tend to have more advanced disease. It was therefore not possible to age‐match controls to the premanifest and manifest HD subjects. Age was included in all analyses as a regressor to control for this.
CAG repeat length, quantified through genetic testing, had a range of 39–48 repeats in the early manifest subjects (mean 42.7) and 40–48 repeats in the premanifest subjects (mean 43.4). Disease burden score, calculated as [CAG repeat length‐35.5] × age [Penney et al., 1997], had a range of 224–493.5 in the early manifest subjects (mean 346.7) and 220–387.5 in the premanifest subjects (mean 308.1).
The study was approved by the Institute of Neurology's combined University College London/University College London Hospital, UK ethics committee. Written informed consent was obtained from every subject.
Data Acquisition
Imaging data
Subjects were scanned on a Siemens Trio 3 Tesla MRI scanner. Diffusion‐weighted images with 64 unique gradient directions (b = 1000 s/mm2) and eight images with minimal diffusion weighting (b = 100 s/mm2) were acquired, all with dimensions of 96 pixels × 96 pixels × 55 slices per volume, TE = 90 ms and TR = 150 ms. Voxel size was 2.3 mm isotropic. Two T1‐weighted structural images were also acquired using a 3D MPRAGE acquisition sequence; these were checked by eye and the best quality scan used for analyses. The structural imaging parameters were: TR = 2200 ms; TE = 2.2 ms; dimensions = 256 pixels × 256 pixels × 208 sagittal slices per volume; slice thickness 1.0 mm with no gap.
Behavioural data
Behavioural data were acquired as part of the Track‐HD testing protocol [Tabrizi et al., 2009, 2011, 2012]. A subset of the Track‐HD behavioural dataset was used in this study. This included a motor score (total Unified Huntington's Disease Rating Scale motor score; a rating scale used to quantify motor features of Huntington's disease such as chorea, dystonia and oculomotor signs [Huntington Study Group, 1996]), cognitive scores (Symbol Digit Modalities Test [Smith, 1968], and assessment of ability to recognise negative emotions using a subset of the Ekman and Friesen facial stimuli [Young et al., 2002]), and behavioural scores (Frontal Systems Behaviour Scale score, a rating scale used to quantify irritability, disinhibition and dysexecutive behaviours [Gioia et al., 2000], Baltimore Apathy and Irritability Scale [Chatterjee et al., 2005], the Hospital Anxiety and Depression Scale [Zigmond and Snaith, 1983], and the Beck Depression Inventory [Beck and Brown, 1996]).
Characterisation of Structural Connectivity
The imaging data processing pipeline was based on methods developed by Draganski et al. [2008]. The details of the pipeline are outlined below and a schematic is provided in Figure 1.
Figure 1.
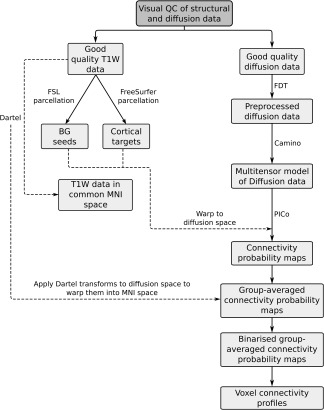
Schematic illustrating imaging data processing pipeline.
Preparation of T1‐weighted (T1W) data: creation of seed and target regions: The T1W data were used to: (a) optimise within‐ and between‐subject registration of the diffusion‐weighted data; (b) create seed and target regions for the tractography; and (c) improve anatomical characterisation of the tractography results by overlaying them on the higher resolution structural images. 54 cortical target regions (27 regions bilaterally; see Table 1 for details) were created from the structural data in subject‐specific native space using standard settings in FreeSurfer (version 5.0) [Desikan et al., 2006]. Ten basal ganglia seed regions (five regions bilaterally) were created using FMRIB's Software Library (FSL) [Smith et al., 2004]: they included the caudates, putamena, nuclei accumbens, globus pallida, and thalami.
Table 1.
Target ROIs used in voxel connectivity profile analysis (generated by FreeSurfer)
| 1 | Hippocampus | 11 | Middle temporal | 21 | Rostral anterior cingulate |
| 2 | Caudal anterior cingulate | 12 | Parahippocampal | 22 | Rostral middle frontal |
| 3 | Caudal middle frontal | 13 | Paracentral | 23 | Superior frontal |
| 4 | Cuneus | 14 | Pars opercularis | 24 | Superior parietal |
| 5 | Inferior parietal | 15 | Pars orbitalis | 25 | Superior temporal |
| 6 | Inferior temporal | 16 | Pars triangularis | 26 | Supramarginal |
| 7 | Isthmus cingulate | 17 | Postcentral | 27 | Frontal pole |
| 8 | Lateral occipital | 18 | Posterior cingulate | ||
| 9 | Lateral orbitofrontal | 19 | Precentral | ||
| 10 | Medial orbitofrontal | 20 | Precuneus |
Preprocessing of diffusion data: Preprocessing of the diffusion data was carried out using FMRIB's Diffusion Toolbox (FDT) in FSL [Smith et al., 2004] (http://www.fmrib.ox.ac.uk/fsl): data were first corrected for distortions caused by eddy currents and motion using eddycorrect, and the gradient directions updated to reflect any rotation of the diffusion‐weighted images; brain tissue was then extracted by stripping the skull from the one of the b = 100 s/mm2 images using the brain extraction tool. FA maps were created from the corrected data using dtifit [Basser et al., 1994; Smith et al., 2004].
At this stage, the diffusion data were in subject‐specific diffusion space, the seed regions were in “FSL T1W space” (i.e., a common space generated by FSL), and the target regions were in “FreeSurfer T1W space” (i.e., a common space generated by FreeSurfer). All data were moved into a common subject‐specific stereotactic space so that the anatomical regions defined in T1W space could be applied to the diffusion data. Specifically, both sets of structural data were warped into subject‐specific diffusion space using the following pipeline: (i) FLIRT, a linear registration algorithm within FSL, was used to warp the FA maps to FSL T1W space [Smith et al., 2004]; (ii) the basal ganglia seed regions were warped into diffusion space using FNIRT, a nonlinear registration algorithm within FSL, using the inverse transforms from FLIRT as the starting point; (iii) FLIRT was then used to warp the original FA maps to FreeSurfer T1W space; and (iv) FNIRT was then used to warp the cortical target regions into subject‐specific diffusion space, again using the inverse transforms from FLIRT as a starting point.
Tractography and creation of individual subject connectivity probability maps: A multitensor model was applied to the diffusion data using the Camino toolkit (http://cmic.cs.ucl.ac.uk/camino/) [Cook et al., 2006]. Once the multitensor model was fitted at every voxel, probabilistic tractography (PICo) [Parker and Alexander, 2003] was used to track streamlines to the cortex from every voxel within each one of the seed regions. 1000 streamlines were initiated from each voxel, and streamlines were terminated when they reached a cortical region. Streamlines were also terminated if fractional anisotropy <0.1 or if they turned more than 80 degrees between steps. The probability of connectivity between every seed voxel and every target region was established for each subject and the data were stored as individual subject connectivity probability maps.
Warping individual subject data into common space: Diffusion data were warped into a common stereotactic space at this point so that group‐averaged probability maps could be created: Dartel [Ashburner, 2007] was used to first warp the T1W data together in Montreal Neurological Institute space, and the transforms which were generated by this process were then applied to the connectivity probability maps to bring them into the same (Montreal Neurological Institute) space.
Creation of binary connectivity probability maps and summary profiles
The individual subject connectivity probability maps were combined to form group‐averaged connectivity probability maps. Both the individual subject and group‐averaged maps were then binarised by thresholding at a connectivity probability of 1%. To illustrate the heterogeneity of subcortical–cortical connectivity both within the basal ganglia and between subject groups, group voxel connectivity profiles (VCPs) were computed. These were generated from the binary group‐averaged connectivity probability maps as described by Draganski et al. [2008]. Voxel connectivity profiles allow voxels with similar connectivity to be identified and seed nuclei to be segmented into subregions with similar connectivity profiles. Colour maps of the basal ganglia voxel connectivity profiles for each subject group are shown in Figures 2, 3, 4, 5. Technical details are given in figure legends.
Figure 2.
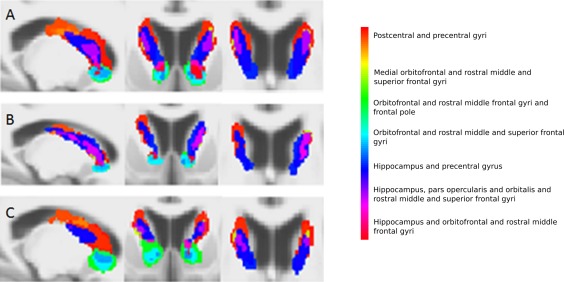
Group‐averaged voxel connectivity profiles in the caudate for controls (A), premanifest Huntington's disease subjects (B) and early manifest Huntington's disease subjects (C). From left to right, sagittal, coronal and axial views are seen. A voxel connectivity profile was defined as the binary pattern of connections at a voxel. Every unique voxel connectivity profile was given a unique label and colour, and every basal ganglia voxel was assigned a colour corresponding to its voxel connectivity profile label. The anatomical regions to which each colour corresponds are displayed alongside. The voxel size of the group‐averaged voxel connectivity profiles is 1 mm isotropic. The voxel connectivity profiles were generated for each subject and averaged within each group separately; each subject group had a different set of voxel connectivity profiles, reflecting the differing patterns of connectivity in the premanifest and manifest Huntington's disease subjects and controls. The voxel connectivity profile labels were therefore reviewed and standardized across the groups after their initial creation to ensure comparability between groups. As shown in Figures 2, 3, 4, 5, colour maps of the basal ganglia voxel connectivity profiles for each subject group were generated from the standardised labels to allow visual comparison of connectivity patterns within the groups. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Figure 3.
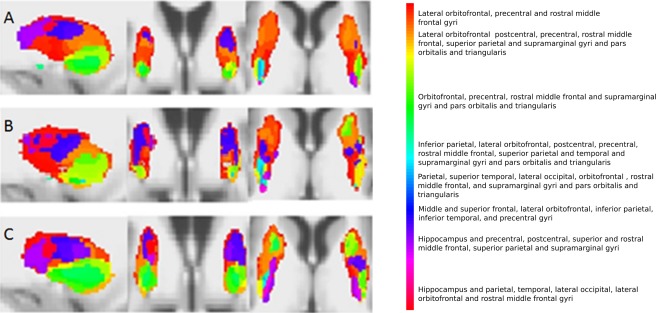
Voxel connectivity profiles in the putamen for controls (A), premanifest Huntington's disease subjects (B) and early manifest Huntington's disease subjects (C). From left to right, sagittal, coronal and axial views are seen. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Figure 4.
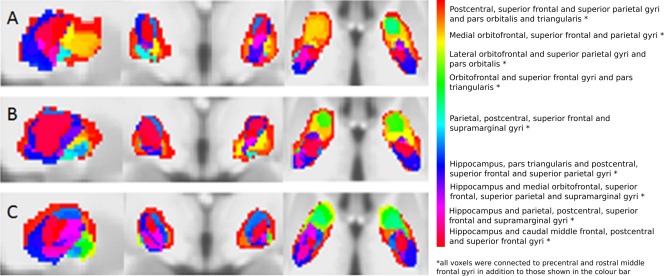
Voxel connectivity profiles in the pallidum for controls (A), premanifest Huntington's disease subjects (B) and early manifest Huntington's disease subjects (C). From left to right, sagittal, coronal and axial views are seen. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Figure 5.
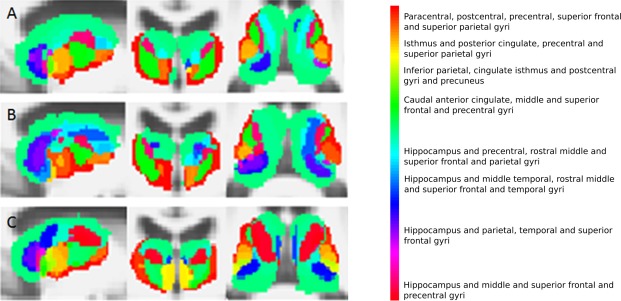
Voxel connectivity profiles in the thalamus for controls (A), premanifest Huntington's disease subjects (B) and early manifest Huntington's disease subjects (C). From left to right, sagittal, coronal and axial views are seen. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Statistical analysis of structural connectivity
Statistical characterisation of group differences—and the putative effects of genetic load and clinical scores—was achieved through analysis of the individual subjects' binarised connection matrices using standard multivariate techniques (canonical variates analysis (CVA), also known as multivariate analysis of variance, or ManCova [Friston et al., 1995, 1996, 2007]). CVA enables one to make statistical inferences about group differences in connectivity and associations between the imaging data and genetic and clinical data that are distributed over connections. It was chosen for analysis of this dataset because it can accommodate statistical dependencies between multivariate predictor variables (genetic and clinical measures) and multivariate outcome variables (reduced connectivity measures). This meant that neither the (reduced) imaging data nor the demographic and clinical data had to be examined in isolation, which had the profound advantage that distributed changes could be identified (while minimising the multiple comparison problem).
The number of connections from each basal ganglia seed region for each subject was first divided by the total number of voxels in that region to control for variation in basal ganglia volumes; this generated normalised data which indicated the percentage of voxels connecting to each cortical target region from each basal ganglia seed region in each individual subject. Out of a potential 270 connections (5 seed regions*27 target regions bilaterally), 249 were non‐zero, that is, in 21 of the potential 270 connections, no connections were seen in any of the subjects. The normalised data were then mean‐corrected across subjects. This resulted in a 249 (connections) × 49 (number of subjects) dataset. The implicit averaging rendered these data approximately normal in their distribution via the central limit theorem. They were, therefore, not subject to any further transformation.
The dimensionality of the dataset at this point was not suitable for multivariate analysis because—as is usual for multivariate analyses of imaging data—there were far more imaging data points than there were subjects. Singular value decomposition (SVD) was therefore used to reduce the dimensionality of the dataset. To significantly reduce the number of data points whilst retaining most of the variability in the data, an a priori decision was made to retain the lowest number of components (singular variates) needed to represent at least 75% of the variance of the imaging dataset. This process was applied twice to generate two sets of components for subsequent analyses: firstly, for the whole imaging dataset, and secondly for the data from the 31 Huntington's disease subjects only.
CVA was then used to assess and characterise the presence of a mapping between the reduced imaging dataset and the genetic and clinical predictors. The objective of the CVA was to find the linear combination of (normalized) connectivity that was best predicted by a linear mixture (contrast) of demographic, genetic and clinical components. The weights of these linear combinations are called canonical vectors. The canonical variates of the outcome (imaging) and predictor (demographic, genetic and/or clinical) variables are the expression of each canonical vector in each subject. Other quantities generated by CVA include Bartlett's approximate chi‐squared statistic for Wilks' Lambda and its associated significance, or P‐value, which test for the significance of a linear mapping or correlation between the canonical variates. In other words, one or more pairs of canonical variates show a significant statistical dependency. To test hypotheses about individual clinical or genetic effects, contrasts of the predictor variables were specified, such that the remainder were treated as confounding or uninteresting variables. In this case, there is only one canonical (connectivity) vector because the dimensionality of the mapping reduces to the dimensionality of the (one‐dimensional) contrast.
Three sets of CVA were carried out. The first CVA was to analyse data from all subjects and was used to test for differences between the subject groups. The second was to analyse data from the Huntington's disease subjects only and was used to test for associations between panels of connectivity and genetic data. The third was to analyse data from all the subjects and was used to test for associations between connectivity and the motor, cognitive and behavioural scores described above. Age and gender were included in all analyses as nuisance variables. The demographic, genetic, clinical, and normalized imaging data were mean‐corrected across subjects.
RESULTS
Group Comparisons: Voxel Connectivity Profile Illustrations
Basal ganglia segmentations based on group‐averaged VCPs are shown in Figures 2, 3, 4, 5. The results of statistical analysis of these data are given in the Section Group Comparisons: Statistical Analyses onward, but Figures 2, 3, 4, 5 broadly illustrate that altered topography can be seen in the Huntington's disease group nuclei in comparison with controls. For example, there is a reduced connectivity between the head of the caudate and the hippocampus in Huntington's disease patients compared to controls (Fig. 2; red‐purple voxels). Similarly, group‐averaged VCPs of the putamen (Fig. 3) suggest reduced connectivity to the lateral orbitofrontal and rostral middle frontal gyri (red voxels) in Huntington's disease. Group‐averaged VCPs of the pallidum (Fig. 4), show reduced medial orbitofrontal and superior frontal gyri (yellow/red voxels in the anterior portion) connectivity in Huntington's disease patients compared to controls. Group‐averaged VCPs of the thalamus are presented in Figure 5. In this figure, an increase in the relative proportion of blue/purple voxels in the VCPs of Huntington's disease patients compared to controls suggests reduced connectivity to the precentral gyrus. However, there is a complex pattern of group differences for all of the subcortical structures, which limits the insights offered by group‐averaged VCPs.
Group Comparisons: Statistical Analyses
For the connectivity data from all subjects, the first 16 components (singular variates) of the singular value decomposition accounted for 75.9% of the variance. For the dataset containing data from the 31 Huntington's disease subjects only, the first 13 components accounted for 77.5% of the variance in the connectivity data. 16 and 13 components the respectively were therefore used to summarise subject‐specific connectivity patterns in subsequent CVA analyses.
Between‐group comparisons revealed significant differences in the connectivity patterns of the combined group of all Huntington's disease subjects and controls (Chi‐squared value = 32.08; P = 0.0098), the early manifest subjects and controls (Chi‐squared value = 43.96; P = 0.0002), and the early manifest and premanifest subjects (Chi‐squared value = 35.54; P = 0.0033).
The contribution of each of the 270 connections to the final correlation can be seen graphically in Figure 6. This contribution is obtained by projecting the principal canonical back into the original connection space, using the appropriate singular vectors. Inferences must be cautious with regard to individual connections because the test is inherently multivariate in nature, but the graphs suggest that altered structural connectivity in Huntington's disease affects a high proportion of basal ganglia‐cortical connections. Figure 7 is an anatomical representation of the data shown in Figure 6A (projected onto meshes derived from canonical FreeSurfer brain images): the mean canonical vector (weight) for each basal ganglion and for each cortical region (calculated across all 270 connections) is projected onto meshes derived from canonical FreeSurfer brain images to localise differences in structural connectivity between all Huntington's disease subjects and controls. Within the basal ganglia, the patterns of connectivity in the putamen, pallidum and thalamus appear most different from those of controls. Of note, the direction of the contribution appears to be the same for most connections in the putamena (i.e., the components of the canonical vector are mainly positive for the putamena; illustrated as yellow‐orange in Fig. 7), which suggests consistently higher putamenal connectivity in Huntington's disease patients compared to controls. In other words, an increase in the normalised volume of connectivity to all cortical regions is seen in the putamena in Huntington's disease patients compared to controls. Figure 7 shows that in the cortex, relative reduction in connectivity in the Huntington's disease subjects is seen most clearly in the superior and inferior frontal gyri and the inferior temporal gyrus, whereas a relative increase in connectivity is seen in much of the parietal cortex (with the exception of the postcentral gyrus).
Figure 6.
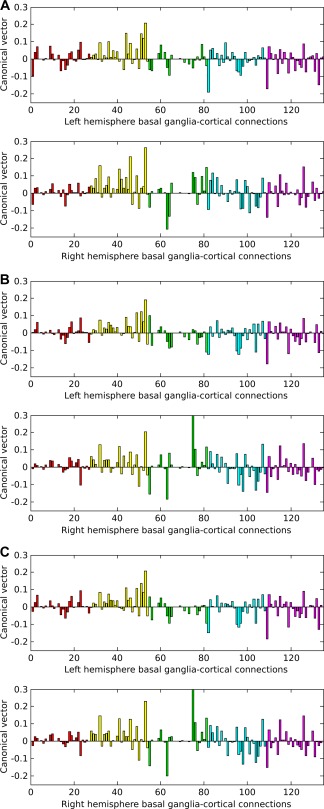
Graphs of the canonical vector (weights) over basal ganglia‐cortical connections that showed differences in structural connectivity between: (A) all Huntington's disease subjects and controls; (B) early manifest Huntington's disease subjects and controls; (C) early manifest and premanifest Huntington's disease subjects. The canonical vector indicates the contribution each connection makes to a pattern that is best predicted by between‐group differences; in these graphs, the canonical vector has been combined with the singular vectors used to reduce the connectivity data so that the contribution of each individual structural connection can be seen. The horizontal axis shows the 270 (zero and non‐zero) connections; each bar of the histogram represents one connection. The vertical axis indicates the weight of the contribution of that connection to the canonical correlation. Positive weights indicate gain or relative preservation of connections in the Huntington's disease subjects in comparison with controls and negative weights indicate loss of connections in the Huntington's disease subjects in (A) and (B). In (C), positive weights indicate gain or relative preservation of connections in the early manifest Huntington's disease subjects in comparison with premanifest subjects and negative weights indicate loss of connections in the early manifest Huntington's disease subjects. In each pair of graphs, left hemisphere connections are shown in the top graph and right hemisphere connections are shown in the bottom graph. The colours of the bars indicate the basal ganglia nucleus from which each connection arises: red=caudate; yellow = putamen; green = nucleus accumbens; blue = pallidum; pink = thalamus. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Figure 7.
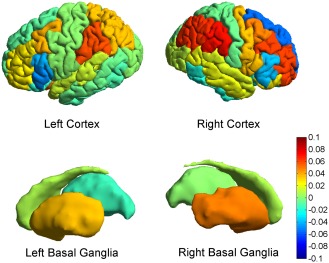
Anatomical projections of the mean canonical vector (weights) over basal ganglia‐cortical connections showing differences in structural connectivity between all Huntington's disease subjects and controls. This is an anatomical representation of the data shown in Figure 6A (projected onto meshes derived from canonical FreeSurfer brain images); the mean canonical vector (weight) for each basal ganglion and for each cortical region is illustrated according to a colour scale where hot (red) colours illustrate relative preservation or gain of connectivity and cold (blue) colours illustrate relative loss of connectivity. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Associations between Imaging Data and Genetic Markers
No significant associations were seen between connectivity and either CAG repeat length or disease burden score (P > 0.05). This was the case both when the premanifest and manifest Huntington's disease data were tested separately, and when the data for both groups were tested together. A trend toward an association between CAG repeat length and connectivity was seen in the premanifest subjects (Chi‐squared value = 22.33; P = 0.0504).
Associations between Imaging Data and Clinical Scores
Associations between connectivity and clinical scores were seen in the early manifest Huntington's disease subject data: associations were present with total motor score (Chi‐squared value = 41.25; P = 0.0005), symbol digit modalities test score (Chi‐squared value = 33.93; P = 0.0055) and Stroop word‐reading score (Chi‐squared value = 46.66; P = 0.0001). There was a trend toward an association between the early manifest imaging data and negative emotion recognition score (Chi‐squared value = 23.70; P value = 0.0962) and between the early manifest imaging data and total Beck depression inventory score (Chi‐squared value = 24.16; P = 0.0860). There was a trend toward a difference in the association with negative emotion recognition score according to subject group (Chi‐squared value = 43.50; P value = 0.0845). There were no significant differences in the associations between imaging data and clinical scores between subject groups.
DISCUSSION
This is the first study to provide a comprehensive framework for statistical analysis of disease‐associated differential patterns of basal ganglia‐cortical structural connectivity. By examining the structural connections of every basal ganglia voxel concurrently, anatomically extensive differences in the basal ganglia‐cortical connections of the brains of early manifest Huntington's disease subjects in comparison with controls have emerged, and altered topographic organisation of the basal ganglia nuclei has been shown. Inferences cannot be drawn about changes in specific basal ganglia‐cortex connections on the basis of these analyses—we cannot state that the loss of any particular connection is statistically significant—but patterns of connectivity are clearly altered. This has not previously been shown. Of note, our analyses suggest apparent (relative) loss and gain of connectivity in the early manifest Huntington's disease subjects (Figs. 6 and 7). One possible explanation for the apparent gain is that new structural connections develop in Huntington's disease brains to compensate for other degenerative pathways; conversely, it should be noted that the connectivity data were normalized and that these differences are relative in nature. (In other words, there has not necessarily been an absolute increase in connectivity). Atrophy of a structure or altered neuronal function, for example, might lead to reorganisation, or loss of connections. However, it is not possible from these data to determine the exact relationship between the two processes. In the CVA, an increase in normalised connectivity volume in the putamen was observed for Huntington's disease patients compared to controls. This increase suggests that the majority of connections are being concentrated into a smaller volume as the putamen atrophies, perhaps as a result of preferential loss of regions with limited connectivity over more densely connected regions.
Our analyses also show that altered structural connectivity in Huntington's disease affects a high proportion of basal ganglia‐cortical connections. Putamen, pallidum, and thalamus connectivity appear to be particularly affected, in contrast with the results of previous studies suggesting predominantly altered caudate connectivity [Kloppel et al., 2008; Marrakchi‐Kacem et al., 2010]. Loss of connectivity appears highest in pallidum and thalamus, linking to evidence in the functional imaging literature of altered basal ganglia function beyond the caudate [Gray et al 1997, Hennenlotter et al 2004, Novak et al 2012, Paulsen et al 2004, Saft et al 2008, Wolf et al 2008a,2008b, Zimbelman et al 2007]. We suggest that allowing tractography streamlines to reach multiple cortical targets (in comparison with the more commonly used “winner takes all” approach) and analysing the subsequent data with multivariate analyses has allowed the detection of complex patterns of altered connectivity which extend beyond the previously determined anatomical limits of such findings.
The patterns of altered connectivity that we describe in this paper clearly have clinical relevance. The associations seen between connectivity and Unified Huntington's Disease Rating Scale total motor score, symbol digit modalities test score and Stroop word‐reading score in the early manifest subjects (and trend toward an association with negative emotion recognition and Beck depression inventory) strongly suggest that altered structural connectivity either directly contributes to clinical phenotype or is linked with it through another causative factor or factors. This is in keeping with previously reported associations between the integrity of structural connections and motor and cognitive function [Bohanna et al., 2011a, 2011b, Kloppel et al., 2008, Poudel et al., 2014]: plausibly, the clinical phenotype of manifest Huntington's disease can be thought of, at least in part, as a disconnection syndrome (Catani and ffytche, 2005). In contrast, the relative preservation of parietal cortex connections is consistent with the paucity of parietal features in the clinical phenotype of Huntington's disease. The trend toward an association with Beck depression inventory is particularly interesting, as associations between brain structure and mood and “behavioural” markers of Huntington's disease in the literature are striking in their absence when considered next to numerous reports of associations with motor and cognitive markers. This lack of evidence can be interpreted as demonstrating that either the scores used to quantify psychiatric symptoms are not sensitive or specific enough, or that psychiatric symptoms are not caused by structural change. Our results, however, suggest a potential additional factor: that multivariate analysis is needed to detect complex relationships between brain structure and the psychiatric manifestations of Huntington's disease. An additional hypothesis is that these symptoms are caused by changes in brain function rather than structure—functional imaging studies provide some evidence for this [Gray et al., 2013; Kloppel et al., 2010] —and a combination of explanatory factors seems likely. We acknowledge that our use of multivariate statistics to analyse the associations between clinical and connectivity data in this way is a novel approach and future work will be important to validate the approach and further explore these early findings.
As well as contributing to understanding of the pathophysiology of Huntington's disease, the findings of this study have a practical bearing on drug development. Potential disease‐modifying drugs include compounds administered through intracerebral infusion and directed at specific anatomical targets; our findings suggest that any directly injected therapy (e.g., HTT gene silencing through striatal administration of siRNAs [Sah and Aronin, 2011]) might have to incorporate multiple regions to have full efficacy. We also suggest that those clinical tests which were most strongly associated with altered structural connectivity patterns in this study might be used to reflect preservation of white matter connections.
Given that Huntington's disease is a degenerative disease, we presume that disease‐driven changes in structural connectivity occur progressively. Our findings suggest that changes in structural connectivity probably occur around the time that the disease becomes symptomatic; the lack of a significant difference between the connectivity patterns of premanifest subjects and controls suggests that premanifest Huntington's disease gene carriers have a pattern of structural connectivity similar to that of controls. It is interesting that no significant association was seen between altered connectivity and disease burden score in either group of Huntington's disease subjects. Disease burden score is a marker of disease progression derived from CAG repeat length and age and is generally accepted as a good marker of Huntington's disease progression (validated by numerous reports of associations with other markers of disease progression in the literature). The lack of a statistically significant association in this study indicates that either CAG repeat length is not the primary driver of altered structural connectivity in Huntington's disease, or that a relationship between the two variables exists but was not captured by the linear modelling of the CVA. There was a trend toward an association between CAG repeat length and the structural connectivity pattern in the premanifest subjects: this suggests that changes in structural connectivity might be to some extent CAG‐driven, at least in the earliest stages of the disease, but the absence of an association in the early manifest subjects, and the absence of an association with disease burden score, suggests the presence of additional or alternative mediators of changing connectivity. Although no additional genetic or environmental modifiers of Huntington's disease progression have yet been identified, the putative existence of factors beyond CAG repeat length alone is generally accepted by Huntington's disease researchers: we propose our work as additional evidence of this.
We acknowledge that a limitation of our present work is the use of the multitensor reconstruction. While the multitensor model recovers multiple fiber‐orientation estimates per voxel, the technique suffers from a model‐selection problem, where we lose accuracy by trying to fit a multitensor model to voxels containing a single fibre population. The approach therefore requires a voxel classification step [Seunarine and Alexander, 2013]. In addition to this, the multitensor reconstruction uses a nonlinear fitting procedure which can be unstable and dependent upon initial conditions. State of the art techniques such as persistent angular structure MRI (PASMRI) [Jansons and Alexander, 2003] and constrained spherical deconvolution (CSD) [Tournier et al. 2007] overcome these limitations; in future work one of these techniques will be incorporated into the VCP processing pipeline.
In summary, however, we have demonstrated extensive changes in patterns of basal ganglia‐cortical structural connections in Huntington's disease and have shown that the methodology used is sensitive to complex differences in cross‐sectional data from patient and control cohorts. Longitudinal studies are now needed to test the sensitivity of this technique for tracking disease‐associated changes in brain structure over time. Our findings indicate that widespread changes in structural connections are strongly associated with clinical symptoms and highlight the utility of multivariate techniques in investigating extensive and complex disease‐related changes.
Marianne J.U. Novak and Kiran K. Seunarine are joint first authors and Chris A. Clark and Sarah J. Tabrizi joint senior authors.
REFERENCES
- Ashburner J (2007): A fast diffeomorphic image registration algorithm. Neuroimage 38:95–113. [DOI] [PubMed] [Google Scholar]
- Aylward EH, Brandt J, Codori AM, Mangus RS, Barta PE, Harris GJ (1994): Reduced basal ganglia volume associated with the gene for Huntington's disease in asymptomatic at‐risk persons. Neurology 44:823–828. [DOI] [PubMed] [Google Scholar]
- Basser PJ, Mattiello J, LeBihan D (1994): Estimation of the effective self‐diffusion tensor from the NMR spin echo. J Magn Reson Series B 103:247–254. [DOI] [PubMed] [Google Scholar]
- Beck ATS, RA ; Brown, GK (1996): Manual for the Beck Depression Inventory‐II, Vol. 1. San Antonio, TX: Psychological Corporation; p 82. [Google Scholar]
- Bohanna I, Georgiou‐Karistianis N, Egan GF (2011a): Connectivity‐based segmentation of the striatum in Huntington's disease: Vulnerability of motor pathways. Neurobiol Dis 42:475–481. [DOI] [PubMed] [Google Scholar]
- Bohanna I, Georgiou‐Karistianis N, Sritharan A, Asadi H, Johnston L, A Churchyard, G Egan (2011b): Diffusion tensor imaging in Huntington's disease reveals distinct patterns of white matter degeneration associated with motor and cognitive deficits. Brain Imaging Behav 5:171–180 [DOI] [PubMed] [Google Scholar]
- Catani M, ffytche DH (2005): The rises and falls of disconnection syndromes. Brain 128:2224–2239. [DOI] [PubMed] [Google Scholar]
- Chatterjee A, Anderson KE, Moskowitz CB, Hauser WA, Marder KS (2005): A comparison of self‐report and caregiver assessment of depression, apathy, and irritability in Huntington's disease. J Neuropsychiatry Clin Neurosci 17:378–383. [DOI] [PubMed] [Google Scholar]
- Cook PA, Bai Y, Nedjati‐Gilani S, Seunarine KK, Hall MG, Parker GJ, Alexander DC (2006): Camino: Open source diffusion‐MRI reconstruction and processing. In: 14th Scientific Meeting of the International Society for Magnetic Resonance in Medicine, Seattle, WA, USA. p 2759
- Della Nave R, Ginestroni A, Tessa C, Giannelli M, Piacentini S, M Filippi, M Mascalchi (2010): Regional distribution and clinical correlates of white matter structural damage in Huntington Disease: A tract‐based spatial statistics study. Am J Neuroradiol 31:1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmaire C, Dumas EM, Sharman MA, van den Bogaard SJ, Valabregue R, Jauffret C, Justo D, Reilmann R, Stout JC, Craufurd D, Tabrizi SJ, Roos RA, Durr A, Lehéricy S (2013): The structural correlates of functional deficits in early huntington's disease. Hum Brain Mapp 34:2141–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan RS, Segonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, Buckner RL, Dale AM, Maguire RP, Hyman BT, Albert MS, Killiany RJ (2006): An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 31:968–980. [DOI] [PubMed] [Google Scholar]
- Di Paola M, Luders E, Cherubini A, Sanchez‐Castaneda C, Thompson PM, Toga AW, Caltagirone C, Orobello S, Elifani F, Squitieri F, Sabatini U (2012): Multimodal MRI analysis of the corpus callosum reveals white matter differences in presymptomatic and early Huntington's disease. Cereb Cortex 22:2858–2866. [DOI] [PubMed] [Google Scholar]
- Douaud G, Behrens TE, Poupon C, Cointepas Y, Jbabdi S, Gaura V, Golestani N, Krystkowiak P, Verny C, Damier P, Bachoud‐Lévi AC, Hantraye P, Remy P (2009): In vivo evidence for the selective subcortical degeneration in Huntington's disease. Neuroimage 46: 958–966. [DOI] [PubMed] [Google Scholar]
- Draganski B, Kherif F, Kloppel S, Cook PA, Alexander DC, Parker GJ, Deichmann R, Ashburner J, Frackowiak RS (2008): Evidence for segregated and integrative connectivity patterns in the human Basal Ganglia. J Neurosci 28:7143–7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas EM, van den Bogaard SJ, Ruber ME, Reilman RR, Stout JC, Craufurd D, Hicks SL, Kennard C, Tabrizi SJ, van Buchem MA, van der Grond J, Roos RA (2012): Early changes in white matter pathways of the sensorimotor cortex in premanifest Huntington's disease. Hum Brain Mapp 33:203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friston KJ (2007): Functional connectivity: Eigenimages and multivariate analyses In: Friston KJ, Ashburner JT, Kiebel S, Nichols T, Penny WD. Statistical Parametric Mapping: The Analysis of Functional Brain Images, Academic Press, London; pp 492–507. [Google Scholar]
- Friston KJ, Frith CD, Frackowiak RS, Turner R (1995): Characterizing dynamic brain responses with fMRI: A multivariate approach. Neuroimage 2:166–172. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Poline JB, Holmes AP, Frith CD, Frackowiak RS (1996): A multivariate analysis of PET activation studies. Hum Brain Mapp 4:140–151. [DOI] [PubMed] [Google Scholar]
- Georgiou‐Karistianis N, Gray MA, Dominguez DJ, Dymowski AR, Bohanna I, Johnston LA, Churchyard A, Chua P, Stout JC, Egan GF (2013): Automated differentiation of pre‐diagnosis Huntington's disease from healthy control individuals based on quadratic discriminant analysis of the basal ganglia: The IMAGE‐HD study. Neurobiol Dis, 51:82–92. [DOI] [PubMed] [Google Scholar]
- Gioia GA, Isquith PK, Guy SC, Kenworthy L (2000): Behavior rating inventory of executive function. Child Neuropsychol 6:235–238. [DOI] [PubMed] [Google Scholar]
- Gray JM, Young AW, Barker WA, Curtis A, Gibson D (1997): Impaired recognition of disgust in Huntington's disease gene carriers. Brain 120 (Pt 11):2029–2038. [DOI] [PubMed] [Google Scholar]
- Gray MA, Egan GF, Ando A, Churchyard A, Chua P, Stout JC, Georgiou‐Karistianis N (2013): Prefrontal activity in Huntington's disease reflects cognitive and neuropsychiatric disturbances: The IMAGE‐HD study. Exp Neurol 239:218–228. [DOI] [PubMed] [Google Scholar]
- Hennenlotter A, Schroeder U, Erhard P, Haslinger B, Stahl R, Weindl A, von Einsiedel HG, Lange KW, Ceballos‐Baumann AO (2004): Neural correlates associated with impaired disgust processing in pre‐symptomatic Huntington's disease. Brain 127:1446–1453. [DOI] [PubMed] [Google Scholar]
- Huntington Study Group (1996): Unified Huntington's Disease Rating Scale: Reliability and consistency. Mov Disord 11:136–142. [DOI] [PubMed] [Google Scholar]
- Jansons KM, Alexander DC (2003): Persistent angular structure: New insights from diffusion magnetic resonance imaging data. Inverse Problems 19:1031–1046. [Google Scholar]
- Kloppel S, Draganski B, Golding CV, Chu C, Nagy Z, Cook PA, Hicks SL, Kennard C, Alexander DC, Parker GJ, Tabrizi SJ, Frackowiak RS (2008): White matter connections reflect changes in voluntary‐guided saccades in pre‐symptomatic Huntington's disease. Brain 131:196–204. [DOI] [PubMed] [Google Scholar]
- Kloppel S, Stonnington CM, Petrovic P, Mobbs D, Tuscher O, Craufurd D, Tabrizi SJ, Frackowiak RSJ (2010): Irritability in pre‐clinical Huntington's disease. Neuropsychologia 48:549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnotta VA, Kim J, Koscik T, Beglinger LJ, Espinso D, Langbehn D, Nopoulos P, Paulsen JS (2009): Diffusion tensor imaging in preclinical Huntington's disease. Brain Imaging Behav 3:77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelli ML, Savoiardo M, Minati L, Mariotti C, Aquino D, Erbetta A, Genitrini S, Di Donato S, Bruzzone MG, Grisoli M (2010): Decreased diffusivity in the caudate nucleus of presymptomatic huntington disease gene carriers: Which explanation? AJNR Am J Neuroradiol 31:706–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrakchi‐Kacem L, Delmaire C, Tucholka A, Roca P, Guevara P, Poupon F, Yelnik J, Durr A, Mangin JF, Lehericy S, Poupon C (2010): Analysis of the striato‐thalamo‐cortical connectivity on the cortical surface to infer biomarkers of Huntington's disease. In: Medical image computing and computer‐assisted intervention: MICCAI. International Conference on Medical Image Computing and Computer‐Assisted Intervention,Vol. 13 pp 217–224. [DOI] [PubMed] [Google Scholar]
- Marrakchi‐Kacem L, Delmaire C, Guevara P, Poupon F, Lecompte S, Tucholka A Roca P, Yelnik J, Durr A, Mangin JF, Lehericy S, Poupon C. (2013): Mapping cortico‐striatal connectivity onto the cortical surface: A new tractography‐based approach to study Huntington disease. PLoS One 8:e53135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascalchi M, Lolli F, Della Nave R, Tessa C, Petralli R, Gavazzi C, Politi LS, Macucci M, Filippi M, Piacentini S (2004): Huntington disease: Volumetric, diffusion‐weighted, and magnetization transfer MR imaging of brain. Radiology 232:867–873. [DOI] [PubMed] [Google Scholar]
- Novak MJ, Warren JD, Henley SM, Draganski B, Frackowiak RS, Tabrizi SJ (2012): Altered brain mechanisms of emotion processing in pre‐manifest Huntington's disease. Brain 135:1165–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker GJ, Alexander DC (2003): Probabilistic Monte Carlo based mapping of cerebral connections utilising whole‐brain crossing fibre information In: Taylor CJ. and Noble JA. (eds) Information Processing in Medical Imaging. LNCS vol 2732, pp684–695. Springer, Heidelberg. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Zimbelman JL, Hinton SC, Langbehn DR, Leveroni CL, Benjamin ML, Reynolds NC, Rao SM (2004): fMRI biomarker of early neuronal dysfunction in presymptomatic Huntington's Disease. AJNR Am J Neuroradiol 25:1715–1721. [PMC free article] [PubMed] [Google Scholar]
- Penney JB, Jr. , Vonsattel JP, MacDonald ME, Gusella JF, Myers RH (1997): CAG repeat number governs the development rate of pathology in Huntington's disease. Ann Neurol 41:689–692. [DOI] [PubMed] [Google Scholar]
- Poudel GR, Stout JC, Dominguez DJ, Salmon L, Churchyard A, Chua P, Georgiou‐Karistianis N , Egan GF (2014): White matter connectivity reflects clinical and cognitive status in Huntington's disease. Neurobiol Dis 65:180–187. [DOI] [PubMed] [Google Scholar]
- Reading SA, Yassa MA, Bakker A, Dziorny AC, Gourley LM, Yallapragada V, Rosenblatt A, Margolis RL, Aylward EH, Brandt J, Mori S, van Zijl P, Bassett SS, Ross CA (2005): Regional white matter change in pre‐symptomatic Huntington's disease: A diffusion tensor imaging study. Psychiatry Res 140:55–62. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Koroshetz WJ, Chen YI, Skeuse C, Vangel M, Cudkowicz ME, Caplan K, Marek K, Seidman LJ, Makris N, Jenkins BG, Goldstein JM (2003): Evidence for more widespread cerebral pathology in early HD: An MRI‐based morphometric analysis. Neurology 60:1615–1620. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Tuch DS, Hevelone ND, Zaleta AK, Vangel M, Hersch SM, Salat DH (2006): Diffusion tensor imaging in presymptomatic and early Huntington's disease: Selective white matter pathology and its relationship to clinical measures. Mov Disord 21:1317–1325. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Lee SY, Bender AC, Zaleta AK, Vangel M, Yu P, Fischl B, Pappu V, Onorato C, Cha JH, Salat DH, Hersch SM (2010): Altered white matter microstructure in the corpus callosum in Huntington's disease: Implications for cortical. Neuroimage 49:2995–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saft C, Schuttke A, Beste C, Andrich J, Heindel W, Pfleiderer B (2008): fMRI reveals altered auditory processing in manifest and premanifest Huntington's disease. Neuropsychologia 46:1279–1289. [DOI] [PubMed] [Google Scholar]
- Sah DW, Aronin N (2011): Oligonucleotide therapeutic approaches for Huntington disease. J Clin Invest 121:500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seppi K, Schocke MF, Mair KJ, Esterhammer R, Weirich‐Schwaiger H, Utermann B, Egger K, Brenneis C, Granata R, Boesch S, Poewe W, Wenning GK (2006): Diffusion‐weighted imaging in Huntington's disease. Mov Disord 21:1043–1047. [DOI] [PubMed] [Google Scholar]
- Seunarine KK, Alexander DC (2013): Multiple fibers: beyond the diffusion tensor In: Behrens, TEJ , Johansen‐Berg, H (eds) Diffusion MRI: From Quantitative Measurement to In vivo Neuroanatomy, 2nd ed., Academic Press, San Diego: pp 105–123. [Google Scholar]
- Smith A (1968): The Symbol Digit Modalities Test: A neuropsychologic test for economic screening of learning and other cerebral disorders. Learn Disord 3:83–91. [Google Scholar]
- Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TE, Johansen‐Berg H, Bannister PR, De Luca M, Drobnjak I, Flitney DE, Niazy RK, Saunders J, Vickers J, Zhang Y, De Stefano N, Brady JM, Matthews PM (2004): Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 23 Suppl 1:S208–S219. [DOI] [PubMed] [Google Scholar]
- Sritharan A, Egan G, Johnston L, Horne M, Bradshaw J, Bohanna I, Asadi H, Cunnington R, Churchyard AJ, Chua P, Farrow M, Georgiou‐Karistianis N (2010): A longitudinal diffusion tensor imaging study in symptomatic Huntington's disease. J Neurol Neurosurg Psychiatry 81:257–262. [DOI] [PubMed] [Google Scholar]
- Stoffers D, Sheldon S, Kuperman J, Goldstein J, Corey‐Bloom J, Aron A (2010): Contrasting gray and white matter changes in preclinical Huntington disease. Neurology 74:1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Langbehn DR, Leavitt BR, Roos RA, Durr A, Craufurd D, Kennard C, Hicks SL, Fox NC, Scahill RI, Borowsky B, Tobin AJ, Rosas HD, Johnson H, Reilmann R, Landwehrmeyer B, Stout JC; TRACK‐HD investigators (2009): Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK‐HD study: Cross‐sectional analysis of baseline data. Lancet Neurol 8:791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Scahill RI, Durr A, Roos RA, Leavitt BR, Jones R, Landwehrmeyer GB, Fox NC, Johnson H, Hicks SL, Kennard C, Craufurd D, Frost C, Langbehn DR, Reilmann R, Stout JC; TRACK‐HD Investigators (2011): Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK‐HD study: The 12‐month longitudinal analysis. Lancet Neurol 10:31–42. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Reilmann R, Roos RA, Durr A, Leavitt B, Owen G, Jones R, Johnson H, Craufurd D, Hicks SL, Kennard C, Landwehrmeyer B, Stout JC, Borowsky B, Scahill RI, Frost C, Langbehn DR; TRACK‐HD investigators (2012): Potential endpoints for clinical trials in premanifest and early Huntington's disease in the TRACK‐HD study: Analysis of 24 month observational data. Lancet Neurol 11:42–53. [DOI] [PubMed] [Google Scholar]
- Tournier JD, Calamante F, Connelly A (2007): Robust determination of the fibre orientation distribution in diffusion MRI: Non‐negativity constrained super‐resolved spherical deconvolution. NeuroImage 35:1459–1472. [DOI] [PubMed] [Google Scholar]
- Vandenberghe W, Demaerel P, Dom R, Maes F (2009): Diffusion‐weighted versus volumetric imaging of the striatum in early symptomatic Huntington disease. J Neurol 256:109–114. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP, Jr. (1985): Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44:559–577. [DOI] [PubMed] [Google Scholar]
- Weaver KE, Richards TL, Liang O, Laurino MY, Samii A, Aylward EH (2009): Longitudinal diffusion tensor imaging in Huntington's Disease. Exp Neurol 216:525–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf RC, Sambataro F, Vasic N, Schonfeldt‐Lecuona C, Ecker D, Landwehrmeyer B (2008a): Aberrant connectivity of lateral prefrontal networks in presymptomatic Huntington's disease. Exp Neurol 213:137–144. [DOI] [PubMed] [Google Scholar]
- Wolf RC, Sambataro F, Vasic N, Schonfeldt‐Lecuona C, Ecker D, Landwehrmeyer B (2008b): Altered frontostriatal coupling in pre‐manifest Huntington's disease: Effects of increasing cognitive load. Eur J Neurol 15:1180–1190. [DOI] [PubMed] [Google Scholar]
- Young AW, Perrett D, Calder AJ; Sprengelmayer R, Ekman P (2002): Facial expressions of emotion: Stimuli and tests (FEEST). Psychology: Thames Valley Test Company, Bury St Edmunds, England. [Google Scholar]
- Zigmond AS, Snaith RP (1983): The hospital anxiety and depression scale. Acta Psychiatr Scand 67:361–370. [DOI] [PubMed] [Google Scholar]
- Zimbelman JL, Paulsen JS, Mikos A, Reynolds NC, Hoffmann RG, Rao SM (2007): fMRI detection of early neural dysfunction in preclinical Huntington's disease. J Int Neuropsychol Soc 13:758–769. [DOI] [PubMed] [Google Scholar]