

Abstract
Persistent neuropathic pain due to peripheral nerve degeneration in diabetes is a stressful symptom; however, the underlying neural substrates remain elusive. This study attempted to explore neuroanatomical substrates of thermal hyperalgesia and burning pain in a diabetic cohort due to pathologically proven cutaneous nerve degeneration (the painful group). By applying noxious 44°C heat stimuli to the right foot to provoke neuropathic pain symptoms, brain activation patterns were compared with those of healthy control subjects and patients with a similar degree of cutaneous nerve degeneration but without pain (the painless group). Psychophysical results showed enhanced affective pain ratings in the painful group. After eliminating the influence of different pain intensity ratings on cerebral responses, the painful group displayed augmented responses in the limbic and striatal structures, including the perigenual anterior cingulate cortex (ACC), superior frontal gyrus, medial thalamus, anterior insular cortex, lentiform nucleus (LN), and premotor area. Among these regions, blood oxygen level‐dependent (BOLD) signals in the ACC and LN were correlated with pain ratings to thermal stimulations in the painful group. Furthermore, activation maps of a simple regression analysis as well as a region of interest analysis revealed that responses in these limbic and striatal circuits paralleled the duration of neuropathic pain. However, in the painless group, BOLD signals in the primary somatosensory cortex and ACC were reduced. These results suggest that enhanced limbic and striatal activations underlie maladaptive responses after cutaneous nerve degeneration, which contributed to the development and maintenance of burning pain and thermal hyperalgesia in diabetes. Hum Brain Mapp 34:2733–2746, 2013. © 2012 Wiley Periodicals, Inc.
Keywords: contact heat‐evoked potential, functional magnetic resonance imaging, neuropathic pain, diabetes mellitus, skin innervation
INTRODUCTION
Peripheral neuropathy is a frequent complication of diabetes, with neuropathic pain affecting up to one‐quarter of patients with diabetes [Davies et al., 2006]. As the prevalence of peripheral neuropathy increases with diabetes duration, neuropathic pain is among the most difficult pain syndromes to treat, thus impairing the quality of life and causing a significant socioeconomic burden [DiBonaventura et al., 2011]. In diabetic neuropathy, degeneration of sensory nerve terminals in the skin is common [Shun et al., 2004] and may lead to a burning pain and thermal hyperalgesia affecting the lower extremities [Devigili et al., 2008]. Although this manifestation is attributed to damage and sensitization of C‐nociceptors [Bostock et al., 2005; Treede et al., 1992] and subsequent changes in the synaptic plasticity of the dorsal horn [Battaglia et al., 2003], supraspinal mechanisms of burning pain and thermal hyperalgesia due to cutaneous nerve degeneration are largely unknown. In addition, even under a comparable degree of cutaneous nerve damage, why some patients with diabetic neuropathy manifest neuropathic pain while others do not remains unknown. The anatomical substrates of the development and maintenance of such neuropathic pain after nerve injury in chronic neuropathy have not been explored extensively.
Traditionally, the processing of experimental acute nociceptive pain in normal subjects is categorized into two parallel systems [Treede et al., 1999]. The lateral pain system, responsible for sensory‐discriminative aspects of pain, consists of the lateral thalamic nucleus and the primary (S1) and secondary (S2) somatosensory cortices. The medial pain system, which contributes to the affective components of pain, includes the medial thalamic nucleus and anterior cingulate cortex (ACC). With capsaicin‐induced pain in healthy subjects, both sensory‐discriminative and affective components of pain became activated [Iannetti et al., 2005; Lee et al., 2008; Lorenz et al., 2002; Zambreanu et al., 2005]. However, recent evidence suggested different patterns of brain activation in chronic pain conditions from that in acute experimental pain [Apkarian et al., 2009]. Two issues remain to be elucidated: (1) whether specific component of pain contributes to chronic pathological pain in patients with diabetes, and (2) whether brain areas additional to the lateral and medial pain systems become activated in chronic pathologic pain due to nerve degeneration.
Previous studies exploring clinical pain syndromes, e.g., chronic low back pain [Baliki et al., 2006], fibromyalgia [Jensen et al., 2009], and irritable bower syndrome [Verne et al., 2003], mainly depended on symptomatology. There is a lack of definite pathologic evidence indicating the anatomical origin of pain. To demonstrate the primary pathology of neurodegeneration in diabetes‐related small‐fiber neuropathy, punch skin biopsy is an established tool to diagnose neuropathy affecting the nociceptive nerve terminals [Lauria et al., 2010]. This approach documents degeneration of cutaneous nerve terminals in chronic neuropathy and provides an objective indicator of the degree of skin denervation by quantifying the intraepidermal nerve‐fiber (IENF) density [Polydefkis et al., 2002; Shun et al., 2004].
In this study, we used functional magnetic resonance imaging (fMRI) to explore brain activations in a diabetic cohort with neuropathic pain. Specifically, we examined how the patterns and degree of activations to noxious stimuli in patients with diabetes with nerve degeneration‐induced chronic neuropathic pain differ from those in normal subjects.
MATERIALS AND METHODS
Subjects
The study protocol was approved by the Ethics Committee of National Taiwan University Hospital, Taipei, Taiwan, and informed consent was obtained from each subject. We recruited a group of patients with neuropathy with thermal pain (painful group), defined as burning pain and thermal hyperalgesia, that was aggravated by heat and relieved by cooling the skin. For comparison, healthy volunteers (control group) and patients with diabetes with painless neuropathy (painless group) were also enrolled. Data of two control subjects were reported previously [Tseng et al., 2010]. Five females were premenopausal, and they were scanned during Days 5–10 of their menstrual cycle.
All patients had Type 2 diabetes mellitus with a symmetrical graded stocking distribution of sensory symptoms involving the feet. The duration of their neuropathic symptoms was at least 6 months, and these symptoms were categorized as spontaneous pain, stimulus‐evoked pain, and paresthesias or dysesthesias [Baron, 2006] (Table 1). Patients were assigned to painful neuropathy group if they had spontaneous pain (burning, electrical shocks, and stabbing) or stimulus‐evoked pain (thermal hyperalgesia and mechanical allodynia) [Woolf and Mannion, 1999]. To ensure that the primary pathology for neuropathic pain lay in small‐diameter sensory nerves [Polydefkis et al., 2002], all recruited patients with diabetes had cutaneous nerve degeneration, which was defined as a reduced IENF density when compared with normative values. Patients with cerebral infarcts, hemorrhage, or subcortical arteriosclerotic encephalopathy on T 2 MRI were excluded. The clinical history of members of the control group was evaluated using questionnaires, and neurological examinations were performed to exclude any neuropsychiatric disorder or pain symptoms.
Table 1.
Demographic data and clinical presentation
| Control | Painful | Painless | |
|---|---|---|---|
| Age (years) (range) | 50.0 ± 16.6 (26–69) | 51.1 ± 9.1 (32–64) | 51.3 ± 10.6 (25–65) |
| Gender (M/F) | 4/7 | 4/7 | 6/5 |
| Duration of diabetes (years) | 6.0 ± 3.2 | 6.6 ± 6.2 | |
| NOD [median (range)] | 1 (0–3) | 1 (0–2) | |
| Symptoms [n, median (range)] | |||
| Spontaneous pain | |||
| Burning | 11, 5 (3–10) | 0, 0 (0) | |
| Electrical shocks/stabbing | 5, 0 (0–5) | 0, 0 (0) | |
| Stimulus‐evoked pain | |||
| Thermal hyperalgesia | 11, 6 (2–10) | 0, 0 (0) | |
| Mechanical allodynia | 1, 0 (0–5) | 0, 0 (0) | |
| Paresthesias/dysesthesias | |||
| Numbness | 8, 5 (0–10) | 11, 5 (3–10) |
HPT, heat pain threshold on the skin of the right foot dorsum; n, number of patients with defined symptoms; NOD, the number of drugs used to treat neuropathy. Score in symptoms: 0, no symptom; 10, worst symptom imaginable.
Skin Biopsy, Immunohistochemistry, and Quantification of Epidermal Innervations
We quantified the IENF density to represent the integrity of small‐diameter sensory nerves. A skin biopsy was taken following established procedures [Tseng et al., 2006], and it was performed in all subjects to confirm normal skin innervation in controls and reduced innervation in patients with diabetes. Details of the procedure, immunohistochemistry, and quantification of the IENF density are provided in the Materials and Methods section of Supporting Information.
Thermal Stimulation
A contact heat‐evoked potential (CHEP) stimulator (Medoc, Ramat Yishai, Israel) was used to deliver thermal stimulation via a 27‐mm‐diameter thermode composed of a heating thermofoil (Minco Products, Minneapolis, MN) covered with thermoconductive plastic. The stimulator directly contacted the skin without causing any pressure, and the stimulus temperatures reported in this study were the temperatures of the thermofoil. Cooling began immediately after a prefixed target temperature was reached. To compare activations among the three groups of subjects, we applied stimuli of 44°C because we previously identified distinct networks responsible for the perception of noxious heat in healthy control subjects [Tseng et al., 2010].
Image Acquisition
The fMRI study was performed using a 3‐T MRI scanner (Trio, Siemens, Erlangen, Germany). The subject's head was comfortably positioned inside a receive‐only eight‐channel head coil, padded with sponges and fixed with a strap across the forehead to minimize head motion. Each subject was provided ear plugs to minimize stimulation from the scanning noise. A T 1‐weighted anatomical image was acquired for each subject using the following parameters: a recovery time (TR) of 581 ms; an echo time (TE) of 6.7 ms; a flip angle of 87°; a matrix size of 256 × 205; field of view (FOV) of 250 × 250 mm; a slice thickness of 3.9 mm; and a number of horizontal slices of 35. Gradient‐echo echo planar imaging was used to acquire blood oxygen level‐dependent (BOLD) contrast data. The acquisition parameters were TR/TE of 3,000/30 ms, a flip angle of 90°, a 64 × 64 matrix, a FOV of 250 × 250 mm, and a slice thickness of 3.9 mm, resulting in a voxel size of 3.9 × 3.9 × 3.9 mm. In total, 35 horizontal slices along the anterior/posterior commissure line were obtained covering the entire brain. The first four images were discarded to account for spin saturation effects.
Experimental Protocol
The paradigms of this block‐designed fMRI followed our established protocols [Tseng et al., 2010]. To avoid pain from the skin biopsy wound, the fMRI study was performed either before the skin biopsy or 7 days after it, and none of the subjects reported pain from the skin biopsy wound at the time of the fMRI study. Drugs used for treating neuropathy in patients with diabetes were suspended for at least 1 week. One hour before fMRI scanning, subjects were brought to a waiting room where they were familiarized with the instructions for the experiment and the rating procedure. They were asked to report the mean intensity of each neuropathic sensory symptom (Table 1) on an 11‐point (0–10) numerical scale, in which 0 was “no symptom” and 10 was “worst symptom imaginable” [Bouhassira et al., 2004]. The heat pain threshold (HPT) was determined using a CHEP stimulator. Temperature stimuli were applied to the skin of the right foot dorsum with a slope of 1°C/s starting from an adaptation temperature of 32°C, which was terminated when subjects first felt a heat pain sensation. The average of three successive tests was recorded as the HPT.
The imaging session consisted of one T 2‐weighted image, one T 1‐weighted anatomical scan, and one functional scanning run. The thermode was strapped to the dorsum of the right foot without causing any pressure, and the stimulation site was fixed during the functional scan. The stimulus sequence during the functional scanning run consisted of five presentations of the same stimulus that ramped from the baseline 32 (36 s) to 44°C (12 s) at 20°C/s and then returned to the baseline temperature at 40°C/s. Right before scanning, subjects were instructed to refrain as much as possible from moving throughout the imaging session, to pay close attention to the stimuli, and to silently keep in mind the sensation they felt and report it after each functional scan was completed. During stimulation, subjects were instructed to keep their eyes closed and face a blank screen. A verbal rating scale (VRS) was used to rate the overall perception produced by the thermal stimulation, which ranged from 0 to 10: with 0 indicating no sensation, 4 just painful, and 10 unbearable pain [Tseng et al., 2010]. The short‐form McGill Pain Questionnaire (SFMPQ) was then used to assess both the sensory‐discriminative and affective dimensions of the perceived pain [Melzack, 1987]. Subjects were asked to verbally rate the overall perception of the five stimuli immediately after the fMRI scan. Using a computer projector via a mirror mounted on the head coil, subjects viewed the descriptors one by one on a screen and reported the rating for each descriptor.
fMRI Data Analysis and Statistics
fMRI data were analyzed using SPM5 (http://www.fil.ion.ucl.ac.uk/spm) implemented on MATLAB (Mathworks, Sherborn, MA) as described previously [Tseng et al., 2010]. Briefly, fMRI data series were realigned to the first volume in each scan sequence and resliced with sinc interpolation to correct for motion artifacts [Friston et al., 1995b]. Subjects enrolled in the current study had no scans with sudden head movements of more than 2 mm. The resulting mean image was coregistered to the corresponding T 1‐weighted anatomical image. To enable intersubject analysis, the T 1‐weighted image of each subject was normalized to the standard Montreal Neurological Institute (MNI) template [Collins et al., 1994], and the normalization parameters were then applied to the functional data of that subject. The resampled voxel volume of the normalized images was 2 × 2 × 2 mm. Subsequently, data were smoothed using an isotropic Gaussian kernel with an 8‐mm full width at half maximum. Condition‐specific effects were estimated using the general linear model in SPM5, which was constructed by convolving a boxcar sequence with the hemodynamic response function [Friston et al., 1995a]. A high‐pass filter with a cutoff period of 128 s was used to remove low‐frequency noise.
In the study, we first compared group activation maps in the three groups to explore cerebral activation patterns with the stimulus temperature. Statistical parametric maps of brain activation for stimuli at the target temperatures were presented as t‐contrasts [Tseng et al., 2010]. We took first‐level contrasts to the second level for the group analysis using one‐sample t‐tests within a random‐effects model [Holmes and Friston, 1998]. The threshold for within‐group activation maps of the three groups was set to uncorrected P value of 0.0001. To further compare their activations with eliminating the potential confounding effect of different ratings to pain stimuli on brain activation among the three groups [Bornhövd et al., 2002; Iannetti et al., 2005], an analysis of covariance (ANCOVA) in SPM5 using the VRS rating as a covariate was performed to examine if brain activation patterns differ across different levels of group. To completely delineate activity of neural circuits enhanced in the painful group, a threshold of P < 0.01 uncorrected was applied to the statistical parametric map for the main effect of group in this ANCOVA analysis, and the results of brain activations correlated with the duration of neuropathic pain were reported at a threshold setting of uncorrected P < 0.005. In all statistical parametric maps, an extent threshold of 10 contiguous voxels was used to reduce possible false positive activation [Forman et al., 1995]. Anatomical regions were identified using the SPM Anatomy Toolbox [Eickhoff et al., 2005] and the Talairach Daemon Client software (http://ric.uthscsa.edu/projects/talairachdaemon.html) [Tseng et al., 2010].Within a cluster, local maxima at least 8.0 mm apart with different anatomical locations were reported.
To further analyze the BOLD signal changes in pain‐related regions of interest (ROI), we used the MarsBaR toolbox (http://marsbar.sourceforge.net/) [Brett et al., 2002] to derive the mean BOLD signal change for each ROI, and signal intensities of five blocks were averaged and analyzed in each ROI. Brain ROIs analyzed in the current study included: (1) the ACC, prefrontal cortex (PFC; superior, middle, and inferior frontal gyri), medial thalamus (medial dorsal and midline nuclei), and anterior insular cortex (IC) to show brain activity of the medial pain pathway; (2) the S1 and S2 to represent the lateral pain pathway [Treede et al., 1999]; and (3) the lentiform nucleus (LN) and premotor area (PMA; see Materials and Methods section in Supporting Information for a detailed definition of each ROI). The coordinates and size of each ROI are shown in Supporting Information Table SI.
Continuous variables following a Gaussian distribution are expressed as the mean ± standard deviation, and measures with a non‐Gaussian distribution are presented as the median (range). To compare between‐group statistical differences, a t‐test and parametric one‐way analysis of variance (ANOVA) test were used to compare parametric data, and the Mann‐Whitney U‐test and Kruskal‐Wallis test were adopted to analyze nonparametric measures. A χ2 test and Fisher's exact test were used to analyze proportions, and a simple regression analysis was performed using Pearson's correlation coefficient test. A P value of <0.05 was considered statistically significant. The statistical analysis was performed using SPSS (Chicago, IL) and GraphPad Prism (GraphPad Software, San Diego, CA).
RESULTS
Clinical Features of Neuropathic Pain
All subjects were right handed, and there were 11 subjects in each group (Table 1). The distributions of age (P = 0.97) and gender (P = 0.61) were similar among the three groups (Table 1). In the painful group, all patients had typical features of burning symptoms with thermal hyperalgesia and burning pain in the foot dorsum. Five of them also had electrical shocks, and one patient reported mechanical allodynia. In contrast, the only symptom in the painless group was numbness (Table 1). Eight patients in the painful group also had numbness; however, there was no statistical difference in the scores of numbness between the painful and painless groups (P = 0.92). For the two diabetic groups, there was no statistical difference in the duration of diabetes (P = 0.75) or the number of drugs used for treating the neuropathy (P = 0.92; Table 1). In the painless group, tricyclic antidepressant (imipramine) was used as monotherapy to attenuate numbness, with additional benzodiazepine (clonazepam) use in two patients. As for the painful group, tricyclic antidepressant and anticonvulsants (gabapentin, carbamazepine, or topiramate) were used alone or in combination to treat painful symptoms.
Psychophysical Characteristics of Neuropathic Pain
Patients with painful neuropathy displayed higher ratings of VRS in addition to significantly lower HPT at the foot dorsum when compared with the other two groups of subjects (Fig. 1A,B). Importantly, the painful group reported higher affective pain ratings rather than sensory‐discriminative ratings on the SFMPQ when compared with the control group (Fig. 1C,D). The ratings of various descriptors of the sensory dimension on the SFMPQ were similar between the two groups (Table 2). When compared with the painless group, the painful group reported significantly higher ratings in both sensory‐discriminative and affective dimensions on the SFMPQ to this stimulus temperature (Fig. 1C,D). In the painful group, the HPTs were comparable between those with or without numbness (P = 0.63). The HPTs and ratings of VRS, sensory‐discriminative, and affective dimensions on the SFMPQ did not statistically differ between the painless and control groups (Fig. 1A–D). These findings indicated the existence of heat hypersensitivity and enhanced affective pain ratings in our patients with neuropathic pain.
Figure 1.
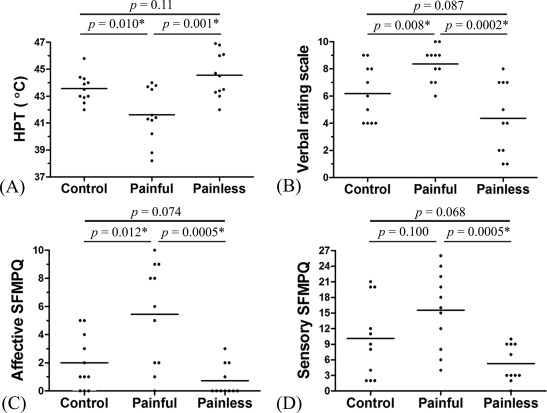
Heat pain thresholds and psychophysical ratings to 44°C stimuli. In the painful group, not only the heat pain threshold (HPT) (A) recorded on the right foot dorsum was significantly reduced (mean ± SD, 41.6°C ± 2.0°C) when compared with those in the control (43.6°C ± 1.1°C) and painless (44.6°C ± 1.7°C) groups but also the ratings of the verbal rating scale (B) were significantly increased. Moreover, the score of the affective dimension (C) but not the sensory‐discriminative dimension (D) on the short‐form McGill Pain Questionnaire (SFMPQ) was increased in the painful group when compared with the control group. The bars show mean values because all data follow Gaussian distribution. *P < 0.05 by unpaired t‐test.
Table 2.
Psychophysical results to 44°C stimuli
| Intensity ratings to stimulus temperature [median (range)] | |||
|---|---|---|---|
| SFMPQ descriptor | Control | Painful | Painless |
| Sensory dimension | |||
| 1. Throbbing | 1 (0–3) | 1 (0–3)* | 1 (0–2) |
| 2. Shooting | 1 (0–3) | 3 (0–3)* | 1 (0–2) |
| 3. Stabbing | 2 (0–3) | 3 (1–3)* | 1 (0–2) |
| 4. Sharp | 0 (0–2) | 0 (0–3) | 0 (0–1) |
| 5. Cramping | 0 (0–1) | 0 (0–1) | 0 (0) |
| 6. Gnawing | 0 (0–2) | 0 (0–2) | 0 (0) |
| 7. Heat | 2 (1–3) | 3 (1–3)* | 1 (0–3) |
| 8. Aching | 1 (0–3) | 3 (0–3)* | 1 (0–2) |
| 9. Heavy | 0 (0–3) | 3 (0–3) | 0 (0–1) |
| 10. Tender | 0 (0–2) | 0 (0–3) | 0 (0–1) |
| 11. Splitting | 0 (0–1) | 0 (0–3) | 0 (0–0) |
| Total scores | 9 (2–21) | 16 (4–26) | 3 (0–10) |
| Affective dimension | |||
| 12. Exhausting | 0 (0–1) | 0 (0–2) | 0 (0) |
| 13. Unpleasant | 1 (0–3) | 3 (0–3)*,† | 0 (0–3) |
| 14. Fearful | 1 (0–2) | 2 (0–3)† | 0 (0) |
| 15. Punishing | 0 (0–2) | 0 (0–3) | 0 (0) |
| Total scores | 1 (0–5) | 6 (0–10) | 0 (0–3) |
Degree of short‐form McGill Pain Questionnaire (SFMPQ) descriptors 1–15: 0, none; 1, mild; 2, moderate; 3, severe. Mann‐Whitney U‐test, P < 0.05 when compared with painless (*) and control (†) groups.
Distinct Activation Patterns on fMRI of Neuropathic Pain Due To Skin Denervation
To investigate the patterns of brain activations in neuropathic pain, we analyzed fMRI data with noxious heat stimulation. The three groups had distinct and different degrees of activations. Consistent with our previous report [Tseng et al., 2010], a group analysis revealed activation of the pain matrix in control subjects to 44°C stimuli, including the S2, ACC, IC, and cerebellum (Fig. 2 and Supporting Information Table SII). The painless group showed no voxels with above‐threshold activation to 44°C stimuli. By contrast, in addition to the brain regions activated in the control group, the painful group displayed additional activations in the S1, perigenual ACC, PMA, supplementary motor area, and LN (Fig. 2 and Supporting Information Table SII).
Figure 2.
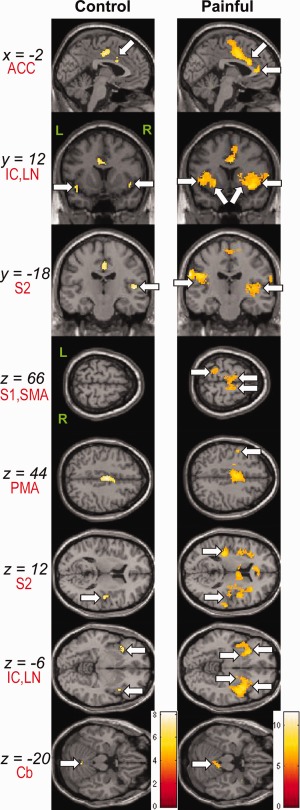
Group activation areas to 44°C stimuli. Results were overlaid on the canonical T 1‐weighted brain image of SPM5. Arrows indicate activated regions thresholded at uncorrected P < 0.0001, with a minimum activation cluster size of 10 voxels. The bar on the right side of group maps shows the range of t‐scores for SPM5. Abbreviations: ACC, anterior cingulate cortex; Cb, cerebellum; IC, insular cortex; L, left; LN, lentiform nucleus; PMA, premotor area; R, right; S1, primary somatosensory cortex; S2, secondary somatosensory cortex; SMA, supplementary motor area.
As the degree of brain activation is affected by perceived pain intensity [Bornhövd et al., 2002; Iannetti et al., 2005], variations of cerebral responses in different groups were potentially explained by their unequal ratings to pain stimuli. To clarify whether brain activation patterns, adjusted for differences in perceived pain intensity, differ across the three groups, we performed a one‐way ANCOVA analysis in SPM5 using the VRS rating as a covariate. After eliminating the influence of perceived pain intensity on cerebral responses, there was a group effect on activations in the contralateral perigenual ACC, medial PFC (superior frontal gyrus and SFG), medial thalamus and PMA, ipsilateral anterior IC, LN and hippocampus, and bilateral temporal gyri (Fig. 3A–F and Table 3). Activations in the S1 and S2 were not discernible (Fig. 3G,H).
Figure 3.
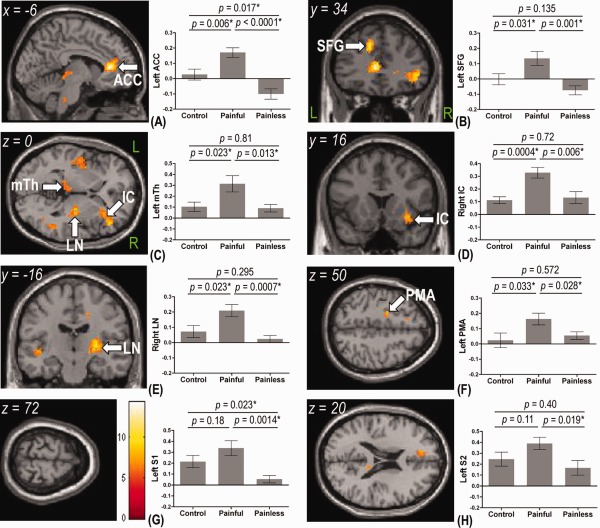
Group activation areas of analysis of covariance in SPM5 using the rating of verbal rating scale as a covariate and their corresponding percent BOLD signal change (%SC) from the regions of interest analysis. Activation maps were overlaid on the canonical T 1‐weighted brain image of SPM5. The color bar in (G) shows the range of F‐values for SPM5. When compared with the control and painless groups, the painful group showed higher %SC to 44°C stimuli in left anterior cingulate cortex (ACC) (A), left superior frontal gyrus (SFG) (B), left medial thalamus (mTh) (C), right anterior insular cortex (IC) (D), right lentiform nucleus (LN) (E), and left premotor area (PMA) (F). Moreover, %SCs in the left primary somatosensory cortex (S1; 0.34 ± 0.07) (G) and left secondary somatosensory cortex (S2; 0.39 ± 0.06) (H) in the painful group were significantly higher than those in the painless group (0.05 ± 0.03 for the S1 and 0.17 ± 0.07 for the S2) but not different from responses in the control group (0.21 ± 0.06 for the S1 and 0.25 ± 0.06 for the S2). When compared with the control group, %SCs in the left ACC (0.03 ± 0.04 versus −0.10 ± 0.03) (A) and left S1 (G) in the painless group were significantly reduced. Data are presented as the mean ± SEM. L, left; R, right. *P < 0.05 by unpaired t‐test.
Table 3.
Group activation areas of analysis of covariance
| Cluster size (voxels) | Region | Side | BA | F [MNI coordinates: x, y, z (mm)] |
|---|---|---|---|---|
| 614 | ACC | L | 24 | 14.34 (−6, 34, 8) |
| SFG | L | 9 | 12.24 (−12, 34, 36) | |
| 569 | IC | R | — | 8.53 (34, 16, −8) |
| 143 | Thalamus | L | — | 6.5 (−6, −22, 0) |
| 512 | LN | R | — | 10.41 (26, −16, 8) |
| Hippocampus | R | — | 9.61 (40, −24, −10) | |
| 74 | PMA | L | 6 | 8.62 (−20, −6, 50) |
| 427 | TG | L | 22 | 8.97 (−46, −6, −6) |
| 161 | TG | R | 22 | 8.58 (52, −50, 6) |
The height threshold for illustrating the clusters was P < 0.01 uncorrected, with a minimum activation cluster size of 10 voxels. Local maxima within the same cluster are those at least 8 mm apart and with different anatomical locations.
Abbreviations: ACC, anterior cingulate cortex; IC, insular cortex; L, left; LN, lentiform nucleus; PMA, premotor area; R, right; SFG, superior frontal gyrus; TG, middle or superior temporal gyrus.
These findings were complemented by another ROI analysis, where we extracted BOLD signals in these medial pain pathway (ACC, SFG, medial thalamus, and anterior IC) and motor‐related pathway (LN and PMA) as well as the lateral pain pathway (S1 and S2) to further characterize activations in pain‐related circuits in neuropathic pain. The painful group displayed significantly higher BOLD signals in the ACC, SFG, medial thalamus, anterior IC, LN, and PMA than the rest of the two groups (Fig. 3A–F). Moreover, although responses in the S1 and S2 in the painful group were significantly higher than those in the painless group, the responses between the painful group and control group were comparable (Fig. 3G,H). These findings are consistent with the above‐mentioned differences in psychophysical ratings evaluating sensory‐discriminative and affective pain dimensions between the painful group and the rest of the two groups. When compared with the control group, the painless group showed significantly lower activation in the ACC and the S1 (Fig. 3A,G).
Correlations of fMRI Activation With Features of Neuropathic Pain
To ascertain that the cerebral activations identified above reflect perceptions of thermal pain, we analyzed correlations of BOLD signals with the intensity of pain ratings. In the control and painless groups, the perceived pain intensity as measured by the VRS and affective pain ratings using the SFMPQ were positively correlated with responses in the somatosensory cortices and ACC, respectively (Table 4). In the painful group, the affective pain ratings were correlated with BOLD signals in both the contralateral ACC and ipsilateral LN, whereas the perceived pain intensity paralleled the cerebral response in the ipsilateral LN (Table 4). In addition, there was no correlation between BOLD signals in brain regions whose activity was enhanced (Fig. 3) and degrees of skin innervation. In another simple regression analysis in SPM5, no voxels were found, in which activity correlated significantly with the degree of spontaneous burning pain or electrical shocks.
Table 4.
Correlation coefficients between pain perception and percent BOLD signal change in regions of interest (ROI) analysis
| Correlation coefficient | ||||
|---|---|---|---|---|
| Perception | ROI | Control | Painful | Painless |
| Intensity of pain (VRS) | Left S1 | 0.82a | 0.50 | 0.86a |
| Left S2 | 0.68a | 0.32 | 0.71a | |
| Right LN | 0.14 | 0.62a | 0.54 | |
| Affective pain ratings (SFMPQ) | Left ACC | 0.70a | 0.76a | 0.75a |
| Right LN | 0.16 | 0.78a | 0.38 | |
Abbreviations: ACC, anterior cingulate cortex; LN, lentiform nucleus; S1, primary somatosensory cortex; S2, secondary somatosensory cortex; SFMPQ, short‐form McGill Pain Questionnaire; VRS, verbal rating scale.
P < 0.05 (Pearson's correlation).
To further explore whether brain regions with enhanced activities in the painful group contributed to the maintenance of neuropathic pain, we correlated their BOLD signals with the duration of neuropathic pain. Intriguingly, both voxelwise and ROI analyses showed that responses in the ACC, SFG, medial thalamus, and LN all correlated with the duration of neuropathic pain (Fig. 4A–D and Table 5). In the ROI analysis, there was also a trend toward correlation between the response in the right anterior IC (r = 0.60, P = 0.053) and the duration of neuropathic pain. By contrast, responses in the S1 and S2 did not parallel the duration of neuropathic pain (Fig. 4E,F).
Figure 4.
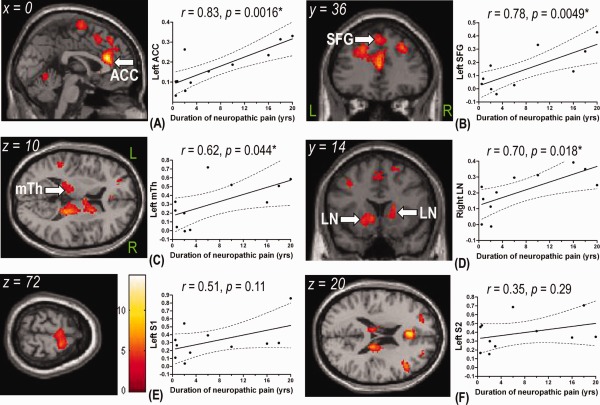
Correlation between the duration of neuropathic pain and cerebral activity during the 44°C stimuli in the painful group. Using the duration of neuropathic pain as covariates, the left perigenual anterior cingulate cortex (ACC) (A), left superior frontal gyrus (SFG) (B), left medial thalamus (mTh) (C), and right lentiform nucleus (LN) (D) showed a positive correlation with the duration of neuropathic pain. There was no correlation in the primary somatosensory cortex (S1) (E) or secondary somatosensory cortex (S2) (F). The color bar in (E) shows the range of t‐scores for SPM5. Correlation between the percent BOLD signal change (%SC) in these regions extracted from the regions of interest analysis were also presented, where the solid lines represent the linear regression lines and dotted lines indicate the 95% confidence intervals. L, left; R, right. *P < 0.05 by Pearson's correlation coefficient test.
Table 5.
Correlation between the duration of neuropathic pain and cerebral activity during the 44°C stimuli in the painful group
| Cluster size (voxels) | Region | Side | BA | t [MNI coordinates: x, y, z (mm)] |
|---|---|---|---|---|
| 1,597 | ACC | L | 24 | 14.37 (0, 32, 20) |
| SFG | L | 8 | 4.75 (−2, 36, 50) | |
| DLPFC | L | 9 | 6.04 (−14, 38, 34) | |
| 759 | DLPFC | R | 9 | 6.24 (28, 32, 42) |
| R | 46 | 8.03 (44, 24, 26) | ||
| 324 | Thalamus | L | — | 4.46 (−16, −28, 14) |
| 722 | Thalamus | R | — | 7.80 (18, −22, 18) |
| 443 | LN | L | — | 6.16 (−22, 18, −4) |
| Caudate | L | 5.90 (−16, 20, −2) | ||
| 97 | LN | R | — | 4.76 (18, 16, 0) |
| Caudate | R | 3.70 (16, 0, 16) | ||
| 696 | SMA | R | 6 | 6.04 (16, −4, 70) |
| 388 | PPC | L | 40 | 4.63 (−34, −54, 54) |
| 1,113 | PPC | R | 40 | 4.02 (44, −42, 54) |
| 109 | TG | R | 37 | 4.66 (56, −58, −4) |
| 366 | Cerebellum | L | — | 4.87 (−16, −52, −20) |
| 319 | Cerebellum | R | — | 4.48 (26, −52, −26) |
The height threshold for illustrating the clusters was P < 0.005 uncorrected, with a minimum activation cluster size of 10 voxels. Local maxima within the same cluster are those at least 8 mm apart and with different anatomical locations.
Abbreviations: ACC, anterior cingulate cortex; BA, Brodmann area; DLPFC, dorsolateral prefrontal cortex; L, left; LN, lentiform nucleus; PPC, posterior parietal cortex; R, right; SFG, superior frontal gyrus; SMA, supplementary motor area; TG, temporal gyrus.
Taken together, these brain regions with an augmented response to heat pain stimuli participated in the perception of thermal pain and were also engaged in the development and maintenance of neuropathic pain.
DISCUSSION
This study demonstrates enhanced cerebral responses to thermal pain after skin degeneration in diabetes. Distinct from previous fMRI study designs, the current study is unique because of the clearly documented primary pathology of neuropathic pain (cutaneous nerve degeneration), a single disease entity in patient groups (diabetes), and no left–right somatotopic differences in the distribution of neuropathic symptoms (symmetry). The most important finding in this study is that the augmented cerebral activities in the limbic (ACC, PFC, medial thalamus, and anterior IC) and striatal (LN) areas determine the phenotype of painful versus painless manifestations after cutaneous nerve degeneration. Furthermore, with neuropathic pain, increased cerebral activities in these areas correlated with the characteristics of neuropathic pain, that is, both evoked pain and the duration of neuropathic pain.
Imaging Representation of Neuropathic Pain Due To Cutaneous Nerve Degeneration
This report identifies brain regions with maladaptive processing of chronic pain by comparing fMRI patterns between neural substrates for clinical chronic pain versus normal nociceptive pain. In control subjects, the sensory‐discriminative pain is related to BOLD signals of the somatosensory cortices [Peyron et al., 2000]. In the painful group, however, there was a significant shift in the pattern of brain activations, that is, augmentation of the limbic and striatal areas, suggesting that these areas became excessively activated. These observations indicated that chronic neuropathic pain due to peripheral nerve degeneration is a type of pathologic pain that is distinct from normal physiological nociceptive pain [Apkarian et al., 2011]. The differences might serve as a neuroimaging biomarker to determine whether or not neuropathic pain develops and persists after cutaneous nerve injury in diabetes [Tracey, 2011] and may also underlie the mechanisms of central sensitization. In addition, this study is the first to document hyperactivities of these circuits in degeneration‐induced pain, the functions of which are related to affective and cognitive processing rather than somatosensory processing.
Augmented Responses in the Limbic System Due To Nerve Degeneration
The augmented BOLD signals in the limbic system after skin denervation in the painful group are in contrast to cerebral responses in control subjects, in which brain areas in both the lateral and medial pain systems were similarly activated. Anatomically, small‐diameter A‐delta and C‐primary afferent fibers responsible for nociceptive‐ and thermoreceptive‐specific inputs terminate in the superficial lamina I layer of the spinal dorsal horn. The medial dorsal nucleus of the thalamus receives sensory inputs from lamina I and then projects to the ACC and anterior IC [Ongür and Price, 2000]. Compelling evidence showed that these limbic circuits are associated with the emotional appraisal of pain [Craig, 2003; Price, 2000]. The ACC modulates somatosensory characters of pain with PFC to add cognitive significance to emotions over long‐term implications of chronic pain states [Price, 2000]. Hence, this study demonstrated that maladaptive responses of the limbic system play key roles in the maintenance of neuropathic pain.
In the ACC, the strongest enhancement of neural activities in the painful group occurred in its perigenual part. Moreover, rather than the posterior part of the insula involved in sensory dimensions of pain [Brooks et al., 2005], the anterior insula also displayed augmented activation, which corroborates with a previous fMRI study investigating neural substrate engaged in brush‐evoked allodynia in neuropathic pain [Schweinhardt et al., 2006]. The underlying biological significance of these results merits discussion because, in contrast to the caudal portion of the ACC participating in pain‐related unpleasantness, the perigenual part of the ACC as well as the anterior IC is more engaged in cognitive and emotional modulation of pain [Rainville et al., 1999], such as placebo analgesia [Petrovic et al., 2002], expectations of decreased pain [Koyama et al., 2005], and pain empathy [Singer et al., 2004]. Therefore, the enhanced activation in both structures suggest alterations in cognitive or emotional processing of pain experience in patients with degeneration‐induced neuropathic pain, such as stronger expectation of pain relief.
Alternatively, electrophysiological studies have shown that pain‐related laser‐evoked potentials originating from the ACC and IC were modulated by the saliency of pain stimulation [Iannetti et al., 2008; Legrain et al., 2011]. As behavioral evidence indicates increased attention and sensitivity to external pain stimulation in chronic pain [Crombez et al., 1998; McCracken, 1997], another implication for the enhanced response in the ACC and IC may be the existence of hypervigilance to painful stimulation in these patients with chronic neuropathic pain. However, in previous neuroimaging studies [Davis et al., 2005; Downar et al., 2000, 2002, 2003], the salience detection mainly resides in the midcingulate area, which is the most commonly activated part of this limbic structure in acute normal physiological pain [Vogt, 2005; Vogt et al., 2003] and participates in response selection and orienting attention to salient events, including pain stimulation [Legrain et al., 2009; Vogt, 2005]. In the current study, two lines of evidence support the perigenual ACC being a major contributor to nerve degeneration‐induced hyperalgesia: (1) it showed the most robust activation between the painful and the control groups, and (2) its neural activity showed the strongest correlation with the duration of neuropathic pain. When compared with the midcingulate cortex, the perigenual ACC has denser connections with the amygdala and higher opiate receptor binding, suggesting its vital role in the emotional component of pain [Vogt, 2005; Vogt et al., 2003]. Neurosurgical lesions in the perigenual ACC resulted in alleviation of the emotional component of chronic pain [Foltz and White, 1962]. On a magnetoencephalographic study, the perigenual ACC displays a strong predominance of C‐fiber‐mediated activation of second pain [Ploner et al., 2002] characterized by prominent unpleasantness and a burning sensation [Price et al., 1977], a condition similar to the enhanced affective pain ratings and presentation of burning pain in the painful group of the current study. Taken together, we propose that, rather than merely a salience detector, the perigenual ACC specifically participates in the affective appraisal of neuropathic pain.
The current report also showed the involvement of another limbic structure, the medial thalamus, in cerebral processing of neuropathic pain due to cutaneous nerve degeneration. In contrast to the extensively studied ventral posterior lateral nucleus of the thalamus in neuropathic pain [Jaggi and Singh, 2011], increased response in the medial thalamus in peripheral neuropathic pain was not previously reported, and its exact role in modulating neuropathic pain is not well established. Nevertheless, in a rat model of peripheral neuropathic pain, blocking the medial thalamic system attenuated manifestations of neuropathic pain, particularly thermal hyperalgesia [Saadé et al., 2007]. Moreover, a previous study indicated that neuropathic pain symptoms could be improved after a medial thalamotomy [Jeanmonod et al., 1996]. Considering that the response in the medial thalamus was further correlated with the duration of neuropathic pain, these observations suggest the participation of the medial thalamus in the state of neuropathic pain due to cutaneous nerve degeneration.
Enhanced Activities of the Striatal System in Neuropathic Pain
This study further demonstrated (1) increased activations of the LN in the painful group, and (2) a correlation between BOLD signals of the LN and ratings of pain perception, which have not been reported in previous brain imaging studies of neuropathic pain [Moisset and Bouhassira, 2007; Schweinhardt et al., 2006; Witting et al., 2006]. Because not only the basal ganglia but also the premotor regions displayed increased activations in the painful group, the responses in these motor control‐related regions may reflect enhanced planning of motor reaction toward painful stimulations in neuropathic pain, such as withdrawal responses [Bingel et al., 2002; Downar et al., 2003]. In addition, several lines of evidence recently expanded functional dimensions of the striatum in the development and maintenance of chronic pain [Borsook et al., 2010]. Basal ganglion neurons respond to somatosensory stimulation and process the sensory‐discriminative dimension of normal pain via the structural reorganization and alterations in dopaminergic functions [Borsook et al., 2010; Nagy et al., 2006; Starr et al., 2011]. In Parkinson's disease with major pathology in the basal ganglia, there is a significant reduction in pain thresholds related to abnormal pain processing [Nolano et al., 2008; Tinazzi et al., 2008]. A significant proportion of primary nociceptive afferent neurons terminate in the globus pallidus, providing structural evidence of the spinopallidal connection that links the striatum to the nociceptive system [Braz et al., 2005]. In the current study, all controls and patients were free of Parkinson's disease. Thus, these observations highlight the previously unrecognized interconnections between the striatal system and chronic pain conditions.
Correlation of Limbic and Striatal Activities With Neuropathic Pain Duration
Intriguingly, in the painful group, we identified that the duration of neuropathic pain paralleled activities of the main structures in the limbic and striatal circuits, which is in contrast to the isolated correlation of the response in the anterior insula with pain duration in a recent chronic pain study [Baliki et al., 2006]. Anatomically, the basal ganglia receive afferent information from the thalamus, frontal lobe and ACC, and its efferent outputs project to frontal areas and the ACC [DeLong and Wichmann, 2007; Ongür and Price, 2000]. Through interconnections, these limbic and striatal structures mediate motivation, emotional processing, and cognition [Haber and Calzavara, 2009; Phillips et al., 2003]. Given that dysregulations in these circuits have been implicated in the development of affective disorder [Bennett, 2011], augmented responses of these circuits in the current study may contribute to the affective component usually described by patients with chronic neuropathic pain [Gormsen et al., 2010]. Moreover, the association between responses in these regions and the length of neuropathic pain corroborates previous observations that the duration of neuropathic pain paralleled neurochemical changes in cingulate, thalamus, and PFC [Grachev et al., 2000], suggesting the contribution of long‐term functional reorganization in these networks to the maintenance of chronic pain due to nerve terminal degeneration.
One potential confounding effect in the current study is the presence of spontaneous pain in the painful group. Spontaneous pain was accompanied by alterations in cognitive processing in chronic pain [Apkarian et al., 2004], and the level of spontaneous pain may affect brain activity [Baliki et al., 2007; Schweinhardt et al., 2006]. As this study did not request patients to record the degree of ongoing pain during scanning, we cannot completely exclude its effect on brain activations. However, as no brain activity was correlated to the level of ongoing pain reported right before scanning, a major impact of spontaneous pain on brain activation observed in our patients seems unlikely. Another potential caveat of the current study is the medications used for the treatment of neuropathic pain. Although patients were asked to withdraw them 1 week prior to scanning, drugs such as gabapentin can modulate pain‐related cerebral response [Iannetti et al., 2005], and the effect of their chronic usage remains to be elucidated in future studies.
In conclusion, the current study provides neuroimaging evidence of central sensitization for the development and maintenance of neuropathic pain after cutaneous nerve degeneration. In control subjects with normal skin innervation, both lateral and medial pain systems were activated on noxious stimulation. In contrast, brain regions in the limbic and striatal systems were exceptionally activated after skin denervation when compared with that of control subjects. This pattern of activation may contribute to the development of burning pain and thermal hyperalgesia. If brain activations in both lateral and medial pain systems are proportionally reduced, patients would not experience neuropathic pain, even though the degree of skin denervation was comparable with that in those with painful symptoms. These observations raise the possibility that distinct brain activations determine the clinical phenotypes after cutaneous nerve degeneration and provide neuroimaging signatures of neuropathic pain.
Supporting information
Supporting Information
REFERENCES
- Apkarian AV, Baliki MN, Geha PY (2009): Towards a theory of chronic pain. Prog Neurobiol 87:81–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apkarian AV, Hashmi JA, Baliki MN (2011): Pain and the brain: Specificity and plasticity of the brain in clinical chronic pain. Pain 152( 3 Suppl):S49–S64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apkarian AV, Sosa Y, Krauss BR, Thomas PS, Fredrickson BE, Levy RE, Harden RN, Chialvo DR (2004): Chronic pain patients are impaired on an emotional decision‐making task. Pain 108:129–136. [DOI] [PubMed] [Google Scholar]
- Baliki MN, Chialvo DR, Geha PY, Levy RM, Harden RN, Parrish TB, Apkarian AV (2006): Chronic pain and the emotional brain: Specific brain activity associated with spontaneous fluctuations of intensity of chronic back pain. J Neurosci 26:12165–12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliki MN, Geha PY, Apkarian AV (2007): Spontaneous pain and brain activity in neuropathic pain: Functional MRI and pharmacologic functional MRI studies. Curr Pain Headache Rep 11:171–177. [DOI] [PubMed] [Google Scholar]
- Baron R (2006): Mechanisms of disease: Neuropathic pain—A clinical perspective. Nat Clin Pract Neurol 2:95–106. [DOI] [PubMed] [Google Scholar]
- Battaglia AA, Sehayek K, Grist J, McMahon SB, Gavazzi I (2003): EphB receptors and ephrin‐B ligands regulate spinal sensory connectivity and modulate pain processing. Nat Neurosci 6:339–340. [DOI] [PubMed] [Google Scholar]
- Bennett MR (2011): The prefrontal‐limbic network in depression: Modulation by hypothalamus, basal ganglia and midbrain. Prog Neurobiol 93:468–487. [DOI] [PubMed] [Google Scholar]
- Bingel U, Quante M, Knab R, Bromm B, Weiller C, Büchel C (2002): Subcortical structures involved in pain processing: Evidence from single‐trial fMRI. Pain 99:313–321. [DOI] [PubMed] [Google Scholar]
- Bornhövd K, Quante M, Glauche V, Bromm B, Weiller C, Büchel C (2002): Painful stimuli evoke different stimulus‐response functions in the amygdala, prefrontal, insula and somatosensory cortex: A single‐trial fMRI study. Brain 125:1326–1336. [DOI] [PubMed] [Google Scholar]
- Borsook D, Upadhyay J, Chudler EH, Becerra L (2010): A key role of the basal ganglia in pain and analgesia—Insights gained through human functional imaging. Mol Pain 6:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostock H, Campero M, Serra J, Ochoa JL (2005): Temperature‐dependent double spikes in C‐nociceptors of neuropathic pain patients. Brain 128:2154–2163. [DOI] [PubMed] [Google Scholar]
- Bouhassira D, Attal N, Fermanian J, Alchaar H, Gautron M, Masquelier E, Rostaing S, Lanteri‐Minet M, Collin E, Grisart J, Boureau F (2004): Development and validation of the Neuropathic Pain Symptom Inventory. Pain 108:248–257. [DOI] [PubMed] [Google Scholar]
- Braz JM, Nassar MA, Wood JN, Basbaum AI (2005): Parallel “pain” pathways arise from subpopulations of primary afferent nociceptor. Neuron 47:787–793. [DOI] [PubMed] [Google Scholar]
- Brett M, Anton JL, Valabregue R, Poline JB (2002): Region of interest analysis using an SPM toolbox. Neuroimage 16 ( Suppl 1):1141–1142. [Google Scholar]
- Brooks JC, Zambreanu L, Godinez A, Craig AD, Tracey I (2005): Somatotopic organisation of the human insula to painful heat studied with high resolution functional imaging. Neuroimage 27:201–209. [DOI] [PubMed] [Google Scholar]
- Collins DL, Neelin P, Peters TM, Evans AC (1994): Automatic 3D intersubject registration of MR volumetric data in standardized Talairach space. J Comput Assist Tomogr 18:192–205. [PubMed] [Google Scholar]
- Craig AD (2003): Pain mechanisms: Labeled lines versus convergence in central processing. Annu Rev Neurosci 26:1–30. [DOI] [PubMed] [Google Scholar]
- Crombez G, Vervaet L, Lysens R, Baeyens F, Eelen P (1998): Avoidance and confrontation of painful, back‐straining movements in chronic back pain patients. Behav Modif 22:62–77. [DOI] [PubMed] [Google Scholar]
- Davis KD, Taylor KS, Hutchison WD, Dostrovsky JO, McAndrews MP, Richter EO, Lozano AM (2005): Human anterior cingulate cortex neurons encode cognitive and emotional demands. J Neurosci 25:8402–8406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies M, Brophy S, Williams R, Taylor A (2006): The prevalence, severity, and impact of painful diabetic peripheral neuropathy in type 2 diabetes. Diabetes Care 29:1518–1522. [DOI] [PubMed] [Google Scholar]
- DeLong MR, Wichmann T (2007): Circuits and circuit disorders of the basal ganglia. Arch Neurol 64:20–24. [DOI] [PubMed] [Google Scholar]
- Devigili G, Tugnoli V, Penza P, Camozzi F, Lombardi R, Melli G, Broglio L, Granieri E, Lauria G (2008): The diagnostic criteria for small fibre neuropathy: From symptoms to neuropathology. Brain 131:1912–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiBonaventura MD, Cappelleri JC, Joshi AV (2011): A longitudinal assessment of painful diabetic peripheral neuropathy on health status, productivity, and health care utilization and cost. Pain Med 12:118–126. [DOI] [PubMed] [Google Scholar]
- Downar J, Crawley AP, Mikulis DJ, Davis KD (2000): A multimodal cortical network for the detection of changes in the sensory environment. Nat Neurosci 3:277–283. [DOI] [PubMed] [Google Scholar]
- Downar J, Crawley AP, Mikulis DJ, Davis KD (2002): A cortical network sensitive to stimulus salience in a neutral behavioral context across multiple sensory modalities. J Neurophysiol 87:615–620. [DOI] [PubMed] [Google Scholar]
- Downar J, Mikulis DJ, Davis KD (2003): Neural correlates of the prolonged salience of painful stimulation. Neuroimage 20:1540–1551. [DOI] [PubMed] [Google Scholar]
- Eickhoff SB, Stephan KE, Mohlberg H, Grefkes C, Fink GR, Amunts K, Zilles K (2005): A new SPM toolbox for combining probabilistic cytoarchitectonic maps and functional imaging data. Neuroimage 25:1325–1335. [DOI] [PubMed] [Google Scholar]
- Foltz EL, White LE (1962): Pain “relief” by frontal cingulumotomy. J Neurosurg 19:89–100. [DOI] [PubMed] [Google Scholar]
- Forman SD, Cohen JD, Fitzgerald M, Eddy WF, Mintun MA, Noll DC (1995): Improved assessment of significant activation in functional magnetic resonance imaging (fMRI): Use of a cluster‐size threshold. Magn Reson Med 33:636–647. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Frith CD, Turner R, Frackowiak RS (1995a): Characterizing evoked hemodynamics with fMRI. Neuroimage 2:157–165. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Holmes AP, Worsley KJ, Poline JP, Frith CD, Frackowiak RSJ (1995b): Statistical parametric maps in functional imaging: A general linear approach. Hum Brain Mapp 2:189–210. [Google Scholar]
- Gormsen L, Rosenberg R, Bach FW, Jensen TS (2010): Depression, anxiety, health‐related quality of life and pain in patients with chronic fibromyalgia and neuropathic pain. Eur J Pain 14:127.e1–127.e8. [DOI] [PubMed] [Google Scholar]
- Grachev ID, Fredrickson BE, Apkarian AV (2000): Abnormal brain chemistry in chronic back pain: An in vivo proton magnetic resonance spectroscopy study. Pain 89:7–18. [DOI] [PubMed] [Google Scholar]
- Haber SN, Calzavara R (2009): The cortico‐basal ganglia integrative network: The role of the thalamus. Brain Res Bull 78:69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes AP, Friston KJ (1998): Generalisability, random effects and population inference. Neuroimage 7:S754. [Google Scholar]
- Iannetti GD, Zambreanu L, Wise RG, Buchanan TJ, Huggins JP, Smart TS, Vennart W, Tracey I (2005): Pharmacological modulation of pain‐related brain activity during normal and central sensitization states in humans. Proc Natl Acad Sci USA 102:18195–18200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannetti GD, Hughes NP, Lee MC, Mouraux A (2008): Determinants of laser‐evoked EEG responses: Pain perception or stimulus saliency? J Neurophysiol 100:815–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaggi AS, Singh N (2011): Role of different brain areas in peripheral nerve injury‐induced neuropathic pain. Brain Res 1381:187–201. [DOI] [PubMed] [Google Scholar]
- Jeanmonod D, Magnin M, Morel A (1996): Low‐threshold calcium spike bursts in the human thalamus. Common physiopathology for sensory, motor and limbic positive symptoms. Brain 119:363–375. [DOI] [PubMed] [Google Scholar]
- Jensen KB, Kosek E, Petzke F, Carville S, Fransson P, Marcus H, Williams SC, Choy E, Giesecke T, Mainguy Y, Gracely R, Ingvar M (2009): Evidence of dysfunctional pain inhibition in fibromyalgia reflected in rACC during provoked pain. Pain 144:95–100. [DOI] [PubMed] [Google Scholar]
- Koyama T, McHaffie JG, Laurienti PJ, Coghill RC (2005): The subjective experience of pain: Where expectations become reality. Proc Natl Acad Sci USA 102:12950–12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauria G, Hsieh ST, Johansson O, Kennedy WR, Leger JM, Mellgren SI, Nolano M, Merkies IS, Polydefkis M, Smith AG, Sommer C, Valls‐Solé J; European Federation of Neurological Societies ; Peripheral Nerve Society (2010): European Federation of Neurological Societies/Peripheral Nerve Society Guideline on the use of skin biopsy in the diagnosis of small fiber neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. Eur J Neurol 17:903–912, e44–e49. [DOI] [PubMed] [Google Scholar]
- Lee MC, Zambreanu L, Menon DK, Tracey I (2008): Identifying brain activity specifically related to the maintenance and perceptual consequence of central sensitization in humans. J Neurosci 28:11642–11649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrain V, Damme SV, Eccleston C, Davis KD, Seminowicz DA, Crombez G (2009): A neurocognitive model of attention to pain: Behavioral and neuroimaging evidence. Pain 144:230–232. [DOI] [PubMed] [Google Scholar]
- Legrain V, Iannetti GD, Plaghki L, Mouraux A (2011): The pain matrix reloaded: A salience detection system for the body. Prog Neurobiol 93:111–124. [DOI] [PubMed] [Google Scholar]
- Lorenz J, Cross DJ, Minoshima S, Morrow TJ, Paulson PE, Casey KL (2002): A unique representation of heat allodynia in the human brain. Neuron 35:383–393. [DOI] [PubMed] [Google Scholar]
- McCracken LM (1997): Attention to pain in persons with chronic pain: A behavioral approach. Behav Ther 28:271–284. [Google Scholar]
- Melzack R (1987): The short‐form McGill Pain Questionnaire. Pain 30:191–197. [DOI] [PubMed] [Google Scholar]
- Moisset X, Bouhassira D (2007): Brain imaging of neuropathic pain. Neuroimage 37 ( Suppl 1):S80–S88. [DOI] [PubMed] [Google Scholar]
- Nagy A, Eordegh G, Paroczy Z, Markus Z, Benedek G (2006): Multisensory integration in the basal ganglia. Eur J Neurosci 24:917–924. [DOI] [PubMed] [Google Scholar]
- Nolano M, Provitera V, Estraneo A, Selim MM, Caporaso G, Stancanelli A, Saltalamacchia AM, Lanzillo B, Santoro L (2008): Sensory deficit in Parkinson's disease: Evidence of a cutaneous denervation. Brain 131:1903–1911. [DOI] [PubMed] [Google Scholar]
- Ongür D, Price JL (2000): The organization of networks within the orbital and medial prefrontal cortex of rats, monkeys and humans. Cereb Cortex 10:206–219. [DOI] [PubMed] [Google Scholar]
- Petrovic P, Kalso E, Petersson KM, Ingvar M (2002): Placebo and opioid analgesia—Imaging a shared neuronal network. Science 295:1737–1740. [DOI] [PubMed] [Google Scholar]
- Peyron R, Laurent B, García‐Larrea L (2000): Functional imaging of brain responses to pain. A review and meta‐analysis. Neurophysiol Clin 30:263–288. [DOI] [PubMed] [Google Scholar]
- Phillips ML, Drevets WC, Rauch SL, Lane R (2003): Neurobiology of emotion perception II: Implications for major psychiatric disorders. Biol Psychiatry 54:515–528. [DOI] [PubMed] [Google Scholar]
- Ploner M, Gross J, Timmermann L, Schnitzler A (2002): Cortical representation of first and second pain sensation in humans. Proc Natl Acad Sci USA 99:12444–12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polydefkis M, Yiannoutsos CT, Cohen BA, Hollander H, Schifitto G, Clifford DB, Simpson DM, Katzenstein D, Shriver S, Hauer P, Brown A, Haidich AB, Moo L, McArthur JC (2002): Reduced intraepidermal nerve fiber density in HIV‐associated sensory neuropathy. Neurology 58:115–119. [DOI] [PubMed] [Google Scholar]
- Price DD (2000): Psychological and neural mechanisms of the affective dimension of pain. Science 288:1769–1772. [DOI] [PubMed] [Google Scholar]
- Price DD, Hu JW, Dubner R, Gracely RH (1977): Peripheral suppression of first pain and central summation of second pain evoked by noxious heat pulses. Pain 3:57–68. [DOI] [PubMed] [Google Scholar]
- Rainville P, Hofbauer RK, Paus T, Duncan GH, Bushnell MC, Price DD (1999): Cerebral mechanisms of hypnotic induction and suggestion. J Cogn Neurosci 11:110–125. [DOI] [PubMed] [Google Scholar]
- Saadé NE, Al Amin H, Abdel Baki S, Chalouhi S, Jabbur SJ, Atweh SF (2007): Reversible attenuation of neuropathic‐like manifestations in rats by lesions or local blocks of the intralaminar or the medial thalamic nuclei. Exp Neurol 204:205–219. [DOI] [PubMed] [Google Scholar]
- Schweinhardt P, Glynn C, Brooks J, McQuay H, Jack T, Chessell I, Bountra C, Tracey I (2006): An fMRI study of cerebral processing of brush‐evoked allodynia in neuropathic pain patients. Neuroimage 32:256–265. [DOI] [PubMed] [Google Scholar]
- Shun CT, Chang YC, Wu HP, Hsieh SC, Lin WM, Lin YH, Tai TY, Hsieh ST (2004): Skin denervation in type 2 diabetes: Correlations with diabetic duration and functional impairments. Brain 127:1593–1605. [DOI] [PubMed] [Google Scholar]
- Singer T, Seymour B, O'Doherty J, Kaube H, Dolan RJ, Frith CD (2004): Empathy for pain involves the affective but not sensory components of pain. Science 303:1157–1162. [DOI] [PubMed] [Google Scholar]
- Starr CJ, Sawaki L, Wittenberg GF, Burdette JH, Oshiro Y, Quevedo AS, McHaffie JG, Coghill RC (2011): The contribution of the putamen to sensory aspects of pain: Insights from structural connectivity and brain lesions. Brain 134:1987–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinazzi M, Del Vesco C, Defazio G, Fincati E, Smania N, Moretto G, Fiaschi A, Le Pera D, Valeriani M (2008): Abnormal processing of the nociceptive input in Parkinson's disease: A study with CO2 laser evoked potentials. Pain 136:117–124. [DOI] [PubMed] [Google Scholar]
- Tracey I (2011): Can neuroimaging studies identify pain endophenotypes in humans? Nat Rev Neurol 7:173–181. [DOI] [PubMed] [Google Scholar]
- Treede RD, Meyer RA, Raja SN, Campbell JN (1992): Peripheral and central mechanisms of cutaneous hyperalgesia. Prog Neurobiol 38:397–421. [DOI] [PubMed] [Google Scholar]
- Treede RD, Kenshalo DR, Gracely RH, Jones AK (1999): The cortical representation of pain. Pain 79:105–111. [DOI] [PubMed] [Google Scholar]
- Tseng MT, Hsieh SC, Shun CT, Lee KL, Pan CL, Lin WM, Lin YH, Yu CL, Hsieh ST (2006): Skin denervation and cutaneous vasculitis in systemic lupus erythematosus. Brain 129:977–985. [DOI] [PubMed] [Google Scholar]
- Tseng MT, Tseng WY, Chao CC, Lin HE, Hsieh ST (2010): Distinct and shared cerebral activations in processing innocuous versus noxious contact heat revealed by functional magnetic resonance imaging. Hum Brain Mapp 31:743–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verne GN, Himes NC, Robinson ME, Gopinath KS, Briggs RW, Crosson B, Price DD (2003): Central representation of visceral and cutaneous hypersensitivity in the irritable bowel syndrome. Pain 103:99–110. [DOI] [PubMed] [Google Scholar]
- Vogt BA (2005): Pain and emotion interactions in subregions of the cingulate gyrus. Nat Rev Neurosci 6:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt BA, Berger GR, Derbyshire SW (2003): Structural and functional dichotomy of human midcingulate cortex. Eur J Neurosci 18:3134–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witting N, Kupers RC, Svensson P, Jensen TS (2006): A PET activation study of brush‐evoked allodynia in patients with nerve injury pain. Pain 120:145–154. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Mannion RJ (1999): Neuropathic pain: Aetiology, symptoms, mechanisms, and management. Lancet 353:1959–1964. [DOI] [PubMed] [Google Scholar]
- Zambreanu L, Wise RG, Brooks JC, Iannetti GD, Tracey I (2005): A role for the brainstem in central sensitisation in humans. Evidence from functional magnetic resonance imaging. Pain 114:397–407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information