

Abstract
Objectives
Investigation of variable response rates to cancer immunotherapies has exposed the immunosuppressive tumor microenvironment (TME) as a limiting factor of therapeutic efficacy. A determinant of TME composition is the tumor location, and clinical data have revealed associations between certain metastatic sites and reduced responses. Preclinical models to study tissue‐specific TMEs have eliminated genetic heterogeneity, but have investigated models with limited clinical relevance.
Methods
We investigated the TMEs of tumors at clinically relevant sites of metastasis (liver and lungs) and their impact on αPD‐1/αCTLA4 and trimAb (αDR5, α4‐1BB, αCD40) therapy responses in the 67NR mouse breast cancer and Renca mouse kidney cancer models.
Results
Tumors grown in the lungs were resistant to both therapies whereas the same tumor lines growing in the mammary fat pad (MFP), liver or subcutaneously could be completely eradicated, despite greater tumor burden. Assessment of tumor cells and drug delivery in 67NR lung or MFP tumors revealed no differences and prompted investigation into the immune TME. Lung tumors had a more immunosuppressive TME with increased myeloid‐derived suppressor cell infiltration, decreased T cell infiltration and activation, and decreased NK cell activation. Depletion of various immune cell subsets indicated an equivalent role for NK cells and CD8+ T cells in lung tumour control. Thus, targeting T cells with αPD‐1/αCTLA4 or trimAb was not sufficient to elicit a robust antitumor response in lung tumors.
Conclusion
Taken together, these data demonstrate that tissue‐specific TMEs influence immunotherapy responses and highlight the importance in defining tissue‐specific response patterns in patients.
Keywords: anti‐CTLA‐4, anti‐PD‐1, tissue‐specific microenvironment, trimAb, tumor microenvironment
Emerging clinical evidence suggests that the location of tumor growth can influence the tumor microenvironment (TME) and immunotherapy responses. In the current study, we investigate the TMEs of tumors at clinically relevant sites of metastasis and their impact on αPD‐1/αCTLA4 and trimAb (αDR5, α4‐1BB, αCD40) therapy responses in murine tumor models. We show that breast tumors growing in the mammary gland were more responsive to immunotherapy and had a less immunosuppressive TME compared to when the same tumor line was growing in the lungs, demonstrating that the tissue of tumor growth is an important consideration in understanding resistance to immunotherapy.
Introduction
Immunotherapies are a highly promising avenue for treatment of certain cancer types. However, the variable response rates in sensitive cancers and insensitivity of others have prompted investigation of factors influencing poor responses.1, 2 The immunosuppressive TME contributes to reducing antitumor immune responses and consequently represents a barrier for effective immunotherapies. Established tumors are enriched in immunosuppressive leucocytes including regulatory T cells (Tregs), myeloid‐derived suppressor cells (MDSCs), tumor‐associated macrophages and a milieu of immunosuppressive cytokines which form the immunosuppressive TME.3, 4 The presence of these cells in human tumors is associated with poor prognosis,5, 6, 7 highlighting their importance in tumor progression. The immunosuppressive TME can limit response to anti‐cancer immunotherapies as evidenced by various preclinical and clinical studies. For example, higher numbers of suppressive cells can dampen responses to checkpoint blockade.8, 9 Therefore, outcomes of immunotherapies are not restricted by the tumor cells themselves, and consideration of the TME is vital for optimal immunotherapies.
An emerging determinant of TME composition is the anatomical site of tumor growth.10, 11, 12 Evidence has recently emerged for the tumor location influencing the response of human tumors to immunotherapies, particularly to αPD‐1 and αPD‐1/αCTLA4.10, 11, 12 In particular, retrospective analysis has revealed reduced responses to checkpoint blockade in tumors in certain metastatic sites compared with primary tumors in melanoma, non‐small‐cell lung cancer (NSCLC), urothelial cancer and triple‐negative breast cancer (TNBC).11, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 Due to the complexity of tumors at different anatomical sites, including tumor genetic heterogeneity, the tissue‐specific impact on therapy responses in human tumors is yet to be elucidated. Previously, we and others used genetically identical murine tumor lines to eliminate genetic heterogeneity and have shown differences in the TME composition and therapy responses of tumors growing in different anatomical sites in murine models of cancer.26, 27, 28, 29 However, these early studies compare tumors in orthotopic and subcutaneous locations or use therapies that are not clinically utilised and have limited relevance to the clinical setting.
In the current study, we utilised more clinically relevant murine models to investigate the tissue‐specific influence to immunotherapy response. We extend comparisons to tumors growing in common metastatic sites, as opposed to subcutaneous models, and investigate therapeutic responses to αPD‐1/αCTLA4 as a clinically relevant therapy in addition to trimAb therapy. We used a genetically homogenous breast cancer cell line to ensure differences observed were not as a result of genetic heterogeneity in the tumor cells. We observed that breast tumors growing in the lungs were more resistant to these immunotherapies than the same tumor cell line growing orthotopically in the mammary fat pad (MFP). We further found that resistant lung tumors were associated with a more immunosuppressive TME. This study provides further evidence for tissue‐specific TMEs having a profound influence on immunotherapy outcomes and has direct implications for understanding immunotherapy responses and resistance in patients as well as the use of appropriate mouse models.
Results
Lung tumors are more resistant to αPD‐1/αCTLA4 and trimAb immunotherapies than tumors growing elsewhere
There is growing evidence that the anatomical site of tumor growth can influence responses to immunotherapy.10, 11, 12 We evaluated responses to αPD‐1/αCTLA4 and trimAb (combination of agonistic antibodies against αDR5, α4‐1BB and αCD40)30 in breast tumors growing in mice in the MFP and common sites of breast cancer metastasis (liver and lungs). Tumors were established using the non‐metastatic31 67NR breast tumor line expressing cherry and luciferase, thus eliminating spontaneous dissemination from the injection site. Tumors in the MFP, liver (whereby tumor cells were surgically injected into the liver) and lungs (whereby tumor cells were injected intravenously and colonise the lungs as micrometastases) were treated with either trimAb or αPD‐1/αCTLA4 when MFP tumors were approximately 30‐50 mm2. In the MFP and liver models, tumors were completely eradicated in 33% of cases (αPD‐1/αCTLA4) and 83‐100% of cases (trimAb) (Figure 1a–d, g). In contrast, when tumors were growing in the lungs, neither therapy resulted in eradication of these tumors (Figure 1c and d; Supplementary figure 1). Surprisingly, this resistance to therapy occurred even though tumor burden at the commencement of treatment was significantly less in the lungs than in the MFP and liver (Figure 1e and h). Our group previously observed differential responses to trimAb when comparing Renca kidney tumor line growing SC or orthotopically.29 As previously, we found that tumors growing SC were responsive to trimAb (Figure 1f). We compared the subcutaneous tumor response to mice bearing lung tumors and found that, as with the 67NR model, mice bearing Renca lung tumors were not responsive to trimAb therapy (Figure 1f). These data indicate that tumors arising in the lungs are resistant to both αPD‐1/αCTLA4 and trimAb compared with tumors growing elsewhere and confirm emerging evidence that tumor location can impact on immunotherapy responses.
Figure 1.
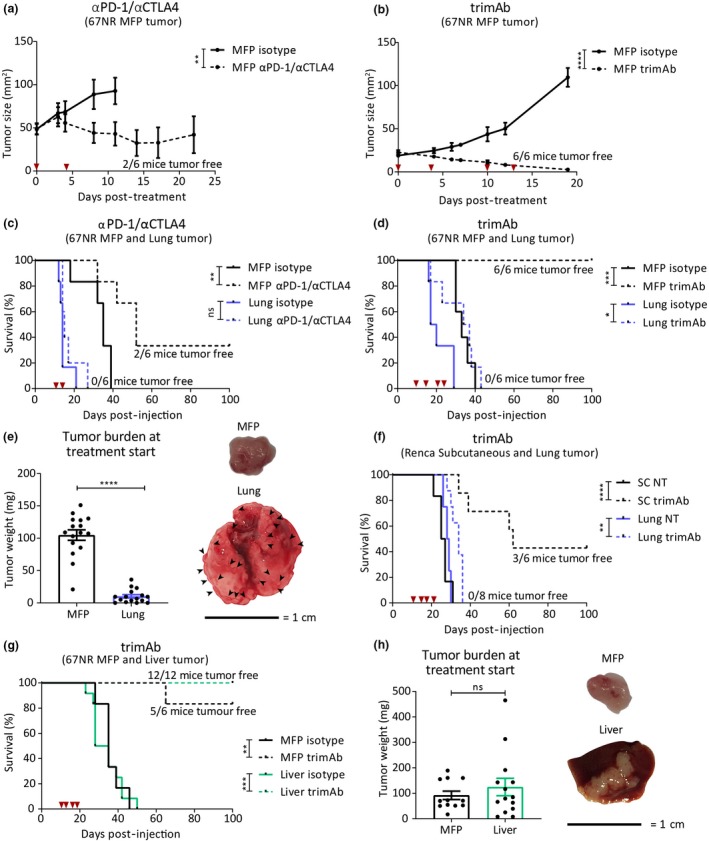
Tumors growing in the lungs have reduced response to trimAb and αPD‐1/αCTLA4 immunotherapies. BALB/c mice were injected with 4 × 105 67NR breast tumor cells in either the mammary fat pad (MFP), intravenously (IV) to produce lung micrometastases (referred to as lung) or intra‐hepatic (referred to as liver). Ten days after tumor inoculation, mice were treated with two doses of αPD‐1/αCTLA4 (200 μg/150 μg) (a and c) or four doses of trimAb (50 μg αDR5, 25 μg αCD40 and 25 μg α41BB) (b and d), or an isotype control antibody (2A3, 200 μg). Timing of dosing is indicated with red arrows. (a, b) Growth of MFP tumors treated as indicated (n = 6 mice/group, representative data of two independent experiments). Two‐way ANOVA. (c, d) Survival of MFP and lung tumor‐bearing mice treated as indicated (n = 6 mice/group, representative data of two independent experiments). Survival was determined by tumor size reaching 150 mm2 (MFP tumor‐bearing mice) or respiratory distress (lung tumor‐bearing mice). Mantel–Cox test. (e) Weight of tumors dissected from MFP or lung on day 10 post‐injection (data pooled from 3 independent experiments n = 16 mice/group) and representative images of tumors. Black arrows indicate lung tumor micrometastases. Data points from lung tumor weight represent the total tumor burden in the lung. Unpaired t‐test. (f) Survival curves for BALB/c mice injected with 5 × 105 Renca kidney tumor cells either SC or IV to produce lung tumors. When subcutaneous tumors reached ~30 mm2 in size, mice were treated with trimAb therapy as indicated (n = 6–8 mice/group). Mantel–Cox test. (g) Survival of mice with MFP and liver tumors. Survival was determined by tumor size approaching 150 mm2 (MFP mice) or bloating and signs of distress (liver mice). Mantel–Cox test. (h) Weight of tumors dissected from MFP or liver on day 10 post‐injection and representative images (data pooled from 2 independent experiments, n = 12–14). Unpaired t‐test. All data represent mean ± SEM. ns P ≥ 0.05; *P < 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
Tumor cells isolated from lung tumors do not have predisposition to therapy resistance
Although the tumor cell lines used have limited genetic heterogeneity, it was still possible that pre‐existing clones within the cell line could grow preferentially in different organs and be predisposed to reduced therapeutic response. To investigate this, we analysed expression profiles of proteins relevant to both therapies on 67NR tumor cells (Cherry+) harvested ex vivo from MFP or lung tumors. We did not find significant differences in frequency of MHCI‐, DR5 (target of trimAb)‐ or PD‐L1‐, CD80‐ and CD86 (ligands of PD‐1 or CTLA4)‐ positive tumor cells between MFP and lung tumors (Figure 2a). Although there was a significant increase in MFI of MHCI and DR5 in tumor cells growing in the lungs, this difference would be expected to enhance rather than dampen response to therapy and therefore does not explain the reduced response of lung tumors (Supplementary figure 3). There was a decrease in CD86 MFI on tumor cells in the lungs; however, this is unlikely to have a major impact on therapy responses as tumor cells in both locations expressed minimal CD86 (Figure 2a; Supplementary figure 2). Additionally, we found no expression of 4‐1BB, CTLA4 and PD‐1 and limited expression of CD40 on tumor cells isolated ex vivo from both tumor sites (Supplementary figure 2). We next performed a cross‐injection experiment where tumor cells were sorted from MFP or lung tumors by their cherry tag, cultured in vitro for 4 weeks to eliminate potential contaminating stroma and reinjected into the same or opposite site from the initial location of growth (Figure 2b). There was no difference in tumor growth, survival or therapy response when tumor cells isolated from MFP or lung were reinjected into either site (Figure 2c and d). Taken together, we did not observe any pre‐existing or induced permanent changes to the tumor cell phenotype in the MFP or lung tumors that confer resistance to αPD‐1/αCTLA4 or trimAb therapies.
Figure 2.
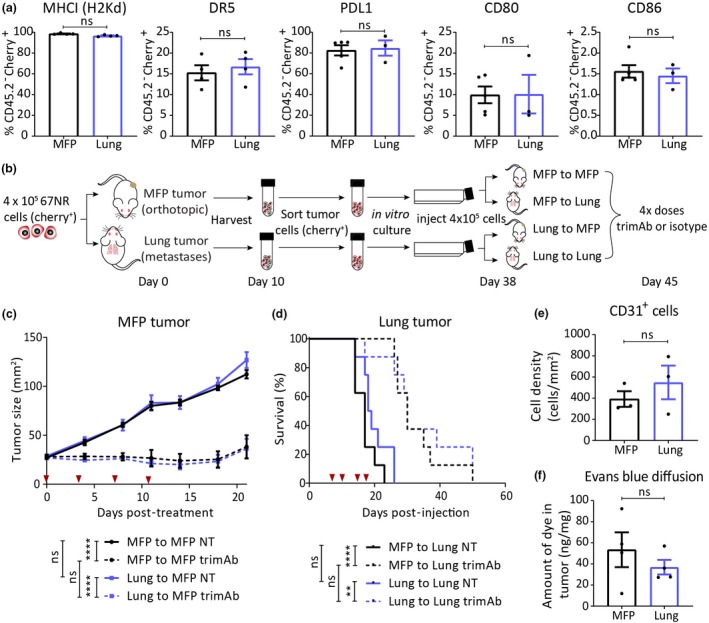
Tumor cells, vasculature or drug diffusion into mammary fat pad (MFP) or lung tumors are not influenced by anatomical site. (a) 67NR tumor cells (CD45.2−Cherry+) extracted ex vivo from either MFP or lung tumors were analysed by flow cytometry for proteins indicated 10 days after tumor inoculation (n = 3–5). Mann–Whitney U‐test. (b) Experimental design for (c, d) 67NR tumor cells from MFP or lung tumors were isolated, cultured, reinjected back into the same (MFP to MFP, lung to lung) or opposite (MFP to lung, lung to MFP) anatomical sites, and established tumors were treated with four doses of trimAb 10 days after reinjection. Doses indicated by red arrows. (c) MFP tumor growth (n = 8 mice/group). Two‐way ANOVA. (d) Survival of mice bearing lung tumors (n = 8 mice/group). Mantel–Cox test. (e) Quantification from immunohistochemistry staining of CD31 or isotype control on FFPE sections from 67NR MFP and lung tumors harvested 10 days after tumor inoculation (n = whole section from 3 tumors/group, representative of the whole tumor). Mann–Whitney U‐test. (f) Evans blue diffusion into mice bearing 67NR MFP or lung tumors that were injected with Evans blue dye 30 min prior to tumor harvest and dye extraction from whole tumor (n = 4 tumors/group). Mann–Whitney U‐test. (e, f) Experiments were performed once. All data represent mean ± SEM. ns P ≥ 0.05; **P ˂ 0.01; ****P ≤ 0.0001.
MFP and lung tumors have equal vascularisation and drug diffusion
Tumor vascularisation can impact on drug delivery32 and could potentially determine therapeutic responses in MFP and lung tumors. To investigate this possibility, we characterised vasculature using IHC staining for CD31, as a marker for endothelial cells. There was no significant difference in the extent of CD31‐positive cell staining in MFP and lung tumors (Figure 2e; Supplementary figure 3). To determine whether there were differences in drug delivery at either site, we performed an Evans blue diffusion assay as described previously.29 Briefly, dye was injected into mice bearing MFP or lung tumors and measured after overnight extraction of the dye from harvested tumors. Using this assay, we found no significant difference in diffusion of Evans blue dye to either MFP or lung tumors (Figure 2f), suggesting that the αPD‐1/αCTLA4 and trimAb antibodies were likely to have similar access to MFP and lung tumors. Given there were no significant differences in the tumor cell expression profile, genetic heterogeneity, vascularisation and drug delivery, and because antibodies used in this study primarily target immune cell populations, we hypothesised that the differential therapeutic responses are because of differences in the immune TME.
MFP and lung tumors have distinct immune gene expression profiles
Previous studies investigating preclinical models of pancreatic, renal cell and prostate cancers showed distinct RNA profiles of tumors growing in different sites.11, 26 To address this in our model, we performed 3’ RNA sequencing of tumors isolated from the MFP and lung. Principal component analysis (PCA) of differentially expressed genes showed a clear distinction between the expression profiles of MFP, lung and liver tumors (Figure 3a). To investigate whether this difference was related to different immune TMEs, we specifically analysed immune‐related genes listed in the curated NanoString immune gene panel. This revealed that tumors in MFP, lung and liver had distinct immune gene expression profiles (Figure 3b). Of the differentially expressed immune‐related genes, various genes involved in cytotoxicity and immune activation were downregulated in lung tumors compared with MFP tumors (Figure 3c). For example, there was significantly less expression in lung tumors of GZMA and GZMB encoding for granzyme A and B cytotoxic effector molecules. Furthermore, EOMES (eomesodermin), TNFRSF9 (4‐1BB/CD137) and SPP1 (secreted phosphoprotein 1/early T lymphocyte activation 1) genes, associated with type I immunity, were downregulated in lung tumors (Figure 3c). Gene set enrichment analysis (GSEA) of all differentially expressed genes revealed CD8+ T cell downstream pathway, NK cell‐mediated cytotoxicity and IL‐12/2 pathway among the top downregulated pathways in lung compared with MFP tumors (Figure 3d–f). Since resistant lung tumors have a gene expression profile associated with less cytotoxicity and reduced immune activation, this could indicate that tumors growing in the lung inherently have dampened antitumor immunity that cannot be rescued by trimAb or αPD‐1/αCTLA4 immunotherapies.
Figure 3.
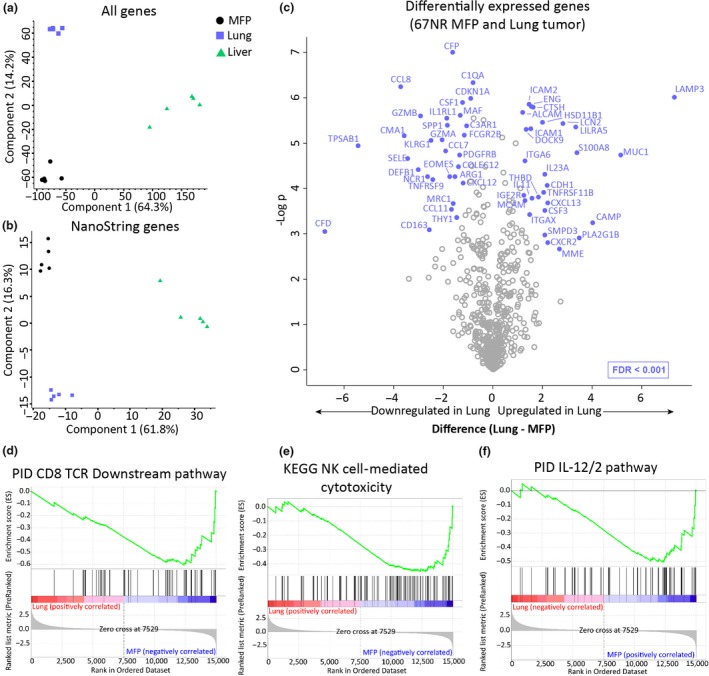
RNA profile of mammary fat pad (MFP) and lung tumors reveals distinct immune‐related differences. RNA was extracted from tumors harvested from BALB/c mice injected with 4 × 105 67NR cells in the MFP, IV (lung mice) or liver, 14 days after injection. (a, b) PCA plot showing separation of samples based on all genes (a) or immune‐related genes (NanoString immune panel gene list) (b) from bulk 3’ RNA sequencing of MFP, lung and liver tumors. (c) Volcano plot displaying significantly differentially expressed immune‐related genes between MFP and lung tumors. (d–f) GSEA plot for all differentially expressed genes in MFP and lung tumors showing enrichment for genes involved in PID CD8 TCR downstream pathway (d), KEGG NK cell‐mediated cytotoxicity (e) and PID IL‐12/2 pathway (f). n = 5 tumors/group.
Immune infiltrate differs between MFP and lung tumors and is differentially changed upon therapy
Given that the immune TME can have a significant impact on immunotherapeutic responses, we characterised the immune cell infiltrates present in the MFP and lungs. We used multiparameter flow cytometry to profile tumors for various lymphoid and myeloid cell markers. For mice with lung tumors, the whole lung was harvested due to practical issues with dissecting micrometastases. To control for this, we harvested MFP and lungs from tumor‐free (naïve) mice in addition to tumors. As expected, there were distinct differences in immune cell populations when comparing naïve lung and MFP tissues (Figures 4a and 5a; Supplementary figures 4 and 5). Predominantly, T cells made up a larger portion of CD45.2+ cells in the MFP than in lungs, which had more NK cells. Potentially, these differences in immune cell populations prior to tumor establishment could impact the established TME of MFP and lung tumors, along with recruitment and polarisation of various other leucocytes through secretion of a host of chemokines by the tumor.
Figure 4.
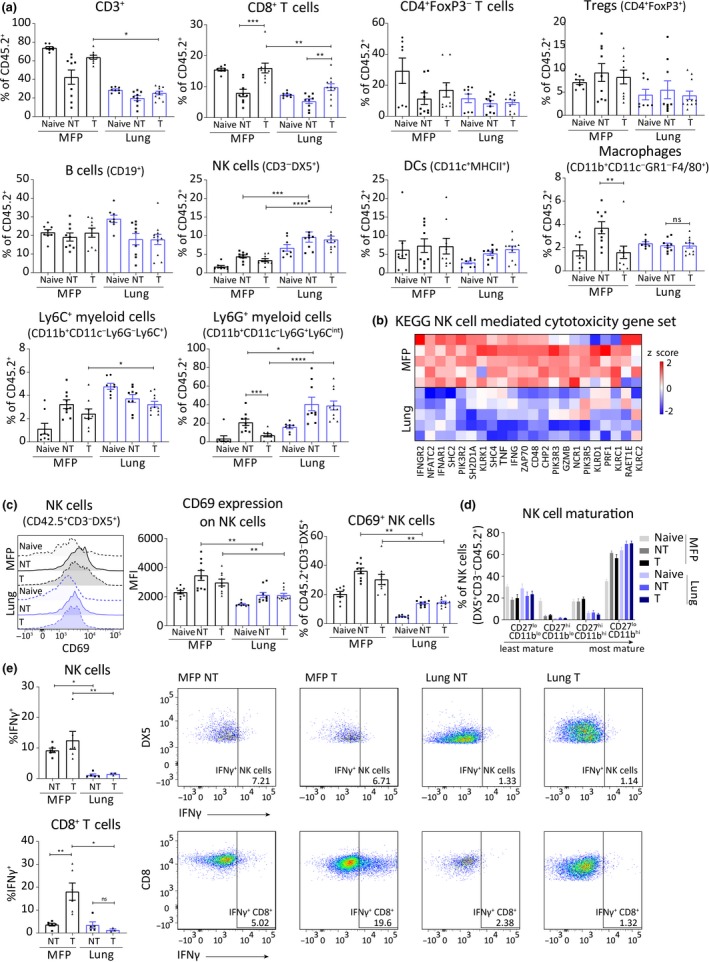
Mammary fat pad (MFP) and lung tumors have distinct TMEs that are altered differentially by αPD‐1/αCTLA4 treatment. BALB/c mice bearing MFP or lung tumors (4 × 105 67NR cells in MFP or IV) were treated with αPD‐1/αCTLA4 and harvested 7 days post‐treatment initiation. (a) Flow cytometry of immune cell populations indicated as a percentage of live CD45.2+ cells within MFP or lung tumors treated with isotype antibody (NT) or αPD‐1/αCTLA4 (T), or organs without tumor (naïve). Pooled data from 2 independent experiments, n = 8–11/group. Mann–Whitney U‐test. (b) Heatmap of gene expression in MFP and lung tumors of genes in KEGG NK cell‐mediated cytotoxicity pathway. RNA was extracted from tumors harvested from BALB/c mice injected with 4 × 105 67NR cells in the MFP or IV (lung mice), 14 days after injection. n = 5 tumors/group. (c) Expression of CD69 on live CD45.2+CD3−DX5+ cells by flow cytometry. Mann–Whitney U‐test. (d) Maturation of NK cells (live CD45.2+CD3−DX5+) assessed by expression of CD11b and CD27 by flow cytometry. (e) Expression of IFNγ by NK cells and CD8+ T cells stimulated ex vivo by flow cytometry. ns P ≥ 0.05 *P < p0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
Figure 5.
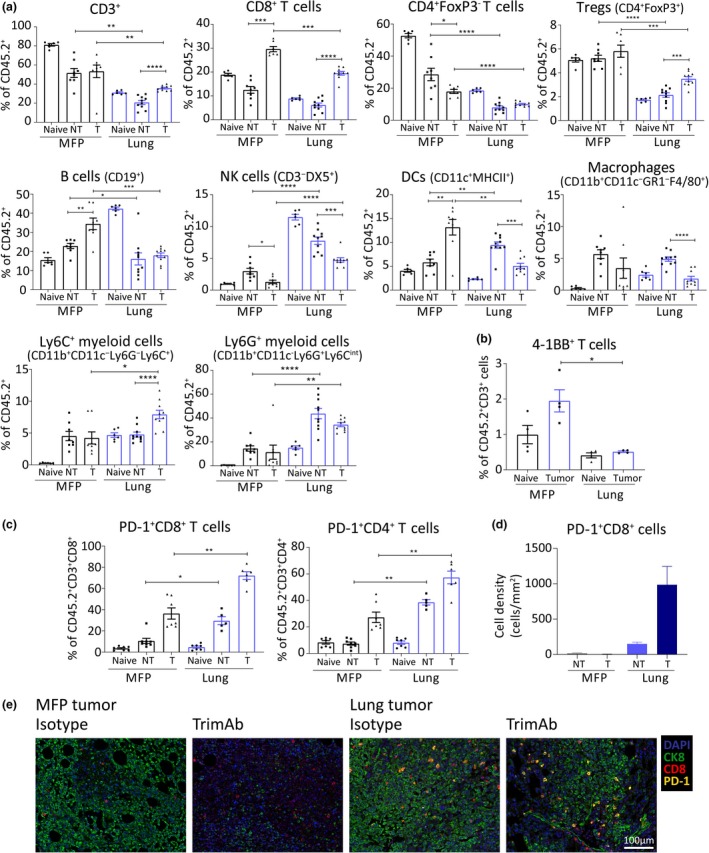
Mammary fat pad (MFP) and lung tumors have distinct TMEs that are altered differentially by trimAb treatment. BALB/c mice bearing MFP or lung tumors (4 × 105 67NR cells in MFP or IV) were treated with trimAb and harvested 7 days post‐treatment initiation. (a) Flow cytometry of immune cell populations indicated as a percentage of live CD45.2+ cells within MFP or lung tumors treated with 2A3 isotype antibody (NT) or trimAb (T), or organs without tumor (naïve). Data are representative from 1 of 3 independent experiments, n = 5‐10/group. Mann–Whitney U‐test. (b) Expression of 4‐1BB on live CD45.2+CD3+ cells by flow cytometry. Mann–Whitney U‐test. (c) Expression of PD‐1 on live CD45.2+CD3+, CD8+ or CD4+ cells by flow cytometry. Mann–Whitney U‐test. (d, e) Representative images and quantitation of multiplexing IHC showing PD‐1‐positive CD8+ cells in MFP and lung tumors, n = 2 or 3 whole tumor section/group. CK8, cytokeratin 8. *P < p0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
Indeed, we found that there was a decrease in total T cell population in lung tumors of mice treated with αPD‐1/αCTLA4 compared with treated MFP tumors (Figure 4a; Supplementary figure 4). The decrease in frequency of T cells was primarily because of a decrease in CD8+ T cell frequencies (Figure 4a). Treatment with αPD‐1/αCTLA4 significantly increased CD8+ T cell frequency in both tumor locations; however, this increase was more pronounced in the MFP tumors (Figure 4a). Regardless of treatment, lung tumors had greater frequencies and numbers of NK cells than MFP tumors, consistent with the lungs being an NK cell‐rich environment (Figures 4a and 5a; Supplementary figures 4 and 5). Despite increased infiltration of NK cells in lung tumors, there was a significant decrease in expression of genes related to NK cell cytotoxicity (Figures 3e and 4b). Many of these genes could be expressed by CD8+ T cells, which are decreased in lung tumors; therefore, we directly assessed NK cell activation. Compared with MFP tumors, NK cells within lung tumors expressed less CD69 (Figure 4c) with no changes in maturation status (Figure 4d), indicating that lung tumor‐infiltrating NK cells are less activated. To assess functionality within the MFP and lung TMEs, we stimulated NK cells ex vivo and stained for IFNγ. While NK cells produced limited IFNγ, a significantly higher percentage of NK cells from MFP tumors were IFNγ+ than those isolated from lung tumors (Figure 4e). Notably, αPD‐1/αCTLA4 therapy had no impact on NK cell activation or IFNγ production in either tumor model (Figure 4c and e). In contrast, CD8+ T cells isolated from αPD‐1/αCTLA4 MFP tumors produced significantly more IFNγ than non‐treated MFP tumors and treated lung tumors (Figure 4e). Thus, αPD‐1/αCTLA4 treatment was insufficient to enhance CD8+ T‐cell function in lung tumors. Treatment with αPD‐1/αCTLA4 promoted a decrease in macrophages and CD11b+CD11c−Ly6CintLy6G+ myeloid population in MFP tumors, but no change in lung tumors (Figure 4a). The CD11b+CD11c−Ly6G+/Ly6C+ myeloid populations were of particular interest as this population can describe MDSCs33; however, functional validation is needed to confirm this. The Ly6CintLy6G+ subset were increased in both non‐treated and treated lung tumors compared with MFP tumors. Given that the lung tumors were less responsive to both therapies and had reduced activation and IFNγ production by effector cells, these potentially suppressive cells could be impacting on the ability of cytotoxic cells within these tumors to respond to therapy.
We next performed comparable analysis on MFP and lung tumors treated with trimAb therapy. Similar to αPD‐1‐/αCTLA4‐treated mice, lung tumors had reduced frequency of T cells compared with MFP tumors (CD8+ and CD4+, both FoxP3+ and FoxP3−) (Figure 5a). In contrast to αPD‐1/αCTLA4, trimAb therapy resulted in decreased NK cells in both MFP and lung tumors (Figure 5a; Supplementary figure 5). In lung tumors, there was a reduced frequency of dendritic cells (DCs) and macrophages upon trimAb treatment, but an increase in DCs and no change in macrophages in MFP tumors (Figure 5a; Supplementary figure 5). The Ly6C+ subset were increased in both frequency and number in the treated lung tumors compared with non‐treated lung tumors and treated MFP tumors (Figure 5a; Supplementary figure 5). As seen previously with αPD‐1/αCTLA4, there was an increase in the Ly6CintLy6G+ myeloid population in lung tumors compared to MFP tumors, with or without trimAb treatment (Figure 5a; Supplementary figure 5).
The RNA profiles indicated a decrease in activation of lung tumor‐infiltrating T cells compared with MFP tumors (Figure 3c and d). Our FACS analysis revealed that the frequency of 4‐1BB+ T cells in lung tumors was decreased (Figure 5b), confirming the decreased expression of TNFRSF9 (4‐1BB) transcript observed (Figure 3c). This indicates that lung tumor‐infiltrating T cells are less activated and, as 4‐1BB is a target of trimAb, suggests that 4‐1BB agonism is less effective on T cells in lung tumors compared with MFP tumors. We also observed a dramatic increase in PD‐1 expression on both CD8+ and CD4+ T cells in lung tumors compared with MFP tumors by flow cytometry and multiplexing IHC (Figure 5c–e). Given the reduced response to therapy and the reduced production of IFNγ, this could be indicative of an exhaustion phenotype, as opposed to activation, where the T cells in lung tumors are unable to perform cytotoxic functions.
Taken together, it seems the immune infiltrate in resistant lung tumors is less geared towards tumor killing and less conducive to immunotherapy. Resistant tumors had more potential suppressor cells, reduced CD8+ T cell infiltration, NK cell activation and IFNγ production by CD8+ T cells and NK cells.
MFP and lung tumors rely on different immune cell populations for tumor control and therapeutic response
To further define how the observed differences in the immune TME contributed to therapy responses and tumor control in MFP and lung tumors, we depleted various immune cell subsets at the time of therapeutic intervention. Depletion of target populations was confirmed by flow cytometry (Supplementary figure 6). These experiments indicated that CD8+ T cells were important in all conditions tested as depletion of CD8+ T cells abrogated any therapeutic effect regardless of therapy or site of tumor growth (Figure 6a, b, e and f). Despite this similarity, there were differences in immune cell populations important in tumor control and therapy response between MFP and lung tumors. CD4+ T cell and NK cell depletion in MFP tumors removed some, but not all, of the therapeutic benefit from αPD‐1/αCTLA4 as demonstrated by the intermediate tumor growth (Figure 6a). This indicates that CD8+ T cells are the predominant cell type involved in response to αPD‐1/αCTLA4 in MFP tumors. Depletion of CD4+ T cells had no effect on the survival of mice with lung tumors (Figure 6c). Although the therapeutic effect of αPD‐1/αCTLA4 in mice with lung tumors was minimal, depleting NK cells in mice with lung tumors that received αPD‐1/αCTLA4 resulted in significantly decreased survival compared to mice treated with αPD‐1/αCTLA4 alone (Figure 6d). This contrasts to the impact of NK cell depletion on αPD‐1/αCTLA4 therapy in MFP tumors which incompletely abrogated the therapeutic effect. Furthermore, the involvement of NK cells in lung tumors is consistent with multiple tumor models where NK cells are vital in the control of lung metastases.34 There was no significant impact on αF4/80 or αLy6G to αPD‐1/αCTLA4 therapy in MFP or lung tumors (Supplementary figure 7a–c).
Figure 6.
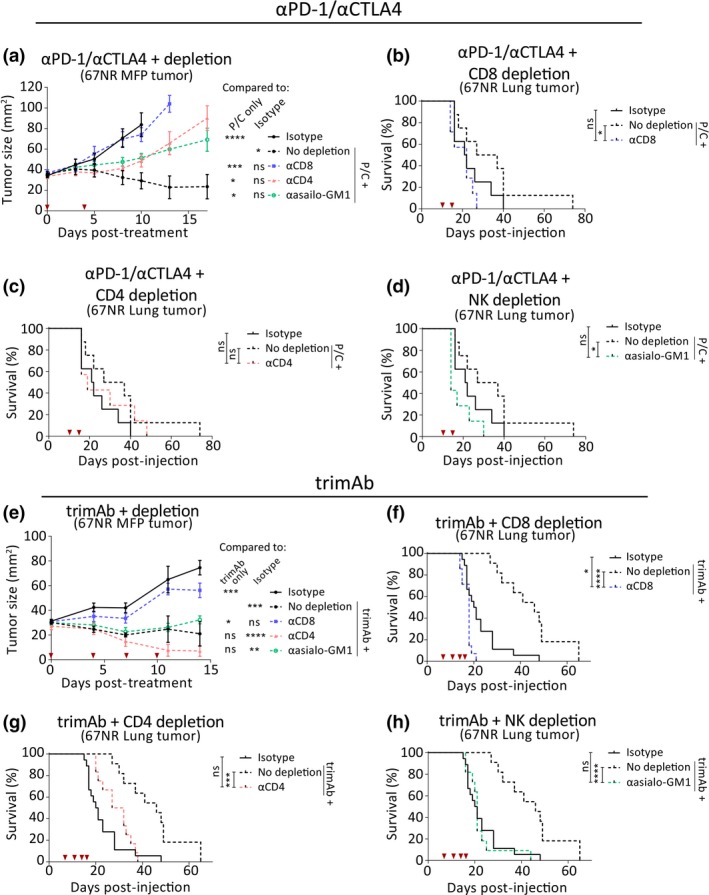
Mammary fat pad (MFP) and lung tumors depend on different immune subsets for therapy response and tumor control. BALB/c mice bearing MFP or lung tumors (4 × 105 67NR cells in MFP or IV) were treated with αPD‐1/αCTLA4 or trimAb alone or with depleting antibodies against CD8, CD4 or asialo‐GM1 (NK cell depletion). (a) Growth of MFP tumors either non‐treated (isotype), treated with αPD‐1/αCTLA4 only (no depletion) or αPD‐1/αCTLA4 and depletion antibodies. Two‐way ANOVA. Data points represent mean ± SEM. n = 6–8 mice/group. (b–d) Survival of all lung tumor‐bearing mice either non‐treated (isotype), treated with αPD‐1/αCTLA4 only (no depletion) or αPD‐1/αCTLA4 and depletion antibodies against CD8 (b), CD4 (c) or asialo‐GM1 (d). n = 6–8 mice/group. Mantel–Cox test. (e) Growth of MFP tumors either non‐treated (isotype), treated with trimAb only (no depletion) or trimAb and depletion antibodies. Two‐way ANOVA. Data points represent mean ± SEM. n = 5 or 6 mice/group, representative of 2 independent experiments. (f–h) Survival of all lung tumor‐bearing mice either non‐treated (isotype), treated with trimAb only (no depletion) or trimAb and depletion antibodies against CD8 (f), CD4 (g) or asialo‐GM1 (h) n = 8–11 mice/group, pooled data from 2 independent experiments. Mantel–Cox test. ns P ≥ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
In the context of trimAb therapy, addition of antibodies against CD4+ T cells, NK cells (αasialo‐GM1), macrophages (αF4/80) and Ly6C/Ly6G+ cells (αGR1) had no significant effect on MFP tumor growth (Figure 6e; Supplementary figure 7d). Thus, the therapeutic effect of trimAb on MFP tumors was entirely dependent on CD8+ T cells. In contrast, antibodies against CD4, asailo‐GM1 (NK cells) and GR1 significantly reduced the survival of lung tumor‐bearing mice treated with trimAb (Figure 6g and h; Supplementary figure 7f). The effect of GR1 depletion is likely because of Ly6C expression on CD8+ T cells, which resulted in significant off‐target depletion using this antibody (Supplementary figure 6). As in MFP tumors, macrophage depletion had no significant effect on trimAb efficacy in lung tumors (Supplementary figure 7e).
These experiments demonstrate that distinct immune cell populations are involved in tumor control and therapy responses between tumors growing in different sites. Specifically, we found that NK cell depletion had a more significant impact in metastatic lung tumors than tumors growing in the MFP, which were predominantly controlled by CD8+ T cells.
Discussion
Despite emerging evidence that the tissue of tumor growth impacts on therapy responses,11 our understanding of this influence is not fully defined. Here, we used a model of breast cancer to show that tumors growing in the lungs were less sensitive to two different immunotherapies compared with the same tumor growing in the MFP. Tumors in these locations had disparate immune microenvironments, and this was hypothesised to influence the differential responses observed.
We found multifactorial changes in the TME depending on the organ of tumor growth, and these changes might explain the differences in the therapeutic response observed between MFP and lung tumors (Figure 7). Notably, lung tumors were resistant to therapy and had a more immunosuppressive TME with decreased frequencies and activation of CD8+ T cells and increased MDSCs compared with MFP tumors, which were responsive to therapy. NK cells were found to be more abundant in the lung tumor infiltrate; however, these NK cells were less activated and produced less IFNγ indicating a potential mechanism of NK cell suppression existing in this setting. Despite the decreased activation of NK cells at the timepoint analysed, NK cell depletion had an equivalent impact to CD8+ T cell depletion on the survival of mice bearing lung tumors, which was reduced compared with treated mice. In contrast, NK cell depletion in the MFP did not completely abrogate the therapeutic response to either therapy as observed with CD8+ T cell depletion. Importantly, neither therapy enhanced NK cell activation at the timepoint analysed, consistent with αPD‐1/αCTLA4 and trimAb primarily targeting T cell antitumor responses. Thus, αPD‐1/αCTLA4 and trimAb may have been less effective in lung tumors because targeting T cell responses alone is not sufficient to elicit an effective antitumor response. Conversely, tumors growing in the MFP had a higher T cell infiltrate, were predominantly dependent on T cells for therapeutic responses and could be eradicated by both αPD‐1/αCTLA4 and trimAb treatments.
Figure 7.
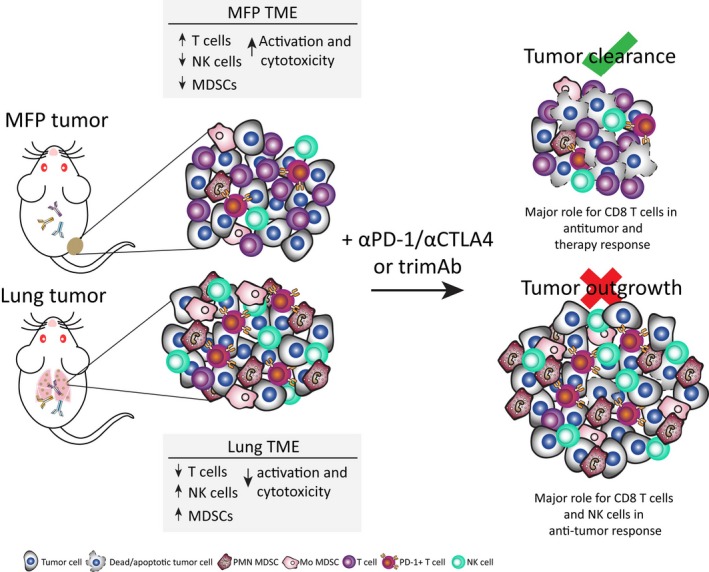
Mammary fat pad (MFP) and lung tumors have distinct TMEs and differential responses to trimAb and αPD‐1/αCTLA4 immunotherapies. Summary schematic of results from the current study of BALB/c mice with established 67NR tumors growing in either the MFP or the lungs. Tumors had distinct TMEs with increased T cells, decreased NK cells and MDSCs and increased activation of T cells and NK cells in MFP tumors compared with lung tumors. Furthermore, depletion of various immune cell subsets revealed a greater role for NK cells in the antitumor immune response in tumors growing in the lungs compared with the MFP. Therefore, upon treatment with trimAb or αPD‐1/αCTLA4 immunotherapies, which primarily target T‐cell antitumor immune responses, MFP tumors were responsive and could be eradicated whereas lung tumors were resistant to therapy and outgrew.
NK cells have been shown to play an important role in controlling lung metastasis in multiple tumor models.34 A study of lung metastasis using the 4T1 parental line evaluated enhancing NK cell activation with IL‐12, which led to induction of PD‐1 expression on NK cells.35 These NK cells were functionally exhausted, but could be rescued by αPD‐1/αCTLA4 therapy. It is possible in our model that the decreased activation of NK cells inhibits induction of PD‐1 expression, and therefore, the infiltrating NK cells do not benefit from αPD‐1/αCTLA4. Future studies are required to investigate whether additional agents targeting NK cells could mount a more durable response in resistant lung tumors. Multiple approaches to block inhibitory signals and enhance activatory signals on NK cells have been developed.36, 37, 38 Cytokines such as IL‐2, IL‐12, IL‐15, IL‐18 and IL‐21 stimulate and increase expression of activating receptors on NK cells.39, 40, 41 Therapeutic antibodies such as α‐killer‐cell immunoglobulin‐like receptors (αKIR) and αNKG2A can remove inhibitory signals on NK cells.42, 43, 44 In fact, lirilumab (an αKIR) has demonstrated a satisfactory safety profile45 and is currently undergoing clinical trial in combination with αPD‐1/αCTLA4 in advanced solid tumors.46 Expression of these activating and inhibitory receptors on NK cells was not investigated in our model; however, using agents to enhance NK cell activation through these receptors could improve trimAb or αPD‐1/αCTLA4 in resistant lung tumors.
Given the immunosuppressive effects of MDSCs, multiple strategies are under investigation to reverse their influence in the TME.47, 48 Agents such as all‐trans retinoic acid (ATRA)49 to differentiate MDSCs to mature myeloid cells, humanised IgG4 mAb targeting Semaphorin 4D.50, 51 to inhibit MDSC expansion and gemcitabine52, 53 to deplete MDSCs have demonstrated antitumor activity in preclinical studies and are currently being tested in clinical trials for synergistic effects with checkpoint inhibitors. Additionally, MDSCs have inhibitory functions against NK cells and their depletion is likely to improve NK cell function.54, 55 Combining trimAb or αPD‐1/αCTLA4 with these strategies to target MDSCs in our model could also improve therapeutic responses in resistant lung tumors.
Despite the characterisation of immune cell subsets within the TME of MFP and lung tumors, we did not investigate the cytokine and chemokine profiles of these tumors in detail. It is likely that cytokine and chemokine expression differences between lung and MFP tumors are involved in mediating immunosuppression and recruitment of immunosuppressive cells to the TME.56, 57 The RNA profiles highlighted differences in some chemokines; for example, inflammatory cytokines CXCL12, CSF1, CCL11, CCL7 and CCL8 were downregulated in lung tumors compared with MFP tumors. The use of 3’ RNA sequencing may underrepresent low abundance or unstable transcripts. Thus, in order to fully understand the cytokine and chemokine profiles of these tumors, other assays, such as CBA or ELISA, could be employed to complement the RNAseq data.
Our study aimed to further investigate the clinical findings that tumors in different anatomical sites have disparate TMEs and responses to immunotherapy. Although the individual agents in trimAb have been tested clinically, the combination of all three has not. In contrast, the variable response rates to checkpoint blockade have prompted retrospective investigation of factors correlating with poor response.2 Multiple studies across various cancer types have found differences between the site of metastatic growth and response rates to αPD‐1 alone or in combination with αCTLA4. It seems that tissue‐specific responses to immunotherapy may differ by cancer type and specific checkpoint therapy used. In our study of the 67NR breast cancer cell line, tumors growing in the lungs were only slightly responsive to trimAb immunotherapy and not responsive to αPD‐1/αCTLA4 whereas tumors growing in the MFP could be eradicated by these therapies. In non‐small‐cell lung cancer (NSCLC), response to nivolumab (αPD‐1) was reduced in patients with secondary lung metastasis in one study,21 and no change in other studies.17, 23 Complete response rates of lung metastases in melanoma are increased to single agent αPD‐1 than at other metastatic sites in some cases,13, 14, 15, 16 but not others,17, 18 and are not associated with responses to αPD‐1/CTLA4.18 There is no observed association of lung metastases with response to pembrolizumab (αPD‐1) in triple‐negative breast cancer (TNBC) patients.20 Unfortunately, limited clinical studies investigating dual αPD‐1/αCTLA4 responses restrict the ability to draw conclusions from our model using αPD‐1/αCTLA4. However, with increasing use, site‐specific responses to αPD‐1/αCTLA4 are likely to be revealed.
A recently revealed clinical trend shows some association with liver metastases and reduced responses to αPD‐1 across cancer types in melanoma,16, 17, 18 urothelial cancer,19 TNBC20 and NSCLC.17, 21, 22, 24 However, there are some conflicting results with some studies reporting no associations with liver metastases.13, 14, 15, 17, 23 Furthermore, data indicating a poor response in patients with liver tumors could be confounded with the aggressive nature of tumors that metastasise to the liver. Some evidence indicates that patients with liver metastases have more metastases in other sites indicating a potentially more advanced stage in the cohort of patients with liver metastases.24, 58
While the injection of genetically identical tumor lines enabled the specific influence of the tissue of tumor growth to be assessed in our mouse models, it should be acknowledged that differences to the human setting need to be considered when drawing parallels to the clinic. In human cancers, the genetics of tumors in distal sites compared with the primary location, and even within the primary tumor, can be highly heterogeneous. Furthermore, the establishment of tumors in the human setting is a much slower process than in our mouse models. Lastly, a point of difference between the published clinical data and murine studies is the pre‐treatment of patients with various chemotherapies that likely results in an impaired immune system which is not reflected in immunocompetent mice used in our study.
Ideally, detailed clinical data outlining responses of each individual metastasis in patients treated with immunotherapy and biopsies of responding and non‐responding tumors at different sites would be instrumental in further understanding of the tissue‐specific TME. These findings could be taken back to preclinical models in order to improve their relevance and assess new strategies in non‐responding settings. For example, our findings that tumors arising in the lungs are more resistant to T‐cell‐targeted immunotherapies as a result of decreased T‐cell infiltration and activation could instruct more effective immunotherapies in NK cell‐dominant TMEs by combining existing immunotherapies with NK cell‐activating agents.
Methods
Cell lines and mice
BALB/c mouse breast carcinoma cell line 67NR was provided by Professor Robin Anderson (Olivia Newton John Cancer Centre, Victoria, Australia). BALB/c mouse kidney adenocarcinoma cell line Renca ch luc line (referred to as Renca) stably transduced with the cherry and luciferase reporter genes was previously generated, and the 67NR ch luc cell line (referred to as 67NR) was generated using the same method.29 All tumor lines were cultured at 37 °C, 10% CO2 in DMEM media supplemented with 10% heat‐inactivated FCS, 2 mm glutamine, 1 mm sodium pyruvate, 0.1 mm nonessential amino acids, 10 mm 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. All lines tested negative for mycoplasma (Peter MacCallum Cancer Centre Genotyping core facility). BALB/c mice were purchased from the Walter and Eliza Hall Institute for Medical Research (Victoria, Australia). Mice were used between 6 and 20 weeks of age, and all work was approved by the Peter MacCallum Cancer Centre Animal Experimentation Ethics Committee.
Mouse experiments
For MFP tumors, mice were injected into the fourth right MFP with 4 × 105 67NR cells in 20 μL PBS. For subcutaneous tumors, 5 × 105 Renca cells were injected subcutaneously in 100 μL PBS. Lung tumors were established by intravenous (IV) injection into the tail vein of 67NR (4 × 105 cells) or Renca (5 × 105 cells) in 200 μL PBS. Liver tumors were established by intra‐hepatic injection of 4 × 105 67NR cells in 5 μL PBS into the left lateral lobe. For MFP and subcutaneous tumors, growth was monitored using digital callipers and survival was defined by tumors reaching the ethical size limit of 150 mm2. For lung and liver tumors, survival was defined as overt signs of stress, confirmed by animal technicians, and autopsied at end point to confirm tumor growth in target organs. For cross‐injection experiments, mice were injected with 4 × 105 67NR cells into the MFP or IV and MFP or lungs were harvested under sterile conditions after 14 days. Tissues were digested in a mixture of 3 mg mL−1 collagenase type IV (Worthington, Lakewood, NJ, USA) and 0.02 mg mL−1 DNase (Sigma‐Aldrich, St Louis, MO, USA) for 40 min at 37 °C. Samples were sorted for Cherry+ cells using BD Fusion 5 sorter. Samples were cultured for 4 weeks before reinjecting back into the MFP or IV with 4 × 105 cells.
Therapeutic and depletion antibodies
All therapeutic anti‐mouse antibodies were injected intra‐peritoneally in 200 μL PBS. Each dose of trimAb consisted of a mixture of 50 μg αDR5 (clone MD5.1), 25 μg αCD40 (clone FGK4.5) and 25 μg α4‐1BB (clone 3H3). One dose of αPD‐1/αCTLA4 consisted 200 μg αPD‐1 (clone RMPI‐14) and 150 μg αCTLA4 (clone 9H10). Mice were dosed every 3‐4 days with a total of 4 doses for trimAb and 2 doses for αPD‐1/αCTLA4. All depletion antibodies were injected at day −1 and 0 (treatment start date) and either weekly for αCD8 (clone YTS 169.4) and αCD4 (clone GK1.5) or twice weekly for αasialo‐GM1 (WAKO, Osaka, Japan), αGR1 (clone RB6‐8C5), αLy6G (clone 1A8) and αF4/80 (clone CI:A3‐1) for up to 4 weeks after treatment commencement. Depletion antibodies dosed weekly were used at 200 μg per dose and twice weekly at 100 μg per dose with the exception of αasialo‐GM1 which was used at 10 μL per mouse as recommended by the manufacturer. All antibodies for in vivo experiments (except αasialo‐GM1) were purchased from BioXcell (Lebanon, NH, USA).
Immunohistochemistry
Tumors were harvested and fixed overnight in 10% neutral buffered formalin (NBF). Fixed samples were embedded in paraffin, and 4 μm sections were cut. Samples were dewaxed in xylene and antigens retrieved at 125 °C for 3 min in 10 mm citrate buffer (Sigma‐Aldrich) pH 6. Endogenous peroxidases were inactivated with 3% H2O2. CD31 staining was performed using DAB IHC. Primary CD31 antibody (abcam, ab28364, 1:1000) was incubated overnight at 4 °C, secondary ImmPress antibody (Vector Laboratories) for 1 h at room temperature, followed by DAB peroxidase (HRP) substrate kit for 6 min. For multiplexing experiments, Perkin Elmer OPAL reagents were used as per kit instructions. The following order was used: CD8 (ebioscience 4SM15, 5 μg mL−1) 570, PD‐1 (abcam EPR20665, 0.8 μg mL−1) 620, CK8 (abcam EP1628Y, 0.2 μg mL−1) 520 and DAPI (Perkin Elmer). DAB IHC was visualised on the Olympus BX51 and VS120 slide scanner and quantified using HALO v2.3 (Indica Labs). Multiplexing IHC was visualised using the Perkin Elmer Vectra 3 microscope and analysed using InForm v2.4.0 (Perkin Elmer) and HALO v2.3.
Evans blue diffusion dye
The Evans blue diffusion dye was described previously.29 Briefly, tumor‐bearing mice were injected IV with 200 μL Evans blue dye (Sigma‐Aldrich) (0.2%; 30 mg kg−1 in PBS) or PBS alone. After 30 min, mice were culled, lungs perfused, tumors excised, washed in PBS, gently blotted and weighed. Dye was extracted from tumors overnight in dimethylformamide (Merck, Melbourne, Australia). Extracted dye was measured at 620 nm (Cytation 3). Quantity of dye was determined by standard curve.
RNA sequencing and analysis
For RNA sequencing, tumors were microdissected and snap frozen. RNA was extracted using RNeasy mini kit (Qiagen, Hilden, Germany). Genome‐wide transcriptomic analyses were performed using 5 µL of DNAse‐treated input RNA for library generation by means of a QuantSeq 3’ mRNA‐Seq library kit (Lexogen, Vienna, Austria) according to the manufacturer’s instructions. NGS sequencing was performed on a NextSeq HO 75SE run at the Molecular Genomics Facility, Peter MacCallum Cancer Centre. The raw FASTQ sequenced samples were trimmed and aligned using TopHat v2.1.1 and quantified using Htseq v0.6.1 in R. In order to identify transcripts with increased or decreased expression, normalisation and differential expression analysis was performed with Limma‐Voom in R v3.3.3.59, 60, 61 In total, 15 040 genes were identified and quantified. Full RNAseq was subsetted based on the NanoString nCounter Pan‐cancer Immune profiling panel of 770 genes, containing 587 genes that overlapped. Principal component analyses (PCA) and heatmaps were generated using the Perseus computational platform62 using normalised log2 counts‐per‐million (CPM) values for relevant transcripts. Two‐sided t‐tests were also performed in Perseus with a false‐discovery rate (FDR) threshold of 0.001, 250 randomisations and S0 = 0.1. Heatmaps of canonical pathways were generated using Perseus using full dataset from MSigDB. Gene set enrichment analysis (GSEA) was performed on full normalised RNAseq data using the GSEA software package and public MSigDB datasets. Parameters for analysis include the following: normalisation meandiv, max probe mode, signal to noise metric, gene set permutation, minimum gene set size of 15, and 100 permutations. Molecular signature datasets used included the C2 curated gene set containing 4762 gene sets in version v6.2. We acknowledge our use of the gene set enrichment analysis, GSEA software, and Molecular Signature Database (MSigDB, Broad Institute, Waltham, MA, USA).63
Analysis of immune cell subsets/ex vivo tumor cells by flow cytometry
Tissues were harvested and digested using a mixture of collagenase described above. After 30‐ to 45‐min digestion at 37 °C, cells were filtered twice (70 μm) and erythrocytes were lysed with ACK (ammonium–chloride–potassium) buffer. For detection of IFNγ production by T cells and NK cells ex vivo, TILs were stimulated with PMA/ionomycin in the presence of GolgiPlug (BD Pharmingen, Franklin Lakes, NJ, USA) and GolgiStop (BD Pharmingen) as described previously.64 Samples were incubated with fluorescently conjugated antibodies in 2.4G2 Fc blocking antibody. Fixable yellow (Invitrogen) was used to determine viable cells. For intracellular staining, cells were fixed/permeabilised using the eBioscience kit (cat. no. 88‐8824‐00). The following antibodies were used: CD45.2 APC Cy7 (eBioscience 104), CD3 BV605 (Biolegend 17A2), CD8 BUV737 (BD 53‐6.7), CD8 BV711 (Biolegend, 53‐6.7), CD4 BUV805 (BD GK1.5), CD4 BV785 (Biolegend GK1.5), FoxP3 e450 (eBioscience FJK16S), PD‐1 BUV395 (BD J43), 4‐1BB PE (BD 1AH2), CD69 PE Cy7 (eBioscience H1.2F3), CD19 BV785 (BD 6D5), CD49b FITC (BD DX5), CD11b BV711 (Biolegend M1/70), Ly6G BV605 (Biolegend, 1A8), Ly6C PE Cy7 (Biolegend HK1.4), F4/80 BV421 (BD T45‐2342), MHCII APC (eBioscience M5/114.1.5.2), CD11c BV785 (Biolegend N418), PD‐L1 BUV395 (BD M1H5), CD80 Biotin (BD 16‐10A1), CD86 Biotin (eBioscience GL1), DR5 Biotin (eBioscience MD5‐1), MHCI Biotin (BD H2Kd), IFNγ APC (Biolegend XMG1.2), CTLA4 APC (BD UC10‐4F10‐11), CD40 PE (BD 3/23) and Streptavidin BUV805 (BD) or e450 (eBioscience). Samples were run on LSRFortessa or Symphony cytometers (Becton Dickinson, Franklin Lakes, NJ, USA) and analysed using FlowJo software V10 (Ashland, Oregon, USA).
Statistical analysis
Statistical analyses in this study for all experiments (except RNAseq) were performed using GraphPad Prism (version 7). Specific types of statistical tests are outlined in the figure captions. Data are presented as mean ± SEM.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Acknowledgments
This work was supported by grants from Cancer Australia (1100199), the Peter MacCallum Cancer Center Foundation, the National Health and Medical Research Council (NHMRC) of Australia (program grant number 1132373), the National Breast Cancer Foundation (NBCF) of Australia (IIRS‐18‐064) and Susan G Komen Breast Cancer Foundation (16376637). AJO was supported by an Australian Postgraduate Award. PKD and MHK were supported by NHMRC Senior Research Fellowships. CYS and PAB were supported by a postdoctoral fellowship from the NBCF. SPK was supported by a Cancer Council Victoria Grant‐in‐aid. JRMVA is a Research fellow of the Research Foundation Flanders (1S32316N).
References
- 1. Sharma P, Hu‐Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017; 168: 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pitt JM, Vetizou M, Daillere R et al Resistance mechanisms to immune‐checkpoint blockade in cancer: tumor‐intrinsic and ‐extrinsic factors. Immunity 2016; 44: 1255–1269. [DOI] [PubMed] [Google Scholar]
- 3. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012; 21: 309–322. [DOI] [PubMed] [Google Scholar]
- 4. Binnewies M, Roberts EW, Kersten K et al Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 2018; 24: 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shou J, Zhang Z, Lai Y, Chen Z, Huang J. Worse outcome in breast cancer with higher tumor‐infiltrating FOXP3+ Tregs : a systematic review and meta‐analysis. BMC Cancer 2016; 16: 687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lan C, Huang X, Lin S et al Expression of M2‐polarized macrophages is associated with poor prognosis for advanced epithelial ovarian cancer. Technol Cancer Res Treat 2013; 12: 259–267. [DOI] [PubMed] [Google Scholar]
- 7. Weide B, Martens A, Zelba H et al Myeloid‐derived suppressor cells predict survival of patients with advanced melanoma: comparison with regulatory T cells and NY‐ESO‐1‐ or melan‐A‐specific T cells. Clin Cancer Res 2014; 20: 1601–1609. [DOI] [PubMed] [Google Scholar]
- 8. Meyer C, Cagnon L, Costa‐Nunes CM et al Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother 2014; 63: 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM‐CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest 2006; 116: 1935–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pao W, Ooi CH, Birzele F et al Tissue‐specific immunoregulation: a call for better understanding of the "Immunostat" in the context of cancer. Cancer Discov 2018; 8: 395–402. [DOI] [PubMed] [Google Scholar]
- 11. Oliver AJ, Lau PKH, Unsworth AS et al Tissue‐dependent tumor microenvironments and their impact on immunotherapy responses. Front Immunol 2018; 9: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Salmon H, Remark R, Gnjatic S, Merad M. Host tissue determinants of tumour immunity. Nat Rev Cancer 2019; 19: 215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Khoja L, Kibiro M, Metser U et al Patterns of response to anti‐PD‐1 treatment: an exploratory comparison of four radiological response criteria and associations with overall survival in metastatic melanoma patients. Br J Cancer 2016; 115: 1186–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lyle MK, Lee JH, Menzies AM et al Lesion‐specific patterns of response and progression with anti‐PD‐1 treatment in metastatic melanoma (MM). J Clin Oncol 2014; 32: 9077–9077. [DOI] [PubMed] [Google Scholar]
- 15. Lee JHJ, Lyle M, Menzies AM et al Metastasis‐specific patterns of response and progression with anti‐PD‐1 treatment in metastatic melanoma. Pigment Cell Melanoma Res 2018; 31: 404–410. [DOI] [PubMed] [Google Scholar]
- 16. Nosrati A, Tsai KK, Goldinger SM et al Evaluation of clinicopathological factors in PD‐1 response: derivation and validation of a prediction scale for response to PD‐1 monotherapy. Br J Cancer 2017; 116: 1141–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tumeh PC, Hellmann MD, Hamid O et al Liver metastasis and treatment outcome with anti‐PD‐1 monoclonal antibody in patients with melanoma and NSCLC. Cancer Immunol Res 2017; 5: 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Davis EJ, Perez MC, Ayoubi N et al Clinical correlates of response to anti‐PD‐1‐based therapy in patients with metastatic melanoma. J Immunother 2019; 42: 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zheng Y, Narwal R, Jin C et al Population modeling of tumor kinetics and overall survival to identify prognostic and predictive biomarkers of efficacy for durvalumab in patients with urothelial carcinoma. Clin Pharmacol Ther 2018; 103: 643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Loi S, Adams S, Schmid P et al LBA13Relationship between tumor infiltrating lymphocyte (TIL) levels and response to pembrolizumab (pembro) in metastatic triple‐negative breast cancer (mTNBC): results from KEYNOTE‐086. Ann Oncol 2017; 28: v605–v649. [Google Scholar]
- 21. Tamiya M, Tamiya A, Inoue T et al Metastatic site as a predictor of nivolumab efficacy in patients with advanced non‐small cell lung cancer: a retrospective multicenter trial. PLoS One 2018; 13: e0192227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Funazo T, Nomizo T, Kim YH. Liver metastasis is associated with poor progression‐free survival in patients with non‐small cell lung cancer treated with nivolumab. J Thorac Oncol 2017; 12: e140–e141. [DOI] [PubMed] [Google Scholar]
- 23. Garde‐Noguera J, Martin‐Martorell P, De Julian M et al Predictive and prognostic clinical and pathological factors of nivolumab efficacy in non‐small‐cell lung cancer patients. Clin Transl Oncol 2018; 20: 1072–1079. [DOI] [PubMed] [Google Scholar]
- 24. Shiroyama T, Suzuki H, Tamiya M et al Clinical characteristics of liver metastasis in nivolumab‐treated patients with non‐small cell lung cancer. Anticancer Res 2018; 38: 4723–4729. [DOI] [PubMed] [Google Scholar]
- 25. Dudnik E, Moskovitz M, Daher S et al Effectiveness and safety of nivolumab in advanced non‐small cell lung cancer: the real‐life data. Lung Cancer 2018; 126: 217–223. [DOI] [PubMed] [Google Scholar]
- 26. Nakamura T, Fidler IJ, Coombes KR. Gene expression profile of metastatic human pancreatic cancer cells depends on the organ microenvironment. Cancer Res 2007; 67: 139–148. [DOI] [PubMed] [Google Scholar]
- 27. Hensel JA, Khattar V, Ashton R, Lee C, Siegal GP, Ponnazhagan S. Location of tumor affects local and distant immune cell type and number. Immun Inflamm Dis 2017; 5: 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhan B, Wen S, Lu J et al Identification and causes of metabonomic difference between orthotopic and subcutaneous xenograft of pancreatic cancer. Oncotarget 2017; 8: 61264–61281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Devaud C, Westwood JA, John LB et al Tissues in different anatomical sites can sculpt and vary the tumor microenvironment to affect responses to therapy. Mol Ther 2014; 22: 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Uno T, Takeda K, Kojima Y et al Eradication of established tumors in mice by a combination antibody‐based therapy. Nat Med 2006; 12: 693–698. [DOI] [PubMed] [Google Scholar]
- 31. Eckhardt BL, Parker BS, van Laar RK et al Genomic analysis of a spontaneous model of breast cancer metastasis to bone reveals a role for the extracellular matrix. Mol Cancer Res 2005; 3: 1–13. [PubMed] [Google Scholar]
- 32. Azzi S, Hebda JK, Gavard J. Vascular permeability and drug delivery in cancers. Front Oncol 2013; 3: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peranzoni E, Zilio S, Marigo I et al Myeloid‐derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol 2010; 22: 238–244. [DOI] [PubMed] [Google Scholar]
- 34. Krasnova Y, Putz EM, Smyth MJ, Souza‐Fonseca‐Guimaraes F. Bench to bedside: NK cells and control of metastasis. Clin Immunol 2017; 177: 50–59. [DOI] [PubMed] [Google Scholar]
- 35. Ohs I, Ducimetiere L, Marinho J, Kulig P, Becher B, Tugues S. Restoration of natural killer cell antimetastatic activity by IL12 and checkpoint blockade. Cancer Res 2017; 77: 7059–7071. [DOI] [PubMed] [Google Scholar]
- 36. Li Y, Sun R. Tumor immunotherapy: new aspects of natural killer cells. Chin J Cancer Res 2018; 30: 173–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol 2016; 17: 1025–1036. [DOI] [PubMed] [Google Scholar]
- 38. Childs RW, Carlsten M. Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: the force awakens. Nat Rev Drug Discov 2015; 14: 487–498. [DOI] [PubMed] [Google Scholar]
- 39. Leclercq G, Debacker V, de Smedt M, Plum J. Differential effects of interleukin‐15 and interleukin‐2 on differentiation of bipotential T/natural killer progenitor cells. J Exp Med 1996; 184: 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kobayashi M, Fitz L, Ryan M et al Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med 1989; 170: 827–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Strengell M, Matikainen S, Siren J et al IL‐21 in synergy with IL‐15 or IL‐18 enhances IFN‐γ production in human NK and T cells. J Immunol 2003; 170: 5464–5469. [DOI] [PubMed] [Google Scholar]
- 42. Romagne F, Andre P, Spee P et al Preclinical characterization of 1–7F9, a novel human anti‐KIR receptor therapeutic antibody that augments natural killer‐mediated killing of tumor cells. Blood 2009; 114: 2667–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. da Silva IP, Gallois A, Jimenez‐Baranda S et al Reversal of NK‐cell exhaustion in advanced melanoma by Tim‐3 blockade. Cancer Immunol Res 2014; 2: 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nguyen S, Beziat V, Dhedin N et al HLA‐E upregulation on IFN‐γ‐activated AML blasts impairs CD94/NKG2A‐dependent NK cytolysis after haplo‐mismatched hematopoietic SCT. Bone Marrow Transplant 2009; 43: 693–699. [DOI] [PubMed] [Google Scholar]
- 45. Vey N, Karlin L, Sadot‐Lebouvier S et al A phase 1 study of lirilumab (antibody against killer immunoglobulin‐like receptor antibody KIR2D; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget 2018; 9: 17675–17688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. A safety study of lirilumab in combination with nivolumab or in combination with nivolumab and ipilimumab in advanced and/or metastatic solid tumors. Available from: https://ClinicalTrials.gov/show/NCT03203876.
- 47. Groth C, Hu X, Weber R et al Immunosuppression mediated by myeloid‐derived suppressor cells (MDSCs) during tumour progression. Br J Cancer 2019; 120: 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu Y, Wei G, Cheng WA et al Targeting myeloid‐derived suppressor cells for cancer immunotherapy. Cancer Immunol Immunother 2018; 67: 1181–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ipilimumab and all‐trans retinoic acid combination treatment of advanced melanoma. Available from: https://ClinicalTrials.gov/show/NCT02403778
- 50. Anti‐SEMA4D monoclonal antibody VX15/2503 with nivolumab or ipilimumab in treating patients with stage III or IV melanoma. Available from: https://ClinicalTrials.gov/show/NCT03425461.
- 51. VX15/2503 and immunotherapy in resectable pancreatic and colorectal cancer. Available from: https://ClinicalTrials.gov/show/NCT03373188.
- 52. Depletion of myeloid derived suppressor cells to enhance anti PD‐1 therapy. Available from: https://ClinicalTrials.gov/show/NCT03302247.
- 53. Combination therapy for patients with untreated metastatic pancreatic ductal adenocarcinoma. Available from: https://ClinicalTrials.gov/show/NCT02754726.
- 54. Hoechst B, Voigtlaender T, Ormandy L et al Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009; 50: 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor‐derived prostaglandin‐e2 blocks the induction of myeloid‐derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res 2014; 20: 4096–4106. [DOI] [PubMed] [Google Scholar]
- 56. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol 2017; 17: 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res 2014; 2014: 149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ren Y, Dai C, Zheng H et al Prognostic effect of liver metastasis in lung cancer patients with distant metastasis. Oncotarget 2016; 7: 53245–53253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high‐throughput sequencing data. Bioinformatics 2015; 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Langmead B, Salzberg SL. Fast gapped‐read alignment with Bowtie 2. Nat Methods 2012; 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ritchie ME, Phipson B, Wu D et al limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 2015; 43: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tyanova S, Temu T, Sinitcyn P et al The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 2016; 13: 731–740. [DOI] [PubMed] [Google Scholar]
- 63. Subramanian A, Tamayo P, Mootha VK et al Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 2005; 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Beavis PA, Henderson MA, Giuffrida L et al Dual PD‐1 and CTLA‐4 checkpoint blockade promotes antitumor immune responses through CD4+Foxp3- cell‐mediated modulation of CD103+ dendritic cells. Cancer Immunol Res 2018; 6: 1069–1081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials