

Abstract
Neurogenic stuttering is an acquired speech disorder characterized by the occurrence of stuttering‐like dysfluencies following brain damage. Because the onset of stuttering in these patients is associated with brain lesions, this condition provides a unique opportunity to study the neural processes underlying speech dysfluencies. Lesion localizations of 20 stroke subjects with neurogenic stuttering and 17 control subjects were compared using voxel‐based lesion symptom mapping. The results showed nine left‐hemisphere areas associated with the presence of neurogenic stuttering. These areas were largely overlapping with the cortico‐basal ganglia‐cortical network comprising the inferior frontal cortex, superior temporal cortex, intraparietal cortex, basal ganglia, and their white matter interconnections through the superior longitudinal fasciculus and internal capsule. These results indicated that stroke‐induced neurogenic stuttering is not associated with neural dysfunction in one specific brain area but can occur following one or more lesion throughout the cortico‐basal ganglia‐cortical network. It is suggested that the onset of neurogenic stuttering in stroke subjects results from a disintegration of neural functions necessary for fluent speech. Hum Brain Mapp 34:2103–2112, 2013. © 2012 Wiley Periodicals, Inc.
Keywords: acquired stuttering, adult stuttering, cerebrovascular accident, brain mapping
INTRODUCTION
Neurogenic stuttering is an acquired speech disorder “characterized by repetition, prolongation or blocking on sounds or syllables in a manner that interrupts the normal rhythm and flow of speech” [Duffy, 2005]. It has recently been shown that neurogenic stuttering affects ∼5% of subjects following stroke [Theys et al., 2011], but it can also occur following traumatic brain injury, neurodegenerative diseases, and other disorders resulting in brain damage [De Nil et al., 2009]. This acquired speech fluency disorder most frequently occurs in adults and is often associated with other deficiencies of speech and language. In a prospective study of stroke subjects with neurogenic stuttering, more than half of the subjects presented with comorbid aphasia and dysarthria. In addition, a minority of the subjects with stroke‐induced neurogenic stuttering were diagnosed with apraxia of speech [Theys et al., 2011].
The manifestation of stuttering following brain lesions provides a unique opportunity to study the neural processes underlying speech dysfluencies. Still, our understanding of the neural correlates of neurogenic stuttering is limited, due primarily to the shortage of systematic lesion analysis studies of patients and controls. To our knowledge, only one such study reported retrospectively on 10 patients with stuttering following traumatic brain injury [Ludlow et al., 1987]. Although this study provided valuable information, the interpretation of the data was limited by the use of templates based on rough outlines of anatomical structures to perform the lesion analysis [Ludlow et al., 1987]. In addition, results obtained on patients with penetrating head injuries cannot be extrapolated to neurogenic stuttering resulting from other etiologies because the characteristics of neurogenic stuttering are known to be etiology‐specific [De Nil et al., 2009; Helm et al., 1980]. Therefore, systematic brain imaging studies in different etiological subgroups of neurogenic stuttering are necessary to further our knowledge on the neural characteristics of neurogenic stuttering.
In this study, the focus was on subjects who experienced a stroke because they represent the largest etiologic subgroup of neurogenic stuttering [Theys et al., 2008]. Furthermore, stuttering in these subjects often has its onset shortly following the stroke [Theys et al., 2008], and generally, the lesions are less diffuse compared with other etiologies. To date, over 50 case studies have been published of subjects with stroke‐induced stuttering. Neurogenic stuttering has been reported following left hemisphere lesions in the frontal lobe [Rosenbek et al., 1978], temporal lobe [Osawa et al., 2006; Rosenbek et al., 1978], parietal lobe [Sahin et al., 2005; Turgut et al., 2002], occipital lobe [Grant et al., 1999], as well as in the basal ganglia [Carluer et al., 2000; Ciabarra et al., 2000; Tani and Sakai, 2011] and thalamus [Abe et al., 1993; Van Borsel and Taillieu, 2001; Van Borsel et al., 2003]. Although less frequently, right hemisphere lesions have also been associated with neurogenic stuttering following stroke, including the temporal [Ardilla and Lopez, 1986] and frontal lobes [Balasubramanian et al., 2003], the basal ganglia [Tani and Sakai, 2011] and the thalamus [Abe et al., 1993]. In addition, cases have been reported with involvement of the supplementary motor area [Ackermann et al., 1996; Van Borsel et al., 1998], corpus callosum [Hamano et al., 2005; Kakishita et al., 2004], cerebellum [Grant et al., 1999; Tani and Sakai, 2010], and brainstem [Abe et al., 1993; Balasubramanian et al., 2003]. These case‐based observations clearly represent a bewildering array of possible lesion sites reported to be associated with the onset of neurogenic stuttering following stroke. Furthermore, the interpretation of these findings is complicated by the possible bias of case studies toward reporting on subjects that represent an atypical case of neurogenic stuttering [e.g., Turgut et al., 2002]. Therefore, case studies may not provide a typical picture of neurogenic stuttering. In addition, case studies all too often have consisted of relatively subjective retrospective descriptions of either the stuttering characteristics or the lesion localization, making it difficult to reach solid inferences on lesion localization in neurogenic stuttering subjects.
For the reasons outlined above, a systematic prospective study on the neural correlates of stroke‐related neurogenic stuttering using brain imaging would be a critical step toward advancing our knowledge of the pathophysiology of stuttering‐like dysfluencies in these subjects. This study was designed to identify brain regions associated with neurogenic stuttering following stroke on a voxel‐by‐voxel basis to provide greater understanding of the neural correlates of stroke‐related neurogenic stuttering and contribute to the development of much needed guidelines for both diagnostic and therapeutic strategies within this subject population.
METHODS
Subjects
Thirty‐seven right‐handed stroke subjects for whom structural brain imaging was available were included in the study. Thirty‐one subjects were recruited as part of a larger prevalence study of neurogenic stuttering following stroke [Theys et al., 2011]. Six additional subjects were referred to the primary author by speech‐language therapists working in clinical settings. All subjects were either identified by a speech pathologist or self‐identified with dysfluencies in their speech following their stroke. Following the referral, each subject received an in‐depth assessment using a battery of speech, language, and cognitive tests to evaluate the presence of stuttering and identify any co‐occurring communication and cognitive deficits. The test battery included tests for aphasia [ScreeLing, Visch‐Brink et al., 2010], anomia [Boston Naming Test, Kaplan et al., 2001], dysarthria [checklist by Dharmaperwira‐Prins, 1996], apraxia of speech [Apraxia Battery for Adults‐2, Dabul, 2000; checklist by Wambaugh et al., 2006], and cognitive problems [Mini Mental State Examination, Folstein et al., 1975; Coloured Progressive Matrices, Raven et al., 1995]. Also, as part of the test battery, speech samples were obtained during spontaneous speech and reading. Analysis of spontaneous speech was based on 300‐word samples collected during both conversation and monologue. To acquire these samples, subjects were prompted to speak about their medical condition, speech and language problems, family, and daily activities. For the collection of speech samples during reading, subjects were instructed to read a phonetically balanced Flemish text [De Kapitein (The Captain), Boey, 2000]. All speech tasks were videotaped with a Panasonic NV‐GS180 camera with external microphone (Sennheiser MKE2‐60Gold‐C) and transcriptions of the speech samples were made based on these video recordings. Subjects were diagnosed with neurogenic stuttering if they had more than 3% stuttering‐like dysfluencies (repetitions of sounds, syllables and monosyllabic words, prolongations, or blocks) during one or more of the three speech tasks [Bloodstein and Bernstein‐Ratner, 2008; Conture, 2000; for further details, see Theys et al., 2011]. Based on these criteria, 20 of the 37 stroke subjects were diagnosed with neurogenic stuttering. The remaining 17 subjects did not meet the 3% stuttering dysfluencies criterion for the diagnosis of neurogenic stuttering and served as the comparison group for the lesion‐symptom analysis (Table 1). All dysfluency analyses were conducted by the first author (CT) and interrater reliability was assessed by independent rescoring of 25% of the conversation samples by a trained second observer. This second observer was a graduate student in speech‐language pathology trained to score stuttering‐like dysfluencies on video recordings of patients with neurological disorders not included in this study. This rater knew that all subjects had a stroke and that some of them might present with neurogenic stuttering, but she had no knowledge of which patients belonged to the neurogenic stuttering or control group. She did not have prior knowledge about any other medical diagnosis besides the stroke nor of any diagnosis of speech‐language problems. The measures of total percentage stuttered syllables were highly correlated (r s = 0.84, P (2‐tailed) = 0.002) between the two raters.
Table 1.
Characteristics of the stroke subjects with neurogenic stuttering and control subjects
| Neurogenic stuttering subjects | Control subjects | |
|---|---|---|
| Number of subjects | 20 | 17 |
| Median age (range) [years] | 72 (45–87) | 69 (50–83) |
| M/F | 13/7 | 11/6 |
| Etiology [N (%)] | 18 (90%) infarcts, 2 (10%) hemorrhages | 15 (88%) infarcts, 2 (12%) hemorrhages |
| Median number of lesioned voxels (range) | 3831 (219–43311) | 11139 (186–29246) |
| Recruited in prevalence study [N (%)] | 16 (80%) | 15 (88%) |
| Subjects with co‐occurring disorders [N (%)] | ||
| Aphasiaa | 14 (70%) | 10 of 15 (67%) |
| Anomia | 13 (65%) | 9 (53%) |
| Dysarthria | 9 (45%) | 4 (24%) |
| Apraxia of speech | 5 (25%) | 2 (12%) |
| Cognitive problemsa | 6 (30%) | 4 of 15 (27%) |
| Median stuttering frequency (range) [%] | ||
| Conversation | 4.5 (1.8–19.4) | 1.5 (0.3–2.6) |
| Monologue | 4.4 (1.8–10.9) | 1.0 (0–2.7) |
| Reading of text | 3.0 (0–13) | 0.5 (0–2.3) |
Test results could not be obtained for two control subjects.
Differences between the neurogenic stuttering and control group were statistically analyzed using a two‐sided nonparametric Mann‐Whitney U‐test. The presence of co‐occurring aphasia, anomia, dysarthria, apraxia of speech, and cognitive problems, as well as group differences concerning gender, stroke etiology, and manner of recruitment, were analyzed using a Fisher Exact test. All statistical analyses were performed with SPSS 16.0.2. The study was completed in accordance with the Declaration of Helsinki and approved by the ethical committee of the University Hospitals Leuven. The nature of the experimental procedures was explained to the subjects, and all subjects or their guardians provided written informed consent.
Brain Imaging and Lesion Analysis
Lesion analysis was performed based on the available clinical MRI (N = 29) or CT‐scans (N = 8) of the subjects. Twenty‐six of the scans (13 in each group) were done within 1 week post stroke‐onset, and all but one of the other scans were performed within 1 month following stroke (Supporting Information Table 1). For those subjects for whom a MRI scan was available, lesion outlines were drawn manually, slice by slice, on the subject's digital poststroke Fluid Attenuation Inversion Recovery (FLAIR) image using MRIcroN [Rorden et al., 2007]. Drawing of lesions was performed by the first author (CT) and reviewed by an experienced neuroradiologist (SS), blind to the subject's behavioral characteristics. By means of the presence of restricted diffusion on the Apparent Diffusion Coefficient map of the Diffusion Weighted scan (DWI) a differentiation could be made between recent lesions and older hyperintensities in the FLAIR image. Only lesions associated with the most recent stroke were included in the analysis. Both the brain scan and the delineated lesion were normalized to a standard brain template (Montreal Neurological Institute's template, MNI, http://www.bic.mni.mcgill.ca/) using affine and nonrigid transformations as described by Friston et al. 2007. Next, lesions were binarized in SPM5 [Friston et al., 2007] implemented in MATLAB. For the six neurogenic stuttering and two control subjects with CT‐scans, the brain lesions were drawn manually on a MNI‐template by the same experienced neuroradiologist (SS) and the first author (CT). The medical records for each of these eight patients indicated that the stroke associated with neurogenic stuttering was their first stroke.
Using FSL 3.1.8 [Smith et al., 2004], we added the normalized binary lesion maps for the groups with and without neurogenic stuttering, creating voxel‐wise lesion prevalence maps (Fig. 1). Next, the lesion maps and behavioral data of the 37 subjects were analyzed using voxel‐based Bayesian Lesion Symptom Mapping [vBLSM, Chen and Herskovits, 2010]. vBLSM is a technique that allows the investigation of structure‐function relationships. It uses a Bayesian approach to lesion symptom mapping which explicitly models the lesion‐proportion differences between two subject groups. Because vBLSM does not involve approximation procedures, it does not require a correction for multiple comparisons [Chen and Herskovits, 2010]. For a detailed description of the Bayesian approach, see Chen and Herskovits 2010.
Figure 1.
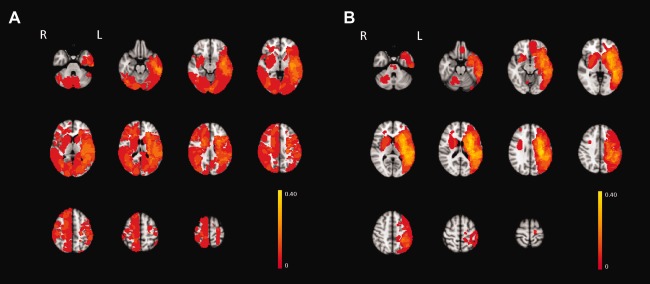
Lesion overlay of the 17 control subjects (A) and 20 neurogenic stuttering subjects (B)
As part of the vBLSM analysis, the subjects' lesion maps were first grouped by the clinical variable of interest, i.e., presence or absence of neurogenic stuttering. Next, for each voxel, the probability (Pr) that the proportion of lesions in the group with neurogenic stuttering ( A) was larger than the proportion of lesions in the control group (
B) was calculated. This calculation of d(δ) = Pr((
A −
B) ≥ δ) was performed using a C++ based analysis program described in Chen and Herskovits 2010. In this analysis, the threshold for proportion differences (δ) was set at 0 because it concerned a first exploratory study, and we were interested in voxels where the proportion of lesions was larger in the neurogenic stuttering group compared with the control group. The resulting probability‐map was visualized in MRIcroN, and the threshold for inclusion of the voxels was set at d(0) ≥ 0.95. This resulted in a map of voxels with 95% or more probability of larger lesion proportions in the neurogenic stuttering group compared with the control group.
A) was larger than the proportion of lesions in the control group (
B) was calculated. This calculation of d(δ) = Pr((
A −
B) ≥ δ) was performed using a C++ based analysis program described in Chen and Herskovits 2010. In this analysis, the threshold for proportion differences (δ) was set at 0 because it concerned a first exploratory study, and we were interested in voxels where the proportion of lesions was larger in the neurogenic stuttering group compared with the control group. The resulting probability‐map was visualized in MRIcroN, and the threshold for inclusion of the voxels was set at d(0) ≥ 0.95. This resulted in a map of voxels with 95% or more probability of larger lesion proportions in the neurogenic stuttering group compared with the control group.
As an additional analysis, we calculated the Spearman correlations between stuttering frequency during conversation, monologue and reading of a text, and the extent of the lesion, measured as number of lesioned voxels and number of lesioned areas identified with the vBLSM analysis. Also, in each of these areas one‐sided Mann‐Whitney U‐tests were performed to test whether the frequency of stuttering was significantly higher in the subjects with lesions to that particular brain area. As the diagnosis of neurogenic stuttering was based on the speech task eliciting the highest frequency of stuttering in a specific patient, the calculation of the Mann‐Whitney test was based on this measure of dysfluency.
RESULTS
Group Characteristics
Analysis of the demographic characteristics revealed no significant differences between the neurogenic stuttering and the control group with regard to age (Table 1, U = 147, P = 0.50) and gender (P = 1.0, Fisher's Exact Test). In both groups, the majority of the subjects suffered from an ischemic stroke, while only two subjects in each group had a hemorrhagic stroke (P = 1.0, Fisher's Exact Test). There was an equal likelihood in both subject groups that they were recruited externally (P = 0.67, Fisher's Exact Test). In addition, the two groups did not differ significantly with respect to the presence of aphasia (P = 1.0, Fisher's Exact Test), anomia (P = 0.52, Fisher's Exact Test), dysarthria (P = 0.30, Fisher's Exact Test), apraxia of speech (P = 0.42, Fisher's Exact Test), or cognitive problems (P = 1.0, Fisher's Exact Test, see also Supporting Information Table 2). The only significant difference between the neurogenic stuttering and the control group was related to the grouping variable, the frequency of stuttering during conversation (Table 1, U = 13, P < 0.001), monologue (U = 16, P < 0.001), and reading (U = 45, P = 0.001).
Lesion Distribution
The voxel‐wise lesion prevalence maps (Fig. 1) showed that lesions in both subject groups were present in the left and right hemispheres as well as the cerebellum. Although the lesions were more widely distributed in the control group, the majority of the subjects in both groups presented with left‐lateralized lesions (Supporting Information Table 1), primarily in the territory of the middle cerebral artery. The number of lesioned voxels did not differ significantly between subjects in the neurogenic stuttering and control groups (Table 1, U = 150, P = 0.56).
Lesion‐Symptom Characteristics
The vBLSM comparison of the lesion maps of the 20 stroke subjects with neurogenic stuttering with those of the 17 control subjects showed that nine areas had a probability of more than 95% to be more frequently affected in the former subject group. These areas comprised both grey and white matter throughout the left hemisphere (Table 2, Fig. 2). In the frontal lobe, these areas included the grey matter of the left inferior frontal gyrus pars triangularis with extension into the inferior frontal sulcus. Additionally, the white matter underlying these two structures as well as the white matter underneath the subcentral gyrus (part of the Rolandic operculum) was involved. In the left temporal lobe, the grey matter and underlying white matter of the posterior part of the superior temporal gyrus and sulcus were significantly associated with neurogenic stuttering. Furthermore, the ascending limb of the inferior temporal sulcus, at the junction of the temporal and occipital lobe, was more lesioned in these subjects. The parietal region which was found to have a higher lesion proportion in the neurogenic stuttering subjects involved the white matter underneath the supramarginal and angular gyri and the grey matter of the intraparietal sulcus. Figure 2 shows that the white matter involvement underneath the above‐described left‐hemisphere cortical structures overlapped extensively with the location of the superior longitudinal fasciculus. Additional areas associated with neurogenic stuttering included the left‐sided head and body of the caudate nucleus and the putamen, together known as the striatum. Finally, the white matter of the anterior and posterior limb of the internal capsule was more frequently affected in the neurogenic stuttering group.
Table 2.
Anatomic localization and MNI‐coordinates of the areas with a probability of 95% or higher to show larger lesion proportions in neurogenic stuttering
| Area number | Anatomic localization | MNI coordinates | Probability |
|---|---|---|---|
| A1 | Grey matter of left inferior frontal gyrus pars triangularis | −46 26 13 | 0.965 |
| A2 | Left subcortical white matter underneath inferior frontal sulcus | −33 25 19 | 0.992 |
| A3 | Left subcortical white matter underneath subcentral sulcus, supramarginal gyrus, and angular gyrus | −37 −18 29 | 0.999 |
| A4 | Grey matter of left superior temporal sulcus/subcortical white matter underneath superior temporal gyrus | −46 −43 13 | 0.981 |
| A5 | Grey matter of left ascending limb of inferior temporal sulcus | −58 −59 4 | 0.983 |
| A6 | Grey matter of anterior end of intraparietal sulcus | −32 −38 42 | 0.992 |
| A7 | Left putamen | −26 13 2 | 0.992 |
| A8 | Left body of nucleus caudatus | −17 −5 21 | 0.983 |
| A9 | Left internal capsule: anterior and posterior limb | −21 −17 14 | 0.965 |
Figure 2.
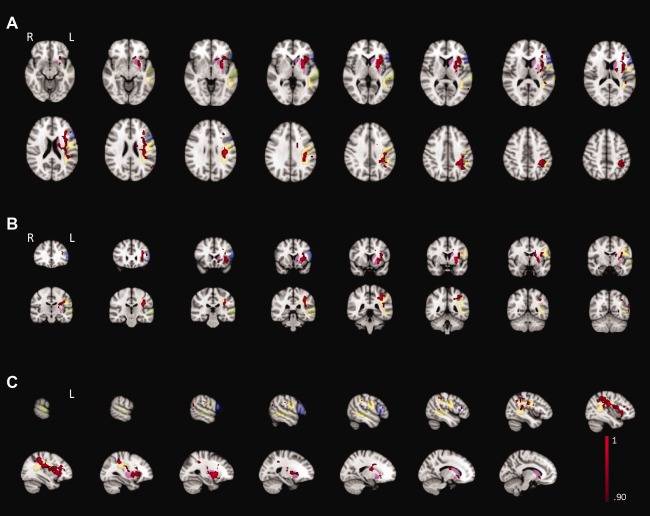
Transversal (A), coronal (B), and sagittal (C) view of the vBLSM results for d(0) for the 20 subjects with neurogenic stuttering compared with 17 controls (red‐black). For visualization purposes, results were smoothed with factor 3 and probability ranges between 0.90 and 1 are shown. The inferior frontal gyrus, superior temporal gyrus, striatum (respectively, blue, green, and pink, Harvard‐Oxford cortical and subcortical structural atlases, http://www.fmrib.ox.ac.uk/fsl/data/atlas-descriptions.html), and superior longitudinal fasciculus (yellow, Verhoeven et al., 2010) are overlaid on the results map.
The lesion contribution of each individual subject to the nine areas identified with the vBLSM‐analysis is shown in Supporting Information Table 1. Fifteen of the neurogenic stuttering subjects (75%) presented with lesions to one or more of these areas. Of the five neurogenic stuttering subjects not showing a lesion in the discussed brain regions, two had lesions in the basal ganglia in the right hemisphere, one in the right‐sided cerebellum, one in the left‐sided pons, and one in the right‐sided pons. In the control group, one subject (6%) presented with a lesion to one of the nine identified areas. Further analysis of the lesion‐symptom characteristics within the two subject groups showed an absence of significant correlations between stuttering frequency and either the number of lesioned voxels or the amount of overlap with the areas identified in the current vBLSM‐analysis (Supporting Information Table 3). Within each identified area, with the exception of the internal capsule, a significantly higher frequency of stuttering was present in the subjects showing a lesion to that specific area (Supporting Information Table 4).
DISCUSSION
This study was designed specifically to investigate the relationship between stroke‐related brain lesions and the presence or absence of neurogenic stuttering. The subject groups with and without neurogenic stuttering were highly comparable with respect to demographic characteristics and co‐occurring speech, language, and cognitive disorders. Although all 37 subjects were identified by a speech‐language pathologist or self‐diagnosed with dysfluencies following their stroke, only 20 subjects met the inclusion criteria for neurogenic stuttering based on the frequency of “stuttering‐like dysfluencies.” The other subjects primarily displayed dysfluencies based on language formulation problems or “normal dysfluencies” such as hesitations, interjections, and phrase repetitions [Bloodstein and Bernstein‐Ratner, 2008] and did not meet the criteria for neurogenic stuttering. Therefore, the presence of stuttering‐like dysfluencies differed quantitatively between the subjects in the neurogenic stuttering and control group. In future studies on neurogenic stuttering, one might also take the frequency of occurrence of “normal dysfluencies” into account to provide additional information with regard to the patient characteristics. Because our two subject groups only differed with regard to the frequency of stuttering‐like dysfluencies, it is reasonable to assume that the lesion localization differences between both groups were associated specifically with the presence of neurogenic stuttering.
The vBLSM‐analysis showed nine areas within the left hemisphere that were significantly more likely to be affected in the subjects with neurogenic stuttering, suggesting that the onset of stroke‐induced neurogenic stuttering is not limited to a lesion in any one particular brain region but emerges following a deficiency in a cortico‐basal ganglia‐cortical network consisting of the inferior frontal cortex, superior temporal cortex, intraparietal cortex, basal ganglia, superior longitudinal fasciculus, and internal capsule (Fig. 2).
The widespread involvement of these grey and white matter structures is consistent with recent neural models of speech production that functionally integrate both sensory and motor aspects of speech [Guenther et al., 2006; Hickok, 2009]. Of the identified grey matter areas, the inferior frontal and superior temporal areas, as well as the basal ganglia are considered core components of the neural network for speech production [Guenther, 2007]. This suggests that the intact functioning of brain areas responsible for articulatory planning [Guenther, 2007; Papoutsi et al., 2009], auditory monitoring of speech output [Edwards et al., 2010], and internal timing [Cunnington et al., 1996] are crucial for the production of fluent speech and that lesions in these areas may result in the manifestation of neurogenic stuttering. The involvement of the intraparietal cortex in our subjects further shows the importance of intact processing of perceptual stimuli for the production of stutter‐free speech [Anderson et al., 2010; Cusack, 2005]. Lesions in the ascending limb of the inferior temporal sulcus were also significantly associated with the presence of neurogenic stuttering. To our knowledge, this is the first report of the potential involvement of this specific brain area in neurogenic stuttering. Studies by Guenther 2007 have shown that the left inferior temporo‐occipital junction is activated during the production of single nonsense monosyllables. While this area typically is associated with visual word form processing, the finding of larger lesion proportions in this brain structure in subjects with neurogenic stuttering suggests that its role in speech production may be more prominent than previously thought, although the precise nature of this role remains unclear.
In addition to the previously described grey matter areas, the network associated with neurogenic stuttering in our subjects substantially involved white matter tracts. The involvement of the anterior limb of the internal capsule, which provides reciprocal connections between the frontal cortex and striatum [Beasley et al., 2009], suggests that intact linkage between frontal cortex and basal ganglia is important for the production of fluent speech. Furthermore, the observation of a high probability (Pr = 0.999) of lesions in the superior longitudinal fasciculus in neurogenic stuttering may indicate that an efficient communication between the cortical areas described above is very important for fluent speech production. The superior longitudinal fasciculus is known to play a role in both speech and language production. The arcuate fasciculus, a subdivision of the superior longitudinal fasciculus, connects temporal receptive and frontal expressive areas and has an indirect pathway passing through the inferior parietal cortex [Catani et al., 2005]. Lesion to this fasciculus can lead to aphasia following stroke [Breier et al., 2008; Catani and Mesulam, 2008]. In addition to its contributions to language processing, the superior longitudinal fasciculus also plays a role in the sensorimotor processing of speech through integration of multimodal proprioceptive information arising from articulatory planning and sensory feedback during speech production [Watkins et al., 2008]. It has previously been suggested that the disconnection between posterior and anterior speech areas may lead to acquired nonfluent speech [Bonilha and Fridriksson, 2009], such as apraxia of speech. However, the subjects diagnosed with neurogenic stuttering in this study did not differ significantly from the control group with respect to co‐occurring aphasia and apraxia of speech. This apparent dissociation between these co‐occurring disorders and neurogenic stuttering emphasizes the distinct nature of these disorders and highlights the need to further investigate the role of the superior longitudinal fasciculus in fluent speech and language production. Indeed, while the functional association of the superior longitudinal fasciculus with aphasic and apraxic speech problems may help to explain the co‐occurrence of these communication problems with neurogenic stuttering in some subjects, the results from our study suggest a specific role for this fasciculus with respect to the occurrence of stuttering dysfluencies following stroke.
Although vBLSM is capable of detecting changes in lesion proportions in relatively small sample sizes, a limitation of this technique concerns the requirement of a binary group‐membership variable. Therefore, we performed additional analyses to further explore the relationship between the percentage syllables stuttered and the lesion data. This data not only showed that the nine identified areas had a significant probability of larger lesion proportions associated with the presence neurogenic stuttering. The additional analyses also revealed that when the subjects were grouped according to the presence or absence of a lesion in a certain voxel, the presence of lesions was associated with a significantly higher frequency of stuttering in all the areas with exception of the internal capsule (Supporting Information Table 4). This confirms that lesions in the identified network do not only occur more frequently in neurogenic stuttering subjects but also that a lesion to one of these areas is also associated with a higher percentage of stuttering‐like dysfluencies. Of additional interest was the observation that in the neurogenic stuttering group, 14 of the 15 stroke subjects with lesions in the identified network presented with lesions in at least two areas within this network (Supporting Information Table 1). In contrast, in the control group, only one of the 17 subjects presented with a lesion to an area within this network. Although the onset of neurogenic stuttering was associated with multiple lesions in 14 neurogenic stuttering subjects, the frequency of stuttering in the neurogenic stuttering group was not correlated with the number of lesioned voxels or number of lesioned areas in the identified cortico‐basal‐ganglia cortical network (Supporting Information Table 3). This suggests that while one or more lesions within this network may trigger the onset of acquired stuttering, such lesions are not directly associated with the severity of stuttering. One could speculate that the severity of stuttering may be more affected by the relative contribution the lesioned area has to the neural network rather than size of the lesion.
The finding that several brain areas throughout the left hemisphere may play a role in the onset of neurogenic stuttering helps to explain why a variety of lesion sites has been reported in case studies of neurogenic stuttering following stroke. The observation of a left‐hemispheric cortico‐basal ganglia‐cortical network involving the inferior frontal, superior temporal, intraparietal areas, the basal ganglia, and their white matter connections is consistent with the majority of the reported cases [for review, see De Nil et al., 2009]. Nevertheless, there have been a number of other case studies [for review, see De Nil et al., 2009] presenting with lesions outside this network. Similarly, five of the 20 neurogenic stuttering subjects in this study also did not show evidence of lesions within the identified left‐sided network. Because the majority of our subjects presented with lesions in the territory of the middle cerebral artery, well known to be closely associated with the occurrence of speech and language problems, the limited lesion coverage outside this territory in this study does not permit us to categorically exclude a potential involvement of other lesion sites, such as the pons, cerebellum, and right‐sided basal ganglia, in the onset of neurogenic stuttering following stroke. The occurrence of neurogenic stuttering following lesions in these areas, however, may still be linked to the network identified in this study in a number of ways. First, the right cerebellar hemisphere, for example, has functional connectivity with the left hemispheric frontal lobe and contributes to the temporal organization of a prearticulatory verbal code [Ackermann et al., 2007]. Thus, a lesion to the cerebellum might influence the functionality of the cortico‐basal ganglia‐cortical network described earlier [distant hodological effect, see Catani and Mesulam, 2008]. Second, other brain regions may not only affect the input to or functioning of the identified cortico‐basal ganglia‐cortical network but also can disrupt its output along the execution pathway for speech articulation. Third, the existence of “crossed aphasias” demonstrated that crucial cortico‐subcortical language networks may be present in the right hemisphere in a small subgroup of right‐handers [Ozeren et al., 1998; Vassal et al., 2010]. Finally, because of limitations of current imaging techniques it cannot be excluded that some subjects may have presented with small lesions to the identified network that remained undetected in this analysis. Nevertheless, although this data provides strong evidence that the cortico‐basal ganglia‐cortical network described above has a crucial role in the onset of neurogenic stuttering in a majority of the subjects, the fact that some of the subjects with neurogenic stuttering in this study had lesions outside the nine identified areas suggests that the role of other regions cannot be excluded and needs further investigation.
These findings in stroke patients are consistent with the reported involvement of the striatum, internal capsule, and frontal white matter in the onset of neurogenic stuttering in head injury patients [Ludlow et al., 1987], as well as with the proposed theoretical model detailing the involvement of the basal ganglia in neurogenic stuttering [Alm, 2004]. These results, however, suggest that this neural model needs to be expanded to include a larger network in addition to the basal ganglia structures. In contrast, these findings challenge the observation that lesions associated with neurogenic stuttering rarely involve the primary speech and language areas in the left hemisphere [Ludlow and Loucks, 2003].
Although this study did not include subjects with developmental stuttering, our observations of lesions associated with neurogenic stuttering in stroke subjects may also contribute to the study of the neural bases of developmental stuttering. Developmental stuttering typically starts in children between 2 and 5 years of age [Bloodstein and Bernstein‐Ratner, 2008], which contrasts with the typical onset of neurogenic stuttering in adulthood. Although some authors have suggested that neurogenic and developmental stuttering constitute two different disorders [Helm‐Estabrooks, 1999; Ringo and Dietrich, 1995], other studies have shown that the speech characteristics of both types of stuttering may be more similar than previously thought [Theys et al., 2008; Van Borsel and Taillieu, 2001]. Interestingly, many of the areas found to be associated with stroke‐related neurogenic stuttering in this study have previously been implicated in brain imaging studies of developmental stuttering [Brown et al., 2005; Lu et al., 2010; Neumann et al., 2003; Sommer et al., 2002; Watkins et al., 2008]. In fact, several researchers [Alm, 2004; Giraud et al., 2008; Lu et al., 2010; Sommer et al., 2002] have proposed that areas similar to those implicated in the cortico‐basal ganglia‐cortical loop in this study may have an important role in the onset and maintenance of developmental stuttering. This overlap of brain areas implicated in both developmental and neurogenic stuttering, as well as the similarities in speech characteristics, suggests that both types of stuttering may share common neural characteristics.
Based on our findings, we suggest that stroke‐related stuttering can occur following one or more lesioned areas spread over the left‐hemispheric cortico‐basal ganglia‐cortical loop. It is possible that interindividual differences in the specific localization of the lesion(s) could give rise to one or more concomitant disorders in subjects with neurogenic stuttering and possibly also in subjects with developmental stuttering, e.g., impaired grammatical skills following a lesion in the intraparietal cortex [Bloodstein and Bernstein‐Ratner, 2008; Varley et al., 2005] or impaired auditory processing following a lesion in the superior temporal lobe [Bloodstein and Bernstein‐Ratner, 2008; Price, 2010]. If supported by further research, this might lead to the identification of subgroups of stuttering subjects based on their specific neural impairment. Identification of subgroups of subjects with neurogenic stuttering, and perhaps also with developmental stuttering, may lead to more refined diagnostic processes and may give rise to specific therapeutic strategies for subjects with dysfunctions in different areas of the cortico‐basal ganglia‐cortical loop.
CONCLUSION
This study is the first to report voxel‐based lesion‐symptom correlates in a large group of stroke subjects with neurogenic stuttering compared with an equivalent group of nonstuttering stroke subjects. The results show involvement of nine areas consisting of both grey and white matter structures throughout the left hemisphere. Based on these findings, we suggest that stroke‐induced neurogenic stuttering is associated with neural dysfunction in the left‐sided cortico‐basal ganglia‐cortical network comprising the inferior frontal cortex, superior temporal cortex, intraparietal cortex, basal ganglia, and their interconnections. It is proposed that this dysfunction results in a disordered integration of neural functions necessary for speech and thus in the occurrence of neurogenic stuttering‐like dysfluencies.
Supporting information
Supporting Information Table 1
Supporting Information Table 2
Supporting Information Table 3
Supporting Information Table 4
Acknowledgments
The authors are grateful to Dr. R. Chen for providing a Windows version of vBLSM and for his assistance with the image conversions. They thank H. Dely for the reassessment of the stuttering scores and Dr. E. Visch‐Brink for providing them a prepublished version of the ScreeLing. They acknowledge Dr. R. Vandenberghe for facilitating their access to the stroke unit at the University Hospitals Leuven.
REFERENCES
- Abe K, Yokoyama R, Yorifuji S (1993): Repetitive speech disorder resulting from infarcts in the paramedian thalami and midbrain. J Neurol Neurosurg Psychiatry 56:1024–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackermann H, Hertrich I, Ziegler W, Bitzer M, Bien S (1996): Acquired dysfluencies following infarction of the left mesiofrontal cortex. Aphasiology 10:409–417. [Google Scholar]
- Ackermann H, Mathiak K, Riecker A (2007): The contribution of the cerebellum to speech production and speech perception: Clinical and functional imaging data. Cerebellum 6:202–213. [DOI] [PubMed] [Google Scholar]
- Alm PA (2004): Stuttering and the basal gangia circuits: A critical review of possible relations. J Commun Disord 37:325–369. [DOI] [PubMed] [Google Scholar]
- Anderson JS, Ferguson MA, Lopez‐Larson M, Yurgelun‐Todd D (2010): Topographic maps of multisensory attention. Proc Natl Acad Sci USA 107:20110–20114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardilla A, Lopez MV (1986): Severe stuttering associated with right hemisphere lesion. Brain Lang 27:239–246. [DOI] [PubMed] [Google Scholar]
- Balasubramanian V, Max L, Van Borsel J, Rayca KO, Richardson D (2003): Acquired stuttering following right frontal and bilateral pontine lesion: A case study. Brain Cogn 53:185–189. [DOI] [PubMed] [Google Scholar]
- Beasley CL, Dwork AJ, Rosoklija G, Mann JJ, Mancevski B, Jakovski Z, Davceva N, Tait AR, Straus SK, Honer WG (2009): Metabolic abnormalities in fronto‐striatal‐thalamic white matter tracts in schizophrenia. Schizophr Res 109:159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloodstein O, Bernstein‐Ratner N (2008): A Handbook on Stuttering, 6th ed. New York: Delmar Learning. [Google Scholar]
- Boey R (2000): Stotteren: Detecteren en meten. Leuven: Garant. [Google Scholar]
- Bonilha L, Fridriksson J (2009): Subcortical damage and white matter disconnection associated with non‐fluent speech. Brain 132:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breier JL, Hasan KM, Zhang W, Men D, Papanicolaou AC (2008): Language dysfunction after stroke and damage to white matter tracts evaluated using diffusion tensor imaging. AJNR Am J Neuroradiol 29:483–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S, Ingham RJ, Ingham JC, Laird AR, Fox PT (2005): Stuttered and fluent speech production: An ALE meta‐analysis of functional neuroimaging studies. Hum Brain Mapp 25:105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carluer L, Marié RM, Lambert J, Defer GL, Coskun O, Rossa Y (2000): Acquired and persistent stuttering as the main symptom of striatal infarction. Mov Disord 15:343–346. [DOI] [PubMed] [Google Scholar]
- Catani M, Jones DK, ffytche DH (2005): Perisylvian language networks of the human brain. Ann Neurol 57:8–16. [DOI] [PubMed] [Google Scholar]
- Catani M, Mesulam M (2008): The arcuate fasciculus and the disconnection theme in language and aphasia: History and current state. Cortex 44:953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Herskovits EH (2010): Voxel‐based Bayesian lesion‐symptom mapping. Neuroimage 49:597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciabarra AM, Elkind MS, Roberts JK, Marshall RS (2000): Subcortical infarction resulting in acquired stuttering. J Neurol Neurosurg Psychiatry 69:546–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conture EG (2000): Stuttering: Its Nature, Diagnosis and Treatment. Needham Heights: Allyn & Bacon. [Google Scholar]
- Cunnington R, Iansek R, Thickbroom GW, Laing BA, Mastaglia FL, Bradshaw JL, Philips JG (1996): Effects of magnetic stimulation over supplementary motor area on movement in Parkinson's disease. Brain 119:815–822. [DOI] [PubMed] [Google Scholar]
- Cusack R (2005): The intraparietal suclus and perceptual organization. J Cogn Neurosci 17:641–651. [DOI] [PubMed] [Google Scholar]
- Dabul B (2000): Apraxia Battery for Adults, 2nd ed. Austin, Texas: Pro‐ed. [Google Scholar]
- De Nil LF, Rochon E, Jokel R (2009): Adult‐onset neurogenic stuttering In: McNeil MR, editor. Clinical management of senorimotor speech disorders, 2nd ed. New York: Thieme; pp 235–248. [Google Scholar]
- Dharmaperwira‐Prins R (1996): Dysartrie en verbale apraxie: Beschrijving, onderzoek en behandeling. Lisse: Swets & Zeitlinger. [Google Scholar]
- Duffy JR (2005): Motor Speech Disorders. Substrates, Differential Diagnosis and Management, 2nd ed. St. Louis, Missouri: Elsevier Mosby. [Google Scholar]
- Edwards E, Nagarajan SS, Dalal SS, Canolty RT, Kirsch HE, Barbaro NM, Knight RT (2010): Spatiotemporal imaging of cortical activation during verb generation and picture naming. Neuroimage 50:291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR (1975): “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12:189–198. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Ashburner JT, Kiebel SJ, Nichols TE, Penny WD, editors (2007): Statistical Parametric Mapping: The Analysis of Functional Brain Images. London: Elsevier. [Google Scholar]
- Giraud A‐L, Neumann K, Bachoud‐Levi A‐C, von Gudenberg AW, Euler HA, Lanfermann H, Preibisch C (2008): Severity of dysfluency correlates with basal ganglia activity in persistent developmental stuttering. Brain Lang 104:190–199. [DOI] [PubMed] [Google Scholar]
- Grant AC, Biousse V, Cook AA, Newman NJ (1999): Stroke‐associated stuttering. Arch Neurol 56:624–627. [DOI] [PubMed] [Google Scholar]
- Guenther FH (2007): Neuroimaging of normal speech production In: Ingham RJ, editor. Neuroimaging in Communication Sciences and Disorders. San Diego: Plural Publishing Inc; pp 1–51. [Google Scholar]
- Guenther FH, Ghosh SS, Tourville JA (2006): Neural modeling and imaging of the cortical interactions underlying syllable production. Brain Lang 96:280–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamano T, Hiraki S, Kawamura Y, Hirayama M, Mutoh T, Kuriyama M (2005): Acquired stuttering secondary to callosal infarction. Neurology 64:1092–1093. [DOI] [PubMed] [Google Scholar]
- Helm NA, Butler RB, Canter GJ (1980): Neurogenic acquired stuttering. J Fluency Disord 5:269–279. [Google Scholar]
- Helm‐Estabrooks NA (1999): Stuttering associated with acquired neurological disorders In: Curlee RF, editor. Stuttering and Related Disorders of Fluency, 2nd ed. New York: Thieme Medical Publishers; pp 255–268. [Google Scholar]
- Hickok G (2009): The functional neuroantaomy of language. Phys Life Rev 6:121–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakishita K, Sekiguchi E, Maeshima S, Okada H, Okita R, Ozaki F, Hiroshi M (2004): Stuttering without callosal apraxia resulting from infarction in the anterior corpus callosum. J Neurol 251:1140–1141. [DOI] [PubMed] [Google Scholar]
- Kaplan E, Goodglass H, Weintraub S (2001): Boston Naming Test, 2nd ed. Austin, Texas: Pro‐ed. [Google Scholar]
- Lu C, Peng D, Chen C, Ning N, Ding D, Li K, Yang Y, Lin C (2010): Altered effective connectivity and anomalous anatomy in the basal ganglia‐thalamocortical circuit of stuttering speakers. Cortex 46:49–67. [DOI] [PubMed] [Google Scholar]
- Ludlow CL, Loucks TM (2003): Stuttering: A dynamic motor control disorder. J Fluency Disord 28:273–295. [DOI] [PubMed] [Google Scholar]
- Ludlow CL, Rosenberg J, Salazar A, Grafman J, Smutok M (1987): Site of penetrating brain lesions causing chronic acquired stuttering. Ann Neurol 22:60–66. [DOI] [PubMed] [Google Scholar]
- Neumann K, Euler HA, von Gudenberg AW, Giraud AL, Lanfermann H, Gall V, Preibisch C (2003): The nature and treatment of stuttering as revealed by fMRI. J Fluency Disord 28:381–409. [DOI] [PubMed] [Google Scholar]
- Osawa A, Maeshima S, Yoshimura T (2006): Acquired stuttering in a patient with Wernicke's aphasia. J Clin Neurosci 13:1066–1069. [DOI] [PubMed] [Google Scholar]
- Ozeren A, Mavi H, Sarica Y, Karatas M (1998): Subcortical crossed aphasia. Acta Neurol Belg 98:204–208. [PubMed] [Google Scholar]
- Papoutsi M, de Zwart JA, Jansma JM, Pickering MJ, Bednar JA, Horwitz B (2009): From phonemes to articulatory codes: An fMRI study of the role of Broca's speech area in speech production. Cereb Cortex 19:2156–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price CJ (2010): The anatomy of language: A review of 100 fMRI studies published in 2009. Ann NY Acad Sci 1191:62–88. [DOI] [PubMed] [Google Scholar]
- Raven JC, Court JH, Raven J (1995): Raven manual: Coloured progressive matrices. Oxford: Oxford Psychologists Press. [Google Scholar]
- Ringo CC, Dietrich S (1995): Neurogenic stuttering: An analysis and critique. J Med Speech Lang Pathol 3:111–122. [Google Scholar]
- Rorden C, Karnath H‐O, Bonilha L (2007): Improving lesion‐symptom mapping. J Cogn Neurosci 19:1081–1088. [DOI] [PubMed] [Google Scholar]
- Rosenbek J, Messert B, Collins M, Wertz RT (1978): Stuttering following brain damage. Brain Lang 6:82–96. [DOI] [PubMed] [Google Scholar]
- Sahin HA, Krespi Y, Yilmaz A, Coban O (2005): Stuttering due to ischemic stroke. Behav Neurol 16:37–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TE, Johansen‐Berg H, Bannister PR, De Luca M, Drobnjak I, Flitney DE, Niazy RK, Saunders J, Vickers J, Zhang Y, De Stefano N, Brady JM, Matthews PM (2004): Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 23:208–219. [DOI] [PubMed] [Google Scholar]
- Sommer M, Koch MA, Paulus W, Weiller C, Büchel C (2002): Disconnection of speech‐relevant brain areas in persistent developmental stuttering. Lancet 360:380–383. [DOI] [PubMed] [Google Scholar]
- Tani T, Sakai Y (2010): Stuttering after right cerebellar infarction: A case study. J Fluency Disord 35:141–145. [DOI] [PubMed] [Google Scholar]
- Tani T, Sakai Y (2011): Analysis of five cases with neurogenic stuttering following brain injury in the basal ganglia. J Fluency Disord 36:1–16. [DOI] [PubMed] [Google Scholar]
- Theys C, van WA, De Nil LF (2008): A clinician survey of speech and non‐speech characteristics of neurogenic stuttering. J Fluency Disord 33:1–23. [DOI] [PubMed] [Google Scholar]
- Theys C, van Wieringen A, Sunaert S, Thijs V, De Nil LF (2011): A one year prospective study of neurogenic stuttering following stroke: Incidence and co‐occurring disorders. J Commun Disord 44:678–687. [DOI] [PubMed] [Google Scholar]
- Turgut N, Utku U, Balci K (2002): A case of acquired stuttering resulting from left parietal infarction. Acta Neurol Scand 105:408–410. [DOI] [PubMed] [Google Scholar]
- Van Borsel J, Taillieu C (2001): Neurogenic stuttering versus developmental stuttering: An observer judgement study. J Commun Disord 34:835–895. [DOI] [PubMed] [Google Scholar]
- Van Borsel J, Van Lierde K, Van Cauwenberghe P, Van Orshoven M (1998): Severe acquired stuttering following injury of the left supplementary motor region: A case report. J Fluency Disord 23:49–58. [Google Scholar]
- Van Borsel J, Van Der Maede S, Santens P (2003): Thalamic stuttering: A distinct clinical entity? Brain Lang 85:185–189. [DOI] [PubMed] [Google Scholar]
- Varley RA, Klessinger NJC, Romanowski CAJ, Siegal M (2005): Agrammatic but numerate. Proc Natl Acad Sci USA 102:3519–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassal M, Le Bars E, Moritz‐Gasser S, Menjot N, Duffau H (2010): Crossed aphasia elicited by intraoperative cortical and subcortical stimulation in awake patients. J Neurosurg 113:1251–1258. [DOI] [PubMed] [Google Scholar]
- Verhoeven JS, Sage CA, Leemans A, Van Hecke W, Callaert D, Peeters R, De Cock P, Lagae L, Sunaert S (2010): Construction of a stereotaxic DTI atlas with full diffusion tensor information for studying white matter maturation from childhood to adolescence using tractography‐based segmentations. Hum Brain Mapp 31:470–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visch‐Brink EG, van de Sandt‐Koenderman M, El Hachioui H (2010): ScreeLing, een linguïstische screeningstest. Houten: Bohn Stafleu van Loghum. [Google Scholar]
- Wambaugh JL, Duffy JR, McNeil MR, Robin DA, Rogers MA (2006): Treatment guidelines for acquired apraxia of speech: A synthesis and evalutation of the evidence. J Med Speech Lang Pathol 14:xv–xxxiii. [Google Scholar]
- Watkins KE, Smith SM, Davis S, Howell P (2008): Structural and functional abnormalities of the motor system in develomental stuttering. Brain 131:50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Table 1
Supporting Information Table 2
Supporting Information Table 3
Supporting Information Table 4