

Abstract
Huntington's disease (HD) is an inherited neurodegenerative disorder associated with motor, cognitive and psychiatric deficits. This study, using a multimodal imaging approach, aims to assess in vivo the functional and structural integrity of regions and regional networks linked with motor, cognitive and psychiatric function. Predicting disease onset in at risk individuals is problematic and thus we sought to investigate this by computing the 5‐year probability of HD onset (p5 HD) and relating it to imaging parameters. Using MRI, 11C‐PK11195 and 11C‐raclopride PET, we have investigated volumes, levels of microglial activation and D2/D3 receptor binding in CAG repeat‐matched groups of premanifest and symptomatic HD gene carriers. Findings were correlated with disease‐burden and UHDRS scores. Atrophy was detected in sensorimotor striatum (SMST), substantia nigra, orbitofrontal and anterior prefrontal cortex in the premanifest HD. D2/D3 receptor binding was reduced and microglial activation increased in SMST and associative striatum (AST), bed nucleus of the stria terminalis, the amygdala and the hypothalamus. In symptomatic HD cases this extended to involve atrophy in globus pallidus, limbic striatum, the red nuclei, anterior cingulate cortex, and insula. D2/D3 receptor binding was additionally reduced in substantia nigra, globus pallidus, limbic striatum, anterior cingulate cortex and insula, and microglial activation increased in globus pallidus, limbic striatum and anterior prefrontal cortex. In premanifest HD, increased levels of microglial activation in the AST and in the regional network associated with cognitive function correlated with p5 HD onset. These data suggest that pathologically activated microglia in AST and other areas related to cognitive function, maybe better predictors of clinical onset and stresses the importance of early cognitive assessment in HD. Hum Brain Mapp, 2011. © 2010 Wiley‐Liss, Inc.
Keywords: PET, MRI, microglia, dopamine, biomarkers, associative striatum, PK, RAC, ROI, VOI
INTRODUCTION
Huntington's disease (HD) is a progressive neurodegenerative disorder with typical onset between 35 and 50 years of age. HD is inherited in an autosomal dominant pattern and the responsible IT15 gene mutation consists of an unstable expansion of CAG repeats within its coding region that is located in the distal portion of the short arm of chromosome 4 (4p16.3) and encodes the production of the protein huntingtin [The Huntington's Disease Collaborative Research Group, 1993]. The toxic gain of function of the mutant huntingtin and its associated proteins contribute to the disruption of multiple intracellular pathways leading to the formation of aggregates that progressively result in the dysfunction and then degeneration of important neuronal pathways and cell loss in regions of the HD brain (for review see Roze et al. [ 2008]).
Twenty‐five years ago, Vonsattel et al. [ 1985] developed a grading system to assess the severity of HD striatal degeneration based on a large post‐mortem study. From that time, although progressive atrophy of the striatum remains one of the main pathological hallmarks of HD, advances in neuroimaging techniques have greatly contributed to a better understanding of HD pathology as more widespread [Tabrizi et al., 2009], complex and challenging for the development of potential treatments.
Structural and functional neuroimaging studies have now established important correlations between morphological and functional brain changes and the development of clinical deficits during the course of the disease. A combined neuroimaging approach incorporating both structural and functional techniques could potentially serve as a biomarker for use in future clinical disease modifying drug trials in HD.
This study has been designed taking into account several issues in HD: (1) The size of CAG repeats expansion inversely correlates with the age at onset [Kieburtz et al., 1994] and positively with the rate of HD progression [Penney et al., 1997]. (2) The extent and localization of atrophy in HD brain remains controversial (for review see Bohanna et al. [ 2008]). (3) Altered dopaminergic neurotransmission is evident from the premanifest stage of the disease in striatal [Andrews et al., 1999; van Oostrom et al., 2009] and extrastriatal areas [Pavese et al., 2003; Politis et al., 2008] and has been implicated as potentially pathogenic, contributing to the neuronal cell death [Jakel and Maragos, 2000]. (4) There is detectable ongoing neuroinflammation and pathogenic immune activation very early in the course of the disease [Björkqvist et al., 2008] and activated microglia have been proved as sensitive reactors to pathological changes in HD brain such as the presence of mutant huntingtin [Sapp et al., 2001; Shin et al., 2005].
11C‐raclopride (RAC) with positron emission tomography (PET) is a marker of dopamine D2 and D3 receptor availability and an indirect marker of synaptic dopamine fluxes in the living human brain. In Huntington's disease, this imaging technique has been largely employed for the assessment of striatal and extrastriatal functional integrity [Andrews et al., 1999; Pavese et al., 2003; Politis et al., 2008; van Oostrom et al., 2009]. Furthermore, in healthy brain, microglia exist in a resting state characterized by ramified morphology, and monitor the brain milieu. In response to a neuronal insult (such as mutant huntingtin), however, microglia become activated and are transformed from their resting ramified state into an amoeboid morphology releasing cytokines and growth factors and having phagic properties [Davalos et al., 2005; Nimmerjahn et al., 2005]. 11C‐PK11195 (PK) is a specific radioligand of the translocator protein (TSPO) previously known as the peripheral benzodiazepine receptor. The TSPO is expressed by the mitochondria of activated microglia and PK PET has been used as a marker for this transition of microglia from a resting to an activated state [Cagnin et al., 2001].
We have measured volumetric and functional (D2/D3 receptor binding and activation of microglia) parameters in HD to investigate the structure and function of individual brain regions and estimated regional networks with known connection to psychiatric, cognitive and motor symptoms. Furthermore, we have looked for associations between imaging findings and clinical scales related to daily living, disease severity, motor dysfunction and in the case of premanifest HD gene carriers, the probability of HD onset. For this imaging paradigm, we have created averaged imaging profiles derived from three cohorts of subjects. Regional volume changes were studied with structural magnetic resonance imaging (MRI) volumetric analysis, whereas for the investigation of the integrity of post‐synaptic dopamine D2/D3 receptors and the presence of altered levels of activated microglia, RAC and PK PET have been employed, respectively.
MATERIALS AND METHODS
Subjects
Thirty‐two subjects participated in this study. Comparisons between the functional and structural imaging profiles of eight premanifest HD brains (4M: 4F; 45 ± 3 CAG repeats, 41 ± 10 years of age, 361 ± 65 disease‐burden score, p5 HD 0.4 ± 0.1; mean ± SD) that all had genetically proven disease, eight premanifest HD brains (4M: 4F; 45 ± 3 CAG repeats, 48 ± 5 years of age, 428 ± 126 disease‐burden score, 5 ± 3 years of disease, 16 ± 11 Unified Huntington's Disease Rating Scale (UHDRS) motor score, Total Functional Capacity (TFC) score 6.5 ± 2, IS score 75% ± 5; mean ± SD) that all had clinically manifest disease and 16 normal brains (MRI: n = 16, 14M, 2F, 46 ± 6 years ± SD; RAC: n = 8, 8M, 44 ± 4 years ± SD; PK: n = 8, 6M, 2F, 48 ± 7 years ± SD) all in good health with no history of neurological or psychiatric disorder, were performed (Table 1). None of these subjects were on any medication.
Table 1.
Clinical profile and characteristics of the subjects formulate the averaged premanifest HD, symptomatic HD, and MRI, RAC and PK normal brains
| Averaged profile | Premanifest HD brain | Symptomatic HD brain | MRI normal brain | RAC normal brain | PK normal brain |
|---|---|---|---|---|---|
| No. of subjects | 8 | 8 | 16 | 8 | 8 |
| Sex | 4M/4F | 4M/4F | 14M/2F | 8M/0F | 6M/2F |
| Age (years ± SD) | 40.8 ± 9.5 | 47.7 ± 5.0 | 45.9 ± 6.1 | 43.7 ± 4.3 | 48.4 ± 7.2 |
| CAG repeats (mean ± SD) | 44.9 ± 2.6 | 44.7 ± 3.1 | – | – | – |
| Disease‐burden scorea (mean ± SD) | 361.2 ± 64.9 | 427.4 ± 126.1 | – | – | – |
| Disease durationb (years ± SD) | – | 5.2 ± 2.7 | – | – | – |
| UHDRS motor scorec (mean ± SD) | 0.0 ± 0.0 | 15.9 ± 11.1 | – | – | – |
| p5 HDd | 0.4 ± 0.1 | – | – | – | – |
| TFCe | 13.0 ± 0.0 | 6.5 ± 2.3 | – | – | – |
| ISf | 100.0% ± 0.0 | 74.3% ± 5.3 | – | – | – |
Disease‐burden score = CAG index = age × (CAG repeats length – 35.5) [Penney et al., 1997].
Disease duration measured from the time where the subject could remember his/her first symptoms.
UHDRS = Unified Huntington's Disease Rating Scale [Huntington Study Group, 1996]. UHDRS motor scores: Range (Best = 0, Worst = 124).
Probability of developing Huntington's disease symptoms in the following 5 years. Taken from the age‐CAG repeats correlation tables [Langbehn et al., 2004].
TFC = Total Functional Capacity scores (Best = 13, Worst = 0).
IS = Independence Scale scores (Best = 100%, Worst = 0%).
Clinical Evaluation
The symptomatic HD and premanifest HD gene carriers were assessed clinically in three major domains: (1) daily living using the TFC and Independence Scale (IS) scores; (2) disease severity using CAG repeat length and the disease‐burden score (CAG index = age × [CAG = 35.5], Penney et al., 1997) and; (3) motor dysfunction using the motor component of the UHDRS. Additionally, the 5‐year probability of symptomatic HD onset (probability of diagnosis calculated based on CAG repeat length and current age, see Langbehn et al. [ 2004]) was calculated in the premanifest HD gene carriers (Table 1).
Imaging Techniques
MRI, RAC and PK PET scanning were performed no more than 2 months apart.
MRI scanning
Subjects had a volumetric T1 MRI with a 1.5 Tesla scanner using a RF‐fast pulse sequence, TR = 30 ms and TE = 3 ms acquisition time and a flip angle of 30° (Philips Medical Systems; Picker Eclipse, Picker International Inc., Highland Heights, OH, USA). This scanner allows the acquisition of high‐resolution RF spoiled T1‐weighted volume datasets. Subjects were positioned supine with their transaxial planes parallel to the line intersecting anterior and posterior commissure (AC–PC line). They were made comfortable and their head position was maintained still with the help of individualized foam holders.
PET scanning
All symptomatic and premanifest HD gene carriers had both RAC and PK PET scan, whereas normal controls had either RAC or PK PET (half‐half). PET was performed using an ECAT EXACT HR++ (CTI/Siemens 966; Siemens, Knoxville, TN) camera with 23.4 cm total axial field of view. The camera has a reconstructed (image) transaxial spatial resolution of 5.1 ± 0.6 mm and an axial resolution of 5.9 ± 0.6 mm over a 10‐cm radius FOV from the centre [Spinks et al., 2000]. This high‐resolution, high‐sensitivity tomograph permits interrogation of the function of small volume structures.
The subjects were positioned supine with their transaxial planes parallel to the line intersecting the AC–PC line. They were made comfortable and their head position maintained with the help of individualised foam holders. In addition, their position relative to the camera's laser light was monitored throughout and was repositioned if movement was detected. Subjects were in a resting state with low light and no noise in the room. Smoking and consumption of alcohol, coffee and other caffeinated beverages was not allowed for 12 h before scanning. Eating and drinking was not allowed for 8 h before PET.
A 5‐min transmission scan was performed before injection of each radiotracer to correct for tissue attenuation of 511 keV gamma radiation. The mean tracer dose for RAC administered was 188 MBq (range: 181–196 MBq) and for PK was 297 MBq (ranging from 293 to 304 MBq). The radiotracers were infused intravenously over 30 s. Scanning began at the start of the tracer injection generating 20 time frames over 65 min for RAC and 18 time frames over 60 min for PK. RAC and PK tracers were supplied by Hammersmith Imanet plc, London, UK and permission to administer RAC and PK was obtained from the Administration of Radioactive Substances Advisory Committee (ARSAC) of the Department of Health, UK.
Data Analysis
MRI
The MRI images after transformation to DICOM format were reoriented parallel to the AC–PC line and then transformed to ANALYZE 7.5 format. Volume‐of‐interest (VOI) analysis was performed on individual MRIs using ANALYZE software (version 8.1, BRU, Mayo Foundation, Rochester, MN). This method allows a direct volume comparison expressed in cubic millimetres between brain regions under investigation after calculating the sum of sampled slices on each MRI.
RAC PET
Region‐of‐interest (ROI) image analysis was performed using ANALYZE software to detect functional changes in dopaminergic D2 and D3 receptor binding. Parametric images of RAC binding potentials (BPND) were generated from the dynamic RAC scan series, using BASIS function implementation of the simplified reference region compartmental model with the cerebellum as the reference tissue for non‐specific tracer binding [Gunn et al., 1997]. Each parametric RAC BPND image was anatomically coregistered and resliced to the corresponding volumetric T1‐weighted MRI. This was done using the Mutual Information Registration algorithm in the SPM2 software package (Wellcome Department of Cognitive Neuroscience, Institute of Neurology, London, UK) implemented in Matlab6 (Version 6.5.1, The Mathworks Inc., Natick, MA). BPND values for the ROIs were obtained by defining ROIs on the individual MR images that were subsequently used to sample the parametric images.
PK PET
Parametric BPND images of PK were generated from the dynamic PK scan series, using cluster analysis to identify a reference cluster of voxels representing non‐specific PK uptake and thus to provide a tissue input function as previously described [Banati, 2002]. In brief, the cluster analysis divides the brain voxels into 10 clusters based on the shape of their time‐activity curves. The cluster with the time–activity curve which most resembles that of the mean for a normal population of cortical voxels, tested for similarity using a χ2 test (P < 0.05), is selected as the reference cluster. Parametric PK BPND images were anatomically co‐registered, resliced and sampled as for RAC.
Regions targeted
The regions selected were based on the study's aims, the sufficiency of PET camera resolution and the post‐mortem [Guverich and Joyce, 1999; Hurd et al., 2001] and imaging [Ito et al., 2008] studies regarding the density of D2 and D3 receptors in human brain.
Regions were defined manually on each subject's volumetric MRI guided by a three‐dimensional sectional atlas [Duvernoy, 1999]. Sensorimotor striatum (SMST), associative striatum (AST), limbic striatum (LST), anterior cingulate cortex (ACC), posterior cingulate cortex (PCC), anterior prefrontal cortex (aPFC), orbitofrontal cortex (OFC), thalamus, hypothalamus, insula, hippocampus, globus pallidus, substantia nigra, red nucleus, superior and inferior colliculus, bed nucleus of the stria terminalis (BNST) and Amygdala were interrogated.
The striatal subregions were examined according to functional criteria previously described [Martinez et al., 2003]. SMST included the post‐commissural putamen; AST included the pre‐commissural dorsal putamen and caudate and the post‐commissural caudate; LST included the Ventral striatum (VST) consisting of nucleus accumbens, and ventral caudate and putamen rostral to the AC.
Volumes and RAC and PK BPND were calculated after PET coregistration to MRI, providing equal structural VOI and functional ROI object maps for each structure. RAC BPND, PK BPND and volume values for left and right hemisphere regions were averaged.
Statistics
Statistical analyses of MRI, PET and clinical data were performed using SPSS software package (Version 16, SPSS Inc, Chicago, IL, USA). The Kruskal‐Wallis test with post‐test (Dunn's) was employed for between‐group comparisons and Spearman's correlation with multiple comparisons for interrogation of correlations between MRI, PET data and clinical parameters. The significance level was set at P < 0.05.
The MRI and PET data were investigated in two ways: first by examining individual regional volumes, RAC BPND and PK BPND values and second by combining the values of regions that are known to be associated with motor, psychiatric and cognitive disorders [Mega and Cummings, 1994].
Brain regions related to motor functions (Regions‐Motor Group) include SMST, globus pallidus, substantia nigra and red nucleus. Regions related to cognitive functions (Regions‐Cognitive Group) include ACC, PCC, AST, aPFC, OFC, insula and hippocampus. Regions related to psychiatric disorders (Regions‐Psychiatric Group) include ACC, LST, OFC, hypothalamus, insula, hippocampus, BNST and amygdala. Volumes and RAC and PK BPND for the regional group investigations were calculated using formula (1) and (2), respectively. Group = Group of regions related to motor or cognitive or psychiatric function; Tracer = PK or RAC; R= Region.
| (1) |
| (2) |
RESULTS
Regional Individual Analyses
MRI‐VOI
Significant decreases in volume were detected in SMST, aPFC, OFC and substantia nigra in premanifest HD compared with normal controls. Additional significant decreases in volume were detected in ACC, AST, LST, insula, globus pallidus and red nucleus in symptomatic HD. A direct comparison of symptomatic and premanifest HD found relative volume decreases in ACC, SMST, insula, globus pallidus and substantia nigra in the former (Table 2; Figures 1, 2, 3).
Table 2.
Comparison of volume losses, RAC BPND decreases and PK BPND increases between the averaged 45‐CAG repeat premanifest HD and symptomatic HD brain, and normal brain
| Area | Premanifest HD vs normal brain | Symptomatic HD vs normal brain | Premanifest HD vs symptomatic HD | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Volume loss (mm3) (%) | RAC BPND decreases (%) | PK BPND increases (%) | Volume loss (mm3) (%) | RAC BPND decreases (%) | PK BPND increases (%) | Volume loss (mm3) (%) | RAC BPND decreases (%) | PK BPND increases (%) | |
| Anterior cingulate cortex (ACC) | 7.7 | 8.4 | NC | 26.9** | 51.1** | NC | 20.9* | 46.5* | NC |
| Posterior cingulate cortex (PCC) | NC | NC | NC | 15.4 | 14.0 | 17.7 | 14.3 | 11.8 | 15.2 |
| Sensorimotor striatum (SMST)a | 26.7* | 35.4** | 54.2*** | 47.0*** | 58.2*** | 57.0*** | 28.1* | 27.4* | NC |
| Associative striatum (AST)b | 14.5 | 28.9* | 46.7** | 32.7** | 54.5*** | 60.0*** | 21.4 | 36.0** | 21.0 |
| Limbic striatum (LST)c | 19.2 | 17.0 | 26.5 | 30.5* | 40.5** | 38.9* | 14.1 | 24.2 | 14.5 |
| Anterior pre‐frontal cortex (aPFC) | 18.2* | 6.0 | 8.2 | 23.5*** | 15.9 | 28.4* | 6.6 | 11.8 | 23.1* |
| Orbitofrontal cortex (OFC) | 23.0* | NC | NC | 31.4** | 13.9 | 9.6 | 10.8 | 9.8 | 14.3 |
| Thalamus | 6.4 | 6.6 | 22.0 | 8.5 | 13.8 | 23.0 | NC | 7.7% | NC |
| Hypothalamus | 9.5 | 22.5* | 36.6** | 16.0 | 28.5** | 40.6*** | 5.5 | NC | NC |
| Insula | 6.6 | 7.4 | 7.9 | 38.5** | 30.9* | 16.7 | 34.2* | 23.2 | 9.6 |
| Hippocampus | 6.0 | 13.4 | 12.8 | 16.5 | 17.8 | 19.9 | 11.2 | 5.1 | 8.1 |
| Globus pallidus | 18.5 | 21.4 | 28.9 | 44.2** | 44.4*** | 51.2* | 31.1* | 24.6 | 24.7 |
| Substantia nigra | 22.1* | 14.7 | NC | 41.4** | 33.9* | 19.6 | 26.8* | 25.8 | 16.3 |
| Red nucleus | 15.4 | 6.6 | 7.7 | 26.5* | 18.0 | 15.0 | 13.1 | 12.2 | 7.9 |
| Superior colliculus | NC | 20.9 | NC | 10.4 | 21.8 | 12.7 | 7.1 | NC | 8.6 |
| Inferior colliculus | NC | 19.9 | 13.4 | NC | 21.1 | 24.3 | NC | NC | 17.2 |
| Bed nucleus of the stria terminalis (BNST) | NC | 34.4* | 31.2* | 7.5 | 54.3** | 36.3* | NC | 22.3 | 6.5 |
| Amygdala | 10.4 | 28.8* | 43.6* | 13.2 | 29.9* | 45.2* | NC | NC | NC |
| Motor groupd | 22.6* | 25.0* | 37.6** | 42.7** | 45.7*** | 43.8** | 26.6* | 23.5* | 11.1 |
| Cognitive groupe | 10.8 | 9.2 | 10.2 | 26.7** | 24.9* | 21.7* | 17.8 | 17.3 | 13.3 |
| Psychiatric groupf | 10.6 | 12.0 | 14.4 | 27.4* | 29.1* | 21.5* | 18.5 | 17.7 | 9.4 |
Sensorimotor striatum (SMST) = post‐commissural putamen.
Associative striatum (AST) = pre‐commissural dorsal putamen + pre‐commissural dorsal caudate + post‐commissural caudate.
Limbic striatum (LST) = ventral striatum (VST) [VST = nucleus accumbens + ventral caudate rostral to the AC + ventral putamen rostral to the AC].
Motor group = SMST + globus pallidus + substantia nigra + red nucleus.
Cognitive group = ACC + PCC + AST + aPFC + OFC + insula + hippocampus.
Psychiatric group = ACC + LST + OFC + hypothalamus + insula + hippocampus + BNST + amygdale.
Levels of significance: *P < 0.05, ** P < 0.01, *** P < 0.001; NC = no change = percentage difference < 5%.
Figure 1.
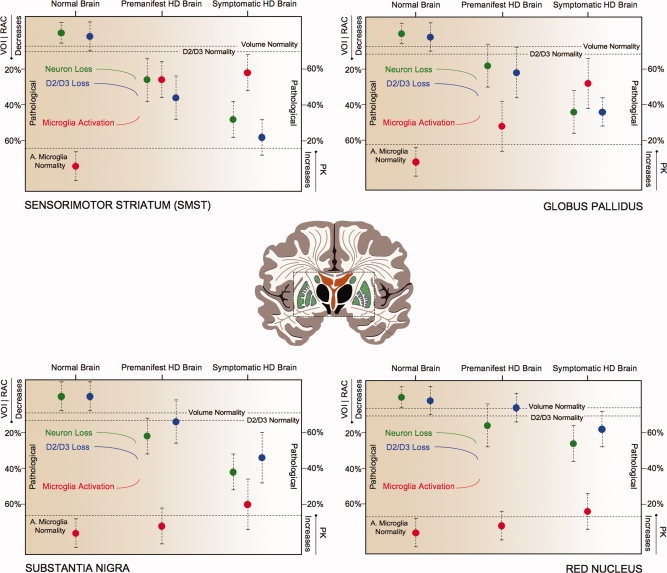
Brain regions involved in motor symptoms of Huntington's disease. Decreases in volume (green), loss of D2/D3 receptors (blue) and increases in activated microglia (red) in the averaged 45‐CAG repeat premanifest HD, the averaged 45‐CAG repeat Symptomatic HD and Normal Brain. Decreases in RAC BPND, mm3 and increases in PK BPND in the premanifest and symptomatic Huntington's disease brain are expressed as percentage changes from the normal brain values. Dotted lines represent normality range defined as mean RAC BPND − SD, mean mm3 − SD, mean PK BPND + SD for D2/D3 receptors, volume and microglia activation, respectively. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 2.
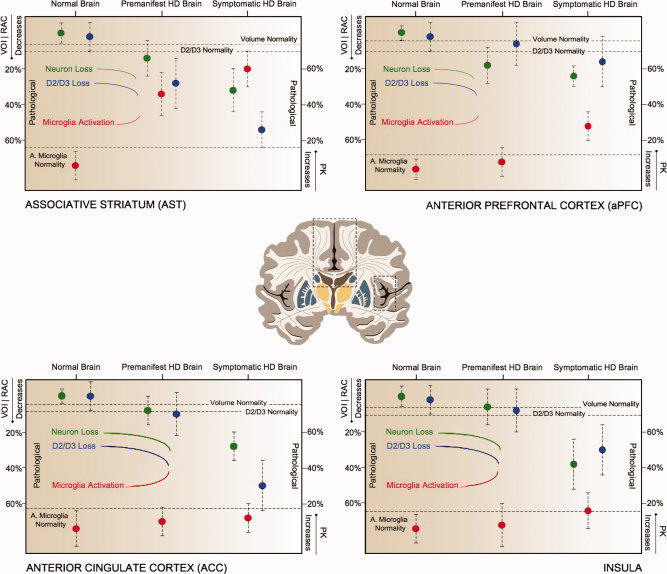
Regions involved in cognitive symptoms of Huntington's disease. Decreases in volume (green), loss of D2/D3 receptors (blue) and increases in activated microglia (red) in the averaged 45‐CAG repeat premanifest HD, the averaged 45‐CAG repeat Symptomatic HD and Normal Brain. Decreases in RAC BPND, mm3 and increases in PK BPND in the premanifest and symptomatic Huntington's disease brain are expressed as percentage changes from the normal brain values. Dotted lines represent normality range defined as mean RAC BPND − SD, mean mm3 − SD, mean PK BPND + SD for D2/D3 receptors, volume and microglia activation, respectively. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 3.
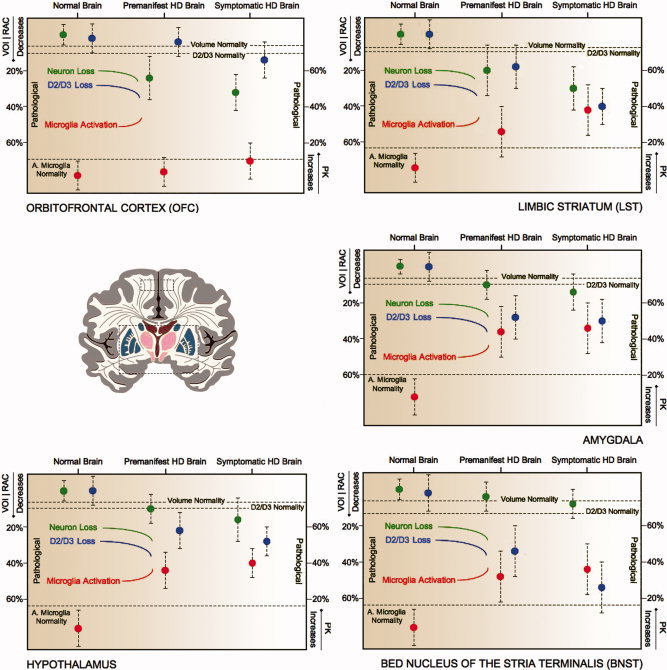
Regions involved in psychiatric symptoms of Huntington's disease. Decreases in volume (green), loss of D2/D3 receptors (blue) and increases in activated microglia (red) in the averaged 45‐CAG repeat premanifest HD, the averaged 45‐CAG repeat Symptomatic HD and Normal Brain. Decreases in RAC BPND, mm3 and increases in PK BPND in the premanifest and symptomatic Huntington's disease brain are expressed as percentage changes from the normal brain values. Dotted lines represent normality range defined as mean RAC BPND − SD, mean mm3 − SD, mean PK BPND + SD for D2/D3 receptors, volume and microglia activation, respectively. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
PET‐RAC ROI
Significant decreases in RAC BPND were detected in SMST, AST, hypothalamus, BNST and amygdala in the premanifest HD with additional decreases in ACC, LST, insula, globus pallidus and substantia nigra in symptomatic HD compared with normal controls. When the symptomatic and premanifest HD groups were directly compared there were relative RAC BPND decreases in ACC, SMST and AST in the former (Table 2; Figures 1, 2, 3).
PET‐PK ROI
Significant increases in PK BPND were detected in SMST, AST, hypothalamus, BNST and amygdala of the premanifest HD cases with additional increases in LST, aPFC and globus pallidus of symptomatic HD compared with normal controls. A direct comparison of symptomatic and premanifest HD groups revealed relative PK BPND increases in aPFC in the former (Table 2; Figures 1, 2, 3).
Regional Group Analyses
Motor group
When the motor group of regions were assessed together, we found significant decreases in volume and RAC BPND and increases in PK BPND in both the premanifest and symptomatic HD groups compared with normal controls. Volume and RAC BPND were relatively decreased in the symptomatic HD compared with the premanifest HD cases (Table 2).
Cognitive group
When the cognitive group of regions were assessed together, we found significant decreases in volume and in RAC BPND and significant PK BPND increases in the symptomatic HD cases compared with normal controls (Table 2).
Psychiatric Group
When the psychiatric group of regions were assessed together, we found significant decreases in volume and in RAC BPND and significant PK BPND increases in the symptomatic HD cases compared with normal controls (Table 2).
Correlations
MRI/PET correlations
No significant correlations are reported.
RAC PET/PK PET correlations
RAC BPND values were negatively correlated with PK BPND values for both HD groups in SMST (premanifest HD: r = −0.6571, P < 0.05; Symptomatic HD: r = −0.68, P < 0.05), hypothalamus (premanifest HD: r = −0.7055, P < 0.05; Symptomatic HD: r = −0.6672, P < 0.01) and BNST (premanifest HD: r = −0.7107, P < 0.05; Symptomatic HD: r = −0.7505, P < 0.05).
Imaging/clinical correlations
For the symptomatic HD, TFC scores negatively correlated with the cognitive group of regions PK BPND values (r = −0.6980, P < 0.05). Disease‐burden scores positively correlated with SMST PK BPND values (r = 0.8176, P < 0.01). UHDRS motor scores correlated positively with the SMST PK BPND values (r = 0.7092, P < 0.05) and negatively with SMST RAC BPND values (r = −0.7465, P < 0.05). The 5‐year probability of developing HD onset positively correlated with PK BPND for both AST (r = 0.8095, P < 0.05) and the cognitive group of regions (r = 0.8810, P < 0.01) in premanifest HD cases (Figures 4 and 5).
Figure 4.
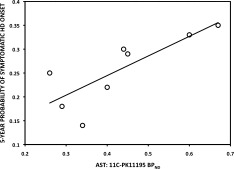
Correlation between AST 11C‐PK11195 BPND and the predicted 5‐year probability of symptomatic HD onset in the averaged 45‐CAG repeat premanifest HD brain (r = 0.8095, P = 0.0218).
Figure 5.
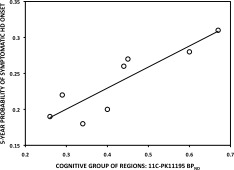
Correlation between Cognitive Group of Regions 11C‐PK11195 BPND and the predicted 5‐year probability of symptomatic HD onset in the averaged 45‐CAG repeat premanifest HD brain (r = 0.8810, P = 0.0072).
DISCUSSION
In this study, we have created averaged brain‐imaging profiles for CAG repeat‐matched premanifest and symptomatic HD groups in order to investigate changes in volume, post‐synaptic dopamine D2/D3 receptor binding and microglial activation in brain regions known to be involved in the motor, cognitive and psychiatric features of HD. The imaging data were interrogated by targeting individual regions as well as combining them into groups according to their broad involvement in motor, cognitive and psychiatric functions. The regional group analyses is an estimation and in some cases overlapped as some, if not all, of the regions share more than one functional component. Where applicable, we have accounted the values for a region in more than one group. For example dysfunctions of the ACC has been connected with both cognitive and psychiatric dysfunction even though the cognitive component seems to be more pronounced [Bush et al., 2000]. With regards to the striatum, in this study, we have applied and sampled a striatal object map that considers the anatomical sections with respect to functionality as described by Martinez et al. [ 2003]. This is a significant improvement in comparison to the traditional way of sampling the striatum by using a strict anatomical definition as it allows us to calculate separately the values for the motor (SMST), cognitive (AST) and psychiatric (LST) components of this brain region. Limitations of the study include the small sample sizes of the groups under investigation and the resolution of PET camera for detecting changes in very small regions.
The Premanifest HD Striatum
We found that in premanifest HD cases, higher levels of activated microglia in the AST correlated with a higher probability of HD onset over the next 5 years. We did not observe any similar correlation in SMST or LST or in any other individual region under investigation and, therefore, it is reasonable to assume that increased levels of activated microglia in the cognitive component of the striatum may be a good predictor of clinical disease onset. Moreover, the functional and structural modalities under investigation were not similarly affected in the striatal functional subdivisions in the premanifest HD cases. SMST and AST seem to be affected whereas none of the changes under investigation in LST reached statistical significance. Taken together these findings suggest, that the more dorsal part of the striatum (related to motor and cognitive function) is affected earliest in HD, whereas the ventral part of the striatum (related to psychiatric function) is affected later, possibly after the onset of motor symptoms.
The Motor Regions
From the regions comprising the motor group, SMST and substantia nigra showed significant changes in the premanifest HD, whereas globus pallidus and red nucleus did not show any changes. In symptomatic HD, we detected volume loss in all four regions, and changes from PET in SMST, globus pallidus and substantia nigra and globus pallidus. Furthermore, in SMST, increased levels of activated microglia correlated with D2/D3 binding loss in both premanifest and symptomatic HD cases and higher disease‐burden scores correlated with higher levels of activated microglia in the symptomatic HD group. The worst UHDRS motor scores correlated with increased levels of activated microglia and D2/D3 receptor binding loss in the symptomatic HD group.
These findings support the primary involvement of SMST dysfunction in the motor component of the disease which appears early and the biological substrate responsible could be related to the close relationship between D2/D3 receptor loss and increased levels of activated microglia. Also, the levels of microglia activation appear to be related to the severity of the clinical disease once the motor symptoms have been established.
The Cognitive Regions
When the cognitive group of regions was assessed together we found significant changes in symptomatic HD for all the imaging modalities under investigation. In the premanifest HD, AST, aPFC and OFC showed significant changes, whereas ACC, PCC, insula and hippocampus did not show any statistical significant differences. The symptomatic HD cases showed additional changes in ACC, insula, in the AST and aPFC.
These findings provide evidence for the central early role of AST pathology in HD, which may explain some of the cognitive dysfunction seen early in the disease [Ho et al., 2003]. Furthermore, the loss of D2/D3 receptors in the ACC may also underlie in part the cognitive dysfunction seen in the disease as in this area D2/D3 receptors modulate executive functioning [Lumme et al., 2007]. Insula, OFC and aPFC are regions involved in many ‘high‐level’ cognitive processes [Peinemann et al., 2005; Ramnani and Owen, 2004; Rosas et al., 2005] and their dysfunction reported in this study could also contribute to the cognitive dysfunction seen in HD.
As discussed earlier, the increased levels of activated microglia in AST, but not in SMST or elsewhere, proved to be a strong regional predictor of developing symptomatic onset over the next 5 years. However, when we investigated for a similar correlation averaging the PK BPND per unit of volume across all the sampled regions with possible connection to cognitive disease, this positive correlation became even stronger. This intriguing finding provides evidence that increased levels of activated microglia in the regional network with possible connection to cognitive disease could serve as a good predictor of clinical onset, highlighting the importance of a regular cognitive assessment from an early premanifest stage.
The importance of the combined assessment of the cognitive group of regions is highlighted even further in the symptomatic HD cases as in these, increased levels of activated microglia significantly correlated with worse TFC scores and consequently with the capacity to maintain a job, with the need for assistance in dealing with finance or domestic chores and with the level of care needed for the patients. However, these findings from functional imaging need to be confirmed with neuropsychological testing in a further study.
The Psychiatric Regions
Assessing the psychiatric regions together, volume and D2/D3 binding were significantly reduced and levels of activated microglia significantly increased only in symptomatic HD cases. The function of ACC, OFC, insula and hippocampus along with their role in cognitive function, share a psychiatric component and OFC is known to be involved in emotion and reward and insula in anger, fear, disgust, happiness and sadness. Also, D2/D3 receptor loss in ACC has been correlated with the positive symptoms in Schizophrenia [Suhara et al., 2002]. Similar symptoms are observed in HD and the D2/D3 receptor loss that we found in the ACC could play a role in the development of some of the psychiatric symptoms observed in these patients.
We also found significant loss of D2/D3 binding and increased levels of activated microglia in the hypothalamus, BNST and amygdala of the premanifest HD cases.
While the hypothalamic involvement in HD has been extensively discussed in a previous report [Politis et al., 2008], BSTN and amygdala are limbic structures well connected to each other and to hypothalamus and brainstem and they mediate many autonomic and behavioural responses to aversive or threatening stimuli [Walker et al., 2003]. Their dysfunction may partly explain the prevalent and early emergence of psychiatric problems in HD.
Interpretation of Atrophy
One of the main issues with PET studies that this combined neuroimaging approach wanted to address, is the interpretation of the functional PET data with regards to volume loss. In other words, are the D2/D3 binding loss and increases in activated microglia a unique pathogenic event or are they reflecting structural changes and the secondary effect of atrophy? To address this important question, grey matter volumes were extracted for each region under investigation. These volumes were the same as used for all MRI, RAC BPND and PK BPND sampling. Hence, we investigated whether MR volume changes associate with PET BPND values changes between normality and disease. Examining each of the regions individually or as a combined group of regions none of these associations reached statistical significance and in many cases the nonparametric correlation coefficient was in the opposite direction providing supporting evidence that the PET BPND values are possibly independent of the reductions in volume.
D2 Receptors
Raised dopaminergic neurotransmission has been implicated in HD as underlying hyperkinetic symptoms and the psychotic features. The cellular localization of dopamine D2 receptors has been demonstrated on both pyramidal neurons and nonpyramidal interneurons, which use GABA as an inhibitory transmitter [Le Moine and Gaspar, 1998]. Unfortunately, RAC cannot discriminate D2 receptors on between different types of neurons. However, loss of inhibitory GABAergic neurons may cause dopaminergic activation, while loss of dopaminergic receptors may lead to compensatory hyperactivity in the remaining neurons and this process, as it is shown in this study, begins in the premanifest stage and intensifies with the progression of the illness affecting regional networks responsible for the motor, cognitive and psychiatric component of the disease.
Activated Microglia
In this study we have also demonstrated a close association between the levels of microglial activation and clinical scales of disease severity and motor dysfunction, measures of daily living and most importantly the 5‐year prediction of HD onset. Pathologically activated microglia could result in neuronal death through the release of a variety of cytokines and other neurotoxic factors [Nakanishi, 2003] and could potentially trigger a cell‐autonomous immune reaction. Also, monocytes are the peripheral counterparts of microglia and it was recently shown that monocytes from HD patients express mutant huntingtin and are dysfunctional even in the premanifest disease stage in both HD patients and mice [Björkqvist et al., 2008]. Our findings that increased levels of activated microglia in premanifest disease and in areas related to motor, cognitive and psychiatric functions along with the aforementioned clinical correlations, enhance the possible pathological importance of activated microglia in the development of clinical signs and symptoms and disease pathogenesis.
CONCLUSIONS
The identification of new biomarkers is a constant need for HD and we believe that a combined neuroimaging approach has the potential to serve this role. This study provides an example of how combined neuroimaging could be used in this way, as we were able first to relate PK PET to MRI findings and show that the increased levels of activated microglia represent a potential pathogenic effect that is not simply secondary to structural changes. Then we were able to correlate the microglial activation effect with the 5‐year probability of premanifest HD gene carriers developing the disease [Langbehn et al., 2004]. However, for such a biomarker to be useful, larger longitudinal studies are needed in order for normality values to be derived and the range of variance to be ascertained in establishing the normal biological range. These biomarkers identify potential pathogenic processes and will be useful for studying the pharmacological responses to novel therapies.
In conclusion, this study provides evidence for the involvement of multiple brain regions in the premanifest and symptomatic HD, although further studies are needed to better understand the relation of these changes to clinical parameters. The use of multimodal imaging has a potential as a biomarker for HD and in this study we showed that imaging activated microglia in the AST and related regions associated with cognitive function provides a prognostic tool for the disease clinical onset. Further studies are needed to investigate the amount of time ahead of disease that activated microglia in these regions can predict clinical disease as well as their prognostic role in the disease progression.
Acknowledgements
We would specially like to thank the patients and their families. Nicola Pavese and David J. Brooks received consultancy fees from GE Healthcare.
REFERENCES
- Andrews TC, Weeks RA, Turjanski N, Gunn RN, Watkins LH, Sahakian B, Hodges JR, Rosser AE, Wood NW, Brooks DJ ( 1999): Huntington's disease progression. PET and clinical observations. Brain 122: 2353–2363. [DOI] [PubMed] [Google Scholar]
- Banati RB ( 2002): Visualising microglial activation in vivo. GLIA 40: 206–217. [DOI] [PubMed] [Google Scholar]
- Björkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, Raibon E, Lee RV, Benn CL, Soulet D, Magnusson A, Woodman B, Landles C, Pouladi MA, Hayden MR, Khalili‐Shirazi A, Lowdell MW, Brundin P, Bates GP, Leavitt BR, Möller T, Tabrizi SJ ( 2008): A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J Exp Med 205: 1869–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohanna I, Georgiou‐Karistianis N, Hannan AJ, Egan GF ( 2008): Magnetic resonance imaging as an approach towards identifying neuropathological biomarkers for Huntington's disease. Brain Res Rev 58: 209–225. [DOI] [PubMed] [Google Scholar]
- Bush G, Luu P, Posner MI ( 2000): Cognitive and emotional influences in anterior cingulate cortex. Trends Cogn Sci 4: 215–222. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB ( 2001): In‐vivo measurement of activated microglia in dementia. Lancet 358: 461–467. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB ( 2005): ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8: 752–758. [DOI] [PubMed] [Google Scholar]
- Duvernoy HM ( 1999): The Human Brain: Surface, Blood Supply, and Three‐Dimensional Sectional Anatomy. New York: Springer‐Verlag Wien. [Google Scholar]
- Gunn RN, Lammertsma AA, Hume SP, Cunningham VJ ( 1997): Parametric imaging of ligand‐receptor binding in PET using a simplified reference region model. Neuroimage 6: 279–287. [DOI] [PubMed] [Google Scholar]
- Gurevich EV, Joyce JN ( 1999): Distribution of dopamine D3 receptor expressing neurons in the human forebrain: Comparison with D2 receptor expressing neurons. Neuropsychopharmacology 20: 60–80. [DOI] [PubMed] [Google Scholar]
- Ho AK, Sahakian BJ, Brown RG, Barker RA, Hodges JR, Ané MN, Snowden J, Thompson J, Esmonde T, Gentry R, Moore JW, Bodner T; NEST‐HD Consortium ( 2003): Profile of cognitive progression in early Huntington's disease. Neurology 61: 1702–1706. [DOI] [PubMed] [Google Scholar]
- Hurd YL, Suzuki M, Sedvall GC ( 2001): D1 and D2 dopamine receptor mRNA expression in whole hemisphere sections of the human brain. J Chem Neuroanat 22: 127–137. [DOI] [PubMed] [Google Scholar]
- Ito H, Takahashi H, Arakawa R, Takano H, Suhara T ( 2008): Normal database of dopaminergic neurotransmission system in human brain measured by positron emission tomography. Neuroimage 39: 555–565. [DOI] [PubMed] [Google Scholar]
- Jakel RJ, Maragos WF ( 2000): Neuronal cell death in Huntington's disease: A potential role for dopamine. Trends Neurosci 23: 239–245. [DOI] [PubMed] [Google Scholar]
- Kieburtz K, MacDonald M, Shih C, Feigin A, Steinberg K, Bordwell K, Zimmerman C, Srinidhi J, Sotack J, Gusella J, Shoulson I. ( 1994): Trinucleotide repeat length and progression of illness in Huntington's disease. J Med Genet 31: 872–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR; International Huntington's Disease Collaborative Group ( 2004): A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clin Genet 65: 267–277. [DOI] [PubMed] [Google Scholar]
- Le Moine C, Gaspar P ( 1998): Subpopulations of cortical GABAergic interneurons differ by their expression of D1 and D2 dopamine receptor subtypes. Brain Res Mol Brain Res 58: 231–236. [DOI] [PubMed] [Google Scholar]
- Lumme V, Aalto S, Ilonen T, Någren K, Hietala J ( 2007). Dopamine D2/D3 receptor binding in the anterior cingulate cortex and executive functioning. Psychiatry Res 156: 69–74. [DOI] [PubMed] [Google Scholar]
- Martinez D, Slifstein M, Broft A, Mawlawi O, Hwang DR, Huang Y, Cooper T, Kegeles L, Zarahn E, Abi‐Dargham A, Haber SN, Laruelle M ( 2003): Imaging human mesolimbic dopamine transmission with positron emission tomography. Part II: Amphetamine‐induced dopamine release in the functional subdivisions of the striatum. J Cereb Blood Flow Metab 23: 285–300. [DOI] [PubMed] [Google Scholar]
- Mega MS, Cummings JL ( 1994): Frontal‐subcortical circuits and neuropsychiatric disorders. J Neuropsychiatry Clin Neurosci 6: 358–370. [DOI] [PubMed] [Google Scholar]
- Nakanishi H ( 2003): Microglial functions and proteases. Mol Neurobiol 27: 163–176. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F ( 2005): Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308: 1314–1318. [DOI] [PubMed] [Google Scholar]
- Pavese N, Andrews TC, Brooks DJ, Ho AK, Rosser AE, Barker RA, Robbins TW, Sahakian BJ, Dunnett SB, Piccini P ( 2003): Progressive striatal and cortical dopamine receptor dysfunction in Huntington's disease: A PET study. Brain 126: 1127–1135. [DOI] [PubMed] [Google Scholar]
- Peinemann A, Schuller S, Pohl C, Jahn T, Weindl A, Kassubek J ( 2005): Executive dysfunction in early stages of Huntington's disease is associated with striatal and insular atrophy: A neuropsychological and voxel‐based morphometric study. J Neurol Sci 239: 11–19. [DOI] [PubMed] [Google Scholar]
- Penney JB Jr, Vonsattel JP, MacDonald ME, Gusella JF, Myers RH ( 1997): CAG repeat number governs the development rate of pathology in Huntington's disease. Ann Neurol 41: 689–692. [DOI] [PubMed] [Google Scholar]
- Politis M, Pavese N, Tai YF, Tabrizi SJ, Barker RA, Piccini P ( 2008): Hypothalamic involvement in Huntington's disease: An in vivo PET study. Brain 131: 2860–2869. [DOI] [PubMed] [Google Scholar]
- Ramnani N, Owen AM ( 2004): Anterior prefrontal cortex: Insights into function from anatomy and neuroimaging. Nat Rev Neurosci 5: 184–194. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Hevelone ND, Zaleta AK, Greve DN, Salat DH, Fischl B ( 2005). Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology 65: 745–747. [DOI] [PubMed] [Google Scholar]
- Roze E, Saudou F, Caboche J ( 2008): Pathophysiology of Huntington's disease: from huntingtin functions to potential treatments. Curr Opin Neurol 21: 497–503. [DOI] [PubMed] [Google Scholar]
- Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, Bhide PG, Vonsattel JP, DiFiglia M ( 2001): Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol 60: 161–172. [DOI] [PubMed] [Google Scholar]
- Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ ( 2005): Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol 171: 1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinks TJ, Jones T, Bloomfield PM, Bailey DL, Miller M, Hogg D, Jones WF, Vaigneur K, Reed J, Young J, Newport D, Moyers C, Casey ME, Nutt R ( 2000): Physical characteristics of the ECAT EXACT 3D positron tomograph. Phys Med Biol 45: 2601–2618. [DOI] [PubMed] [Google Scholar]
- Suhara T, Okubo Y, Yasuno F, Sudo Y, Inoue M, Ichimiya T, Nakashima Y, Nakayama K, Tanada S, Suzuki K, Halldin C, Farde L ( 2002): Decreased dopamine D2 receptor binding in the anterior cingulate cortex in schizophrenia. Arch Gen Psychiatry 59: 25–30. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Langbehn DR, Leavitt BR, Roos RA, Durr A, Craufurd D, Kennard C, Hicks SL, Fox NC, Scahill RI, Borowsky B, Tobin AJ, Rosas HD, Johnson H, Reilmann R, Landwehrmeyer B, Stout JC; the TRACK‐HD investigators ( 2009): Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK‐HD study: Cross‐sectional analysis of baseline data. Lancet Neurol 8: 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group ( 1993): A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72: 971–983. [DOI] [PubMed] [Google Scholar]
- van Oostrom JC, Dekker M, Willemsen AT, de Jong BM, Roos RA, Leenders KL ( 2009): Changes in striatal dopamine D2 receptor binding in pre‐clinical Huntington's disease. Eur J Neurol 2009 16: 226–231. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr ( 1985): Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44: 559–577. [DOI] [PubMed] [Google Scholar]
- Walker DL, Toufexis DJ, Davis M ( 2003): Role of the bed nucleus of the stria terminalis versus the amygdala in fear, stress, and anxiety. Eur J Pharmacol 463: 199–216. [DOI] [PubMed] [Google Scholar]