

Abstract
Brain morphometry measures derived from magnetic resonance imaging (MRI) are important biomarkers for Alzheimer's disease (AD). The objective of the present study was to test whether we could improve morphometry‐based detection and prediction of disease state by use of white matter/gray matter (WM/GM) signal intensity contrast obtained from conventional MRI scans. We hypothesized that including WM/GM contrast change along with measures of atrophy in the entorhinal cortex and the hippocampi would yield better classification of AD patients, and more accurate prediction of early disease progression. T 1‐weighted MRI scans from two sessions approximately 2 years apart from 78 participants with AD (Clinical Dementia Rating (CDR) = 0.5–2) and 71 age‐matched controls were used to calculate annual change rates. Results showed that WM/GM contrast decay was larger in AD compared with controls in the medial temporal lobes. For the discrimination between AD and controls, entorhinal WM/GM contrast decay contributed significantly when included together with decrease in entorhinal cortical thickness and hippocampal volume, and increased the area under the curve to 0.79 compared with 0.75 when using the two morphometric variables only. Independent effects of WM/GM contrast decay and improved classification were also observed for the CDR‐based subgroups, including participants converting from either a non‐AD status to very mild AD, or from very mild to mild AD. Thus, WM/GM contrast decay increased diagnostic accuracy beyond what was obtained by well‐validated morphometric measures alone. The findings suggest that signal intensity properties constitute a sensitive biomarker for cerebral degenration in AD. Hum Brain Mapp 34:2775–2785, 2013. © 2012 Wiley Periodicals, Inc.
Keywords: cerebral cortex, tissue contrast, MRI T1‐weighted signal intensity, cortical thickness, disease prediction, entorhinal, hippocampus
INTRODUCTION
Structural biomarkers derived from magnetic resonance imaging (MRI) in vivo have proven sensitive in detecting cerebral tissue alterations in individuals with Alzheimer's disease (AD) [Barnes et al.,2009; Devanand et al.,2007; Dickerson et al.,2001; Fjell et al.,2010; Jack et al.,1992], providing pertinent information aiding early diagnosis [Dubois et al.,2007]. However, even though noninvasive MRI‐based biomarkers perform at least as good as established cerebrospinal fluid markers in classification and prediction of cognitive deterioration [Vemuri et al.,2009], there is still need for improved accuracy in the early phase of AD [Frisoni et al.,2010], when the potential for intervention is highest. Hence, additional structural markers improving diagnosis and prediction of disease progression, especially without additional examinations of the patients, would be highly valuable.
The signal intensity of T 1‐weighted MRI scans is related to tissue integrity [Davatzikos and Resnick,2002; Salat et al.,2009; Westlye et al.,2010], potentially serving as a putative biomarker of structural degenerative changes. T 1‐weighted signal intensity reflects the underlying tissue proton relaxation times and stems mainly from myelin, whereas cell density and cell properties contribute to a lesser extent [Barbier et al.,2002; Clark et al.,1992; Eickhoff et al.,2005; Walters et al.,2003]. In AD, the neuropathological characteristics include in particular the presence of somatic neurofibrillary tangles and dendritic neuropil threads [Braak and Braak,1991], and may be related to premature oligendrocyte dysfunction [Braak and Braak,1996]. Recently, two cross‐sectional studies have demonstrated tissue signal properties alterations in AD, specifically in the signal intensity contrast between white (WM) and gray (GM) matter [Salat et al.,2011; Westlye et al.,2009]. However, no studies have investigated whether longitudinal tissue contrast changes can be used to distinguish AD patients from controls, and to differentiate subgroups of AD patients. Furthermore, although longitudinal brain imaging have been very useful in detecting morphological changes associated with AD [de Leon et al.,2006; Fjell et al.,2010; Fotenos et al.,2005; Jack et al.,2009; McDonald et al.,2009], reports using WM/GM contrast to track degeneration over time are lacking.
In the present study, the aim was to test whether WM/GM contrast changes can be used to detect early signs of AD and to predict disease progression beyond what can be obtained with standard and well‐validated morphometric measures of entorhinal cortical thickness and hippocampal volume alone. The entorhinal and hippocampal regions of interests (ROIs) were selected based on a substantial body of evidence pointing to the sensitivity of these measures in detecting early and significant changes in AD [e.g., Fjell et al.,2010]. We employed an automated segmentation procedure (FreeSurfer) to first reconstruct representations of the WM/GM boundary and the pial surface at two different time points. Intensity values were sampled from WM and GM tissue at a given distance from the WM/GM boundary, before we calculated WM/GM contrast at each vertex across the surface.
MATERIALS AND METHODS
Participants
Data from 150 participants were obtained from the publicly accessible Open Access Series of Imaging Studies (OASIS, http://www.oasis-brains.org). Recruitment, screening, MRI acquisition details, and previous reports using the current patient population are described and referred to in depth elsewhere [Marcus et al.,2010]. Subjects participated in accordance with guidelines of the Washington University Human Studies Committee. Clinical assessment yielded Clinical Dementia Rating (CDR) scores for all participants [Berg,1988]; a global CDR of 0 was taken to indicate absence of dementia, whereas a CDR of 0.5, 1, and 2 represents very mild, mild, and moderate dementia, respectively (all referred to as ‘participants with AD’ or simply ‘AD’ in the present article, in coherence with the official naming convention used in OASIS). One participant from the control group was excluded due to poor image quality at the second scanning session. A final sample of 149 participants (87 females), ranging 60–96 years of age (mean: 75.4 years, standard deviation (SD) =7.6 years), all right‐handed, was included. Demographic details for the 149 participants, including the different subgroupings based on CDR scores, are presented in Table 1. Appropriate statistical tests revealed no significant differences in age and socioeconomic status between AD (n = 64) and controls (n = 71), but the number of women, education, Mini‐mental State Examination scores, and time between scanning occasions were lower in AD.
Table 1.
Demographics of participants at baseline
| Group | N | % F | Age (SD/range) | Education | SES | MMSE | Time between scans |
|---|---|---|---|---|---|---|---|
| Controls | 71 | 69 | 75.4 (8.3/60–93) | 15.1 (2.7) | 2.4 (1.1) | 29.2 (.9) | 2.0 (.7) |
| AD | 64 | 44 | 75.1 (6.7/61–96) | 13.7 (2.9) | 2.8 (1.2) | 25.3 (3.3) | 1.8 (.5) |
| AD subgroups | |||||||
| CDR = 1 | 10a | 50 | 73.5 (7.1/61–83) | 13.1 (2.8) | 2.8 (1.4) | 23 (3.5) | 1.6 (0.6) |
| CDR = 0.5 | 38 | 39 | 75.7 (6.6/62–90) | 13.5 (3.1) | 2.9 (1.2) | 26.3 (2.9) | 1.9 (0.6) |
| Convertedb | 29c | 59 | 76.1 (7.2/64–96) | 14.9 (2.6) | 2.1 (1.1)d | 26.8 (3.3) | 2.0 (0.9) |
Means with standard deviations in parentheses unless otherwise specified.
Two participants change CDR score from 1 to 0.5.
This group consists of persons changing CDR score (a) from 0 to 0.5 (n = 13, 71% female, mean age (SD) 77.1 (7.7), (b) from 0.5 to 1 [n = 13, 46% female, mean age (SD) = 72.7 (4.6)]. The last of these two groups is also included in the AD group.
One participant changing CDR score from 0.5 to 0 was excluded.
Four persons missing SES scores.
% F = percent females. “Time between scans” and “Education” values are in years. SES = Socioeconomic status, MMSE = Mini‐Mental State Examination. Analysis of variance was performed testing differences in means between groups as follows: AD versus controls, CDR = 0.5 versus controls, and converted versus controls (mean difference not tested between controls and CDR = 1). Bold = significantly different from controls at P < 0.05.
MRI Acquisition and Analysis
Three to four individual T 1‐weighted magnetization prepared rapid gradient‐echo (MPRAGE) images were acquired on a 1.5‐T Vision scanner (Siemens, Erlangen, Germany) at each of the two imaging sessions using the following parameters: repetition time/echo time/inversion time/delay = 9.7/4/20/200 msec, flip angle = 10°, 256 × 256 (1 × 1 mm2) resolution, 128 sagittal 1.25 mm slices without gaps.
The datasets were processed with FreeSurfer 4.5 (http://surfer.nmr.mgh.harvard.edu); the various methods for automated tissue segmentation, surface‐based cortical thickness estimations, automated whole‐brain segmentation procedure, and spherical interindividual surface alignment are described in detail elsewhere [Dale et al.,1999; Fischl and Dale,2000; Fischl et al.,2002, 2004a, 1999, 2004b].
The volumes were motion corrected and averaged across acquisitions within each time point using the three MPRAGEs of highest quality in all but three instances in which two volumes were used, and processed according to FreeSurfer's longitudinal processing scheme [Reuter and Fischl,2011; Reuter et al.,2010]. Briefly, the data from both time points are initially processed cross‐sectionally, before a base template is created from an average of the two time points. The first and second time points are then registered to this unbiased template, reducing the random variation in the processing procedure and increasing the robustness and sensitivity of the longitudinal analysis. Volumes from the longitudinally processed time points 1 and 2 were corrected for intensity nonuniformity prior to sampling tissue intensities to reduce the influence of inhomogeneity [Sled et al.,1998]. Intensity values were sampled 1 mm subjacent to the WM/GM boundary, and at a distance from the WM/GM boundary extending into the GM at a point reaching 35% of the cortical thickness, respectively. These sampling parameters have proved sensitive in cross‐sectional analyses of this sample [Salat et al.,2011]. The WM/GM contrast was computed by dividing the WM by the GM values and projecting the ratios onto a common surface. The use of a ratio metric normalizes tissue values with regards to local imaging environment as closely neighboring WM and GM voxel intensities are expected to be similarly influenced by scanner and sequence‐related noise. The contrast maps at each time point were smoothed with a circularly symmetric Gaussian kernel across the surface with a full width at half maximum of 5 mm. To account for nonidentical intervals between scans, surface maps representing the annual rate of change in WM/GM contrast was calculated (the difference in the contrast measurements between the two time points, divided by the time between scan sessions in years). The change rate maps were mapped to a common surface, smoothed with a kernel of full width at half maximum of 15 mm [Fischl et al.,1999], and fed to statistical analyses. The same preprocessing procedure was performed on the cortical thickness maps.
An automated whole‐brain segmentation procedure [Fischl et al.,2002] was employed to obtain volumetric measures of the hippocampal formation, and an automated surface‐based labeling algorithm yielded entorhinal cortex parcellations [Desikan et al.,2006; Fischl et al.,2004b]. Annual atrophy of hippocampal volumes were computed as above.
Statistical Analysis
Degree of annual change in WM/GM contrast and thickness, respectively, were first tested separately in AD and controls, and then combined assessing the effect of group on change, using General Linear Models at each vertex with age and sex as covariates. The data were tested against an empirical null distribution of maximum cluster size across 10,000 iterations using Z Monte Carlo simulations as implemented in FreeSurfer [Hagler et al.,2006; Hayasaka and Nichols,2003] synthesized with a cluster‐forming threshold of P < 0.05 (two‐tailed), yielding clusters corrected for multiple comparisons across the surface.
We extracted annual changes in WM/GM contrast and cortical thickness from the entorhinal cortex, as well as changes in hippocampal volume, for each participant. Left and right hemisphere ROI‐values were averaged and Z transformed. To assess the associations between these ROIs, Pearson correlation coefficients were computed. We tested three different logistic regression models discriminating AD from controls, as well as CDR‐based subgroups from controls, were tested yielding standardized regression coefficients, P values, and odds ratios: first, model 1 consisted of decrease in entorhinal cortical thickness and hippocampal volume only; second, to assess the effect of WM/GM contrast changes, we first tested model 2 consisting of WM/GM contrast decay from the entorhinal cortex, and the predictor in model 1 with the lowest odds ratio (hippocampal atrophy in all instances) in order to keep the number of predictors constant. Finally, in model 3, we included all three predictors. Age and sex were included as covariates in all models. To assess the discrimination accuracy between AD and controls, and between CDR‐based subgroups and controls across the three models, the logistic regressions were rerun with a leave‐one‐out cross‐validation scheme using Matlab (Mathworks, Nattick, Mass). Based on the predicted values from the leave‐one‐out cross‐validation, we plotted receiver operating characteristic (ROC) curves and calculated the area under the curve (AUC), as well as sensitivity and specificity using a cutoff value of 0.5. The independence of WM/GM contrast change as a predictor of AD classification was assessed in the multivariable logistic regression model and differences in AUC were not additionally tested [Vickers et al.,2011]. Ninety‐five percent confidence intervals of AUCs were computed by methods described by DeLong et al. 1988 using the pROC package in R [Robin et al.,2011]. P < 0.05 (two‐tailed) was considered to indicate significant effects. To evaluate regional differences in classification accuracy outside the predefined ROIs, we repeated the logistic regression for the three models in a vertex‐wise manner, calculating AUC at each vertex across the cortical mantle, and displaying the resulting AUC maps superimposed on semi‐inflated brains.
RESULTS
Longitudinal WM/GM Contrast Decay, and Cortical and Hippocampal Atrophy in AD versus Controls
Mean annual WM/GM contrast change (SD and percent change in parenthesis) in the entorhinal cortex label was −0.01 arbitrary units (0.015, −0.7%) per year for AD and 8 × 10−4 arbitrary units (0.017, 0.06%) for controls, respectively. For entorhinal cortical thickness, mean annual atrophy (SD and percent change) was −0.06 mm (0.07, −2.1%) and −0.02 mm (0.07, −0.6%) for AD and controls, respectively. The annual hippocampal atrophy across hemispheres was −84 mm3 (69, −3.2%) and −37 mm3 (56, −1.1%) for AD and controls, respectively. The hippocampal atrophy was significantly different from zero in both groups (P < 0.001), and also significantly different between groups (P < 0.001). The WM/GM contrast changes and cortical atrophy were tested in a voxelwise manner, and Figure 1A shows clusters of significant annual WM/GM contrast change and cortical atrophy for AD and controls separately (P < 0.05, corrected). For AD, there was a significant annual WM/GM contrast decay in large parts of the temporal lobe, bilaterally, and extending laterally across insula into inferior and middle frontal gyri, and supramarginal gyrus in the left hemisphere, where also a separate medial superior frontal cluser was found. One smaller cluster comprising parts of the right occipital, lingual, and posterior parts of the fusiform cortex showed a significant annual increase in WM/GM contrast in the controls. Significant clusters of annual cortical thinning was observed in large parts of the brain in both hemispheres for AD, including medial temporal lobe, precuneus, and the cingulate, as well as large parts of lateral cortices in all four lobes. In controls, no significant clusters of cortical thinning were observed after correction for multiple comparisons.
Figure 1.
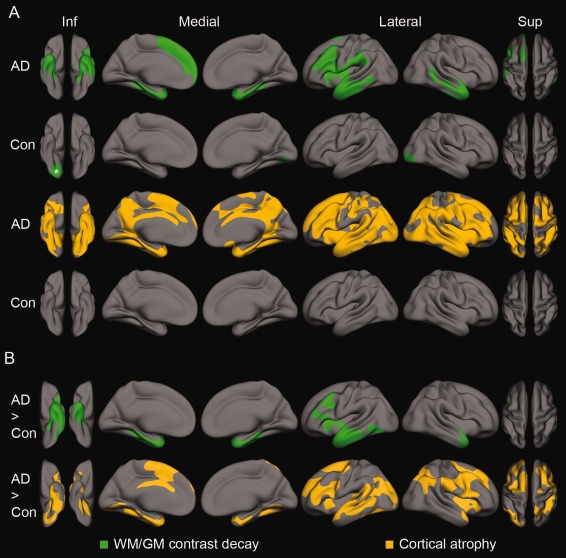
Spatial clusters from surface‐based General Linear Models testing degree of annual change in WM/GM contrast and cortical thickness (A), and the effects of group (AD versus controls, B). A: WM/GM contrast (upper row, green) and cortical thickness (bottom row, orange) change for AD and controls, respectively. All clusters indicate a significant annual decay, except the WM/GM cluster in the controls marked with an asterisk, which showed an significant increase. B: WM/GM contrast (upper row, green) and cortical thickness (bottom row, orange). All clusters indicate areas of larger annual decay in AD than controls. Inf, inferior view; Sup, superior view; Con, controls. P < 0.05, corrected for multiple comparisons.
Figure 1B (upper part, in green) reveals clusters of significantly larger annual WM/GM contrast decay in AD compared with controls in the anterior temporal lobes, bilaterally (P < 0.05, corrected). In the left hemisphere, the significant effects comprised the entire entorhinal cortex and the temporal pole, all but the posterior most part of the parahippocampal and inferior temporal areas, and the anterior parts of the fusiform cortex, middle, and superior temporal cortices, extending across parts of insula and the inferior and middle frontal gyrus. The right hemisphere cluster also covered the entorhinal cortex and the temporal pole, and all but the posterior most part of the parahippocampal gyrus, as well as anterior parts of the fusiform cortex, and inferior, middle, and superior temporal cortices. The clusters remained significant (P < 0.05) when including cortical thickness at baseline as a covariate to control for initial differences in thickness between groups due to either atrophy or segmentation errors following a local blurring of the WM/GM boundary [Westlye et al.,2009], or both.
Figure 1B (lower part, in orange) displays six clusters of more pronounced thinning in AD (P < 0.05, corrected). For the left hemisphere, the effect was observed in large parts of the temporal lobe (entirely covering the entorhinal cortex), parts of the inferior parietal and supramarginal areas, and medial and lateral parts of the frontal lobe (including the cingulate gyrus). In the right hemisphere, the clusters comprised parts of the medial temporal lobe (including the entorhinal cortex) and posterior parts of the inferior temporal gyrus, insula and parts of the superior temporal gyrus, the inferior parietal lobe and supramarginal areas, and lateral frontal areas. These results were in accordance with expectations from the previous literature.
A significant positive relationship between WM/GM contrast decay and cortical thinning in AD was found for the entorhinal cortex (r = 0.40, P < 0.01; Table 2), while there was no such relationship in controls (−0.01, P > 0.5). A significant negative correlation was found between WM/GM contrast decay and hippocampal atrophy (r = −0.38, P < 0.01) in the control group, while no relationship was seen in AD (r = 0.14, P > 0.5). The correlations between cortical and hippocampal atrophy were 0.21 and 0.16 in AD and controls, respectively (both P > 0.5).
Table 2.
Correlations of annual change in entorhinal WM/GM contrast, entorhinal cortical thickness and hippocampal volume
| Cortical thickness | Hippocampal volume | |||
|---|---|---|---|---|
| AD | Controls | AD | Controls | |
| WM/GM contrast | 0.40 | −0.01 | 0.14 | −0.38 |
| Cortical thickness | 0.21 | 0.16 | ||
Bold: P < 0.05.
Assessing the Predictive Value of Longitudinal Changes in Entorhinal WM/GM Contrast, Entorhinal Cortical Thickness, and Hippocampal Volume
Diagnostic accuracy of AD versus controls
Table 3 shows the results of the logistic regression and the leave‐one‐out cross‐validation from the three models (model 1: hippocampal and entorhinal cortical atrophy; model 2: the best predictor from model 1 (hippocampal atrophy in all instances) and WM/GM contrast change; model 3: adding WM/GM contrast change as a third predictor to model 1). ROCs (from model 1 and either model 2 or 3 depending on which showing the highest AUC) based on these analyses are shown in Figure 2A–C.
Table 3.
Logistic regression assessing the predictive value of changes in entorhinal WM/GM contrast, entorhinal cortical thickness, and hippocampal volume
| β | S.E. | OR | AUC (CI) | Sensitivity (CI) | Specificity (CI) | |
|---|---|---|---|---|---|---|
| A. Controls versus AD | ||||||
| Model 1 | ||||||
| Cortical thickness | −0.61 | 0.22 | 0.54 | |||
| Volume | −0.77 | 0.24 | 0.46 | |||
| 0.746 (CI = 0.66–0.83) | 65.6% (53–76%) | 73.2% (62–82%) | ||||
| Model 2 | ||||||
| Volume | −1.02 | 0.26 | 0.36 | |||
| WM/GM contrast | −0.93 | 0.25 | 0.39 | |||
| 0.775 (CI = 0.70–0.85) | 65.6% (53–76%) | 70.4% (59–80%) | ||||
| Model 3 | ||||||
| Cortical thickness | −0.46 | 0.24 | 0.63 | |||
| Volume | −0.95 | 0.27 | 0.39 | |||
| WM/GM contrast | −0.84 | 0.26 | 0.43 | |||
| 0.787 (CI = 0.71–0.86) | 65.6% (53–76%) | 74.6% (63–83%) | ||||
| Model 4 | ||||||
| Cortical thickness | −0.62 | 0.22 | 0.54 | |||
| WM/GM contrast | −0.68 | 0.23 | 0.50 | |||
| 0.718 (CI = 0.63–0.81) | 59.4% (47–71%) | 69.0% (58–79%) | ||||
| B. Controls versus CDR = 0.5 (stable) | ||||||
| Model 1 | ||||||
| Cortical thickness | −0.34 | 0.24 | 0.72 | |||
| Volume | −0.71 | 0.25 | 0.49 | |||
| 0.706 (CI = 0.61–0.81) | 34.2% (21–50%) | 83.1% (73–90%) | ||||
| Model 2 | ||||||
| Volume | −0.89 | 0.27 | 0.41 | |||
| WM/GM contrast | −0.60 | 0.25 | 0.55 | |||
| 0.734 (CI = 0.64–0.83) | 39.5% (26–55%) | 87.3% (78–93%) | ||||
| Model 3 | ||||||
| Cortical thickness | −0.21 | 0.26 | 0.81 | |||
| Volume | −0.85 | 0.27 | 0.43 | |||
| WM/GM contrast | −0.56 | 0.26 | 0.57 | |||
| 0.721 (CI = 0.62–0.82) | 42.1% (28–58%) | 84.5% (74–91%) | ||||
| C. Controls versus converted | ||||||
| Model 1 | ||||||
| Cortical thickness | −0.39 | 0.27 | 0.68 | |||
| Volume | −0.74 | 0.28 | 0.48 | |||
| 0.652 (CI = 0.53–0.78) | 17.9% (8–36%) | 90.1% (81–95%) | ||||
| Model 2 | ||||||
| Volume | −0.95 | 0.33 | 0.39 | |||
| WM/GM contrast | −1.07 | 0.37 | 0.34 | |||
| 0.731 (CI = 0.62–0.85) | 35.7% (21–54%) | 91.5% (83–96%) | ||||
| Model 3 | ||||||
| Cortical thickness | −0.27 | 0.31 | 0.76 | |||
| Volume | −0.87 | 0.34 | 0.42 | |||
| WM/GM contrast | −1.03 | 0.37 | 0.36 | |||
| 0.708 (CI = 0.59–0.83) | 35.7% (21–54%) | 91.5% (83–96%) | ||||
β = standardized regression coefficient, S.E. = standard error of β, OR = odds ratio, AUC = area under the curve, CI = 95% confidence interval. Bold: P < 0.05, italic: P < 0.01.
Figure 2.
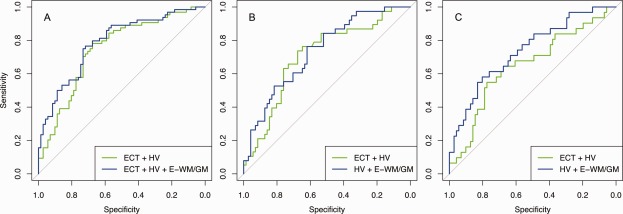
Receiver operating characteristics curves of classification with and without WM/GM contrast change. A: Controls versus AD; B: Controls versus CDR = 0.5 stable; C: Controls versus converted (CDR = 0 ‐‐> 1 and CDR = 0.5 ‐‐> CDR 1). ECT, entorhinal cortical thickness; HV, hippocampal volume; E‐WM/GM, entorhinal WM/GM contrast.
For the classification of AD compared with controls (Table 3), the logistic regression revealed significant effects of WM/GM contrast change in both models 2 and 3 (P = 0.0002 and P = 0.001, respectively), and increased the predictive classification performance as measured by the AUC from 0.746 in the model 1 to 0.775 and 0.787 in models 2 and 3, respectively. The specificity increased from 73.2% in model 1 to 74.6% in model 3, whereas the sensitivity was 65.6% in both models. In model 3, the entorhinal cortical atrophy showed only a trend toward significance. To test if the lack of significance when including WM/GM contrast changes depended on shared variance with hippocampal atrophy, we finally tested a fourth model with entorhinal cortical atrophy and WM/GM contrast changes. The analysis showed a significant effect of entorhinal cortical atrophy (standardized regression coefficient = −0.62, standard error = 0.22, P = 0.005, odds ratio = −0.54) and WM/GM contrast changes (standardized regression coefficient = −0.68, standard error = 0.23, P = 0.003, odds ratio = −0.50), with an AUC of 0.718, a sensitivity of 59.4%, and a specificity of 69%.
To evaluate regional differences in classification accuracy not restricted by the predefined ROIs, we repeated the logistic regression for the three models in a vertex‐wise manner across the cortical mantle. Figure 3 displays the resulting AUC maps superimposed on a semi‐inflated brain, revealing that when including WM/GM contrast change (model 2), highest AUCs are seen in the anterior parts of the medial and inferior temporal lobe, more pronounced in the left hemisphere than the right. For cortical thinning (model 1), the highest AUCs are observed in medial‐frontal areas (medial superior frontal gyrus, cingulate and precuneus), as well as lateral frontal areas in the right hemisphere. Model 3 shows the combined effect of including WM/GM contrast change to model 1. Although not formally tested in a vertex‐wise manner, the AUCs are generally higher in model 3 than model 1, indicating that inclusion of WM/GM contrast change improves diagnostic accuracy of AD.
Figure 3.
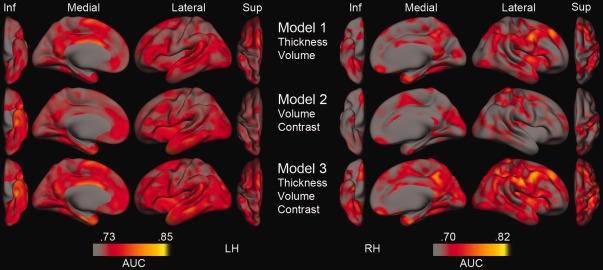
Area under the curve (AUC) estimates at each vertex derived from vertex‐wise logistic regression. Inf, inferior view; Sup, superior view; Thickness, Cortical thickness change; Volume, Hippocampal volume change; Contrast, WM/GM contrast change; LH, left hemisphere; RH = right hemisphere. Please note that the current thresholding was set to highlight differences, thus possibly making relatively small differences seem more pronounced, and that no rigorous statistical testing of differences has been done.
Diagnostic subgroups
To test the diagnostic performance of WM/GM contrast at early stages of AD, we first restricted the AD group to participants with very mild AD (stable CDR of 0.5, n = 38) and performed a comparison with controls (n = 71). WM/GM contrast change contributed significantly in both models 2 and 3, whereas the AUC was highest in model 2 (AUC = 0.734), an increase from 0.706 in model 1 and 0.721 in model 3 (Table 3 and Figure 2B). Without WM/GM contrast change (model 1) the sensitivity was 34.2%, whereas inclusion of WM/GM contrast change increased the sensitivity to 39.5 and 42.1% for models 2 and 3, respectively. The corresponding specificity was 83.1, 87.7, and 84.5%.
To test whether prediction of disease progression could be improved in earliest phases of AD by including WM/GM contrast change, participants converting from a nondemented status to very mild AD (CDR = 0–0.5, n = 13), and from very mild to mild AD (CDR = 0.5–1, n = 13) were combined and compared first with controls (all stable at CDR = 0) and then with patients stable at very mild AD (CDR = 0.5, n = 38). In the classification of the converters versus controls, WM/GM contrast change contributed significantly in both models (P = 0.004 and 0.005, respectively), whereas the AUC was highest in model 2 (AUC = 0.731). The AUCs of models 1 and 3 were 0.652 and 0.708, respectively (Table 3 and Figure 2C). The sensitivity without WM/GM contrast change (model 1) was 17.9%, and increased to 35.7% for both the models 2 and 3 when including WM/GM contrast change. The corresponding specificity was 90.1 and 91.5%. In the discriminations of the combined converters and the patients stable at very mild AD (CDR = 0.5), none of the predictors showed any significant effects in any of the models.
DISCUSSION
The main implication of the present results is that changes in WM/GM contrast derived from conventional T 1‐weighted MRI scans can be used to increase the accuracy of AD diagnosis and prognosis. AD patients and healthy controls differed in the rate of annual WM/GM contrast change especially in the anterior and medial temporal lobe bilaterally. Including WM/GM contrast in the models in addition to the well‐validated morphometric measures of entorhinal cortical and hippocampal atrophy increased classification accuracy as measured by the AUC from 0.75 to 0.79, with a final sensitivity and specificity of 66 and 75%, respectively. Thus, WM/GM contrast changes provided independent and valuable information to diagnostic classification beyond what was obtained by morphometric measures alone. A significant effect of WM/GM contrast change and an increase in prediction accuracy was also observed for participants in the earliest phases of AD, both very mild AD compared with controls, and a combined group of patients converting from either a nondementia status to very mild AD, or from very mild to mild AD versus controls. The results presented here suggest that WM/GM contrast changes may provide a sensitive metric related to early pathological processes in AD, partly independent of atrophy detected by MRI morphometry. Although morphometric measures are sensitive to macroscopic changes in brain structure, signal‐ and contrast measures are likely related to microstructural properties of brain tissue. Thus, WM/GM contrast changes has the potential to be a powerful early biomarker of AD, complementary to established measures of temporal lobe morphometry.
In a recent study, Salat et al. 2011 observed decreased contrast between WM and GM throughout portions of medial and lateral temporal cortex, the precuneus, and the cingulate in AD compared with healthy controls using baseline scans drawn from the same database as in the present study. Our longitudinal findings supported the strong effects in the medial temporal cortex. However, their cross‐sectional results also showed more widespread effects extending beyond the temporal lobes. This discrepancy likely reflects that cross‐sectional data are influenced by accumulated structural changes over longer periods of time, while only ongoing effects will be detected longitudinally over time intervals as short as 2 years reported here.
We observed accelerated changes in WM/GM tissue contrast in anterior medial temporal cortex even in the earliest stages of AD. This finding indicates that longitudinal changes in WM/GM tissue contrast are sensitive to ongoing pathological microstructural alterations in areas known to be affected in early AD. T 1‐weighted signal intensity stems partly from myelin [Barbier et al.,2002; Clark et al.,1992; Eickhoff et al.,2005; Walters et al.,2003]. In AD, the neurofibrillary changes first manifest in the transentorhinal region before proceeding to the neighboring entorhinal region, and may be related to premature oligendrocyte dysfunction [Braak and Braak,1991, 1996]. The observed differences in WM/GM contrast changes thus concur very well with the anatomical distribution of histopathological events in early phases of AD. Based on the findings presented here, WM/GM contrast changes may be more sensitive to these processes that are especially active in and around the entorhinal cortex in early AD, while cortical atrophy might be a less anatomically specific marker of disease‐related atrophy at this stage. In support of the latter hypothesis, the cortical atrophy differences between groups encompassed a larger area compared with the WM/GM contrast change effects, also suggesting that cortical thickness atrophy might be a more sensitive measure of change in other parts of the cortex.
The relations between changes in tissue contrast and morphology were further informed by the logistic regression models. In the entorhinal area, the effect of AD on contrast change was significant when controlling for cortical atrophy. This was not the case for cortical atrophy when contrast change was controlled for. Statistical independence between tissue contrast and thickness has previously been observed in both normal aging and AD with cross‐sectional data [Salat et al.,2011; Westlye et al.,2009]. As the two measures showed similar effects when excluding hippocampal atrophy from the predictors, their respective interrelationship with hippocampal atrophy may at least partially explain why WM/GM contrast change outperformed cortical atrophy in diagnostic accuracy. Further, a positive correlation between entorhinal contrast and thickness changes in AD was found, while no correlation was observed in the controls. The positive correlation in AD may suggest that the histopathological processes that give rise to the contrast changes are at least partially related to the concurrent atrophy as indicated by cortical thinning. At the same time, the lack of a relationship in the controls indicates that changes in tissue contrast and thickness are sensitive to different neurobiological properties [Westlye et al.,2010]. Thus, contrast and thickness changes may be sensitive to both different and overlapping neurobiological properties.
Similar complex relationships were found between entorhinal tissue contrast and hippocampal volume changes. Although we observed no correlation in AD, there was a negative relationship in healthy controls. Again, these results suggest that longitudinal WM/GM contrast changes reflect microstructural properties partly independent of and partly related to a well‐validated morphological measure. In the cross‐sectional study of Salat et al. 2011, associations between tissue contrast and hippocampal volume in AD group was reported in the temporal lobe. The lack of similar associations in the present longitudinal study probably relates to, as mentioned above, differences between the change scores used here detecting ongoing processes compared with the cross‐sectional absolute scores stemming from years of accumulated change. Still, interpretations about the relationships between tissue contrast and more classical imaging indices of AD should be made with caution pending further investigations into the neurobiological mechanisms underlying changes in tissue intensity and morphometry. Such studies should aim at increasing the knowledge about the degree to which the measures reflect independent biological processes, and further how WM/GM contrast relates to other imaging and nonimaging biomarkers in ageing and disease.
The spatial differences observed between effects of AD contrast and cortical thickness changes may relate to differences in sensitivity to the various stages of degeneration in AD. Salat et al. 2011 suggested that WM/GM contrast changes could occur prior to cortical atrophy. In line with this hypothesis, WM/GM contrast showed the lowest odds ratio (in this case indicating the largest decrease in the probability of being in the AD group when increasing the WM/GM contrast scores with 1 SD) when jointly distinguishing those converting from nondementia to very mild AD, and very mild AD to mild AD, from controls. However, the relatively small number of subjects in different CDR‐based subgroups warrants cautious interpretations. Further studies are needed to disentangle this possibility of temporal pattern differences, including analyses of baseline scans grouped according to longitudinal clinical information [Bakkour et al.,2009], with an increased number of subjects in the different CDR‐based subgroups, and ideally analysis of several follow‐up examinations. A higher number of patients in each CDR‐based subgroup would increase subgroup homogeneity, permit finer discrimination between subgroups, and potentially increase the number of correctly classified patients, which now is relatively low in the subgroups compared with the high percentage of correctly classified controls.
A potential caveat in this study regards the potential influence of cortical atrophy differences between groups on the measured WM/GM contrast change differences. However, as mentioned above, the inclusion of entorhinal cortical atrophy as a predictor with WM/GM contrast change statistically controls for the cortical atrophy differences between groups, and we therefore believe that the cortical atrophy does not act as a confounding variable for the WM/GM contrast effects. Further, the cortical thickness differences between groups [Salat et al.,2011] could cause more pronounced partial voluming effects in AD when sampling the initial WM and GM signal intensity values, particularly in areas primarily afflicted in AD such as the medial temporal lobes. When controlling for baseline cortical thickness, however, the effect of WM/GM contrast change was still significant. In addition, on visual inspection of both WM and GM signal intensity values from several cases including moderate AD patients, the medial temporal lobe values were similar to other regions not showing WM/GM contrast change differences between groups. This indicate that the GM and WM values were affected by sampling distance from the WM/GM boundary in a similar manner across groups, without any indication of differential contribution of tissue types in AD compared with controls. Still, as previously noted by Westlye et al. 2009, the mapped intensity values are dependent on the native resolution of the original images, precluding valid inferences on a level of cortical laminae or axonal architecture even though the surface‐based mapping procedure used in the present study enables submillimeter morphometric inferences [Fischl and Dale,2000]. Further studies investigating the complex relationship between tissue microstructure and intrinsic MRI properties [Sigalovsky et al.,2006], as well as the effects of various normalization procedures, field strength, and other scanner‐related parameters [Han et al.,2006], are needed to allow inferences on causative factors of the MRI‐derived brain properties. Similarly, although increased sensitivity to both normal aging and AD with WM/GM contrast adjusted cortical thickness estimates has been observed in a multisample study including cross‐sectional AD data from the same sample used in the present study [Westlye et al.,2009], the current results should be replicated in order to generalize the findings to other samples, scanners, and pulse sequences.
In conclusion, the present results show that WM/GM contrast changes significantly improved diagnostic accuracy beyond what was obtained with entorhinal cortical and hippocampal atrophy alone. These accuracy values, though still short of an absolute diagnostic criterion [Frisoni et al.,2010], demonstrate the potential utility of including changes in signal intensity contrast measures in prediction models. In a research setting, the gains of such an approach may be especially prominent in multicenter studies where different MRI scanners are used [Westlye et al.,2009]. A substantial benefit with this measure is that it involves no extra costs for the patient or the hospital associated with additional examinations. Future research should establish the relationship between this putative imaging biomarker and other established markers of AD, such as PET, cerebrospinal fluid proteomics, APOE genotyping, and neuropsychological performance [Landau et al.,2010; Walhovd et al.,2010].
ACKNOWLEDGMENTS
The authors thank the Washington University Alzheimer Disease Research Center and the developers of the OASIS database for access to the MRI data, as well as all the participants.
REFERENCES
- Bakkour A, Morris JC, Dickerson BC (2009): The cortical signature of prodromal AD: Regional thinning predicts mild AD dementia. Neurology 72:1048–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbier EL, Marrett S, Danek A, Vortmeyer A, van Gelderen P, Duyn J, Bandettini P, Grafman J, Koretsky AP (2002): Imaging cortical anatomy by high‐resolution MR at 3.0T: Detection of the stripe of Gennari in visual area 17. Magn Reson Med 48:735–738. [DOI] [PubMed] [Google Scholar]
- Barnes J, Bartlett JW, van de Pol LA, Loy CT, Scahill RI, Frost C, Thompson P, Fox NC (2009): A meta‐analysis of hippocampal atrophy rates in Alzheimer's disease. Neurobiol Aging 30:1711–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg L (1988): Clinical dementia rating (CDR). Psychopharmacol Bull 24:637–639. [PubMed] [Google Scholar]
- Braak H, Braak E (1991): Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E (1996): Development of Alzheimer‐related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathol 92:197–201. [DOI] [PubMed] [Google Scholar]
- Clark VP, Courchesne E, Grafe M (1992): In vivo myeloarchitectonic analysis of human striate and extrastriate cortex using magnetic resonance imaging. Cereb Cortex 2:417–424. [DOI] [PubMed] [Google Scholar]
- Dale AM, Fischl B, Sereno MI (1999): Cortical surface‐based analysis. I. Segmentation and surface reconstruction. Neuroimage 9:179–194. [DOI] [PubMed] [Google Scholar]
- Davatzikos C, Resnick SM (2002): Degenerative age changes in white matter connectivity visualized in vivo using magnetic resonance imaging. Cereb Cortex 12:767–771. [DOI] [PubMed] [Google Scholar]
- de Leon MJ, DeSanti S, Zinkowski R, Mehta PD, Pratico D, Segal S, Rusinek H, Li J, Tsui W, Saint Louis LA, Clark CM, Tarshish C, Li Y, Lair L, Javier E, Rich K, Lesbre P, Mosconi L, Reisberg B, Sadowski M, DeBernadis JF, Kerkman DJ, Hampel H, Wahlund LO, Davies P (2006): Longitudinal CSF and MRI biomarkers improve the diagnosis of mild cognitive impairment. Neurobiol Aging 27:394–401. [DOI] [PubMed] [Google Scholar]
- DeLong ER, DeLong DM, Clarke‐Pearson DL (1988): Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44:837–845. [PubMed] [Google Scholar]
- Desikan RS, Segonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, Buckner RL, Dale AM, Maguire RP, Hyman BT, Albert MS, Killiany RJ (2006): An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 31:968–980. [DOI] [PubMed] [Google Scholar]
- Devanand DP, Pradhaban G, Liu X, Khandji A, De Santi S, Segal S, Rusinek H, Pelton GH, Honig LS, Mayeux R, Stern Y, Tabert MH, de Leon MJ (2007): Hippocampal and entorhinal atrophy in mild cognitive impairment: Prediction of Alzheimer disease. Neurology 68:828–836. [DOI] [PubMed] [Google Scholar]
- Dickerson BC, Goncharova I, Sullivan MP, Forchetti C, Wilson RS, Bennett DA, Beckett LA, deToledo‐Morrell L (2001): MRI‐derived entorhinal and hippocampal atrophy in incipient and very mild Alzheimer's disease. Neurobiol Aging 22:747–754. [DOI] [PubMed] [Google Scholar]
- Dubois B, Feldman HH, Jacova C, Dekosky ST, Barberger‐Gateau P, Cummings J, Delacourte A, Galasko D, Gauthier S, Jicha G, Meguro K, O'Brien J, Pasquier F, Robert P, Rossor M, Salloway S, Stern Y, Visser PJ, Scheltens P (2007): Research criteria for the diagnosis of Alzheimer's disease: Revising the NINCDS‐ADRDA criteria. Lancet Neurol 6:734–746. [DOI] [PubMed] [Google Scholar]
- Eickhoff S, Walters NB, Schleicher A, Kril J, Egan GF, Zilles K, Watson JD, Amunts K (2005): High‐resolution MRI reflects myeloarchitecture and cytoarchitecture of human cerebral cortex. Hum Brain Mapp 24:206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Dale AM (2000): Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci USA 97:11050–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, van der Kouwe A, Killiany R, Kennedy D, Klaveness S, Montillo A, Makris N, Rosen B, Dale AM (2002): Whole brain segmentation: Automated labeling of neuroanatomical structures in the human brain. Neuron 33:341–355. [DOI] [PubMed] [Google Scholar]
- Fischl B, Salat DH, van der Kouwe AJ, Makris N, Segonne F, Quinn BT, Dale AM (2004a): Sequence‐independent segmentation of magnetic resonance images. Neuroimage 23( Suppl 1):S69–S84. [DOI] [PubMed] [Google Scholar]
- Fischl B, Sereno MI, Dale AM (1999): Cortical surface‐based analysis. II: Inflation, flattening, and a surface‐based coordinate system. Neuroimage 9:195–207. [DOI] [PubMed] [Google Scholar]
- Fischl B, van der Kouwe A, Destrieux C, Halgren E, Segonne F, Salat DH, Busa E, Seidman LJ, Goldstein J, Kennedy D, Caviness V, Makris N, Rosen B, Dale AM (2004b): Automatically parcellating the human cerebral cortex. Cereb Cortex 14:11–22. [DOI] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Fennema‐Notestine C, McEvoy LK, Hagler DJ, Holland D, Brewer JB, Dale AM (2010): CSF biomarkers in prediction of cerebral and clinical change in mild cognitive impairment and Alzheimer's disease. J Neurosci 30:2088–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotenos AF, Snyder AZ, Girton LE, Morris JC, Buckner RL (2005): Normative estimates of cross‐sectional and longitudinal brain volume decline in aging and AD. Neurology 64:1032–1039. [DOI] [PubMed] [Google Scholar]
- Frisoni GB, Fox NC, Jack CRJr, Scheltens P, Thompson PM (2010): The clinical use of structural MRI in Alzheimer disease. Nat Rev Neurol 6:67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagler DJJr, Saygin AP, Sereno MI (2006): Smoothing and cluster thresholding for cortical surface‐based group analysis of fMRI data. Neuroimage 33:1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Jovicich J, Salat D, van der Kouwe A, Quinn B, Czanner S, Busa E, Pacheco J, Albert M, Killiany R, Maguire P, Rosas D, Makris N, Dale A, Dickerson B, Fischl B (2006): Reliability of MRI‐derived measurements of human cerebral cortical thickness: The effects of field strength, scanner upgrade and manufacturer. Neuroimage 32:180–194. [DOI] [PubMed] [Google Scholar]
- Hayasaka S, Nichols TE (2003): Validating cluster size inference: Random field and permutation methods. Neuroimage 20:2343–2356. [DOI] [PubMed] [Google Scholar]
- Jack CRJr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, Weiner M, Petersen RC (2009): Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: Implications for sequence of pathological events in Alzheimer's disease. Brain 132( Pt 5):1355–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CRJr, Petersen RC, O'Brien PC, Tangalos EG (1992): MR‐based hippocampal volumetry in the diagnosis of Alzheimer's disease. Neurology 42:183–188. [DOI] [PubMed] [Google Scholar]
- Landau SM, Harvey D, Madison CM, Reiman EM, Foster NL, Aisen PS, Petersen RC, Shaw LM, Trojanowski JQ, Jack CRJr, Weiner MW, Jagust WJ (2010): Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 75:230–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus DS, Fotenos AF, Csernansky JG, Morris JC, Buckner RL (2010): Open access series of imaging studies: Longitudinal MRI data in nondemented and demented older adults. J Cogn Neurosci 22:2677–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald CR, McEvoy LK, Gharapetian L, Fennema‐Notestine C, Hagler DJJr, Holland D, Koyama A, Brewer JB, Dale AM (2009): Regional rates of neocortical atrophy from normal aging to early Alzheimer disease. Neurology 73:457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter M, Fischl B (2011): Avoiding asymmetry‐induced bias in longitudinal image processing. NeuroImage 57:19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter M, Rosas HD, Fischl B (2010): Highly accurate inverse consistent registration: A robust approach. Neuroimage 53:1181–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Muller M (2011): pROC: An open‐source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salat DH, Chen JJ, van der Kouwe AJ, Greve DN, Fischl B, Rosas HD (2011): Hippocampal degeneration is associated with temporal and limbic gray matter/white matter tissue contrast in Alzheimer's disease. Neuroimage 54:1795–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salat DH, Lee SY, van der Kouwe AJ, Greve DN, Fischl B, Rosas HD (2009): Age‐associated alterations in cortical gray and white matter signal intensity and gray to white matter contrast. Neuroimage 48:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigalovsky IS, Fischl B, Melcher JR (2006): Mapping an intrinsic MR property of gray matter in auditory cortex of living humans: A possible marker for primary cortex and hemispheric differences. Neuroimage 32:1524–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sled JG, Zijdenbos AP, Evans AC (1998): A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans Med Imaging 17:87–97. [DOI] [PubMed] [Google Scholar]
- Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW, Knopman DS, Petersen RC, Jack CRJr (2009): MRI and CSF biomarkers in normal, MCI, and AD subjects: Diagnostic discrimination and cognitive correlations. Neurology 73:287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers AJ, Cronin AM, Begg CB (2011): One statistical test is sufficient for assessing new predictive markers. BMC Med Res Methodol 11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhovd KB, Fjell AM, Brewer J, McEvoy LK, Fennema‐Notestine C, Hagler DJJr, Jennings RG, Karow D, Dale AM (2010): Combining MR imaging, positron‐emission tomography, and CSF biomarkers in the diagnosis and prognosis of Alzheimer disease. AJNR Am J Neuroradiol 31:347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters NB, Egan GF, Kril JJ, Kean M, Waley P, Jenkinson M, Watson JD (2003): In vivo identification of human cortical areas using high‐resolution MRI: An approach to cerebral structure‐function correlation. Proc Natl Acad Sci USA 100:2981–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westlye LT, Walhovd KB, Dale AM, Bjornerud A, Due‐Tonnessen P, Engvig A, Grydeland H, Tamnes CK, Ostby Y, Fjell AM (2010): Differentiating maturational and aging‐related changes of the cerebral cortex by use of thickness and signal intensity. Neuroimage 52:172–185. [DOI] [PubMed] [Google Scholar]
- Westlye LT, Walhovd KB, Dale AM, Espeseth T, Reinvang I, Raz N, Agartz I, Greve DN, Fischl B, Fjell AM (2009): Increased sensitivity to effects of normal aging and Alzheimer's disease on cortical thickness by adjustment for local variability in gray/white contrast: A multi‐sample MRI study. Neuroimage 47:1545–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]