

Abstract
In this report, we present the first regional quantitative analysis of age‐related differences in the heritability of cortical thickness using anatomic MRI with a large pediatric sample of twins, twin siblings, and singletons (n = 600, mean age 11.1 years, range 5–19). Regions of primary sensory and motor cortex, which develop earlier, both phylogenetically and ontologically, show relatively greater genetic effects earlier in childhood. Later developing regions within the dorsal prefrontal cortex and temporal lobes conversely show increasingly prominent genetic effects with maturation. The observation that regions associated with complex cognitive processes such as language, tool use, and executive function are more heritable in adolescents than children is consistent with previous studies showing that IQ becomes increasingly heritable with maturity(Plomin et al. 1997: Psychol Sci 8:442–447). These results suggest that both the specific cortical region and the age of the population should be taken into account when using cortical thickness as an intermediate phenotype to link genes, environment, and behavior. Hum Brain Mapp, 2009. © 2007 Wiley‐Liss, Inc.
INTRODUCTION
Debates over the relative influence of genetic factors versus environmental influences in determining the course of an individual's developmental trajectory have given way to acceptance that brain structure and function is created through complex interactions, which change over the life of an individual [Rutter et al., 2006]. Identifying factors which most strongly affect brain development at a given time is important to our understanding of what drives developmental trajectories to unfold in a particular way. Quantitative genetics provides a method of estimating the relative contributions of genetic and nongenetic sources in generating individual differences in traits. Quantification is possible by examining covariance patterns between family members of different levels of genetic relatedness, such between as monozygotic and dizygotic twins. Heritability is the proportion of the variance in a trait due to genetic factors. It has been known for over a decade that brain size in adults is highly heritable [Baare et al., 2001; Geschwind et al., 2002; Posthuma et al., 2000; Reveley et al., 1984; Tramo et al., 1998; White et al., 2002]. More recent studies have demonstrated that differences in brain size between children are also strongly influenced by genetic factors [Pennington et al., 2000] and that heritability of gray and white matter volumes changes over childhood and adolescence [Wallace et al., 2006].
Cortical thickness is of interest as a measure of brain anatomy potentially sensitive to a variety of developmental and functional differences between cortical regions [Roland and Zilles, 1998]. The thickness of the cortical sheet is determined by cytoarchitectural characteristics such as laminar structure and cellular size and density [Rakic et al., 2004]. Until recently, technical challenges in measurement of the geometrically complex cortex have limited quantitative analyses of the heritability of regional cortical thickness. Wright et al. measured heritability of average cortical thickness in 92 cortical regions in 10 pairs of monozygotic and 10 pairs of dizygotic healthy adult twins. This study found the strongest genetic effects (heritabilities greater than 0.50) in areas within the frontal, temporal, and superior parietal lobes [Wright et al., 2002]. In the first study at the voxel level of resolution, Thompson et al. [2001] measured cortical density in 10 MZ and 10 DZ adult twin pairs. They found evidence of significant heritability in bilateral frontal and superior temporal regions, with greater values in language‐associated areas of the left hemisphere. Hulshoff Pol et al. [2006] found evidence for significant heritability of gray and white matter density within several brain regions in a twin study of 258 adult subjects. There have been no previous studies of developmental changes in heritability of cortical features in humans, despite evidence of developmental changes in postnatal cortical structure [Gogtay et al., 2004] and postnatal gene expression [Plomin and Craig, 1997; Sun et al., 2005].
The present study extends our understanding of genetic and environmental influences on brain development by using magnetic resonance imaging (MRI) to examine the heritability of cerebral cortical thickness in a large pediatric sample of twin, twin sibling, and singleton subjects. We also explore changes in heritability over the course of childhood and adolescence using two different methods: first by modeling the interaction of heritability and age, and second through direct comparison of estimated variance components between older and younger groups.
MATERIALS AND METHODS
Subjects
Six‐hundred normally developing same‐sex monozygotic (MZ) and dizygotic (DZ) twins, siblings of twins, and unrelated singletons were recruited as part of an ongoing longitudinal brain imaging project being conducted at the Child Psychiatry Branch of the National Institute of Mental Health (NIMH) (see Tables I and II for demographic characteristics). Heritability of brain volumes was previously reported from a subset of this sample [Wallace et al., 2006] (MRI data from 86 of the MZ twin pairs, 37 of the DZ twin pair, and 153 of the singletons reported here were included in the previous study). Parents of prospective participants were interviewed by phone and asked to report their child's health, developmental, and educational history. During their visit to the NIMH, subjects underwent a clinical interview and physical examination. Subjects were excluded if they had taken psychiatric medications, had been diagnosed with a psychiatric disorder, had undergone brain trauma, or had any condition known to affect gross brain development. Inclusion criteria were a minimum gestational age of 29 weeks and a minimum birth weight of 1,500 g for both members of each twin pair. Approximately 80% of families responding to the ads met inclusion criteria. Socio‐economic status was rated using the Hollingshead scale [Hollingshead and Redlich, 1958]. Zygosity was determined by DNA analysis of buccal cheek swabs using 9–21 unlinked short tandem repeat loci for a minimum certainty of 99%, by BRT Laboratories and Proactive Genetics. We obtained verbal or written assent from the child and written consent from the parents for their participation in the study. The NIMH Institutional Review Board approved the protocol.
Table I.
Demographic characteristics of samplea
| Subject type | Number of subjects | Age (yrs) (s.d.) Range | Sex | Ethnicity | Handedness | SES (s.d.) Range | Mean global cortical thickness (mm) (s.d.) |
|---|---|---|---|---|---|---|---|
| Monozygotic twins | 214 | 11.03 (3.2) 5.4–18.7 | 117 M, 97 F | 202 W, 6 B, 2 A, 4 M, 0 U | 183 R, 12 L, 13 M, 6 U | 43.5 (18.4) 20–89 | 4.20 (0.34) M, 4.19 (0.34) F, 4.20 (0.34) C |
| Dizygotic twins | 94 | 11.20 (3.8) 5.6–19.3 | 53 M, 41 F | 92 W, 0 B, 0 A, 2 M, 0 U | 76 R, 7 L, 9 M, 2 U | 42.9 (13.9) 20–70 | 4.14 (0.32) M, 4.14 (0.32) F, 4.13 (0.32) C |
| Siblings of twins | 64 | 11.62 (3.5) 5.0–19.1 | 31 M, 33 F | 63 W, 0 B, 0 A, 1 M, 0 U | 51 R, 6 L, 3 M, 4 U | 40.1 (16. 9) 20–77 | 4.11 (0.48) M, 4.14 (0.23) F, 4.12 (0.37) C |
| Singletons | 228 | 10.92 (3.5) 5.2–18.9 | 132 M, 96 F | 172 W, 36 B, 8 A, 11 M, 1 U | 203 R, 12 L, 11 M, 2 U | 40.7 (20.6) 20–95 | 4.15 (0.37) M, 4.08 (0.39) F, 4.12 (0.38) C |
| Total | 600 | 11.08 (3.4) 5.0–19.3 | 332 M, 268 F | 529 W, 42 B, 10 A, 18 M, 1 U | 513 R, 37 L, 36 M, 14 U | 42.0 (18.5) 20–95 | 4.17 (0.36) M, 4.12 (0.35) F, 4.15 (0.36) C |
Abbreviations: Sex: M = male, F = female, C = male and female combined; Ethnicity: W = white, B = black, A = Asian, M = mixed, U = unknown; Handedness: R = right, L = left, M = mixed, U = unknown.
Socioeconomic status (SES) assessed using the Hollingshead scale (Hollingshead and Redlich, 1958), which ranges from 20 (highest SES) to 134 (lowest SES).
Table II.
Description of family structures
| Subject type | Number of individuals |
|---|---|
| Monozygotic twins (data from one twin was missing for six pairs) | 220 |
| Dizygotic twins (data from one twin was missing for two pairs) | 96 |
| Single‐birth siblings of twins | |
| One single‐birth sibling in family | 34 |
| Two or more single‐birth siblings in family | 22 |
| Single‐birth sibling groups from families without twins | |
| Siblings in pairs | 68 |
| Siblings in trios | 27 |
| Siblings in quartets | 16 |
| Siblings in quintet | 5 |
| Single birth individuals without siblings | 112 |
| Total | 600 |
MRI Acquisition
All MRI images were acquired on the same General Electric 1.5 Tesla Signa Scanner located at the National Institutes of Health Clinical Center in Bethesda, Maryland. A three‐dimensional spoiled gradient recalled echo sequence in the steady state sequence, designed to optimize discrimination between gray matter, white matter, and cerebrospinal fluid, was used to acquire 124 contiguous 1.5‐mm thick slices in the axial plane (TE/TR = 5/24 ms; flip angle = 45 degrees, matrix = 256 × 192, NEX = 1, FOV = 24 cm, acquisition time 9.9 min). A Fast Spin Echo/Proton Density weighted imaging sequence was also acquired for clinical evaluation.
Image Analysis
The native MRI scans were registered into standardized stereotaxic space using a linear transformation [Collins et al., 1994] and corrected for nonuniformity artifacts [Sled et al., 1998]. The registered and corrected volumes were segmented into white matter, gray matter, cerebrospinal fluid, and background using a neural net classifier [Zijdenbos et al., 2002]. The white and gray matter surfaces were fitted using deformable surface‐mesh models and nonlinearly aligned toward a standard template surface [Kim et al., 2005; MacDonald et al., 2000; Robbins et al., 2004]. The white and gray matter surfaces were resampled into native space, and cortical thickness was measured in native‐space millimeters using the linked distance between the white and pial surfaces at each of 40,962 cortical points throughout the cortex [Lerch and Evans, 2005; MacDonald et al., 2000]. To improve the ability to compare populations, each subject's cortical thickness map was blurred using a 30‐mm surface‐based diffusion blurring kernel, chosen to maximize statistical power while minimizing false positives [Lerch and Evans, 2005]. Cortical points were assigned to specific regions using a probabilistic atlas [Collins et al., 1999]. These methods have been validated using both manual measurements [Kabani et al., 2001] and a population simulation [Lerch and Evans, 2005], and have been used in studies of Alzheimer's disease [Lerch et al., 2005] and normal development [Shaw et al., 2005], among others. Statistical results from structural equation modeling analyses of cortical thickness at each point (see Statistical Analysis section below) were projected upon the smoothed brain template using in‐house software developed by the Montreal Neurological Institute.
Statistical Analysis
Since MZ twins are genetically identical, while on average only one‐half of the genes in DZ twins are identical by descent (IBD), the classical twin design allows for the parsing of observed variability in phenotype into additive genetic factors (A), environmental factors shared between family members (C), and nongenetic factors unique to individuals (E) (Fig. 1A) [Neale et al., 1992]. The addition of single‐birth siblings (including siblings of twins and a large number of siblings of families with no twins), half of whose genes are IBD to those of their twin and nontwin siblings, provides substantially increased power to detect genetic signal due to a greater number of observed covariance statistics [Posthuma and Boomsma, 2000; Posthuma et al., 2000]. This “extended twin design” assumes that the shared environment operates similarly in both twins and singleton births with respect to the phenotype of interest (Fig. 1B). In our sample, families contained either a twin pair and up to three additional siblings, or no twins and up to five nontwin siblings.
Figure 1.
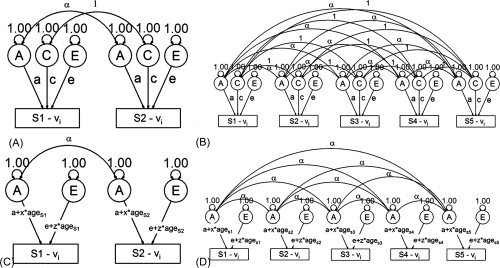
Path diagram of genetic models. Panels A and C depict the classical ACE twin model and an AE twin age‐moderated model, respectively, while panels B and D depict the extensions to these models used in the present study to accommodate between zero and three additional subjects per family. Panels A and B: Latent factors A, C, and E are allowed to influence the observed causal paths a, c, and e, with latent shared environmental factors correlated at unity, but additive genetic correlations correlated 1 or [1/2] depending on familial relationship. Panels C and D: In the moderated model, parameter estimates are allowed to vary based on individual subject ages. For both models, age and sex regressions on mean CT are not shown for the sake of simplicity, though these parameters were estimated simultaneously with the variance components. S = Subject number (1–5), vi = CT for the ith vertex.
We constructed structural equation models of expected variance–covariance matrices for each cortical point in an iterative fashion using Mx, a statistical package designed for the analysis of genetically‐informative multi‐group data [Neale et al., 1999]. Three variance components parameters were estimated that quantified the relationship between A, C, E, and the observed cortical thickness measures. The proportion of variance due to each component was then derived (a 2, c 2, and e 2) by dividing each variance component by the total variance. The model also contained parameters to account for the effects of age and sex on mean cortical thickness, which were estimated concurrently with the variance components. Sex effects were estimated using a linear model, and age was estimated using a cubic model based on prior evidence of age interactions with cortical thickness [Lenroot et al., 2005]. Optimum model fit was determined using maximum likelihood [Edwards, 1972], which produces unbiased parameter estimates and allows the identification of statistically significant parameters in the model [Neale and Miller, 1997]. Statistical significance of variance components was determined by comparing the likelihood from models with or without the parameter; the difference in −2 times the log likelihood (−2LL) asymptotically follows a 50:50 mixture of zero and χ2 distribution with one degree of freedom. The model‐fitting procedure was iterated for all positions in the brain using the R statistical package [R Development Core Team, 2006], which generated a dataset for a given spatial location and subsequently initiated Mx statistical analysis. Effects of handedness were assessed by dividing the population into right‐handed and nonright‐handed individuals and comparing variance components in the two groups. To determine whether heritability estimates were biased by scaling issues related to differences in mean cortical thickness, variance components were converted to coefficients of variation and age interactions modeled on both measures for comparison.
The relationship of age to heritability was explored both by modeling age as a continuous variable and by dichotomizing the sample into younger and older groups [Purcell, 2002]. We limited this division to two groups to maximize power to detect variance components within each group. The younger sample consisted of children under 12, and the older sample of children and adolescents 12 or more years of age (see Table III for demographic characteristics and comparison of the two groups). The age cut‐off was chosen as an approximate division between childhood and adolescence. For a classical twin model, the usual approach would be to run two sets of univariate models in parallel and calculate parameter estimates separately for the two groups. However, the use of the extended twin model complicates the analytic design, since nontwin siblings do not have identical ages, and as a result any two siblings of a particular family might not belong to the same age group.
Table III.
Demographic characteristics of younger and older subsamples
| Younger (s.d.) | Older (s.d.) | Differences | |
|---|---|---|---|
| Groups | P = 0.59 | ||
| MZ | 134 | 80 | |
| DZ | 62 | 32 | |
| Siblings | 36 | 29 | |
| Singletons | 143 | 84 | |
| Age | 8.88 (2.00) | 14.72 (0.82) | P < 0.0001 |
| Sex | P = 0.10 | ||
| Male | 198 | 91 | |
| Female | 177 | 135 | |
| Ethnicity | P = 0.67 | ||
| White | 333 | 197 | |
| Black | 26 | 16 | |
| Asian | 6 | 4 | |
| Mixed | 10 | 8 | |
| Unknown | 0 | 1 | |
| Handedness | P = 0.82 | ||
| Right | 322 | 192 | |
| Mixed | 21 | 15 | |
| Left | 22 | 15 | |
| Unknown | 10 | 4 | |
| SESa | 42.63 (18.54) | 40.93 (18.39) | P = 0.29 |
| Mean cortical thicknessb | 4.17 (.38) | 4.11 (.31) | P = 0.03 |
Total number of younger subsamples is 375, and the total number of older subsamples is 226.
Socioeconomic status (SES) assessed using the Hollingshead scale(Hollingshead and Redlich, 1958), which ranges from 20 (highest SES) to 134 (lowest SES).
Unequal variances between groups; results are reported using one‐way ANOVA for consistency; Welch ANOVA provides a similar outcome.
To overcome this problem, we combined an extended twin design with moderator models for the examination of gene by age interaction [Purcell, 2002]. Path diagrams for this model are shown in Figure 1C,D; since our original models found no evidence for a significant shared environmental contribution to cortical thickness (CT) variability, it was not modeled to increase the power to detect genetic effects. Each subject was dummy coded 0 or 1 depending on whether age was below or above the age threshold, respectively. Two additional free parameters (x and z) were included in the model that allowed for the magnitude of each variance component to change depending on age grouping. For the ith vertex, the estimation of heritability was calculated as
where agejn represents the dichotomized age value for nth family member of the jth family. From this equation, the heritability for the younger (Y) and older (O) age groups were calculated as
To test whether heritability differed between groups, we tested submodels that constrained
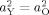 for each vertex. Under the null hypothesis of no heritability difference between groups, this simplification was expected to produce a difference in −2LL following a χ2 distribution with one degree of freedom, which was confirmed via simulation of 1,000 datasets with identical sample size and family structure compared to the present study.
for each vertex. Under the null hypothesis of no heritability difference between groups, this simplification was expected to produce a difference in −2LL following a χ2 distribution with one degree of freedom, which was confirmed via simulation of 1,000 datasets with identical sample size and family structure compared to the present study.
The resultant output for all models consisted of maximum likelihood variance components parameter estimates for all cortical points as well as P‐values of the statistical significance of A and C. Brain maps were reconstructed from these data to visualize the maximum likelihood estimates of variance components owed to additive genetic (A), shared environmental (C), and unique environmental (E) factors and of proportional variability for each of these factors (a
2, c
2, and e
2). Probability maps also were constructed to assess the significance of genetic factors on individual differences in cortical thickness. An α <0.05 was set as the threshold for statistical significance. A false discovery rate (FDR) adjustment was applied to control for Type I error [Genovese et al., 2002]. Unless otherwise specified, FDR was set to allow for a 5% chance of false positives. A likelihood‐ratio χ2 test was used to test the statistical significance of age interactions by comparing how well a model incorporating the age interaction fit the data compared with a model that did not include the age interaction. For the moderator models with dichotomized age variables, difference maps also were constructed by subtracting
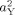 from
from 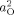 to estimate how heritability changed between early and late childhood.
to estimate how heritability changed between early and late childhood.
RESULTS
Cross‐Sectional Analysis
Cross‐sectional analysis showed heritability estimates at individual cortical points ranging from 0 to 0.61. Values over 0.50 were found in regions of the frontal lobes, temporal lobes, postcentral gyri, and supramarginal gyri. Areas in which genetic effects were statistically significant included the frontal pole, dorsolateral and orbital prefrontal cortices, prefrontal gyrus, angular and superior temporal gyri, and the superior parietal region (see Figs. 2 and 3 for maps of heritability values and statistical significance, and Table IV for numerical ranges of values in individual cortical regions). Unique environmental factors and other elements within E were the primary determinants of variance in the remaining regions. Shared environmental effects (C) ranging in value up to 0.25 were seen in regions including the left prefrontal cortex, right superior medial gyrus, right superior posterior gyrus, bilateral inferior postcentral gyrus, left posterior medial temporal gyrus, left insular, bilateral medial cingulate regions, and bilateral occipital lobes, but did not reach significance in any cortical region, consistent with what has been reported elsewhere [Pennington et al., 2000; Wright et al., 2002]. Heritability estimates were not significantly affected by sex or handedness. There were no differences in heritability values modeled after variance components were converted to coefficients of variation.
Figure 2.
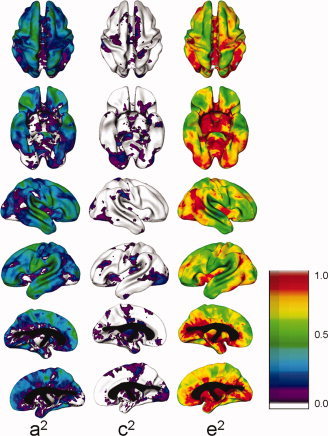
Estimated proportional variance components. The proportions of total variance due to genetic (a 2), shared environmental (c 2), and unique environmental (e 2) variance are shown at voxels corresponding to each cortical vertex.
Figure 3.
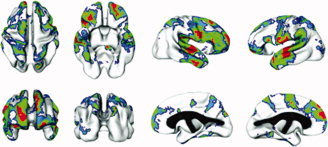
Regions of significant heritability. Voxels are color‐coded for level of significance following application of FDR threshold. Red voxels are significant at P ≤ 0.05; green = 0.05–0.10; blue = 0.10–0.15 (corresponding to uncorrected P values of 0.002, 0.016, and 0.042, respectively).
Table IV.
Maximum likelihood parameter estimates and P‐values from hypothesis testing of univariate ACE models
| Variance components (95% CI) | Hypothesis test (P‐values*) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| a 2 | c 2 | e 2 | A | C | A and C | ||||
| Superior Frontal Gyrus ‐R | 0.45 | (0.20, 0.60) | 0.00 | (0.00, 0.15) | 0.55 | (0.40, 0.72) | 0.004 | 1.000 | 0.00 |
| Superior Frontal Gyrus ‐L | 0.51 | (0.24, 0.64) | 0.00 | (0.00, 0.17) | 0.49 | (0.36, 0.65) | 0.002 | 1.000 | 0.00 |
| Middle Frontal Gyrus ‐R | 0.43 | (0.21, 0.59) | 0.00 | (0.00, 0.10) | 0.57 | (0.41, 0.76) | 0.002 | 1.000 | 0.00 |
| Middle Frontal Gyrus ‐L | 0.38 | (0.05, 0.52) | 0.00 | (0.00, 0.21) | 0.62 | (0.48, 0.80) | 0.027 | 1.000 | 0.00 |
| Inferior Frontal Gyrus ‐R | 0.52 | (0.32, 0.66) | 0.00 | (0.00, 0.09) | 0.48 | (0.34, 0.67) | 0.000 | 1.000 | 0.00 |
| Inferior Frontal Gyrus ‐L | 0.44 | (0.15, 0.58) | 0.00 | (0.00, 0.19) | 0.56 | (0.42, 0.73) | 0.007 | 1.000 | 0.00 |
| Precentral Gyrus ‐R | 0.43 | (0.20, 0.58) | 0.00 | (0.00, 0.13) | 0.57 | (0.42, 0.75) | 0.004 | 1.000 | 0.00 |
| Precentral Gyrus ‐L | 0.52 | (0.27, 0.65) | 0.00 | (0.00, 0.15) | 0.48 | (0.35, 0.65) | 0.001 | 1.000 | 0.00 |
| Lateral Orbitofrontal Gyrus ‐R | 0.38 | (0.12, 0.54) | 0.00 | (0.00, 0.15) | 0.62 | (0.46, 0.81) | 0.010 | 1.000 | 0.00 |
| Lateral Orbitofrontal Gyrus ‐L | 0.34 | (0.03, 0.49) | 0.00 | (0.00, 0.19) | 0.66 | (0.51, 0.84) | 0.037 | 1.000 | 0.00 |
| Medial Orbitofrontal Gyrus ‐R | 0.22 | (0.00, 0.38) | 0.00 | (0.00, 0.17) | 0.78 | (0.62, 0.96) | 0.099 | 1.000 | 0.05 |
| Medial Orbitofrontal Gyrus ‐L | 0.27 | (0.00, 0.43) | 0.00 | (0.00, 0.21) | 0.73 | (0.57, 0.91) | 0.087 | 1.000 | 0.01 |
| Cingulate ‐R | 0.36 | (0.16, 0.53) | 0.00 | (0.00, 0.09) | 0.64 | (0.47, 0.83) | 0.003 | 1.000 | 0.00 |
| Cingulate ‐L | 0.40 | (0.18, 0.56) | 0.00 | (0.00, 0.10) | 0.60 | (0.44, 0.80) | 0.003 | 1.000 | 0.00 |
| Medial Frontal Gyrus ‐R | 0.38 | (0.10, 0.54) | 0.00 | (0.00, 0.17) | 0.62 | (0.46, 0.80) | 0.016 | 1.000 | 0.00 |
| Medial Frontal Gyrus ‐L | 0.50 | (0.27, 0.64) | 0.00 | (0.00, 0.13) | 0.50 | (0.31, 0.68) | 0.001 | 1.000 | 0.00 |
| Superior Parietal Gyrus ‐R | 0.44 | (0.20, 0.59) | 0.00 | (0.00, 0.13) | 0.56 | (0.41, 0.76) | 0.004 | 1.000 | 0.00 |
| Superior Parietal Gyrus ‐L | 0.30 | (0.04, 0.47) | 0.00 | (0.00, 0.15) | 0.70 | (0.54, 0.89) | 0.032 | 1.000 | 0.01 |
| Supramarginal Gyrus ‐R | 0.39 | (0.00, 0.53) | 0.00 | (0.00, 0.28) | 0.61 | (0.47, 0.79) | 0.056 | 1.000 | 0.00 |
| Supramarginal Gyrus ‐L | 0.51 | (0.22, 0.63) | 0.00 | (0.00, 0.21) | 0.49 | (0.37, 0.64) | 0.003 | 1.000 | 0.00 |
| Angular Gyrus ‐R | 0.20 | (0.00, 0.39) | 0.00 | (0.00, 0.18) | 0.80 | (0.61, 0.99) | 0.171 | 1.000 | 0.12 |
| Angular Gyrus ‐L | 0.24 | (0.00, 0.41) | 0.00 | (0.00, 0.18) | 0.76 | (0.59, 0.95) | 0.113 | 1.000 | 0.04 |
| Precuneus ‐R | 0.19 | (0.00, 0.36) | 0.00 | (0.00, 0.21) | 0.81 | (0.64, 0.98) | 0.227 | 1.000 | 0.09 |
| Precuneus ‐L | 0.12 | (0.00, 0.28) | 0.00 | (0.00, 0.16) | 0.88 | (0.72, 1.00) | 0.367 | 1.000 | 0.32 |
| Postcentral Gyrus ‐R | 0.57 | (0.36, 0.68) | 0.00 | (0.00, 0.13) | 0.43 | (0.32, 0.58) | 0.000 | 1.000 | 0.00 |
| Postcentral Gyrus ‐L | 0.48 | (0.25, 0.61) | 0.00 | (0.00, 0.14) | 0.52 | (0.39, 0.68) | 0.001 | 1.000 | 0.00 |
| Superior Temporal Gyrus ‐R | 0.41 | (0.13, 0.56) | 0.00 | (0.00, 0.17) | 0.59 | (0.44, 0.77) | 0.010 | 1.000 | 0.00 |
| Superior Temporal Gyrus ‐L | 0.40 | (0.14, 0.55) | 0.00 | (0.00, 0.16) | 0.60 | (0.45, 0.77) | 0.007 | 1.000 | 0.00 |
| Middle Temporal Gyrus ‐R | 0.33 | (0.00, 0.49) | 0.00 | (0.00, 0.21) | 0.67 | (0.51, 0.86) | 0.047 | 1.000 | 0.00 |
| Middle Temporal Gyrus ‐L | 0.39 | (0.04, 0.54) | 0.00 | (0.00, 0.22) | 0.61 | (0.46, 0.80) | 0.031 | 1.000 | 0.00 |
| Inferior Temporal Gyrus ‐R | 0.38 | (0.17, 0.53) | 0.00 | (0.00, 0.12) | 0.62 | (0.47, 0.70) | 0.003 | 1.000 | 0.00 |
| Inferior Temporal Gyrus ‐L | 0.47 | (0.18, 0.60) | 0.00 | (0.00, 0.20) | 0.53 | (0.40, 0.69) | 0.004 | 1.000 | 0.00 |
| Uncus ‐R | 0.01 | (0.00, 0.16) | 0.00 | (0.00, 0.09) | 0.99 | (0.84, 1.00) | 1.000 | 1.000 | 1.00 |
| Uncus ‐L | 0.05 | (0.00, 0.24) | 0.00 | (0.00, 0.10) | 0.95 | (0.76, 1.00) | 0.584 | 1.000 | 0.86 |
| Medial Occipitotemporal Gyrus ‐R | 0.31 | (0.00, 0.47) | 0.01 | (0.00, 0.30) | 0.68 | (0.53, 0.87) | 0.196 | 0.938 | 0.00 |
| Medial Occipitotemporal Gyrus ‐L | 0.26 | (0.00, 0.42) | 0.00 | (0.00, 0.22) | 0.74 | (0.59, 0.92) | 0.128 | 1.000 | 0.01 |
| Lateral Occipitotemporal Gyrus ‐R | 0.33 | (0.00, 0.48) | 0.00 | (0.00, 0.22) | 0.67 | (0.52, 0.85) | 0.052 | 1.000 | 0.00 |
| Lateral Occipitotemporal Gyrus ‐L | 0.28 | (0.00, 0.44) | 0.00 | (0.00, 0.20) | 0.72 | (0.56, 0.90) | 0.074 | 1.000 | 0.01 |
| Parahippocampal Gyrus ‐R | 0.06 | (0.00, 0.24) | 0.01 | (0.00, 0.16) | 0.93 | (0.76, 1.00) | 0.808 | 0.929 | 0.61 |
| Parahippocampal Gyrus ‐L | 0.10 | (0.00, 0.33) | 0.06 | (0.00, 0.25) | 0.84 | (0.67, 0.98) | 0.651 | 0.705 | 0.07 |
| Occipital Pole ‐R | 0.30 | (0.00, 0.50) | 0.05 | (0.00, 0.32) | 0.65 | (0.50, 0.84) | 0.183 | 0.764 | 0.00 |
| Occipital Pole ‐L | 0.47 | (0.09, 0.60) | 0.00 | (0.00, 0.27) | 0.53 | (0.40, 0.70) | 0.018 | 1.000 | 0.00 |
| Superior Occipital Gyrus ‐R | 0.37 | (0.01, 0.52) | 0.00 | (0.00, 0.24) | 0.63 | (0.48, 0.81) | 0.045 | 1.000 | 0.00 |
| Superior Occipital Gyrus ‐L | 0.31 | (0.00, 0.48) | 0.00 | (0.00, 0.30) | 0.69 | (0.52, 0.91) | 0.216 | 1.000 | 0.01 |
| Middle Occipital Gyrus ‐R | 0.26 | (0.00, 0.43) | 0.00 | (0.00, 0.22) | 0.74 | (0.57, 0.94) | 0.136 | 1.000 | 0.02 |
| Middle Occipital Gyrus ‐L | 0.33 | (0.04, 0.51) | 0.00 | (0.00, 0.16) | 0.67 | (0.49, 0.87) | 0.032 | 1.000 | 0.01 |
| Inferior Occipital Gyrus ‐R | 0.23 | (0.00, 0.40) | 0.00 | (0.00, 0.21) | 0.77 | (0.60, 0.96) | 0.193 | 1.000 | 0.04 |
| Inferior Occipital Gyrus ‐L | 0.12 | (0.00, 0.49) | 0.21 | (0.00, 0.39) | 0.67 | (0.50, 0.83) | 0.580 | 0.200 | 0.00 |
| Cuneus ‐R | 0.35 | (0.02, 0.50) | 0.00 | (0.00, 0.22) | 0.65 | (0.50, 0.83) | 0.039 | 1.000 | 0.00 |
| Cuneus ‐L | 0.15 | (0.00, 0.32) | 0.00 | (0.00, 0.19) | 0.85 | (0.68, 1.00) | 0.307 | 1.000 | 0.25 |
| Lingual Gyrus ‐R | 0.01 | (0.00, 0.33) | 0.13 | (0.00, 0.25) | 0.86 | (0.67, 0.98) | 1.000 | 0.349 | 0.06 |
| Lingual Gyrus ‐L | 0.22 | (0.00, 0.39) | 0.00 | (0.00, 0.24) | 0.78 | (0.61, 0.97) | 0.325 | 1.000 | 0.06 |
| Insula ‐R | 0.30 | (0.00, 0.46) | 0.00 | (0.00, 0.20) | 0.70 | (0.54, 0.88) | 0.059 | 1.000 | 0.01 |
| Insula ‐L | 0.26 | (0.00, 0.43) | 0.00 | (0.00, 0.20) | 0.74 | (0.57, 0.95) | 0.120 | 1.000 | 0.03 |
P‐values test the hypotheses of no genetic (A), shared environmental (C), or familial (A and C) effects on phenotypic variance; statistically significant effects (at an α=0.05) are shown in boldface.
Differences in Heritability Associated With Development
In the first part of the analysis of effects of development upon heritability, in which age was considered as a continuous variable, we found that developmental changes in heritability during the age range of our sample (5–18) were markedly different across the brain (see Fig. 4). Heritability decreased with age in dorsal regions of primary motor and somatosensory cortex in the pre‐ and post‐central gyri, posterior temporal and inferior parietal regions, and occipital regions. Heritability increased over the same period in other regions, most markedly in dorsolateral prefrontal cortex, superior parietal cortex, and temporal cortex, and in language‐related regions in the left hemisphere including Broca and Wernicke's areas. Inferior regions of primary motor and sensory cortex in the prefrontal and postgyral cortices that are associated with function within the facial region also showed increased heritability with age. Estimated heritability values at age 18 increased to greater than 0.90 in regions of the prefrontal cortex, superior parietal cortex, temporal cortex, and left inferior pre‐ and post‐central gyri and left angular gyrus.
Figure 4.
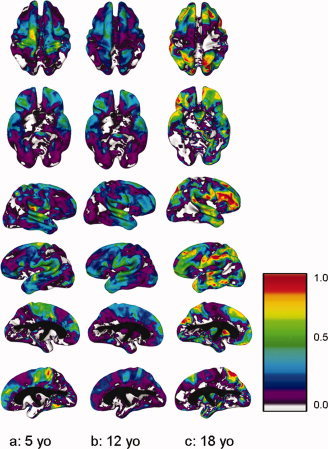
Heritability (a 2) at ages 5, 12, and 18 years for superior, inferior, right and left cortical surfaces. Values at each age were derived from the modeled interaction of age as a continuous variable with estimates of genetic and environmental variance components. Color bar shows scale of heritability values from 0.0 to 1.0.
Changes in heritability were driven by regionally specific alterations of both the genetic and environmental components (Supplemental Fig. 1). The total variance averaged across the brain decreased with age. Regions of increasing heritability were typically characterized by stable genetic variance in the presence of a decreasing environmental component, although in some regions increases in absolute genetic variance also contributed, particularly within the anterior frontal and dorsal parietal regions. The areas of decreasing heritability within the superior medial frontal gyrus, primary motor, and somatosensory cortices showed decreases in the genetic component. Environmental variance increased in the primary somatosensory regions, decreased within the superior medial frontal gyrus, and decreased or stayed stable in other areas of the brain.
We further explored developmental effects on heritability by dividing the sample into an older and younger group and comparing variance components between the two groups. Shared environmental factors were again removed from the model because they had not shown a significant effect in the cross‐sectional analyses. Figure 5 shows areas in which the proportion of variance due to genetic factors is statistically significant for the younger and older age groups and the differences in heritability between the two groups. Younger children showed heritability in the pre‐ and post‐central sulcus, cingulate gyri, left superior temporal gyrus and right inferior temporal gyrus, which was not present in the older group. Areas that were heritable in the older but not younger groups included bilateral areas of the dorsal prefrontal cortex, orbitofrontal cortex, bilateral superior parietal cortex, inferior occipital cortex, and left inferior temporal gyri (see Supplemental Fig. 2 for differences in variance components between the two age groups).
Figure 5.
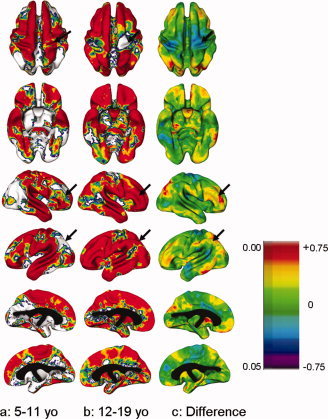
Age‐related changes in heritability for younger and older children. Variance component estimates are calculated using the AE model, since the shared environment component did not significantly affect results. Estimates of variance components were computed separately for younger children (aged 5–11) and for older children and adolescents (aged 12–19). Columns (a) and (b) show areas that are significantly heritable for younger (a) and older (b) age groups. Column (c) is a map of differences in heritability, created by subtracting the values for the younger group from those of the older group. Arrows indicate regions in which heritability changes between age groups. Numbers to the left of the scale bar indicate degree of statistical significance. Numbers to the right of the scale bar indicate the magnitude of the difference in heritability: light green, yellow, and red indicate heritability increasing with age; dark green, blue, and purple regions indicate decreasing heritability. A FDR threshold of q = 0.05 was applied to significance maps.
DISCUSSION
In this report, we have demonstrated for the first time that heritability of cortical thickness shows both regional and age‐related variation in children and adolescents. The sensitivity of brain structures to genetic or environmental factors is a topic of intense current interest as it may provide a means to link genetic or environmental causes with downstream behavioral and cognitive function. For example, regions that show significant heritability may be useful as intermediate phenotypes in the search for the genes responsible for neuropsychiatric disorders [Boomsma et al., 2002; Glahn et al., 2007]. Regions that instead show significant plasticity in response to environmental factors may represent areas with the potential for adaptive response.
In the cross‐sectional analyses, we found regions within the dorsal frontal and temporal cortices to be significantly heritable, consistent with previous studies (Figs. 2 and 3) [Hulshoff Pol et al., 2006; Thompson et al., 2001]. However we additionally found areas of significant heritability in the orbitofrontal cortex, superior parietal regions, and inferior surfaces of the temporal lobes, possibly related to increased power afforded by the larger sample size, or to dynamic effects of age as discussed in more detail below.
It is noteworthy that many of the gyri with the highest heritability estimates have well‐documented roles in cognition, speech, sociality, and language; functions thought to have developed or been enhanced in humans relatively recently in evolutionary time [Fisher and Marcus, 2006]. Comparative anatomic studies have demonstrated that many of the most prominent anatomic differences between humans and nonhuman primates lie within these regions as well, including the gyri encompassing Brodmann's areas 9, 10, and 11 (prefrontal and orbitofrontal cortices), 44 and 45 (Broca's area), and areas 21, 22, 37, 39, and 40 (superior temporal and supramarginal cortex) [Carroll, 2003]. Though studies in animal models clearly demonstrate the universal importance of genes in the patterning of all brain regions [Grove and Fukuchi‐Shimogori, 2003; Monuki and Walsh, 2001], the present findings show that genetically‐mediated variance is topologically variable, at least with respect to cortical thickness.
An ongoing evolutionary process might also explain why high genetic variability persists in relatively novel cortical regions, but not in others, as genes influencing evolutionarily “older” regions have had more time to reach allelic fixation (i.e., an elimination of genetic variance at relevant loci over time due to natural selection, genetic drift, or other evolutionary processes). An alternate, albeit related, explanation would be that regions with low genetic variance have greater functional constraints on their determinants of cortical thickness, such that genetic mutations influencing these regions will typically be eliminated quickly from the population through purifying selection. Comparative genomic experiments have shown that a subset of neurally‐expressed genes have evolved more rapidly in humans than in other primates [Chimpanzee Sequencing and Analysis Consortium, 2005; Dorus et al., 2004; Khaitovich et al., 2005]; both gene expression changes and protein sequence modification have accelerated in humans relative to nonhuman primates [Caceres et al., 2003; Enard et al., 2002; Gu and Gu, 2003; Hsieh et al., 2003; Uddin et al., 2004]. The findings of increased genetic variance in evolutionarily recent structures may represent a remnant of these rapid neurogenetic changes that accompanied our divergence from other primates.
The ACE model allows separation of familial similarity due to shared environment (C) from that due to genetic factors (A). In the few available studies of heritability of brain volumes which employed an ACE model, only the lateral ventricles have demonstrated significant contributions from shared environmental factors [Baare et al., 2001; Wallace et al., 2006]. A previous study using ACE modeling together with VBM to measure heritability of gray and white matter density in a sample of adult twins found common environmental factors to have limited effects within small regions of the left amygdala, left anterior corpus callosum, right optic radiation, and right corticospinal tract [Hulshoff Pol et al., 2006]. In the present study, we found that shared environmental factors had minimal effect on cortical thickness (see Fig. 2). It has been demonstrated that a large sample is necessary to detect C [Posthuma and Boomsma, 2000], and the present study may lack sufficient power. Studies of heritability of cognition have found that there is a gene × environment interaction such that shared environmental factors become more prominent relative to genetic factors as socioeconomic conditions worsen [Harden et al., 2007; Turkheimer et al., 2003]. The subjects in the current study came primarily from middle to high socioeconomic settings. It is possible that if twins were recruited from across a broader range of environmental conditions, shared environmental effects would also show stronger influence on brain structure.
Areas in which nongenetic factors were the chief contributors to variance were extensive. It is not possible with this study design to separate true unique environmental influences from other nongenetic sources of variance, including measurement error, making interpretation more difficult. However, it is intriguing that these areas included those regions associated with primary motor and sensory functions, whereas stronger genetic effects were seen more prominently in regions of association cortex. One interpretation may be that increased plasticity within these regions reflects their relatively more direct interaction with the external environment.
The discussion of the cross‐sectional findings given above should be considered in light of the other conclusion of this study, which is that heritability of brain regions during development in children and adolescents is a moving target. Age effects showed a similar pattern regardless of whether changes were modeled using age as a continuous variable or if heritability values were calculated separately in younger and older children. Regions in the dorsolateral prefrontal cortex, orbitofrontal cortex, inferior postcentral gyrus, bilateral anterior and lateral superior temporal lobes, and left inferior temporal lobe showed greater heritability in older children, whereas regions of the superior pre‐ and postcentral gyrus, medial frontal lobe, anterior cingulate, posterior superior temporal lobes, and right inferior temporal lobe showed greater heritability in younger children. The modeled heritability at the oldest ages appear strikingly similar to those reported in the previously described voxel‐based study in adults [Thompson et al., 2001].
Current theories describe the creation of cortical areas as occurring through the establishment of a series of genetically controlled anchor points, which serve as loci for overlapping gradients of growth factors [Grove and Fukuchi‐Shimogori, 2003]. Characteristics of specific cortical areas develop over time in response to the local combination of growth factors and activation. It has been argued that primary motor and sensory cortices may serve as core anchor regions, which develop early in ontogeny through clear, strongly genetically‐mediated steps [Rosa and Tweedale, 2005]. The pattern observed here of genetic effects predominating in these core regions early in childhood may be consistent with their relatively early development.
Conversely, later maturing areas such as the prefrontal cortex, superior temporal gyri, and superior parietal lobes tend to become increasingly heritable during development. Increased heritability over childhood and adolescence has been observed in cognitive traits such as IQ [Plomin et al., 1997] and prosocial behavior [Knafo and Plomin, 2006]. One likely contributor to changing heritability is age‐ dependent gene expression [Plomin et al., 1997; Sun et al., 2005; Weickert et al., 2007]. Another potential factor is gene‐environment correlation, which occurs when the same genes affect both a trait and relevant features of the environment, and also acts to increase heritability values [Kendler and Baker, 2007; Scarr and McCartney, 1983]. This effect may become stronger during adolescence, as children become more independent and able to choose environments based on their genetic predispositions.
Total variance decreased over much of the cortex, and in many areas increasing heritability was due to a stable genetic contribution in the presence of decreasing environmental variance. One potential explanation for the decreasing variance is that it represents an example of canalization, the frequently observed robustness of phenotypes against minor genetic or environmental perturbations [Flatt, 2005; Schmalhausen, 1949; Tanner, 1963; Waddington, 1942]. Gene‐environment interactions have been proposed as a mechanism by which variation can be decreased over the course of development. Genetic determinants of plasticity in response to the environment may constrain structures to develop along a heritable trajectory from an undifferentiated beginning to a genetically determined mature state [Garlick, 2002]. Repetitive patterns of activity may also sculpt plastic developing structures. Zelditch et al. [2004] found that variance in murine skull morphometry decreased during early postnatal development. They hypothesized that high initial variance was due to the random stresses placed on skull tissues from relatively unorganized muscular activity early in life, and that variance decreased as patterns of activity took on the predictable characteristics of maturity [Zelditch et al., 2004]. Such a process could be relevant to activity‐dependent changes in the cerebral cortex.
Another constraint upon phenotypic variation is through morphological integration. Components of complex structures that are closely spatially or functionally connected tend to show increased covariance. In a previous study of a subset of this population, we found that variance decreased and strength of intracortical correlations increased in several of the same association areas, which demonstrate increasing heritability in the current study [Lerch et al., 2006]. It is tempting to speculate that these observations may be related to the increasing functional integration of these areas in maturing cortical networks.
The contrast between findings of increased heritability of cortical thickness with age seen here and decreased heritability of lobar gray matter volumes reported in a subset of this population [Wallace et al., 2006] appears to be driven chiefly by differences in the interaction of environmental variance with age. In the earlier report, unique environmental variance increased more than was the case for additive genetic variance, leading to a proportional decrease of genetic variance and thus of heritability. In the present study, the effects of unique environmental factors on average decreased, causing an increase in the proportion of variance explained by genetic factors.
Cortical gray matter volume is affected both by the thickness of the cortex and its surface area. Cortical surface area shows much more variation both between and within species than cortical thickness. The thickness of the cortex and its area are determined by different types of cell division during the original formation of the cortex [Rakic, 1988], suggesting that cortical thickness and area may be affected by different factors during both evolution and individual development [Rakic, 1995]. Although developmental changes in both cortical thickness and surface topology during childhood and adolescence have been reported [Gogtay et al., 2004; Sowell et al., 2002], little is known about how longitudinal changes in these measurements may relate to each other. These findings suggest that variations in cortical thickness and lobar volumes may be controlled by different factors, and that caution should be observed in interpreting gray matter volume and thickness as equivalent measures.
We used a completely automated method to assess cortical thickness. An advantage of an automated method is that rater error is not a factor. A limitation common to current structural neuroimaging techniques is that the location of specific cytoarchitecturally unique regions is estimated based on sulcal and gyral landmarks. It has been shown that the actual placement of Brodmann's areas with respect to cortical topological landmarks shows only partial overlap between individuals [Uylings et al., 2005]. This implies that when brain MRIs are coregistered to a common template using surface topology, the alignment of cytoarchitecturally similar regions between individuals is necessarily incomplete, and consequently that heritability values for cortical thickness also reflect variation in the location of the boundaries of these regions.
In summary, cross‐sectional analysis showed that more recently evolved regions such as the dorsal prefrontal cortex and orbitofrontal cortex, temporal lobes, and superior parietal lobes showed stronger genetic influences than phylogenetically older and earlier developing areas of the cortex. However, an exploration of age effects found that heritability values in these areas were affected by development. The regions which developed earlier also showed stronger genetic influences earlier. Conversely, later‐developing regions associated with complex cognitive functions became more heritable with maturation, consistent with previous studies showing that cognitive abilities such as IQ become more heritable with maturity [Plomin et al., 1997]. These findings suggest that some areas of the cortex are likely to be more useful as intermediate phenotypes for relating genes with behavioral features, and that studies of the effects of specific genes or environmental influences on cortical structure may be influenced by the age of the population under study.
Supporting information
Additional Supporting Information may be found in the online version of this article.
Supporting Information Figure 1
Supporting Information Figure 2
REFERENCES
- Chimpanzee Sequencing and Analysis Consortium ( 2005): Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437: 69–87. [DOI] [PubMed] [Google Scholar]
- Baare WF,Hulshoff Pol HE,Boomsma DI,Posthuma D,de Geus EJ,Schnack HG,van Haren NE,van Oel CJ,Kahn RS ( 2001): Quantitative genetic modeling of variation in human brain morphology. Cereb Cortex 11: 816–824. [DOI] [PubMed] [Google Scholar]
- Boomsma D,Busjahn A,Peltonen L ( 2002): Classical twin studies and beyond. Nat Rev Genet 3: 872–82. [DOI] [PubMed] [Google Scholar]
- Caceres M,Lachuer J,Zapala MA,Redmond JC,Kudo L,Geschwind DH,Lockhart DJ,Preuss TM,Barlow C ( 2003): Elevated gene expression levels distinguish human from non‐human primate brains. Proc Natl Acad Sci USA 100: 13030–13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SB ( 2003): Genetics and the making of Homo sapiens. Nature 422: 849–857. [DOI] [PubMed] [Google Scholar]
- Collins DL,Neelin P,Peters TM,Evans AC ( 1994): Automatic 3D intersubject registration of MR volumetric data in standardized Talairach space. J Comput Assist Tomogr 18: 192–205. [PubMed] [Google Scholar]
- Collins DL,Zijdenbos AP,Baare WFC,Evans AC ( 1999): ANIMAL+INSECT: Improved Cortical Structure Segmentation. Proceedings of the Annual Conference on Information Processing in Medical Imaging (IPMI). Visegrad, Hungary: Springer; pp 210–223. [Google Scholar]
- Dorus S,Vallender EJ,Evans PD,Anderson JR,Gilbert SL,Mahowald M,Wyckoff GJ,Malcom CM,Lahn BT ( 2004): Accelerated evolution of nervous system genes in the origin of Homo sapiens. Cell 119: 1027–1040. [DOI] [PubMed] [Google Scholar]
- Edwards AWF. 1972. Likelihood; an account of the statistical concept of likelihood and its application to scientific inference. Cambridge [Eng.]: University Press; xv, 235 pp. [Google Scholar]
- Enard W,Khaitovich P,Klose J,Zollner S,Heissig F,Giavalisco P,Nieselt‐Struwe K,Muchmore E,Varki A,Ravid R,Doxiadis GM,Bontrop RE,Paabo S( 2002): Intra‐ and interspecific variation in primate gene expression patterns. Science 296: 340–343. [DOI] [PubMed] [Google Scholar]
- Fisher SE,Marcus GF ( 2006): The eloquent ape: Genes, brains and the evolution of language. Nat Rev Genet 7: 9–20. [DOI] [PubMed] [Google Scholar]
- Flatt T ( 2005): The evolutionary genetics of canalization. Q Rev Biol 80: 287–316. [DOI] [PubMed] [Google Scholar]
- Garlick D ( 2002): Understanding the nature of the general factor of intelligence: The role of individual differences in neural plasticity as an explanatory mechanism. Psychol Rev 109: 116–136. [DOI] [PubMed] [Google Scholar]
- Genovese CR,Lazar NA,Nichols T ( 2002): Thresholding of statistical maps in functional neuroimaging using the false discovery rate. Neuroimage 15: 870–878. [DOI] [PubMed] [Google Scholar]
- Geschwind DH,Miller BL,DeCarli C,Carmelli D ( 2002): Heritability of lobar brain volumes in twins supports genetic models of cerebral laterality and handedness. Proc Natl Acad Sci USA 99: 3176–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn DC,Thompson PM,Blangero J ( 2007): Neuroimaging endophenotypes: Strategies for finding genes influencing brain structure and function. Hum Brain Mapp 28: 488–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogtay N,Giedd JN,Lusk L,Hayashi KM,Greenstein D,Vaituzis AC,Nugent TF 3rd,Herman DH,Clasen LS,Toga AW,Rapoport JL,Thompson PM ( 2004): Dynamic mapping of human cortical development during childhood through early adulthood. Proc Natl Acad Sci USA 101: 8174–8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove EA,Fukuchi‐Shimogori T ( 2003): Generating the cerebral cortical area map. Annu Rev Neurosci 26: 355–380. [DOI] [PubMed] [Google Scholar]
- Gu J,Gu X ( 2003): Induced gene expression in human brain after the split from chimpanzee. Trends Genet 19: 63–65. [DOI] [PubMed] [Google Scholar]
- Harden KP,Turkheimer E,Loehlin JC ( 2007): Genotype by environment interaction in adolescents' cognitive aptitude. Behav Genet 37: 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingshead AdB,Redlich FC (1958): Social Class and Mental Illness; A Community Study. New York: Wiley; ix, 442 p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh WP,Chu TM,Wolfinger RD,Gibson G ( 2003): Mixed‐model reanalysis of primate data suggests tissue and species biases in oligonucleotide‐based gene expression profiles. Genetics 165: 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulshoff Pol HE,Schnack HG,Posthuma D,Mandl RC,Baare WF,van Oel C,van Haren NE,Collins DL,Evans AC,Amunts K,Burgel U,Zilles K,de Geus E,Boomsma DI,Kahn RS ( 2006): Genetic contributions to human brain morphology and intelligence. J Neurosci 26: 10235–10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabani N,Le Goualher G,MacDonald D,Evans AC ( 2001): Measurement of cortical thickness using an automated 3‐D algorithm: A validation study. Neuroimage 13: 375–380. [DOI] [PubMed] [Google Scholar]
- Kendler KS,Baker JH ( 2007): Genetic influences on measures of the environment: A systematic review. Psychol Med 37: 615–626. [DOI] [PubMed] [Google Scholar]
- Khaitovich P,Hellmann I,Enard W,Nowick K,Leinweber M,Franz H,Weiss G,Lachmann M,Paabo S ( 2005): Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science 309: 1850–1854. [DOI] [PubMed] [Google Scholar]
- Kim JS,Singh V,Lee JK,Lerch J,Ad‐Dab'bagh Y,MacDonald D,Lee JM,Kim SI,Evans AC ( 2005): Automated 3‐D extraction and evaluation of the inner and outer cortical surfaces using a Laplacian map and partial volume effect classification. Neuroimage 27: 210–221. [DOI] [PubMed] [Google Scholar]
- Knafo A,Plomin R ( 2006): Prosocial behavior from early to middle childhood: Genetic and environmental influences on stability and change. Dev Psychol 42: 771–786. [DOI] [PubMed] [Google Scholar]
- Lenroot RK,Gogtay N,Greenstein D,Molloy E,Wallace GL,Vaituzis AC,Clasen LS,Blumenthal J,Lerch J,van Zijdenbos A,Evans AC,Thompson PM,Giedd JN ( 2005): Sexual dimorphism of brain developmental trajectories during childhood and adolescence. NeuroImage, published online, April 6, 2007, doi: 10.1016/j.neuroimage.2007.03/053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerch JP,Evans AC ( 2005): Cortical thickness analysis examined through power analysis and a population simulation. Neuroimage 24: 163–173. [DOI] [PubMed] [Google Scholar]
- Lerch JP,Pruessner JC,Zijdenbos A,Hampel H,Teipel SJ,Evans AC ( 2005): Focal decline of cortical thickness in Alzheimer's disease identified by computational neuroanatomy. Cereb Cortex 15: 995–1001. [DOI] [PubMed] [Google Scholar]
- Lerch JP,Worsley K,Shaw WP,Greenstein DK,Lenroot RK,Giedd J,Evans AC ( 2006): Mapping anatomical correlations across cerebral cortex (MACACC) using cortical thickness from MRI. Neuroimage 31: 993–1003. [DOI] [PubMed] [Google Scholar]
- MacDonald D,Kabani N,Avis D,Evans AC ( 2000): Automated 3‐D extraction of inner and outer surfaces of cerebral cortex from MRI. Neuroimage 12: 340–356. [DOI] [PubMed] [Google Scholar]
- Monuki ES,Walsh CA ( 2001): Mechanisms of cerebral cortical patterning in mice and humans. Nat Neurosci 4 ( Suppl): 1199‐1206. [DOI] [PubMed] [Google Scholar]
- Neale MC,Boker SM,Xie G,Maes HH ( 1999): Statistical Modeling, 5th ed. Richmond, VA: Department of Psychiatry, Virginia Commonwealth University. [Google Scholar]
- Neale MC,Cardon LR, North Atlantic Treaty Organization. Scientific Affairs Division ( 1992): Methodology for Genetic Studies of Twins and Families. Dordrecht, The Netherlands: Kluwer; xxv, 496 pp. [Google Scholar]
- Neale MC,Miller MB ( 1997): The use of likelihood‐based confidence intervals in genetic models. Behav Genet 27: 113–120. [DOI] [PubMed] [Google Scholar]
- Pennington BF,Filipek PA,Lefly D,Chhabildas N,Kennedy DN,Simon JH,Filley CM,Galaburda A,DeFries JC ( 2000): A twin MRI study of size variations in human brain. J Cogn Neurosci 12: 223–232. [DOI] [PubMed] [Google Scholar]
- Plomin R,Craig I ( 1997): Human behavioural genetics of cognitive abilities and disabilities. Bioessays 19: 1117–1124. [DOI] [PubMed] [Google Scholar]
- Plomin R,Fulker D,Corley R,DeFries J ( 1997): Nature, nurture, and cognitive development from 1 to 16 years: A parent‐offspring adoption study. Psychol Sci 8: 442–447. [Google Scholar]
- Posthuma D,Boomsma DI ( 2000): A note on the statistical power in extended twin designs. Behav Genet 30: 147–158. [DOI] [PubMed] [Google Scholar]
- Posthuma D,De Geus EJ,Neale MC,Hulshoff Pol HE,Baare WEC,Kahn RS,Boomsma D ( 2000): Multivariate genetic analysis of brain structure in an extended twin design. Behav Genet 30: 311–319. [DOI] [PubMed] [Google Scholar]
- Purcell S ( 2002): Variance components models for gene‐environment interaction in twin analysis. Twin Res 5: 554–571. [DOI] [PubMed] [Google Scholar]
- R Development Core Team ( 2006): R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Rakic P ( 1988): Specification of cerebral cortical areas. Science 241: 170–176. [DOI] [PubMed] [Google Scholar]
- Rakic P ( 1995): A small step for the cell, a giant leap for mankind: A hypothesis of neocortical expansion during evolution. Trends Neurosci 18: 383–388. [DOI] [PubMed] [Google Scholar]
- Rakic P,Ang E,Breunig J. 2004. Setting the stage for cognition: genesis of the primate cerebral cortex In: Gazzaniga MS, editor. The Cognitive Neurosciences, 3rd ed. Cambridge, Massachusetts: MIT Press; pp 33–49. [Google Scholar]
- Reveley AM,Reveley MA,Chitkara B,Clifford C ( 1984): The genetic basis of cerebral ventricular volume. Psychiatry Res 13: 261–266. [DOI] [PubMed] [Google Scholar]
- Robbins S,Evans AC,Collins DL,Whitesides S ( 2004): Tuning and comparing spatial normalization methods. Med Image Anal 8: 311–323. [DOI] [PubMed] [Google Scholar]
- Roland PE,Zilles K ( 1998): Structural divisions and functional fields in the human cerebral cortex. Brain Res Brain Res Rev 26(2/3): 87–105. [DOI] [PubMed] [Google Scholar]
- Rosa MG,Tweedale R ( 2005): Brain maps, great and small: Lessons from comparative studies of primate visual cortical organization. Philos Trans R Soc Lond B Biol Sci 360: 665–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter M,Moffitt TE,Caspi A ( 2006): Gene‐environment interplay and psychopathology: Multiple varieties but real effects. J Child Psychol Psychiatry 47(3/4): 226‐261. [DOI] [PubMed] [Google Scholar]
- Scarr S,McCartney K ( 1983): How people make their own environments: A theory of genotype greater than environment effects. Child Dev 54: 424–435. [DOI] [PubMed] [Google Scholar]
- Schmalhausen II ( 1949): Factors of evolution: The Theory of Stabilizing Selection. Chicago: University of Chicago Press. [Google Scholar]
- Shaw P,Greenstein D,Lerch J,Clasen L,Lenroot RK,Gogtay N,Evans A,Rapoport JL,Giedd J ( 2006): Intellectual ability and cortical development in children and adolescents. Nature 440: 676–679. [DOI] [PubMed] [Google Scholar]
- Sled JG,Zijdenbos AP,Evans AC ( 1998): A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans Med Imaging 17: 87–97. [DOI] [PubMed] [Google Scholar]
- Sowell ER,Thompson PM,Rex D,Kornsand D,Tessner KD,Jernigan TL,Toga AW ( 2002): Mapping sulcal pattern asymmetry and local cortical surface gray matter distribution in vivo: Maturation in perisylvian cortices. Cereb Cortex 12: 17–26. [DOI] [PubMed] [Google Scholar]
- Sun T,Patoine C,Abu‐Khalil A,Visvader J,Sum E,Cherry TJ,Orkin SH,Geschwind DH,Walsh CA ( 2005): Early asymmetry of gene transcription in embryonic human left and right cerebral cortex. Science 308: 1794–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner JM ( 1963): The regulation of human growth. Child Dev 34: 817–847. [DOI] [PubMed] [Google Scholar]
- Thompson PM,Cannon TD,Narr KL,van Erp T,Poutanen VP,Huttunen M,Lonnqvist J,Standertskjold‐Nordenstam CG,Kaprio J,Khaledy M,Dail R,Zoumalan CI,Toga AW ( 2001): Genetic influences on brain structure. Nat Neurosci 4: 1253–1258. [DOI] [PubMed] [Google Scholar]
- Tramo MJ,Loftus WC,Stukel TA,Green RL,Weaver JB,Gazzaniga MS ( 1998): Brain size, head size, and intelligence quotient in monozygotic twins. Neurology 50: 1246–1252. [DOI] [PubMed] [Google Scholar]
- Turkheimer E,Haley A,Waldron M,D'Onofrio B, Gottesman,II ( 2003): Socioeconomic status modifies heritability of IQ in young children. Psychol Sci 14: 623–628. [DOI] [PubMed] [Google Scholar]
- Uddin M,Wildman DE,Liu G,Xu W,Johnson RM,Hof PR,Kapatos G,Grossman LI,Goodman M ( 2004): Sister grouping of chimpanzees and humans as revealed by genome‐wide phylogenetic analysis of brain gene expression profiles. Proc Natl Acad Sci USA 101: 2957–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uylings HB,Rajkowska G,Sanz‐Arigita E,Amunts K,Zilles K ( 2005): Consequences of large interindividual variability for human brain atlases: Converging macroscopical imaging and microscopical neuroanatomy. Anat Embryol (Berl) 210(5/6): 423–431. [DOI] [PubMed] [Google Scholar]
- Waddington CH ( 1942): Canalization of development and the inheritance of acquired characters. Nature 150: 563–565. [DOI] [PubMed] [Google Scholar]
- Wallace GL,Schmitt JE,Lenroot RK,Viding E,Ordaz S,Rosenthal MA,Molloy E,Clasen L,Kendler KS,Neale MC,Giedd JN ( 2006): A pediatric twin study of brain morphometry. Journal of Child Psychology and Psychiatry 47: 987–993. [DOI] [PubMed] [Google Scholar]
- Weickert CS,Webster MJ,Gondipalli P,Rothmond D,Fatula RJ,Herman MM,Kleinman JE,Akil M ( 2007): Postnatal alterations in dopaminergic markers in the human prefrontal cortex. Neuroscience 144: 1109–1119. [DOI] [PubMed] [Google Scholar]
- White T,Andreasen NC,Nopoulos P ( 2002): Brain volumes and surface morphology in monozygotic twins. Cereb Cortex 12: 486–493. [DOI] [PubMed] [Google Scholar]
- Wright IC,Sham P,Murray RM,Weinberger DR,Bullmore ET ( 2002): Genetic contributions to regional variability in human brain structure: Methods and preliminary results. Neuroimage 17: 256–271. [DOI] [PubMed] [Google Scholar]
- Zelditch ML,Lundrigan BL,Garland T Jr ( 2004): Developmental regulation of skull morphology. I. Ontogenetic dynamics of variance. Evol Dev 6: 194–206. [DOI] [PubMed] [Google Scholar]
- Zijdenbos AP,Forghani R,Evans AC ( 2002): Automatic “pipeline” analysis of 3‐D MRI data for clinical trials: Application to multiple sclerosis. IEEE Trans Med Imaging 21: 1280–1291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article.
Supporting Information Figure 1
Supporting Information Figure 2