

Abstract
The association of white matter (WM) lesions and grey matter (GM) atrophy is a feature in relapsing‐remitting multiple sclerosis (RRMS). The spatiotemporal distribution pattern of WM lesions, their relations to regional GM changes and the underlying dynamics are unclear. Here we combined parametric and non‐parametric voxel‐based morphometry (VBM) to clarify these issues. MRI data from RRMS patients with progressive (PLV, n = 45) and non‐progressive WM lesion volumes (NPLV, n = 44) followed up for 12 months were analysed. Cross‐sectionally, the spatial WM lesion distribution was compared using lesion probability maps (LPMs). Longitudinally, WM lesions and GM volumes were studied using FSL‐VBM and SPM5‐VBM, respectively. WM lesions clustered around the lateral ventricles and in the centrum semiovale with a more widespread pattern in the PLV than in the NPLV group. The maximum local probabilities were similar in both groups and higher for T2 lesions (PLV: 27%, NPLV: 25%) than for T1 lesions (PLV: 15%, NPLV 14%). Significant WM lesion changes accompanied by cortical GM volume reductions occured in the corpus callosum and optic radiations (P = 0.01 corrected), and more liberally tested (uncorrected P < 0.01) in the inferior fronto‐occipital and longitudinal fasciculi, and corona radiata in the PLV group. Not any WM or GM changes were found in the NPLV group. In the PLV group, WM lesion distribution and development in fibres, was associated with regional GM volume loss. The different spatiotemporal distribution patterns of patients with progressive compared to patients with non‐progressive WM lesions suggest differences in the dynamics of pathogenesis. Hum Brain Mapp, 2010. © 2010 Wiley‐Liss, Inc.
Keywords: MRI, longitudinal, voxel‐based morphometry, lesion‐probability maps, parametric, non‐parametric, grey matter, relapsing‐remitting multiple sclerosis
INTRODUCTION
Multiple sclerosis is an inflammatory‐mediated demyelinating disease of the central nervous system implicating degenerative processes and progressive cerebral atrophy of both, white matter (WM) and grey matter (GM) [Pirko et al., 2007]. Quantitative MRI indicates that GM atrophy develops faster than WM atrophy [Chard et al., 2004], occurs even in the earliest stages of the disease [Chard et al., 2002; Chard et al., 2004; Dalton et al., 2004] and is more related to physical disability and cognitive impairment [Amato et al., 2004; Chard et al., 2002; Dalton et al., 2004; De Stefano et al., 2003; Ge et al., 2001; Quarantelli et al., 2003; Sailer et al., 2003; Sanfilipo et al., 2005; Sanfilipo et al., 2006; Tiberio et al., 2005] than WM‐lesion volumes. To date, however, the spatiotemporal relations between regional WM and GM changes are less well understood.
The assumption that WM lesions and GM volumes are associated with each other is supported by a positive correlation between WM lesion load and atrophy of total GM [Chard et al., 2002; De Stefano et al., 2003; Furby et al., 2009; Ge et al., 2001; Sanfilipo et al., 2005]. In contrast, regression analyses of regional GM atrophy and WM lesions have revealed conflicting results ranging from low [Morgen et al., 2006; Prinster et al., 2006] to moderate correlation [Charil et al., 2007; Sailer et al., 2003]. Regional measures in principle are more sensitive to subtle inhomogenously distributed cerebral volume changes than global measures. In contrast to WM lesions, however, the majority of regional GM changes are not visible on conventional MRI [Pirko et al., 2007]. Therefore, measurement of GM volume loss as an alternative, indirect measure of regional pathology has been used in numerous neuroimaging studies of relapsing and progressive MS [Chard et al., 2002; Charil et al., 2007; De Stefano et al., 2003; Ge et al., 2001; Morgen et al., 2006; Prinster et al., 2006; Sailer et al., 2003; Sanfilipo et al., 2005]. Longitudinal MRI studies relating regional GM volume changes to WM lesions in MS [Battaglini et al., 2009a; Bendfeldt et al., 2009a; Pagani et al., 2005; Sepulcre et al., 2006] (Table I) suggested an association between increasing WM lesion volumes and decreasing GM volumes in patients with RRMS. The presence of WM lesions, however, only partially accounted for variance changes of GM atrophy, suggesting that other neurodegenerative processes are probably involved as well [Sepulcre et al., 2009].
Table I.
Association of WM lesions and regional GM volumes
| n | Age (years) | DD (years) | EDSS | Method | MRI findings (GM/WM association) | |
|---|---|---|---|---|---|---|
| [Battaglini et al., 2009] | 59 (RRMS) 25 (NC) bs vs. y3 | 41.0 (10.6) | 1.8 (0.1–17) | Baseline 1.5 (0–5) Follow‐up 1.5 (1–5) | FSL‐VBM/ randomize SIENAr | RRMS: ‐baseline: significant GMV reductions in the frontal, temporal, parietal, occipital lobes and insula compared to NC; follow‐up: further GMV reductions in lateral frontal and parietal cortices, correlated with increasing T2‐LV |
| [Bendfeldt et al., 2009a] | 151 (RRMS) bs vs. y1 | 35 (17–60) | 10 (6–17) | Baseline 2.0 (1.5–3.0); Follow‐up 2.3 (1.5–3.0) | VBM/SPM5 | RRMS (N = 151): follow‐up: significant GMV reductions in fronto‐temporal regions RRMS subgroup analysis (N = 45): follow‐up: additional GMVreductions in frontal and parietal cortical regions of patients with increasing T2‐plusT1‐LV, but not in patients without increasing T2plusT1‐LV |
| [Sepulcre et al., 2006] | 31 (PPMS) 15 (NC) bs vs. y1 | 43.2 43.7 | 3 (2–5) | 4.5 | VBM/SPM2 | PPMS: baseline: bilateral thalamic atrophy in association with lesion load (number of voxels, 893; corrected P = 0.003); follow‐up: GMV reductions in putamen, caudate, thalami, cortical and infratentorial areas |
| [Pagani et al., 2005] | 70 (MS): 20 (RRMS) 19 (SPMS) 31 (PPMS) 15 months | 49.9 (25–68); 35.9 (25–43); 49.3 (35–58) | 10 (0–34); 5 (0–20); 16 (6–34) | 5 (0–8); 3 (0–5); 6 (4–8) | SIENA/SPM99 | RRMS: ‐follow‐up: enlargement of the ventricular system correlated with changes in T2‐LV (r = −0.72 to −0.81) and T1‐LV (r = −0.69 to −0.76); ‐SPMS: ‐follow‐up: changes in T2‐LV correlated with development of atrophy in the left cingulate sulcus (r = −0.74), left inferior frontal gyrus (r = −0.71), left orbital gyrus (r = −0.75), left insula (r = −0.71), and right inferior portion of the lateral fissure (r = −0.76).; PPMS:‐follow‐up: changes in T1‐LV correlated with development of atrophy of the bilateral central sulci (r = −0.58 to −0.65), left angular gyrus (r = −0.69), and head of the left caudate nucleus (r = −0.61). |
In this study, we examined the spatiotemporal relations between individual WM lesions and regional GM volume changes. We focused on patients with RRMS and (a) increasing, i.e. ‘progressive’ total T2 and T1 lesion volumes (PLV; n = 45) and newly developed WM lesions and (b) with ‘non‐progressive’ total T2 and T1 lesion volumes without newly developed WM lesions (NPLV; n = 44) followed up for a mean period of one year. The regional distribution pattern of T2‐ as well as T1 lesions was studied in both groups at baseline and one‐year follow‐up using lesion probaility maps (LPMs) which represent consistent image changes across groups of subjects. The LPM is a suitable tool for studying in vivo MS lesion distribution, providing indirect clues regarding the mechanisms of lesion development of the entire brain, while avoiding WM aprioristic biases [Sepulcre et al., 2009]. Here, two different VBM analyses (FSL/VBM, based on the FMRIB Software Library tools and SPM5/VBM, based on the Statistical Parametric Mapping software package) were used to compare longitudinally voxel‐wise regional WM lesion volume changes and GM volume changes. The anatomical association of regional GM atrophy progression in relation to the extent of focal white matter lesions was studied. On the basis of our own and other previous longitudinal MRI studies (Table I), we hypothesized that the distribution patterns of focal WM lesions and GM volume changes are associated in RRMS. In particular, we hypothesized that regional WM lesion volume changes are anatomically associated with GM volume reductions in related cortical brain areas in the PLV but not in the NPLV group of patients.
MATERIALS AND METHODS
Patients
We analysed pairs of MRI data from 89 patients of the case‐controlled study for genotype‐phenotype associations in MS (GeneMSA; GSK, UK) with the diagnosis of relapsing‐remitting MS [McDonald et al., 2001] (64 women, 25 men; mean age at baseline, 41.5 years; SD 10.0 years). Seventy‐three patients received immunomodulatory‐immunosuppressive drugs (interferon‐β‐1a, interferon‐β‐1b, glatiramer acetate) during the entire study (Table I). No change of medication occurred between baseline and follow‐up scan. During follow‐up, 23 RRMS patients had received corticosteroid therapy to treat acute relapse. At time of baseline and follow up MR scan, all patients had been relapse‐ and steroid‐free for at least one month. 45 RRMS patients had increasing T2‐ as well as T1‐lesion burden ('progressive lesion volumes', PLV) with a minimum of 1% change in total lesion volume, whereas 44 patients had neither T2‐ nor T1‐lesion volume increase ('non‐progressive lesion volumes', NPLV) during follow‐up. The study was approved by the local ethical standards committee and written informed consent was obtained from each subject.
MR Image Acquisition
All subjects were scanned twice (baseline and one‐year follow‐up) using a MAGNETOM Avanto 1.5 T scanner (SIEMENS, Erlangen, Germany) at the University Hospital Basel. A three‐dimensional (3D) magnetization prepared rapid acquisition gradient echo sequence (MPRAGE) generated 160 contiguous, 1 mm thick sagittal slices. Imaging parameters were: echo time, 3 ms; time‐to‐repetition, 20.8 ms; flip angle, 12; matrix size, 240 × 256; field of view, 24.0 × 25.6 cm2 matrix; voxel dimensions, 1 × 1 × 1 mm3. Transaxial contiguous 3‐mm dual‐echo 3D T2‐weighted images were acquired using turbo spin echo. T2 image parameters were: echo time 14 ms; time‐to‐repetition 3,980 ms; flip angle 180; turbo factor 7; matrix size 192 × 256; field of view 18.75 × 25 cm2; voxel dimension 0.9766 × 0.9766 × 3.0 mm3. T1 image parameters were: TE 8.1 ms, TR 400 ms, matrix size 192 × 256 mm, field of view 250 mm, voxel dimension 0.9766 × 0.9766 × 3.0 mm3.
MR Imaging Data Analysis
VBM protocol
MR images for all subjects were analysed on a commercially available Intel‐based workstation running Debian Linux 3.1 using voxel‐based morphometry (VBM). Images were processed with Statistical Parametric Mapping software (SPM5, Wellcome Department of Imaging Neurosciences, University College London, [http://www.fil.ion.ucl.ac.uk/spm]. version 958, last updated Dec13, 2007 running under the MATLAB 7.00 (R14) environment.
The images were processed using the VBM toolbox v1.03 (http://dbm.neuro.uni-jena.de/vbm/, last updated Dec 6, 2006 as described before [Bendfeldt et al., 2009a]. In brief: the method was modified to reduce the influence of MS lesions in the process, which could alter the normalization and segmentation procedures. For any fitted model outliers and problem subjects were identified with diagnostic statistics (SPMd toolbox, http://www.sph.umich.edu/~nichols/SPMd/version 5beta, last downloaded 03/01/2008). The essential diagnostics checked were the spatial outlier rate (expressed as a percentage of the expected number of outliers per image) and the image of Shapiro‐Wilk normality test statistics.
To prevent WM lesions from being misclassified as GM, lesions identified on T2w images were masked from the 3D MPRAGE images. The 3D binary masks of the MS lesions of the T2w scans were co‐registered to the MPRAGE using the SPM5 Co‐register function. In the segmentation step, images were spatially normalized into the same stereotactic space. The normalization was performed by first estimating the optimum 12‐variable affine transformation for matching images and then optimizing the normalization using 16 nonlinear iterations. Segmentation accuracy was assessed by examining axial slices of each subject's GM, WM and cerebrospinal fluid (CSF) image in the individual's space. The warping accuracy was assessed by displaying axial slices from each subject with edges from the atlas image.
To preserve the total within‐voxel volume, which may have been affected by the nonlinear transformation, every voxel's signal intensity in the segmented GM images was multiplied by the Jacobian determinants derived from the spatial normalization. The analysis of these modulated datasets was used to detect regional differences in absolute tissue volume. Finally, to increase the signal‐to‐noise ratio and to account for variations in normal gyral anatomy all images were smoothed using a 5 mm full‐width‐at half‐maximum isotropic gaussian kernel as done before [Bendfeldt et al., 2009a; Borgwardt et al., 2007a, b, 2008; Fusar‐Poli et al., 2007]. A small smoothing kernel, allowed us to detect volume changes in small structures such as the medial temporal lobes, parahippocampal gyrus (PHG) and anterior cingulate cortex (ACC). Also, according to the matched filter theorem, the width of the smoothing kernel determines the scale at which morphological changes are most sensitively detected [White et al., 2001].
White Matter LPMs
To obtain WM LPMs and total lesion volumes, WM lesions were outlined at voxel level by four independent observers on each of the transverse sections of the T2‐weighted MR images and T1‐weighted MR images by means of the commercially available semi‐automatic thresholding contour software AMIRA 3.1.1 (Mercury Computer Systems Inc) in accordance with common practice in brain imaging studies [Kappos et al., 2006]. Interrater reliability was excellent (intraclass correlation coefficient = 0.99; P < 0.001).
To create LPMs of the T2‐ and T1‐weighted images, all baseline and follow‐up images were linearly registered and normalized from native space to a template in standard space (MNI152_T1_1mm_brain, 1 × 1 × 1 mm3/voxel) with an affine transformation using FMRIB Software library's (FSL) FLIRT (fast linear interpolation and registration tool; with the following settings: 12‐parameter, correlation ratio, trilinear interpolation). The resulting transformation matrices were then co‐registered and averaged by using the SPM5 software, resulting in maps with a value for each voxel ranging from zero to one, indicating the proportion of patients with a lesion in that particular voxel. According to [Roosendaal et al., 2009] the probability maps were thresholded at 0.1, thus showing voxels, in which in at least 10% of the total of 45 and 44 patients, respectively, a lesion was present.
Statistical Analysis
Demographic data
The median and interquartile range, or the mean and standard deviation were used to describe clinical and MRI characteristics. We used chi‐squared test, paired t‐test, Mann‐Whitney‐U and Wilcoxon test for non‐parametric data to compare demographic and clinical variables. For these tests, a significance level of P < 0.05 was considered. Statistical analysis was performed with SPSS software, version 15 (SPSS Inc, Chicago, IL).
Longitudinal GM and WM Changes
Regional GM changes
Summary from previous publication
Between‐group differences (baseline vs. one‐year follow‐up) in GM volume were estimated by fitting an analysis of covariance (ANCOVA) model at each intracerebral voxel in standard space [Bendfeldt et al., 2009a]. For each subject follow‐up minus baseline difference images were created, and then analysed with a regression model with an intercept (parameter of interest) and centred covariates of age, gender and disease duration.
To assess additional nuisance variation due to head size differences the analysis was adjusted for each subject's global GM volume (GMV) by entering the global values as additional covariate. GMV (mean value) was calculated by SPM5. Statistical maps were assessed for significance with cluster size inference adjusted for non‐stationarity [Hayasaka et al., 2004; Moorhead et al., 2005] (http://dbm.neuro.uni-jena.de/vbm/non-stationary-cluster-extent-correction/). A cluster‐defining threshold of P = 0.001 uncorrected was used, and clusters were considered significant at P < 0.01 cluster level, corrected for a whole‐brain search (though for completeness our tables also report family‐wise error (FWE)‐corrected voxel‐wise P‐values as well). Finally, the MNI (Montreal Neurological Institute) coordinates corresponding to areas of significant differences were converted to Talairach coordinates. Significant clusters were anatomically localised using the atlas of Talairach and Tournoux, except for foci in and close to the cerebellum, which were localised using the atlas of Schmahmann et al. [1999].
Regional WM Lesion Changes
Statistical analysis of total WM lesion volume change (baseline vs. one‐year follow‐up) was performed with SPSS software, version 15 (SPSS Inc, Chicago, IL).
We tested for regionally specific differences in the expression of lesions in the PLV and NPLV groups in the course of one year. This involved comparison of the mean lesion load at each voxel from the baseline and year one scans using non‐parametric FSL‐VBM analysis.
For statistical analysis of the individual lesion volume changes during follow‐up, a single 4D image file was created from the registered 3D lesion files. To approximate the neuropathological characteristics of MS with greater tissue damage in the centre of the lesions, the 4D image file was smoothed by applying an isotropic gaussian kernel (5‐mm FWHM). A WM regional lesion frequency mask was created with the mean of the individual 3D binary masks. The use of this mask ensured that only voxels of WM were included in the analysis. Subsequently, paired differences were determined using ‘randomise’ implemented in FMRIB's Software Library (FSL), a permutation program, which enables modelling and inference on statistic maps (with an unknown null distribution) using standard GLM design setup [Nichols et al., 2002].
To analyse WM lesions data, we used Threshold‐Free Cluster Enhancement (TFCE), a newly proposed alternative to cluster‐based thresholding, which enhances cluster‐like structures in an image without having to define an initial cluster‐forming threshold in a binary way or carry out a large amount of data smoothing. Because of the exploratory nature of this analysis a rather liberal cluster‐defining threshold (P = 0.01 uncorrected) was used. To avoid false positive results only clusters >50 voxels were reported. Cluster‐like structures are enhanced but the image remains fundamentally voxelwise. Projection, commissural and association fibres are shown in Table III. Projection and commissural fibres refer to the anatomy toolbox of SPM 5, Wellcome Department of Imaging Neurosciences, University College London, [http://www.fil.ion.ucl.ac.uk/spm], version 958, last updated Dec 13, 2007]. Association fibres refer to the Jülich Histological Atlas (JHU) and the JHU WM tractography atlas, implemented in FMRIB's Software Library (FSL).
Table III.
Location and LPM measures of ‘progressive’ T2 lesions
| Projection‐and commissural fibres | Association fibres | Hemi‐sphere | MNI coordinates (x y z) | Clustersize (voxels) | P uncorrected | P corrected |
|---|---|---|---|---|---|---|
| Callosal body and Optic radiation | Anterior thalamic radiation; Forceps major; Cingulum (hippocampus) | Right | 22 −51 26 | 2535 | <0.001 | <0.01 |
| ILF Inferior occipitofrontal fasciculus | Left | −35 −43 −9 | 227 | <0.01 | 0.079 | |
| Forceps major Cingulum (hippocampus) | Left | −22 −55 19 | 52 | <0.01 | 0.079 | |
| Callosal body | n.l.f | Right | 18 −16 31 | 305 | <0.001 | 0.056 |
| Superior occipitofrontal fasciculus | Left | −27 −33 22 | 73 | <0.01 | 0.077 | |
| n.l.f | Left | −17 −33 26 | 61 | <0.01 | 0.094 | |
| n.l.f | Right | 18 −29 34 | 53 | <0.01 | 0.087 | |
| Corticospinal tract | SLF | Left | −31 −17 26 | 598 | <0.01 | 0.058 |
| SLF | Right | 31 −11 19 | 67 | <0.01 | 0.081 | |
| SLF | Right | 31 −5 26 | 59 | <0.01 | 0.094 | |
| Superior occipitofrontal fasciculus; Anterior thalamic radiation | Right | 22 11 21 | 389 | <0.01 | 0.056 | |
| SLF | Left | −25 9 19 | 155 | <0.001 | 0.063 | |
| ITG, optic radiation | ILF, SLF | Left | −44 −34 −15 | 61 | <0.01 | 0.089 |
Projection and commissural fibres refer to the anatomy toolbox of SPM5, Wellcome Department of Imaging Neurosciences, University College London, [http://www.fil.ion.ucl.ac.uk/spm]. Association fibres refer to the Jülich Histological Atlas (JHU) and the JHU WM tractography atlas, implemented in FMRIB's Software Library (FSL). ITG, inferior temporal gyrus; ILF, inferior longitudinal fasciculus; SLF, superior longitudinal fasciculus; n.l.f, no label found.
Association of WM LPMs and GM Volumes in the PLV Group
To determine the impact of T2 lesion volumes on cortical GM volume changes, we searched for correlations between both of them by using the framework of the general linear model implemented in the SPM5 software and applied voxel‐by‐voxel multiple linear regressions. This analysis strategy enabled us to screen the WM of the entire brain for baseline lesion volumes associated with further longitudinal WM lesion progression and increasing GM volumes, while avoiding the introduction of any a priori knowledge of possible WM anatomic target locations. All correlations were adjusted by age, gender and disease duration.
To ensure that only voxels of significant WM lesion volume changes were included in the analysis, a WM lesion mask was applied created with the TFCE image of the regional WM changes. The mean signal intensity values (SPM eigenvalues) of each significant cluster were extracted with the VOI (volume of interest) toolbox of SPM5. To minimize type I error, the level of significance for the results was set at stringent P < 0.05 after correction for multiple comparisons (false discovery rate method) [Grossman et al., 2004].
RESULTS
Clinical and MRI Characteristics
Of the 89 patients with RRMS, 45 subjects with PLV did not differ significantly from the 44 RRMS subjects with NPLV with respect to ethnicity, gender, mean inter‐scan interval, expanded disability status scale (EDSS) scores, medication, and lesion load neither at baseline nor at follow‐up (Table II).
Table II.
Clinical and MRI characteristics
| ‘Progressive’ N = 45 | ‘Non‐progressive’ N = 44 | Statistics: ‘progressive’ versus ‘non‐progressive’ (P value) | |
|---|---|---|---|
| Age at bs in years, mean (SD) | 39.0 (9.1) | 44.0 (10.3) | 0.01 |
| Male/Female (ratio) | 16/29 (1:1.8) | 9/35 (1:3.8) | 0.13 |
| Disease duration:Time since first symptoms at bs in years, median (IQR) | 8 (6–14) 80 | 10 (8–19) | 0.07 |
| Drug treatmenta (% of patients) | 77 | 0.91 | |
| Scan interval (months), mean, SD, p value | 12.6 (0.7) | 12.6 (1.1) | 0.99 |
| EDSS at bs, median (IQR) | 2.5 (1.5–3.0) | 2.5 (2.0–3.13) | 0.51 |
| EDSS at y1, median (IQR) | 2.5 (1.5–3.5) | 2.5 (2.0–3.13) | 0.26 |
| Statistics: EDSS change, bs versus y1 (P value) | 0.71 | 0.91 | |
| T2 lesion load in ml, median (IQR) at bs | 3.9 (2.4–10.1) | 3.3 (1.3–7.6) | 0.25 |
| T2 lesion load in ml, median (IQR) at y1 | 4.6 (2.6–11.3) | 3.1 (1.1–7.1) | 0.06 |
| T2 lesion volume change, bs versus y1: P‐value | <0.001 | <0.001 | |
| T1 lesion load in ml, median (IQR) at bs | 1.2 (0.3–3.5) | 0.9 (0.2–2.6) | 0.37 |
| T1 lesion load in ml, median (IQR) at y1 | 1.4 (0.5–4.1) | 0.8 (0.2–2.4) | 0.06 |
| T1 lesion volume change, bs versus y1: p‐value | <0.001 | <0.001 | |
| New T2‐Lesions at y1 (count) | 1 (0–4) | 0 (0–0) | |
| New Gd‐enhancing lesions at y1 (count) | 0 (0–1) | 0 (0–0) | 0.014 |
| T1/T2 at bs (mean, SD) | 0.32 (0.19) | 0.29 (0.16) | 0.45 |
| T1/T2 at y1 (mean, SD) | 0.33 (0.18) | 0.27 (0.15) | 0.06 |
| GMV in cm3, (mean SD) at bs | 656 (78) | 637 (74) | 0.42 |
| GMV in cm3, (mean SD) at y1 | 643 (80) | 638 (75) | 0.78 |
| PGMVC, bs versus y1: (mean, SD), | −1.28 (0.3) | 0.17 (0.1)0.59 | |
| P‐value | 0.001 | 0.59 |
No changes in medication during follow‐up.
SD, standard deviation; IQR, interquartile range; EDSS, expanded disability status scale; bs, baseline; y1, year1; GMV, global grey matter volume; PGMVC, percentage grey matter volume change.
The mean inter‐scan interval of all MS subjects was 12 months. Changes of the T2 and T1 lesion volumes in the PLV and NPLV groups were highly significant (P < 0.001). Global GM volume decreased significantly in the PLV group, and remained unchanged in the NPLV group. Age and disease duration, however, were slightly increased in the NPLV group as compared with the PLV group.
Summary of the Results From Previous Publication
Longitudinal changes of regional cortical grey matter volumes
In patients with relapsing remitting MS (RRMS) (n = 151), significant cortical GM volume reductions between baseline and follow‐up scans were found in the anterior and posterior cingulate, the temporal cortex, and cerebellum [Bendfeldt et al. 2009a]. Within the RRMS group, those patients with PLV (n = 45) showed additional GM volume loss during follow‐up in the frontal and parietal cortex, and precuneus. In contrast, NPLV patients (n = 44) did not show any significant GM volume changes.
Results From the Present Study
Spatial deployment of T2‐hyperintense and T1‐hypointense lesions
We characterised the spatial deployment of WM lesions in the PLV and NPLV groups. LPMs revealed a typical distribution pattern of T2‐hyperintense lesions in the cerebral white matter, with lesion clusters mainly in the periventricular regions around the anterior and posterior horns of the lateral ventricles and in the centrum semiovale in the PLV (left column) and NPLV groups of patients (right column).
The T2‐ and T1‐lesion probability maps were thresholded to show voxels in which in at least 10% of the patients a lesion was present. The maximum local probabilities for both the T2‐ and T1‐lesions in the PLV group (peak probability 27% for T2 and 15% for T1; Fig. 1, upper panel) and in the NPLV group (peak probability 25% for T2 and 14% for T1; Fig. 1, lower panel) were almost the same. The peak probability for T2‐lesions was twice as high as compared to T1‐lesions in both groups. T2 as well as T1 lesions were localised more widespread in the PLV group, in particular in the optic radiations, the corona radiata and in the centrum semiovale (see Fig. 1).
Figure 1.
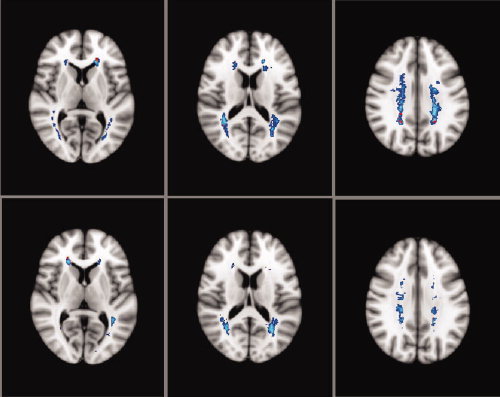
Baseline lesion probability maps for patients with ‘progressive’ (PLV) and ‘non‐progressive’ (NPLV) lesion volumes. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Regional WM Lesion Changes in PLV and NPLV
Non‐parametric FSL‐VBM analysis comparing the proportion of WM lesions at each voxel between baseline and year one identified specific T2 hyperintense areas, but not T1 hypointense areas, to be significantly more frequently involved at year one relative to baseline in the PLV group (Table III, Figs. 2 and 3). These areas corresponded anatomically to parts of the corticospinal tract, the corpus callosum, the inferior fronto‐occipital fasciculus, the ILF, both optic radiations and the corona radiata. Figure 3 illustrates WM lesion changes occuring in the PLV group in relation to baseline LPM and GM changes.
Figure 2.
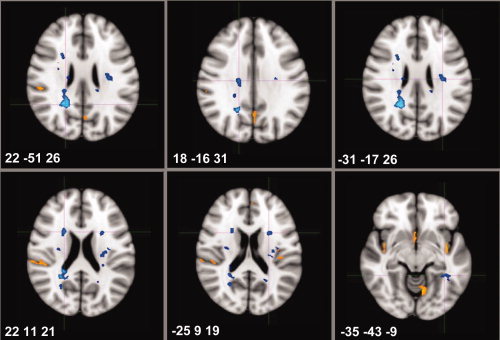
Areas of GM and WM volume changes in RRMS patients and ‘progressive’ WM lesion volume (n = 45). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 3.
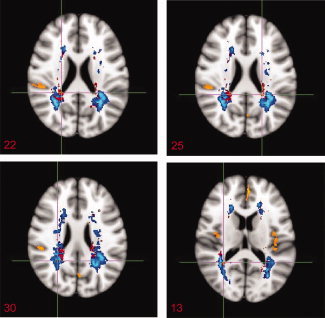
Overlay map of significant T2 lesion volume increases (red) in the PLV group of patients (n = 45) on a lesion probability map at baseline (LPM, blue) and GM volume changes (orange). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
No significant differences neither for T2 nor T1 lesions were found in the NPLV group.
Associations between WM LPMs and GM volume changes
Although significant focal WM lesion volume changes appeared in the PLV group (Table I), and not in the NPLV group, no direct anatomical overlap was found between areas of significant GM volume reduction and significant focal WM lesion volume changes in the PLV group (see Fig. 1). Reduced cortical GM volumes of the PLV group did not correlate with baseline WM LPMs associated with further longitudinal WM lesion progression in any of the areas described above.
DISCUSSION
To develop a better understanding of the spatiotemporal relations between regional GM atrophy and WM lesion changes in RRMS, here we studied their spatiotemporal relations in two patient groups with either ‘progressive’ or ‘non‐progressive’ WM lesion load. Voxelwise regional brain volume changes were assessed using VBM, a structural image analysis method that avoids an aprioristic knowledge about the relationship among these anatomic structures, and queries the entire brain [Sepulcre et al., 2009]. We used LPMs, obtained from T2‐weighted or T1‐weighted structural MRI of a large sample of MS patients and applied ‘optimized’ VBM to compare WM and GM changes.
Spatial Deployment and Development of WM Lesions
Baseline differences in the spatial deployment of cerebral WM lesions in the PLV and NPLV groups were shown using LPMs, which provide a quantitative description of the spatial distribution of the different lesion types. T2 lesions have a predilection for specific areas around the lateral ventricles and within the corpus callosum [Lee et al., 1999; Narayanan, 1997; Vrenken, 2006]. In this study, the majority of WM lesions in patients with RRMS were located in the periventricular regions as reported previously for patients with clinical isolated syndrome (CIS) and secondary progressive MS (SPMS) [Ceccarelli et al. 2008]. Typical distributions of lesion clusters with peaks at the anterior and posterior horns of the lateral ventricles and in the centrum semiovale were found, with a twice as high peak probability for T2 hyperintense lesions than for T1 hypointense lesions (black holes) in both patient groups.
Generally, both T2 and T1 lesions were localised more widespread in the PLV group as compared with the NPLV group. Except for age, which was lower in patients with increasing lesion volumes, the clinical and MRI data, including baseline WM lesion load, did not differ between the PLV and NPLV groups. Thus, the pattern of a more widespread spatial distribution of WM injury at baseline might be a central issue predicting further lesion development as well as development of GM atrophy in the PLV group. In comparison to the NPLV group, the PLV group consisted of the more active patients in terms of new lesions formation after one year follow‐up [Bendfeldt et al., 2009a], implicating the development of more brain atrophy in active than in non‐active patients. The underlying dynamics of new lesion development and the development of cortical pathology might therefore differ between the NPLV and PLV patients.
T2 lesion volume changes in the corticospinal tract, the corpus callosum, the inferior fronto‐occipital fasciculus, the ILF, both optic radiations, and the corona radiata in the PLV group, but not in the NPLV group of patients, was in accordance with the more widespread distribution of WM lesions in the PLV group at baseline. In comparison to the T2 lesions, however, no significant longitudinal differences were found for the T1 lesions. Total T1 lesion volume significantly increased in the PLV group during follow‐up. The peak probability of 15% for the T1 lesions, however, was relatively low as compared to a peak probability of 27% for the T2 lesions. Therefore, the variance between the spatial distribution patterns of the focal T1 lesions of the individual patients could have limited power of statistical group analysis. No significant longitudinal changes neither for T2 nor T1 lesions were found in the NPLV group, consistent with the finding that total lesion volume did not increase during follow‐up in this patient sample [Bendfeldt et al., 2009a].
Associations of WM Lesion Changes and GM Atrophy
Cerebral atrophy of both the WM and the GM of the central nervous system is a hallmark of MS pathology [Kutzelnigg et al., 2005] and has become an important marker for charting disease progression. Recently, research has focused on the tissue compartments and regions within which brain atrophy occurs [Chard et al., 2002; Chen et al., 2004; Dalton et al., 2004; De Stefano et al., 2003; Jasperse et al., 2007; Pagani et al., 2005; Prinster et al., 2006; Quarantelli et al., 2003; Sailer et al., 2003; Sepulcre et al., 2006; Tedeschi et al., 2005; Tiberio et al., 2005]. Previous VBM studies, as well as studies using other image analysis methods, have shown GM changes in MS patients in both cortical and deep GM regions [Audoin et al., 2006; Battaglini et al., 2009; Bendfeldt et al., 2009a; Chen et al., 2004; Pagani et al., 2005; Sepulcre et al., 2006].
Longitudinal MRI studies [Battaglini et al., 2009; Bendfeldt et al., 2009a; Pagani et al., 2005; Sepulcre et al., 2006] showed an association between increasing WM lesion volumes and increasing GM volume loss in the frontal and parietal cortex of patients with RRMS. In addition, WM lesion volumes were shown to correlate with ventricular enlargement [Pagani et al., 2005] and focal WM damage in the optic radiations with upstream GM atrophy of the lateral geniculate nucleus and visual cortex in patients with RRMS [Sepulcre et al., 2009]. The presence of WM lesions, however, only accounted for less than half of the variance changes for GM atrophy, suggesting that other neurodegenerative processes are probably involved as well [Sepulcre et al., 2009]. Battaglini et al. [2009] showed in 59 RRMS patients decreasing GM in several cortical regions (frontal, temporal, parietal, occipital lobes and insula). After three‐year follow‐up, a further reduction of GM volume in the lateral frontal and parietal cortices was observed, which generally correlated with increasing T2‐hyperintense lesion volume over the follow‐up period. Our own prior longitudinal VBM study demonstrated significant cortical GM volume reductions between baseline and follow‐up scans in the PLV but not in the NPLV group [Bendfeldt et al., 2009a, b]. Overall, longitudinal MRI studies support the hypothesis that the progression of regional GM volume reductions is associated with global WM lesion volume progression, predominantly in fronto‐temporal cortical areas.
Spatiotemporal Relationship of WM Lesion and Regional GM Volumes in RRMS
In this study, regionally specific significant differences in the expression of voxelwise T2 lesion changes in the PLV group occurred in different pathways interconnecting several cortical areas. Of all the cortical regions showing reduced GM volumes in the PLV group, none was in close spatial relationship to those WM lesions showing increasing T2 lesion volume during follow‐up. The pattern of GM reductions in the PLV group, however, might also originate from degeneration of axons damaged by progressive changes in WM lesions located more distantly in these pathways, e.g. in large periventricular lesions.
Different mechanisms may underlie the observed GM changes, including primary damage of the GM, such as GM lesions or transsynaptic degeneration [Brück, 2005; Hauser and Oksenberg, 2006; Perry and Anthony, 1999]. GM changes could also be secondary to axonal transection and retrograde neurodegeneration in the WM lesions of related WM tracts [Chard et al., 2002; Evangelou et al., 2000]. A recent cross‐sectional study using MRI‐based lesion probability maps (LPM) supports the hypothesis, that retrograde damage of the perykarya from axonal injury within MS plaques might be crucial to the genesis of GM atrophy [Sepulcre et al., 2009]. In this study, however, of all the cortical regions showing reduced GM volumes in the PLV group, none was anatomically closely adjacent to the WM lesions. Significant WM changes appeared in a number of fibre tracts only in the PLV, but not in the NPLV group. Thus, retrograde damage of the neuronal perikarya could at least partly be related to injury of their axons in remote lesions and could contribute to the observed regional GM atrophy in the PLV group.
WM injury, however, not only involves focal lesions but also the dirty appearing white matter (DAWM) [Moore et al., 2008] and normal appearing white matter (NAWM) [Ciccarelli et al., 2003; Vrenken, 2006; Werring et al., 1999]. In autopsy tissue from MS patients, degenerating axons were present throughout the whole NAWM, with an increase around demyelinated lesions and within defined fibre tracts emerging from the lesions [Kutzelnigg et al., 2005]. Diffuse tissue changes in the NAWM have in part been explained by axonal destruction in the MS lesions followed by secondary Wallerian degeneration [Evangelou et al., 2000]. Thus, in addition to the more widespread distribution of WM lesions in the PLV group of our study population, the NAWM in particular in the vicinity of the focal lesions might also be damaged to a larger extent as compared with the NPLV group, thereby contributing to regional cortical GM changes.
Pathways of Progressive WM Lesion Changes and Their Effect on Regional GM
Location of GM and WM pathology is an important factor in determining the nature of the deficits in MS [Charil et al., 2003]. The GM volume reductions in the PLV group occured in a variety of highly interconnected cortical areas, e.g. the cingulate gyrus and the insula, prone to be affected by axonal degeneration in the WM [Charil et al. 2007]. Those areas seemed to be more vulnerable to GM volume reductions than regions with relatively fewer connections. Multiple disconnections between different areas of cortical networks, could relate to widespread cortical atrophy and cognitive impairment, commonly observed in MS [Charil et al., 2003; Rao et al., 1991; Mesulam, 1998 ; Goodale, 1997; Fox et al., 2008; ffytche, 2008; ffytche and Catani, 2005; Ross, 2008; Catani and Mesulam, 2008; Catani et al., 2003; Plant, 2008]. A small number of non‐progressive lesions located in WM tracts, i.e. of the associative cortex, might have interrupted relatively few connections with little or no effect on regional GM volumes in the NPLV group. A larger number of progressive lesions, however, accompanied by a widespread spatial distribution, could have interrupted a higher number of associative connections, thereby contributing to the progression of GM atrophy in the PLV group.
Methodological Issues
In this study, voxel‐wise statistical non‐parametric (FMRIB's randomize utility) [Nichols and Holmes, 2002] and parametric mapping technique (SPM5/VBM) were combined to compare longitudinally voxel‐wise regional WM lesion volume changes and GM volume changes. A number of studies have used voxel‐wise techniques to compare lesion distributions across populations [Charil et al., 2003; DeCarli et al., 2005; Di Perri et al., 2008; Di Perri et al., 2009; Enzinger et al., 2006; Ghassemi et al., 2008; Lee et al., 1998; Narayanan 1997; Wen and Sachdev 2004] and the resulting statistical parametric and non‐parametric probability maps have been shown to be powerful tools for studying lesion distribution in vivo. The non‐parametric permutation approach gives results similar to those obtained from a comparable SPM approach using GLM with multiple comparisons corrections derived from random field theory [Nichols and Holmes, 2002], with a close correlation between the numbers of significant voxels obtained with the two different procedures [Battaglini et al., 2009].
An ‘optimized’ and modulated VBM method [Good et al., 2001] was used to minimize the potentially confounding effects of errors in stereotactic normalization. To prevent WM lesions from being misclassified as GM [Nakamura and Fisher, 2009], lesions identified on T2w images were masked from the 3D MPRAGE images [Bendfeldt et al., 2009a]. The potential bias coming from errors in registration has also been minimized by visually checking all GM and WM lesion registrations analyses to ensure that there were no failures of alignment and consequent misclassification of tissues. The latter was in particular important, because in order to provide for modest group sizes, we used a relatively small cut‐of of 1% change in T2 lesion volume (LV) over a year of follow up to distinguish the PLV and NPLV groups. Despite our excellent interrater reliability (intraclass correlation coefficient = 0.999; P < 0.001), changes in the order of such a magnitude may be simply due to images mis‐alignment or mis‐registration over time and may not necessarily mean disease progression. However, there was a significantly higher number of new T2 as well as gadolinium‐enhancing lesions in the PLV group compared to the NPLV group, which contributes to the definition of progressive versus non‐progressive cases. Overall there were no differences in the T2 LV at either the baseline or the one‐year time point between the PLV and NPLV groups. Conversely, T2 LV changed significantly during the one‐year follow‐up within each group. However, the changes were relatively low and part of the lack of significant association between GM and WM disease changes we failed in finding might be due to the small changes observed in the WM lesion volume. Similar considerations can be applied to the T1 lesion volume.
There might be other methodological limitations in the present study, leading to a lack of regional correspondence, e.g. the parametric SPM5/VBM approach we used to search for correlations between WM and GM data, the size of the WM lesion mask or the smoothing factor used.
Generally, it would have been interesting to validate the lesions on T2WI or T1WI with those seen on FLAIR images, and to compare the spatiotemporal distribution patterns of WM lesions obtained with the different sequences. Genuine WML, are best seen with T2‐weighted sequences such as long TR dual‐echo spin‐echo or FLAIR (fluid‐attenuated inversion recovery [Barkhof and Scheltens, 2002] and is therefore a promising technique for quantifying cerebral pathology in MS which is not accessible to 2D CSE [Tubridy et al., 1998]. However, in this study, we were more interested in the comparison of the spatiotemporal distribution patterns of the MS lesions between the PLV and the NPLV groups, than the absolute number of detectable lesions.
Medication might possibly influence T2W lesion volume and grey matter volume (for review see: Smieskova et al., 2009). The changes in regional GM volume that we observed, however, are unlikely to be related to medication, as medication did not differ between the groups. Further on, there was no evidence for volumetric GM changes in the different treatment groups during the follow‐up period (data not shown). Although the rates of atrophy progression in the first year of treatment with interferons are higher than in the following years ('pseudoatrophy') it remains controversial whether there is an overall treatment effect on atrophy. In several treatment trials with different agents, no significant effects on grey matter volume were reported [Molyneux et al., 2000; Rovaris et al., 2001], although whole brain volume seems to be affected [Hardmeier et al., 2003; Rudick et al., 1999].
CONCLUSION
By using LPM's, here we have demonstrated a more widespread regional distribution pattern of WM lesions in the PLV group as compared to the NPLV group of RRMS patients. This might be a central issue predicting further lesion development as well as development of GM atrophy. Furthermore, the longitudinal VBM analysis revealed spatial T2 lesion changes in parts of the cerebral projection, commissural and association fibre system. These changes were accompanied by GM volume reductions in specific cortical regions predilected for atrophy. Multiple disconnections between different areas of cortical networks, could relate to widespread cortical atrophy and cognitive impairment, commonly observed in MS.
Acknowledgements
The authors thank the Head of GSK Clincal Imaging Centre, Professor Paul Matthews and the Swiss MS Society. They also acknowledge Peter Schaerli, Danilo Marzetti, Petra Huber and Markus Colussi for their expert technical assistance.
REFERENCES
- Amato MP, Bartolozzi ML, Zipoli V, Portaccio E, Mortilla M, Guidi L, Siracusa G, Sorbi S, Federico A, De Stefano N ( 2004): Neocortical volume decrease in relapsing‐remitting MS patients with mild cognitive impairment. Neurology 63: 89–93. [DOI] [PubMed] [Google Scholar]
- Audoin B, Davies GR, Finisku L, Chard DT, Thompson AJ, Miller DH ( 2006): Localization of grey matter atrophy in early RRMS. J Neurol 253: 1495–1501. [DOI] [PubMed] [Google Scholar]
- Barkhof F, Scheltens P ( 2002): Imaging of white matter lesions. Cerebrovascular Dis 13 ( Suppl 2): 21–30. [DOI] [PubMed] [Google Scholar]
- Battaglini M, Giorgio A, Stromillo ML, Bartolozzi ML, Guidi L, Federico A, De Stefano N ( 2009): Voxel‐wise assessment of progression of regional brain atrophy in relapsing‐remitting multiple sclerosis. J Neurol Sci 282( 1–2): 55–60. [DOI] [PubMed] [Google Scholar]
- Bendfeldt K, Kuster P, Traud S, Egger H, Winklhofer S, Mueller‐Lenke N, Naegelin Y, Gass A, Kappos L, Matthews PM, Nichols TE, Radue EW, Borgwardt SJ. ( 2009a): Association of regional gray matter volume loss and progression of white matter lesions in multiple sclerosis—A longitudinal voxel‐based morphometry study. NeuroImage 45: 60–67. [DOI] [PubMed] [Google Scholar]
- Bendfeldt K, Radue EW, Borgwardt SJ, Kappos L (2009b): Progression of gray matter atrophy and its association with white matter lesions in relapsing‐remitting multiple sclerosis. J Neurol Sci 2009; 285: 268–269. Epub 2009. [DOI] [PubMed] [Google Scholar]
- Borgwardt SJ, McGuire P, Fusar‐Poli P, Radue EW, Riecher‐Rossler A ( 2008): Anterior cingulate pathology in the prodromal stage of schizophrenia. Neuroimage 39: 553–554. [DOI] [PubMed] [Google Scholar]
- Borgwardt SJ, McGuire PK, Aston J, Berger G, Dazzan P, Gschwandtner U, Pfluger M, D'Souza M, Radue EW, Riecher‐Rossler A ( 2007a): Structural brain abnormalities in individuals with an at‐risk mental state who later develop psychosis. Br J Psychiatry Suppl 51: s69–s75. [DOI] [PubMed] [Google Scholar]
- Borgwardt SJ, Riecher‐Rossler A, Dazzan P, Chitnis X, Aston J, Drewe M, Gschwandtner U, Haller S, Pfluger M, Rechsteiner E, D'Souza M, Stieglitz RD, Radue EW, McGuire PK ( 2007b): Regional gray matter volume abnormalities in the at risk mental state. Biol Psychiatry 61: 1148–1156. [DOI] [PubMed] [Google Scholar]
- Brück W ( 2005): Inflammatory demyelination is not central to the pathogenesis of multiple sclerosis. J Neurol 252: v10–v15. [DOI] [PubMed] [Google Scholar]
- Catani M, Jones DK, Donato R, ffytche DH ( 2003): Occipito‐temporal connections in the human brain. Brain 126: 2093–2107. [DOI] [PubMed] [Google Scholar]
- Catani M, Mesulam M ( 2008): The arcuate fasciculus and the disconnection theme in language and aphasia: History and current state. Cortex 44: 953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli A, Rocca MA, Pagani E, Colombo B, Martinelli V, Comi G, Filippi M ( 2008): A voxel‐based morphometry study of grey matter loss in MS patients with different clinical phenotypes. NeuroImage 42: 315–322. [DOI] [PubMed] [Google Scholar]
- Chard DT, Griffin CM, Parker GJM, Kapoor R, Thompson AJ, Miller DH ( 2002): Brain atrophy in clinically early relapsing‐remitting multiple sclerosis. Brain 125: 327–337. [DOI] [PubMed] [Google Scholar]
- Chard DT, Griffin CM, Rashid W, Davies GR, Altmann DR, Kapoor R, Barker GJ, Thompson AJ, Miller DH ( 2004): Progressive grey matter atrophy in clinically early relapsing‐remitting multiple sclerosis. Mult Scler 10: 387–391. [DOI] [PubMed] [Google Scholar]
- Charil A, Dagher A, Lerch JP, Zijdenbos AP, Worsley KJ, Evans AC ( 2007): Focal cortical atrophy in multiple sclerosis: Relation to lesion load and disability. NeuroImage 34: 509–517. [DOI] [PubMed] [Google Scholar]
- Charil A, Zijdenbos AP, Taylor J, Boelman C, Worsley KJ, Evans AC, Dagher A ( 2003): Statistical mapping analysis of lesion location and neurological disability in multiple sclerosis: Application to 452 patient data sets. NeuroImage 19: 532–544. [DOI] [PubMed] [Google Scholar]
- Chen JT, Narayanan S, Collins DL, Smith SM, Matthews PM, Arnold DL ( 2004): Relating neocortical pathology to disability progression in multiple sclerosis using MRI. NeuroImage 23: 1168–1175. [DOI] [PubMed] [Google Scholar]
- Ciccarelli O, Werring DJ, Barker GJ, Griffin CM, Wheeler‐Kingshott CAM, Miller DH, Thompson AJ ( 2003): A study of the mechanisms of normal‐appearing white matter damage in multiple sclerosis using diffusion tensor imaging. J Neurol 250: 287–292. [DOI] [PubMed] [Google Scholar]
- Dalton CM, Chard DT, Davies GR, Miszkiel KA, Altmann DR, Fernando K, Plant GT, Thompson AJ, Miller DH ( 2004): Early development of multiple sclerosis is associated with progressive grey matter atrophy in patients presenting with clinically isolated syndromes. Brain 127: 1101–1107. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Matthews PM, Filippi M, Agosta F, De Luca M, Bartolozzi ML, Guidi L, Ghezzi A, Montanari E, Cifelli A, Federico A, Smith SM ( 2003): Evidence of early cortical atrophy in MS: Relevance to white matter changes and disability. Neurology 60: 1157–1162. [DOI] [PubMed] [Google Scholar]
- DeCarli C, Fletcher E, Ramey V, Harvey D, Jagust WJ ( 2005): Anatomical mapping of white matter hyperintensities (WMH): Exploring the relationships between periventricular WMH, deep WMH, and total WMH burden. Stroke 36: 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Perri C, Battaglini M, Stromillo ML, Bartolozzi ML, Guidi L, Federico A, De Stefano N ( 2008): Voxel‐based assessment of differences in damage and distribution of white matter lesions between patients with primary progressive and relapsing‐remitting multiple sclerosis. Arch Neurol 65: 236–243. [DOI] [PubMed] [Google Scholar]
- Di Perri C, Dwyer MG, Wack DS, Cox JL, Hashmi K, Saluste E, Hussein S, Schirda C, Stosic M, Durfee J, Poloni GU, Nayyar N, Bergamaschi R, Zivadinov R ( 2009): Signal abnormalities on 1.5 and 3 Tesla brain MRI in multiple sclerosis patients and healthy controls. A morphological and spatial quantitative comparison study. NeuroImage 47: 1352–1362. [DOI] [PubMed] [Google Scholar]
- Enzinger C, Smith S, Fazekas F, Drevin G, Ropele S, Nichols T, Behrens T, Schmidt R, Matthews P ( 2006): Lesion probability maps of white matter hyperintensities in elderly individuals. J Neurol 253: 1064–1070. [DOI] [PubMed] [Google Scholar]
- Evangelou N, Konz D, Esiri MM, Smith S, Palace J, Matthews PM ( 2000): Regional axonal loss in the corpus callosum correlates with cerebral white matter lesion volume and distribution in multiple sclerosis. Brain 123: 1845–1849. [DOI] [PubMed] [Google Scholar]
- ffytche DH ( 2008): The hodology of hallucinations. Cortex 44: 1067–1083. [DOI] [PubMed] [Google Scholar]
- ffytche DH, Catani M ( 2005): Beyond localization: From hodology to function. Philos Trans R Soc B Biol Sci 360: 767–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox CJ, Iaria G, Barton JJS ( 2008): Disconnection in prosopagnosia and face processing. Cortex 44: 996–1009. [DOI] [PubMed] [Google Scholar]
- Furby J, Hayton T, Altmann D, Brenner R, Chataway J, Smith K, Miller D, Kapoor R ( 2009): Different white matter lesion characteristics correlate with distinct grey matter abnormalities on magnetic resonance imaging in secondary progressive multiple sclerosis. Mult Scler 15: 687–694. [DOI] [PubMed] [Google Scholar]
- Fusar‐Poli P, Perez J, Broome M, Borgwardt S, Placentino A, Caverzasi E, Cortesi M, Veggiotti P, Politi P, Barale F, MCGuire P ( 2007): Neurofunctional correlates of vulnerability to psychosis: A systematic review and meta‐analysis. Neurosci Biobehav Rev 31: 465–484. [DOI] [PubMed] [Google Scholar]
- Ge Y, Grossman RI, Udupa JK, Babb JS, Nyul LG, Kolson DL ( 2001): Brain atrophy in relapsing‐remitting multiple sclerosis: Fractional volumetric analysis of gray matter and white matter. Radiology 220: 606–610. [DOI] [PubMed] [Google Scholar]
- Ghassemi R, Antel BS, Narayanan S, Francis JS, Bar‐Or A, Sadovnick AD, Banwell BL, Arnold D; Canadian Pediatric Demyelinating Disease Study G ( 2008): Lesion distribution in children with clinically isolated syndromes. Ann Neurol 63: 401–405. [DOI] [PubMed] [Google Scholar]
- Good CD, Johnsrude IS, Ashburner J, Henson RNA, Friston KJ, Frackowiak RSJ ( 2001): A voxel‐based morphometric study of ageing in 465 normal adult human brains. NeuroImage 14: 21–36. [DOI] [PubMed] [Google Scholar]
- Grossman M, McMillan C, Moore P, Ding L, Glosser G, Work M, Gee J ( 2004): What's in a name: Voxel‐based morphometric analyses of MRI and naming difficulty in Alzheimer's disease, frontotemporal dementia and corticobasal degeneration. Brain 127: 628–649. [DOI] [PubMed] [Google Scholar]
- Hardmeier M, Wagenpfeil S, Freitag P, Fisher E, Rudick RA, Kooijmans‐Coutinho M, Clanet M, Radue EW, Kappos L ( 2003): Atrophy is detectable within a 3‐month period in untreated patients with active relapsing remitting multiple sclerosis. Arch Neurol 60: 1736–1739. [DOI] [PubMed] [Google Scholar]
- Hauser SL, Oksenberg JR ( 2006): The neurobiology of multiple sclerosis: Genes, inflammation, and neurodegeneration. Neuron 52: 61–76. [DOI] [PubMed] [Google Scholar]
- Hayasaka S, Phan KL, Liberzon I, Worsley KJ, Nichols TE ( 2004): Nonstationary cluster‐size inference with random field and permutation methods. NeuroImage 22: 676–687. [DOI] [PubMed] [Google Scholar]
- Jasperse B, Vrenken H, Sanz‐Arigita E, de Groot V, Smith SM, Polman CH, Barkhof F ( 2007): Regional brain atrophy development is related to specific aspects of clinical dysfunction in multiple sclerosis. NeuroImage 38: 529–537. [DOI] [PubMed] [Google Scholar]
- Kappos L, Antel J, Comi G, Montalban X, O'Connor P, Polman CH, Haas T, Korn AA, Karlsson G, Radue EW, et al. ( 2006): Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med 355: 1124–1140. [DOI] [PubMed] [Google Scholar]
- Kutzelnigg A, Lucchinetti CF, Stadelmann C, Bruck W, Rauschka H, Bergmann M, Schmidbauer M, Parisi JE, Lassmann H ( 2005): Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 128: 2705–2712. [DOI] [PubMed] [Google Scholar]
- Lee MA, Smith S, Palace J, Matthews PM ( 1998): Defining multiple sclerosis disease activity using MRI T2‐weighted difference imaging. Brain 121: 2095–2102. [DOI] [PubMed] [Google Scholar]
- Lee MA, Smith S, Palace J, Narayanan S, Silver N, Minicucci L, Filippi M, Miller DH, Arnold DL, Matthews PM ( 1999): Spatial mapping of T2 and gadolinium‐enhancing T1 lesion volumes in multiple sclerosis: Evidence for distinct mechanisms of lesion genesis? Brain 122: 1261–1270. [DOI] [PubMed] [Google Scholar]
- McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD, McFarland HF, Paty DW, Polman CH, Reingold SC, et al. ( 2001): Recommended diagnostic criteria for multiple sclerosis: Guidelines from the International panel on the diagnosis of multiple sclerosis. Ann Neurol 50: 121–127. [DOI] [PubMed] [Google Scholar]
- Mesulam MM ( 1998): From sensation to cognition. Brain 121: 1013–1052. [DOI] [PubMed] [Google Scholar]
- Molyneux PD, Kappos L, Polman C, Pozzilli C, Barkhof F, Filippi M, Yousry T, Hahn D, Wagner K, Ghazi M, Beckmann K, Dahlke F, Lossef N, Barker GJ, Thompson AJ, Miller DH ( 2000): The effect of interferon beta‐1b treatment on MRI measures of cerebral atrophy in secondary progressive multiple sclerosis. Brain 123: 2256–2263. [DOI] [PubMed] [Google Scholar]
- Moore G, Laule C, MacKay A, Leung E, Li D, Zhao G, Traboulsee A, Paty D ( 2008): Dirty‐appearing white matter in multiple sclerosis. J Neurol 255: 1802–1811. [DOI] [PubMed] [Google Scholar]
- Moorhead TWJ, Job DE, Spencer MD, Whalley HC, Johnstone EC, Lawrie SM ( 2005): Empirical comparison of maximal voxel and non‐isotropic adjusted cluster extent results in a voxel‐based morphometry study of comorbid learning disability with schizophrenia. NeuroImage 28: 544–552. [DOI] [PubMed] [Google Scholar]
- Morgen K, Sammer G, Courtney SM, Wolters T, Melchior H, Blecker CR, Oschmann P, Kaps M, Vaitl D ( 2006): Evidence for a direct association between cortical atrophy and cognitive impairment in relapsing‐remitting MS. NeuroImage 30: 891–898. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Fisher E ( 2009): Segmentation of brain magnetic resonance images for measurement of gray matter atrophy in multiple sclerosis patients. NeuroImage 44: 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan S, Fu L, Pioro E, De Stefano N, Collins DL, Francis GS, Antel JP, Matthews PM, Arnold DL ( 1997): Imaging of axonal damage in multiple sclerosis: Spatial distribution of magnetic resonance imaging lesions. Ann Neurol 41: 385–391. [DOI] [PubMed] [Google Scholar]
- Nichols TE, Holmes AP ( 2002): Nonparametric permutation tests for functional neuroimaging: A primer with examples. Hum Brain Mapp 15: 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani E, Rocca MA, Gallo A, Rovaris M, Martinelli V, Comi G, Filippi M ( 2005): Regional brain atrophy evolves differently in patients with multiple sclerosis according to clinical phenotype. AJNR Am J Neuroradiol 26: 341–346. [PMC free article] [PubMed] [Google Scholar]
- Perry VH, Anthony DC ( 1999): Axon damage and repair in multiple sclerosis. Philos Trans R Soc Lond B Biol Sci 354: 1641–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirko I, Lucchinetti CF, Sriram S, Bakshi R ( 2007): Gray matter involvement in multiple sclerosis. Neurology 68: 634–642. [DOI] [PubMed] [Google Scholar]
- Plant GT ( 2008): Optic neuritis and multiple sclerosis. Curr Opin Neurol 21: 16–21. 10.1097/WCO.0b013e3282f419ca. [DOI] [PubMed] [Google Scholar]
- Prinster A, Quarantelli M, Orefice G, Lanzillo R, Brunetti A, Mollica C, Salvatore E, Morra VB, Coppola G, Vacca G, Alfano B, Salvatore M ( 2006): Grey matter loss in relapsing‐remitting multiple sclerosis: A voxel‐based morphometry study. NeuroImage 29: 859–867. [DOI] [PubMed] [Google Scholar]
- Quarantelli M, Ciarmiello A, Morra VB, Orefice G, Larobina M, Lanzillo R, Schiavone V, Salvatore E, Alfano B, Brunetti A ( 2003): Brain tissue volume changes in relapsing‐remitting multiple sclerosis: Correlation with lesion load. NeuroImage 18: 360–366. [DOI] [PubMed] [Google Scholar]
- Rao SM, Leo GJ, Bernardin L, Unverzagt F ( 1991): Cognitive dysfunction in multiple sclerosis. I. Frequency, patterns, and prediction. Neurology 41: 685–691. [DOI] [PubMed] [Google Scholar]
- Roosendaal SD, Geurts JJG, Vrenken H, Hulst HE, Cover KS, Castelijns JA, Pouwels PJW, Barkhof F ( 2009): Regional DTI differences in multiple sclerosis patients. NeuroImage 44: 1397–1403. [DOI] [PubMed] [Google Scholar]
- Ross ED ( 2008): Sensory‐specific amnesia and hypoemotionality in humans and monkeys: Gateway for developing a hodology of memory. Cortex 44: 1010–1022. [DOI] [PubMed] [Google Scholar]
- Rovaris M, Comi G, Rocca MA, Wolinsky JS, Filippi M ( 2001): Short‐term brain volume change in relapsing‐remitting multiple sclerosis: Effect of glatiramer acetate and implications. Brain 124: 1803–1812. [DOI] [PubMed] [Google Scholar]
- Rudick RA, Fisher E, Lee JC, Simon J, Jacobs L ( 1999): Use of the brain parenchymal fraction to measure whole brain atrophy in relapsing‐remitting MS. Neurology 53: 1698–1704. [DOI] [PubMed] [Google Scholar]
- Sailer M, Fischl B, Salat D, Tempelmann C, Schonfeld MA, Busa E, Bodammer N, Heinze H‐J, Dale A ( 2003): Focal thinning of the cerebral cortex in multiple sclerosis. Brain 126: 1734–1744. [DOI] [PubMed] [Google Scholar]
- Sanfilipo MP, Benedict RHB, Sharma J, Weinstock‐Guttman B, Bakshi R ( 2005): The relationship between whole brain volume and disability in multiple sclerosis: A comparison of normalized gray vs. white matter with misclassification correction. NeuroImage 26: 1068–1077. [DOI] [PubMed] [Google Scholar]
- Sanfilipo MP, Benedict RHB, Weinstock‐Guttman B, Bakshi R ( 2006): Gray and white matter brain atrophy and neuropsychological impairment in multiple sclerosis. Neurology 66: 685–692. [DOI] [PubMed] [Google Scholar]
- Schmahmann JD, Doyon J, McDonald D, Holmes C, Lavoie K, Hurwitz AS, Kabani N, Toga A, Evans A, Petrides M ( 1999): Three‐dimensional MRI atlas of the human cerebellum in proportional stereotaxic space. NeuroImage 10: 233–260. [DOI] [PubMed] [Google Scholar]
- Sepulcre J, Goni J, Masdeu JC, Bejarano B, Velez de Mendizabal N, Toledo JB, Villoslada P ( 2009): Contribution of white matter lesions to gray matter atrophy in multiple sclerosis: Evidence from voxel‐based analysis of T1 lesions in the visual pathway. Arch Neurol 66: 173–179. [DOI] [PubMed] [Google Scholar]
- Sepulcre J, Sastre‐Garriga J, Cercignani M, Ingle GT, Miller DH, Thompson AJ ( 2006): Regional gray matter atrophy in early primary progressive multiple sclerosis: A voxel‐based morphometry study. Arch Neurol 63: 1175–1180. [DOI] [PubMed] [Google Scholar]
- Smieskova R, Fusar‐Poli P, Allen P, Bendfeldt K, Stieglitz RD, Drewe J, Radue EW, McGuire PK, Riecher‐Rössler A, Borgwardt SJ (2009) The effects of antipsychotics on the brain: what have we learnt from structural imaging of schizophrenia?—a systematic review. Curr Pharm Des 15: 2535–2549. [DOI] [PubMed] [Google Scholar]
- Tedeschi G, Lavorgna L, Russo P, Prinster A, Dinacci D, Savettieri G, Quattrone A, Livrea P, Messina C, Reggio A, et al. ( 2005): Brain atrophy and lesion load in a large population of patients with multiple sclerosis. Neurology 65: 280–285. [DOI] [PubMed] [Google Scholar]
- Tiberio M, Chard DT, Altmann DR, Davies G, Griffin CM, Rashid W, Sastre‐Garriga J, Thompson AJ, Miller DH ( 2005): Gray and white matter volume changes in early RRMS: A 2‐year longitudinal study. Neurology 64: 1001–1007. [DOI] [PubMed] [Google Scholar]
- Tubridy N, Barker GJ, Macmanus DG, Moseley IF, Miller DH ( 1998): Three‐dimensional fast fluid attenuated inversion recovery (3D fast FLAIR): A new MRI sequence which increases the detectable cerebral lesion load in multiple sclerosis. Br J Radiol 71: 840–845. [DOI] [PubMed] [Google Scholar]
- Vrenken H, Pouwels, Petra JW, Geurts JJG, Knol DL, Polman, CH , Barkhof F, Castelijns JA ( 2006): Altered diffusion tensor in multiple sclerosis normal‐appearing brain tissue: Cortical diffusion changes seem related to clinical deterioration. J Magn Reson Imaging 23: 628–636. [DOI] [PubMed] [Google Scholar]
- Wen W, Sachdev P ( 2004): The topography of white matter hyperintensities on brain MRI in healthy 60‐ to 64‐year‐old individuals. NeuroImage 22: 144–154. [DOI] [PubMed] [Google Scholar]
- Werring DJ, Clark CA, Barker GJ, Thompson AJ, Miller DH ( 1999): Diffusion tensor imaging of lesions and normal‐appearing white matter in multiple sclerosis. Neurology 52: 1626–1632. [DOI] [PubMed] [Google Scholar]
- White T, O'Leary D, Magnotta V, Arndt S, Flaum M, Andreasen NC ( 2001): Anatomic and functional variability: The effects of filter size in group fMRI data analysis. NeuroImage 13: 577–588. [DOI] [PubMed] [Google Scholar]