

Abstract
Childhood psychiatric disorders are rarely static; rather they change over time and longitudinal studies are ideally suited to capture such dynamic processes. Using longitudinal data, insights can be gained into the nature of the perturbation away from the trajectory of typical development in childhood disorders. Thus, some disorders may reflect a delay in neurodevelopmental trajectories. Our studies in children with attention‐deficit/hyperactivity disorder (ADHD) suggest that cortical development is delayed with a rightward shift along the age axis in cortical trajectories, most prominent in prefrontal cortical regions. Other disorders may be characterized by differences in the velocity of trajectories: the basic shape of neurodevelopmental curves remains intact, but with disrupted tempo. Thus, childhood onset schizophrenia is associated with a marked increase during adolescence in the velocity of loss of cerebral gray matter. By contrast, in childhood autism there is an early acceleration of brain growth, which overshoots typical dimensions leading to transient cerebral enlargement. Finally, there may be more profound deviations from typical neurodevelopment, with a complete “derailing” of brain growth and a loss of the features which characterize typical brain development. An example is the almost complete silencing of white matter growth during adolescence of patients with childhood onset schizophrenia. Adopting a longitudinal perspective also readily lends itself to the understanding of the neural bases of differential clinical outcomes. Again taking ADHD as an example, we found that remission is associated with convergence to the template of typical development, whereas persistence is accompanied by progressive divergence away from typical trajectories. Hum Brain Mapp, 2010. © 2010 Wiley‐Liss, Inc.
Keywords: magnetic resonance imaging, child development, childhood psychiatric disorders, modeling
INTRODUCTION: HOW TRAJECTORIES CAN GO AWRY
Structural brain development in healthy children follows regionally heterochronous, complex trajectories [Giedd et al., 1999]. In gray matter development, whether measured by cortical volume or thickness, there is a phase of early increase, followed by a late childhood/adolescent phase of decrease, before the cortex settles into adult dimensions. White matter has a more sustained pattern of expansion persisting through adolescence. Given the complexities of these trajectories, it is perhaps not surprising that they can go awry, resulting in disturbances in cognition, affect, and behavior.
What can go wrong with a trajectory? First, the trajectory can be intact in the sense that it has the same general shape as a typical neurodevelopmental curve, but the curve is shifted along the age axis. An example is given in Figure 1A. Here the curve is shifted rightward along the age axis so that the age of reaching key transition points is later; in this example, peak values of neuroanatomic variables are attained at a later age. This implies that the disorder is characterized by a delay in the pattern of typical development. Alternatively, disorders may be associated with differences in the tempo of neural change; Figure 1B shows primarily the acceleration of the course of typical development. Another possibility is more profound deviance, with the trajectory lacking the basic shape of a typical developmental trajectory (Fig. 1C). In this selective review, anomalies in developmental trajectories are linked with various childhood mental disorders. It is important to stress that this suggested categorization of developmental trajectory anomalies is preliminary and the anomalies are not mutually exclusive—a trajectory could incorporate elements of delay (in the sense that points of curve inflection occur later) but also be altered in velocity.
Figure 1.
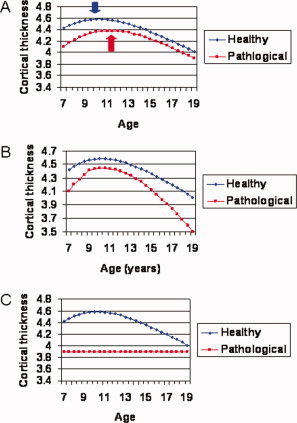
How developmental trajectories can go awry. In all examples hypothetical data representing the change in cortical thickness of a cerebral point is given. (A) The pathological trajectory has the same form as the typical trajectory but is displaced rightward along the age axis and so key characteristics such as the age of peak thickness, shown in the bold arrows, is attained later. (B) The pathological trajectory has the same form, but changes at a higher velocity. (C) The pathological trajectory loses the form or shape of a typical trajectory.
The focus throughout this selective review is on neuroanatomic findings as this constitutes the bulk of longitudinal neuroimaging in childhood disorders, but findings from other modalities—of brain function (e.g., functional MRI, magnetoencephalography, positron emission tomography) and ultrastructure (e.g., magnetic resonance spectroscopy and diffusion tensor imaging)—will undoubtedly yield rich insights. The examples of childhood attention‐deficit/hyperactivity disorder (ADHD) and childhood‐onset schizophrenia (COS) predominate, but a similar longitudinal approach has been used by many other research groups to give insights into autism [Courchesne et al., 2007; Hazlett et al., 2005] and neurodegenerative disorders [Vemuri et al., 2009].
DELAY: SHIFTS ALONG THE AGE AXIS
In research on ADHD, cross‐sectional studies have established that there is global cerebral and cerebellar volumetric reduction in the disorder [Ellison‐Wright et al., 2008; Krain and Castellanos, 2006; Seidman et al., 2005], with the basal ganglia [Ellison‐Wright et al., 2008] and prefrontal cortex being most compromised [Valera et al., 2007]. At least some of these differences are not epiphenomena of symptoms as they are found in unaffected relatives [Durston et al., 2004, 2005] and do not seem to be due to medication [Bledsoe et al., 2009; Semrud‐Clikeman et al., 2006; Shaw et al., 2009]. But what can longitudinal studies add?
At the NIMH we have collected a large group of children and adolescents with ADHD the majority of whom have had repeated neuroanatomic imaging in tandem with ongoing clinical assessments. This data allows us to define neurodevelopmental trajectories and to relate these to cognitive and clinical variables. We have started by examining cortical and cerebellar trajectories. This is merely a first step and will be complemented in future work by a delineation of white matter and subcortical trajectories, with an emphasis on defining the interconnections between growth patterns of different brain regions.
Before discussing the findings, it is worthwhile to note that longitudinal studies are not without their limitations: foremost are problems with retention, and the possibility of nonrandom loss to follow‐up. Gaps in data sets can appear, often with poorer coverage of younger age ranges (especially under 6) reflecting the challenges associated with acquiring high‐quality structural neuroimaging data on the very young, especially those who have problems lying still. In studies conducted over years, continual improvements in technology can lead to the complex, but not insurmountable, problem of integrating data acquired on different scanners over time.
Despite these issues, longitudinal data are well poised to capture developmental processes. As an example, in our studies on cortical development, we have studied cortical thickness, partly as this can be determined by computational neuroanatomic methods at thousands of points throughout the cerebrum allowing an exquisite spatial resolution surpassing that of most volumetric studies (which are conducted either at the level of the entire lobe or use specific regions of interest). We compared the change in the thickness of the cortex—estimated at over 40,000 cerebral points using computational techniques—in 223 children and adolescents with ADHD against matched healthy controls, with a total of 823 neuroanatomic scans [Shaw et al., 2007]. Most participants had scans acquired at least twice, with an average of a 2‐year interval between scans. The age of the initial scan ranged from 4 to 20 years of age, with most data between the ages of 8 and 16, and the resulting data set thus afforded good coverage from childhood through adolescence. From this data the growth trajectory of each cortical point was determined using mixed model polynomial regression. This method is chosen as it permits the inclusion of multiple measurements per person and irregular intervals between measurements, thereby increasing statistical power [Diggle et al., 1994]. Many types of developmental trajectories can be determined using this method. In this study, a trajectory which had a childhood phase of increase followed by a phase of decrease was appropriate. From this trajectory, we defined the age at which peak cortical thickness was attained, that is, the age at which childhood increase in cortical thickness gives way to adolescent decrease. Cortical maturation, as indexed by this marker, in children with ADHD lagged behind that of typically developing children by ∼3 years overall and the delay was most prominent in the lateral prefrontal cortex (Fig. 2). However, the ordered sequence of regional development, with primary sensory and motor areas attaining their peak cortical thickness before high‐order association areas, was similar in both groups, suggesting that ADHD is characterized by delay rather than deviation in cortical maturation. The primary motor cortex was the only cortical area where the ADHD group showed slightly earlier maturation. These findings imply that the neuroanatomic signature of childhood ADHD is dynamic and that the disorder is associated with cortical developmental curves which retain many typical features, but are shifted along the age axis. Also note, for much of the cortex, the developmental curve for ADHD nestles within the curve of typical development. This implies that in many regions the peak cortical thickness is smaller, reflecting the general thinning of much of the cortex seen in ADHD. This integrates previous findings of reduction in cortical dimensions in ADHD with the current finding of a prominent delay in cortical maturation.
Figure 2.
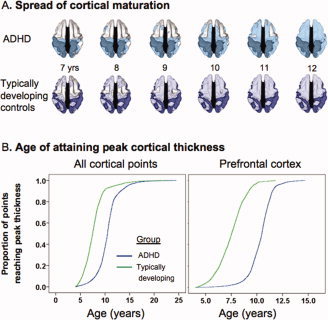
(A) Dorsal view of the cortical regions where peak thickness was attained at each age. The darker color indicates regions where a peak age could not be calculated or the peak age was estimated to lie outside the age range covered. Both groups showed a similar sequence of the regions which attained peak thickness, but the ADHD group showed considerable delay in reaching this developmental marker. (B) Kaplan Meier curves illustrating the proportion of cortical points which had attained peak thickness at each age for (a) all cerebral cortical points and (b) the prefrontal cortex. The median age by which 50% of cortical points had attained their peak differed significantly between the groups (all P<1.0 × 10−20).
The combination of frontotemporal delay in cortical maturation in ADHD, but with a sequence of development which mirrored that of typical development, supports the concept that childhood ADHD may reflect a “maturational delay.” This is in keeping with reports that the brain activity at rest and in response to cognitive probes is similar between children with ADHD and their slightly younger but healthy peers [El‐Sayed et al., 2003; Fernández et al., 2009; Rubia, 2002]. In one of the few studies to link cognitive measures and neuroanatomy McAlonan et al. [ 2009] found that 7‐ to 12‐year‐olds with ADHD lagged several years behind their typically developing peers in a measure of response inhibition and that improvement with age correlated with increasing volumes in a network compromising the anterior cingulate, striatum, and medial temporal lobes. Thus both the McAlonan study and our work found increasing cortical dimensions during late childhood. The McAlonan study suggests that this increase may ameliorate cognitive deficits in ADHD. It is also notable that the only cortical region where we found the ADHD group to have earlier maturation was the primary motor cortex, and perhaps the combination of early maturation of the primary motor cortex with late maturation of higher order motor control regions may reflect or even drive the excessive and poorly regulated motor activity cardinal to the syndrome. It should also be noted that the maturational delay is most prominent in the lateral prefrontal cortex, the neuroanatomic substrate most frequently implicated in neuropsychological models of ADHD as a result of deficits in core cognitive processes such as response inhibition, temporal processing, and working memory [Barkley, 1997; Castellanos and Tannock, 2002; Nigg and Casey, 2005; Sonuga‐Barke, 2002; Toplak et al., 2006].
DISRUPTED VELOCITY OF A TRAJECTORY
Developmental curves can also differ in their velocity (the first‐order derivative of a curve) or acceleration/deceleration (the second‐order derivative). In a study of healthy children of differing intellectual ability, we found that the main difference in cortical development was the velocity of change. Thus, while all children showed the same basic developmental trajectory, (a childhood phase of cortical thickening gave way to a phase of adolescent thinning which eventually settled into adult cortical dimensions); all of these changes happened at a greater velocity in more intelligent children [Shaw et al., 2006a]. Might similar differences in the rate of neurodevelopment characterize childhood mental health disorders?
Meta‐analysis of studies of brain size in children and adults with autism suggest that the disorder may indeed be associated with a change in the velocity of neurodevelopmental curves [Redcay and Courchesne, 2005]. The most anomalous period of growth is very early in life. Two longitudinal studies examined brain volumes as determined by MRI at age 2 and related these to head circumference measures (taken as a proxy for brain volume) made at birth and during the early years [Courchesne et al., 2003; Hazlett et al., 2005]. They found that while head circumference at birth was normal or low‐normal in infants who would later be diagnosed with autism, there was an infantile rapid acceleration of brain growth both in gray matter and white matter. In one study, this spurt started around 6 months; in the other study, the acceleration started around 1 year. The accelerated growth was dysregulated as by 2 years of age the children with autism had greater brain volumes than typically developing and developmentally delayed comparison subjects. It appears that this accelerated growth plateaus after early childhood and so by late childhood brain volumes regress to normal ranges [Redcay and Courchesne, 2005]. A similar pattern in early childhood was found for the amygdalae: at both age 2 and age 4, children with autism had large amygdalae consistent with a pattern of early overgrowth leading to bigger volumes [Mosconi et al., 2009]. Enlarged amygdalae were associated with deficits in a core aspect of social cognition—the ability to engage in joint attention.
The pattern of accelerated growth resulting in abnormally large volumes in autism contrasts with reports of increased velocity in gray matter loss during adolescence in patients with COS. Much of this work has stemmed from a longitudinal study at NIMH of over 100 patients with COS, who have had repeated combined neuroimaging and clinical assessments, and is reviewed more fully elsewhere [Gogtay, 2008; Rapoport and Gogtay, 2008]. As discussed earlier in typical development, cortical gray matter appears to mature in a parietofrontal (back to front) direction and medially in a centripetal (“top–down”) fashion. In patients with COS, the sequence of gray matter development during adolescence resembled that of healthy controls but had a greater velocity [Thompson et al., 2001]. Thus, the adolescent parietofrontal wave of cortical thinning which characterizes typical development was more pronounced in adolescents with COS. Similarly in the medial cortical wall, centripetal thinning in adolescents with COS resembled that of typical development but again occurred with greater velocity [Vidal et al., 2006]. This increased rate of cortical thinning did not persist into adulthood rather it “stopped” in late adolescence in the parietal cortex leading to a relative normalization of this region [Greenstein et al., 2006]. However, in frontotemporal regions, the anomalous trajectories persisted resulting in frontotemporal cortical deficits resembling those reported in adult‐onset schizophrenia.
Interestingly, the younger, healthy siblings of patients with COS in childhood showed significant gray matter deficits in the left prefrontal and bilateral temporal cortices and smaller deficits in the right prefrontal and inferior parietal cortices compared with the controls, suggesting that the prefrontal and temporal deficits may be familial/trait markers [Gogtay et al., 2007]. However, in the frontotemporal regions, a slower rate of cortical thinning during adolescence led to a correction of the initial cortical deficits by adulthood. Could such disruptions in the velocity of cortical developmental trajectories act as a endophenotype in future studies, as they are present in unaffected first‐degree relatives and may lie close to the neurobiology of the disorder?
An altered pace of neurodevelopmental trajectories thus characterizes autism and COS. Autism shows pronounced early life acceleration of growth leading to a transient overshoot of brain volumes. By contrast, in young adolescents with COS, there is increased velocity of gray matter thinning, which rectifies itself in parietal regions, but partly persists in frontotemporal regions leading to the cortical deficit pattern found in adult‐onset schizophrenia.
DEVIANT TRAJECTORIES
Another possibility is that a developmental curve can lose much of its form or shape. Again an example comes from COS. During typical development there appears to be an increase in white matter volumes during the adolescent years [Giedd et al., 1999]. However, this volumetric gain is almost completely absent during adolescence in patients with COS [Gogtay et al., 2008]. Instead of steady increase, these patients show a loss of white matter expansion throughout the entire right hemisphere with “growth” rates not differing significantly from zero and only a trend to white matter growth in the left hemisphere (see Fig. 3).
Figure 3.
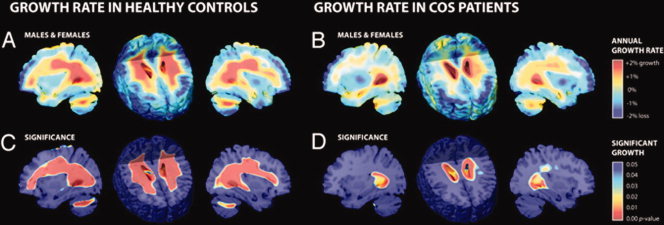
Tissue growth rates mapped in healthy controls and COS patients. (A, B) These maps show the average rates of tissue growth (red) and tissue loss (blue) throughout the brain in percentage per year, for healthy controls (A) and COS patients (B). (C, D) These corresponding maps show the significance of the tissue growth in (A, B), respectively. There is almost no significant growth in the COS patients compared to the healthy controls.
There are many other disorders that are likely to show a loss of the features which typify development, such as some of the syndromes associated with moderate and severe global intellectual disability. For example, extrapolating from the cross‐sectional studies of people who have Down's syndrome (DS; trisomy 21), there appears to be a complex mix of deviant trajectories (particularly of the cerebellum and frontal lobes) and intact trajectories (of the basal ganglia and thalamus) [Pinter et al., 2001]. Given the known genetic lesion in this disorder, it may thus give insights into distinct control mechanisms for cortical, cerebellar, and basal ganglia growth. People with DS also have a high rate of early onset Alzheimer's disease and thus can give rich insights into this common dementia. For example, even prior to the onset of clinical symptoms, people with DS most at risk for dementia show volume loss in the hippocampus and caudate, structures which other studies find to be most compromised in those with DS with established Alzheimer's disease [Beacher et al., 2009; Haier et al., 2008].
It is also possible that nestling within the syndromes such as autism and schizophrenia there are subgroups which may have particularly pronounced deviant developmental trajectories. There are many possible variables which might define these subgroups, such as clinical severity, poor clinical outcome (a point we will return to later), and genetic characteristics. Of particular interest are recent demonstrations that a high percentage of patients with autism [around 10%; Bucan et al., 2009; Cusco et al., 2009; Sebat et al., 2007] and schizophrenia [around 20% for COS; Rapoport et al., 2009; Walsh et al., 2008] may have large‐scale DNA deletions and duplications or copy number variants (CNVs). In this vein, people who have a microdeletion on 22q have a constellation of congenital defects and greatly increased risk for schizophrenia. One of the genes deleted is the catechol‐O‐methyl‐transferase (COMT), which has both low and high activity forms and is critical in the regulation of cortical dopamine. As subjects with 22q deletion have only one copy of the COMT gene, they may be particularly prone to the fluctuations in cortical dopamine associated with the gene variants. Indeed, in a longitudinal study of 18 people with the syndrome, extreme deficiency of COMT activity was associated both with increased velocity of prefrontal cortex volume loss, more rapid decline in intelligence, and a higher rate of emergence of psychotic symptoms [Gothelf et al., 2005]. Initial observations from our group suggest that early subtle developmental anomalies are prominent in patients with CNVs, possibly regardless of diagnosis, and they may have particularly anomalous brain structures [Addington and Rapoport, 2009]. It is possible that CNVs may also segregate with deviant developmental trajectories, which might cut across traditional diagnostic categories.
USING TRAJECTORIES TO UNDERSTAND CLINICAL OUTCOME
Longitudinal data is also ideal for studying the neuroanatomic correlates of one of the most important features of childhood psychiatric disorders, namely their variable clinical outcome. To take ADHD as an example, the disorder has a tendency to improve with age in most, but certainly not all, subjects. A recent meta‐analysis shows only 15–20% of those with childhood ADHD retained the diagnosis in adulthood and a further ∼50% had residual symptoms which were impairing [Faraone et al., 2006]. This is apparent in our cohort: while at baseline (mean age 10) all had combined‐type ADHD, but by mid‐adolescence (mean age 15.5) 18 (21%) of those with DSM‐IV re‐interview data had a complete remission, 37 (42%) had a partial remission, and 32 (37%) showed no improvement (retaining the diagnosis of combined type ADHD) [Shaw et al., 2006b].
Using our cohort's yoked, prospectively acquired neuroanatomic and clinical data, we defined neuroanatomic trajectories which reflected varying clinical outcome. Because of the smaller sample size of those with outcome data and the restricted age range we could only delineate the phase of cortical thinning which characterizes adolescent cortical development. We first examined cortical development in ADHD subjects with full or partial remission at last follow‐up. This group differed significantly in the trajectory of cortical change in the right parietal cortex, most prominently in the superior parietal lobule (where the trajectory “normalization” reached significance). Here, there was a convergence between the trajectories of the remitted ADHD group and the typically developing cohort such that by late adolescence there was “normalization” of cortical thickness in those who remit from ADHD (see Fig. 4). A similar trend held throughout lateral prefrontal cortex. By contrast, the group of ADHD subjects with persistent combined type ADHD showed either fixed nonprogressive cortical deficits or a slight tendency to diverge away from typical trajectories. These deficits localized to the medial prefrontal wall, especially the left posterior cingulate and regions in the superior frontal gyri. An independent study of adults with ADHD, who by definition have a poor outcome of their childhood ADHD, found cortical thinning in similar regions [Makris et al., 2007].
Figure 4.
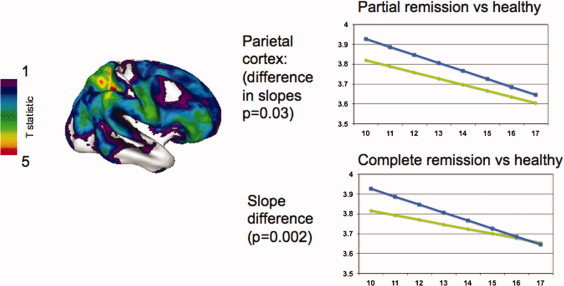
Cortical normalization in ADHD with a good clinical outcome. The brain template shows regions where good outcome ADHD converges with the trajectory of typically developing children. Regions in blue and green show where this occurs at a trend level, and regions in yellow and red show where the convergence is significant, namely the right superior parietal lobule. The graphs show the cortical change in the right superior parietal lobule for the groups showing partial remission (top) and full remission (bottom).
We found similar links between clinical outcome and developmental trajectories at the cerebellar level [Mackie et al., 2007]. Using a semiautomated method to measure ten cerebellar hemispheric and vermal compartments, we found that trajectories of the inferior posterior hemispheres, the largest single cerebellar compartment, differed between the outcome groups as the subjects with persistent ADHD diverged from both their peers with remitting ADHD and typically developing controls. A theme emerges: remission in ADHD may be characterized by a normalization of initial delays and deficits (more prominent in the cortex than the cerebellum), whereas persistent ADHD may be characterized by a more deviant trajectory (more prominent in the cerebellum than cortex).
What is the significance of the different cortical trajectories we find in those with different clinical outcome? A reasonable hypothesis is that deviation away from the trajectory of typical development is associated with persistent or worsening cognitive deficits, whereas convergence toward the template of typical development may be associated with the amelioration of cognitive deficits. In a study of 98 adolescents with a history of childhood ADHD, Halperin et al. [ 2008] found that those with persistent and remitted ADHD both showed deficient perceptual processing and increased movement, but only those with persistent ADHD showed deficits in effortful executive processing relative to healthy controls. Earlier this research group reported that behavioral trends of performance on a task of response inhibition were accompanied by neural activation gradients such that those with persistent ADHD with most errors on a response inhibition tasks showed greatest activation of the ventrolateral prefrontal cortex, followed by those with remitted ADHD (who had an intermediate level of performance in response inhibition) and then healthy controls [Schulz et al., 2005]. While this activation pattern of increased inferior frontal activation in ADHD is controversial, the study is one of the few to examine the neural mechanisms of recovery in ADHD and supports the concept of a convergence toward typical brain activity accompanying cognitive improvement in remitting ADHD. Other brain regions may be involved: for example might the prominent right parietal cortical “normalization” we found in the remitting ADHD be the structural correlate of cognitive compensation, with the parietal region supporting attentional functions typically mediated by the prefrontal cortex which does not show such marked cortical normalization? Similarly, the link between enlargement of the hippocampus and fewer ADHD symptoms reported by Plessen et al. [ 2006] is compatible with a hippocampal compensatory response which in turn could plausibly be linked to improvement in temporal sequencing and perception of time. In those with a poor clinical outcome, progressive deviation away from the template of typical development in cerebellar volumes, especially of the inferior posterior hemispheres, could perhaps translate into persistent, even progressive deficits in executive function and verbal working memory [Castellanos and Tannock, 2002; Nigg and Casey, 2005; Sonuga‐Barke, 2002; Toplak et al., 2006].
ETIOLOGICAL CONSIDERATIONS
Thinking of childhood psychiatric disorders as reflecting anomalies in the tempo of developmental trajectories prompts us to look at etiological factors driving these disruptions. Most childhood psychiatric disorders are highly heritable, implicating genetic factors and their interaction with the environment. The relative weighing of genetic, shared, and unique environmental factors in shaping developmental trajectories can be defined using a twin design (comparing mono‐ and dizygotic twins). Lenroot et al. [ 2009] have adopted this approach to demonstrate age‐related differences in cortical heritability with later developing regions (both in terms of phylogeny and ontogeny) such as the dorsolateral prefrontal cortex showing increasing genetic effects with age. A similar “twins” approach defining the heritability of key brain structures using cross‐sectional data has been applied in other disorders including ADHD [van't Ent et al., 2007] and bipolar affective disorder [van der Schot et al., 2009], and an extension of this work using longitudinal data would be welcome.
There is already considerable interest in mechanisms controlling the developmental sequencing of the activation and deactivation of genes which sculpt brain architecture. In this context, neurotrophins, essential for the proliferation, differentiation, and survival of neuronal and non‐neuronal cells, emerge as promising candidates, and indeed polymorphisms within the brain‐derived neurotrophic factor and nerve growth‐factor 3 genes have already been tentatively linked with ADHD [Kent et al., 2005; Syed et al., 2007]. Similarly, disrupted trajectories call attention to the dynamic nature of underlying cellular events. For example, it has been speculated that the early brain overgrowth in autism may reflect excess neuron numbers, which demonstrate excessive local connectivity at the cost of the long‐distance interactions between different brain regions which are necessary for the development of normal language and social cognition [Courchesne et al., 2007].
Environmental factors, both as main effects and by virtue of their interaction with genetic factors, must also be considered. Studies such as the National Children's Study are ideally placed to define prospectively the effect of key environmental exposures on later brain development. There are also a host of other individual differences such as sex [Lenroot et al., 2007] and intellectual ability [Shaw et al., 2006a], which appear to influence developmental trajectories in typically developing children. How these factors act to confer vulnerability to certain disorders remains unclear, but is a focus of current work.
CONCLUSIONS
Using neuroanatomic trajectories may not only provide one part of the pathogenic puzzle of childhood disorders but also help us move away from a purely syndromic categorization to one incorporating features such as the pattern of brain growth. This will require the reliable identification at the individual level of diagnostic signals in neurodevelopmental trajectories. This is complicated by the subtle nature of the neural changes detectable with current technology, as there are generally large overlaps in the distributions of neuroanatomic variables in healthy and ill populations. Perhaps a move to new technologies, novel combinations of existing ones, and advances in statistical analysis may help us realize this goal. Finally, longitudinal studies throw light into the neural bases of differential clinical outcome in disorders, which promises better, more targeted treatments.
REFERENCES
- Addington A, Rapoport J ( 2009): The genetics of childhood‐onset schizophrenia: When madness strikes the prepubescent. Curr Psychiatry Rep 11: 156–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkley RA ( 1997): Behavioral inhibition, sustained attention, and executive functions: Constructing a unifying theory of ADHD. Psychol Bull 121: 65–94. [DOI] [PubMed] [Google Scholar]
- Beacher F, Daly E, Simmons A, Prasher V, Morris R, Robinson C, Lovestone S, Murphy K, Murphy DGM ( 2009): Alzheimer's disease and Down's syndrome: An in vivo MRI study. Psychol Med 39: 675–684. [DOI] [PubMed] [Google Scholar]
- Bledsoe J, Semrud‐Clikeman M, Pliszka SR ( 2009): A magnetic resonance imaging study of the cerebellar vermis in chronically treated and treatment‐naïve children with attention‐deficit/hyperactivity disorder combined type. Biol Psychiatry 65: 620–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucan M, Abrahams BS, Wang K, Glessner JT, Herman EI, Sonnenblick LI, Alvarez Retuerto AI, Imielinski M, Hadley D, Bradfield JP, Kim C, Gidaya NB, Lindquist I, Hutman T, Sigman M, Kustanovich V, Lajonchere CM, Singleton A, Kim J, Wassink TH, McMahon WM, Owley T, Sweeney JA, Coon H, Nurnberger JI, Li M, Cantor RM, Minshew NJ, Sutcliffe JS, Cook EH, Dawson G, Buxbaum JD, Grant SFA, Schellenberg GD, Geschwind DH, Hakonarson H ( 2009): Genome‐wide analyses of exonic copy number variants in a family‐based study point to novel autism susceptibility genes. PLoS Genet 5: e1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellanos FX, Tannock R ( 2002): Neuroscience of attention‐deficit/hyperactivity disorder: The search for endophenotypes. Nat Rev Neurosci 3: 617–628. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Carper R, Akshoomoff N ( 2003): Evidence of brain overgrowth in the first year of life in autism [see comment]. JAMA 290: 337–344. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Pierce K, Schumann CM, Redcay E, Buckwalter JA, Kennedy DP, Morgan J ( 2007): Mapping early brain development in autism. Neuron 56: 399–413. [DOI] [PubMed] [Google Scholar]
- Cusco I, Medrano A, Gener B, Vilardell M, Gallastegui F, Villa O, Gonzalez E, Rodriguez‐Santiago B, Vilella E, Del Campo M, Perez‐Jurado LA ( 2009): Autism‐specific copy number variants further implicate the phosphatidylinositol signaling pathway and the glutamatergic synapse in the etiology of the disorder. Hum Mol Genet 18: 1795–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggle PJ, Liang KY, Zeger SL ( 1994): Analysis of Longitudinal Data. Oxford: Oxford University Press. [Google Scholar]
- Durston S, Hulshoff Pol HE, Schnack HG, Buitelaar JK, Steenhuis MP, Minderaa RB, Kahn RS, van Engeland H ( 2004): Magnetic resonance imaging of boys with attention‐deficit/hyperactivity disorder and their unaffected siblings. J Am Acad Child Adolesc Psychiatry 43: 332–340. [DOI] [PubMed] [Google Scholar]
- Durston S, Fossella JA, Casey BJ, Hulshoff Pol HE, Galvan A, Schnack HG, Steenhuis MP, Minderaa RB, Buitelaar JK, Kahn RS van Engeland H. ( 2005): Differential effects of DRD4 and DAT1 genotype on fronto‐striatal gray matter volumes in a sample of subjects with attention deficit hyperactivity disorder, their unaffected siblings, and controls. Mol Psychiatry 10: 678–685. [DOI] [PubMed] [Google Scholar]
- Ellison‐Wright I, Ellison‐Wright Z, Bullmore E ( 2008): Structural brain change in attention deficit hyperactivity disorder identified by meta‐analysis. BMC Psychiatry 8: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Sayed E, Larsson JO, Persson HE, Santosh PJ, Rydelius PA ( 2003): “Maturational lag” hypothesis of attention deficit hyperactivity disorder: An update. Acta Paediatr 92: 776–784. [PubMed] [Google Scholar]
- Faraone SV, Biederman J, Mick E ( 2006): The age‐dependent decline of attention deficit hyperactivity disorder: A meta‐analysis of follow‐up studies. Psychol Med 36: 159–165. [DOI] [PubMed] [Google Scholar]
- Fernández A, Quintero J, Hornero R, Zuluaga P, Navas M, Gómez C, Escudero J, García‐Campos N, Biederman J, Ortiz T ( 2009): Complexity analysis of spontaneous brain activity in attention‐deficit/hyperactivity disorder: Diagnostic implications. Biol Psychiatry 65: 571–577. [DOI] [PubMed] [Google Scholar]
- Giedd JN, Blumenthal J, Jeffries NO, Castellanos FX, Liu H, Zijdenbos A, Paus T, Evans AC, Rapoport JL ( 1999): Brain development during childhood and adolescence: A longitudinal MRI study. Nat Neurosci 2: 861–863. [DOI] [PubMed] [Google Scholar]
- Gogtay N ( 2008): Cortical brain development in schizophrenia: Insights from neuroimaging studies in childhood‐onset schizophrenia. Schizophr Bull 34: 30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogtay N, Greenstein D, Lenane M, Clasen L, Sharp W, Gochman P, Butler P, Evans A, Rapoport J ( 2007): Cortical brain development in nonpsychotic siblings of patients with childhood‐onset schizophrenia. Arch Gen Psychiatry 64: 772–780. [DOI] [PubMed] [Google Scholar]
- Gogtay N, Lu A, Leow AD, Klunder AD, Lee AD, Chavez A, Greenstein D, Giedd JN, Toga AW, Rapoport JL, Thompson PM ( 2008): Three‐dimensional brain growth abnormalities in childhood‐onset schizophrenia visualized by using tensor‐based morphometry. Proc Natl Acad Sci USA 105: 15979–15984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gothelf D, Eliez S, Thompson T, Hinard C, Penniman L, Feinstein C, Kwon H, Jin S, Jo B, Antonarakis SE, Morris MA, Reiss AL ( 2005): COMT genotype predicts longitudinal cognitive decline and psychosis in 22q11.2 deletion syndrome. Nat Neurosci 8: 1500–1502. [DOI] [PubMed] [Google Scholar]
- Greenstein D, Lerch J, Shaw P, Clasen L, Giedd J, Gochman P, Rapoport J, Gogtay N ( 2006): Childhood onset schizophrenia: Cortical brain abnormalities as young adults. J Child Psychol Psychiatry 47: 1003–1012. [DOI] [PubMed] [Google Scholar]
- Haier RJ, Head K, Head E, Lott IT ( 2008): Neuroimaging of individuals with Down's syndrome at‐risk for dementia: Evidence for possible compensatory events. Neuroimage 39: 1324–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halperin JM, Trampush JW, Miller CJ, Marks DJ, Newcorn JH ( 2008): Neuropsychological outcome in adolescents/young adults with childhood ADHD: Profiles of persisters, remitters and controls. J Child Psychol Psychiatry 49: 958–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazlett HC, Poe M, Gerig G, Smith RG, Provenzale J, Ross A, Gilmore J, Piven J ( 2005): Magnetic resonance imaging and head circumference study of brain size in autism: Birth through age 2 years. Arch Gen Psychiatry 62: 1366–1376. [DOI] [PubMed] [Google Scholar]
- Kent L, Green E, Hawi Z, Kirley A, Dudbridge F, Lowe N, Raybould R, Langley K, Bray N, Fitzgerald M, Owen MJ, O'Donovan MC, Gill M, Thapar A, Craddock N ( 2005): Association of the paternally transmitted copy of common Valine allele of the Val66Met polymorphism of the brain‐derived neurotrophic factor (BDNF) gene with susceptibility to ADHD. Mol Psychiatry 10: 939–943. [DOI] [PubMed] [Google Scholar]
- Krain AL, Castellanos FX ( 2006): Brain development and ADHD. Clin Psychol Rev 26: 433–444. [DOI] [PubMed] [Google Scholar]
- Lenroot RK, Gogtay N, Greenstein DK, Wells EM, Wallace GL, Clasen LS, Blumenthal JD, Lerch J, Zijdenbos AP, Evans AC, Thompson PM, Giedd JN ( 2007): Sexual dimorphism of brain developmental trajectories during childhood and adolescence. Neuroimage 36: 1065–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenroot RK, Schmitt JE, Ordaz SJ, Wallace GL, Neale MC, Lerch JP, Kendler KS, Evans AC, Giedd JN ( 2009): Differences in genetic and environmental influences on the human cerebral cortex associated with development during childhood and adolescence. Hum Brain Mapp 30: 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie S, Shaw P, Lenroot R, Pierson R, Greenstein DK, Nugent TF, 3rd , Sharp WS, Giedd JN, Rapoport JL ( 2007): Cerebellar development and clinical outcome in attention deficit hyperactivity disorder [see comment]. Am J Psychiatry 164: 647–655. [DOI] [PubMed] [Google Scholar]
- Makris N, Biederman J, Valera EM, Bush G, Kaiser J, Kennedy DN, Caviness VS, Faraone SV, Seidman LJ ( 2007): Cortical thinning of the attention and executive function networks in adults with attention‐deficit/hyperactivity disorder. Cereb Cortex 17: 1364–1375. [DOI] [PubMed] [Google Scholar]
- McAlonan GM, Cheung V, Chua SE, Oosterlaan J, Hung S‐f, Tang C‐p, Lee C‐c, Kwong S‐l, Ho T‐p, Cheung C, Suckling J, Leung PWL ( 2009): Age‐related grey matter volume correlates of response inhibition and shifting in attention‐deficit hyperactivity disorder. Br J Psychiatry 194: 123–129. [DOI] [PubMed] [Google Scholar]
- Mosconi MW, Cody‐Hazlett H, Poe MD, Gerig G, Gimpel‐Smith R, Piven J ( 2009): Longitudinal study of amygdala volume and joint attention in 2‐ to 4‐year‐old children with autism. Arch Gen Psychiatry 66: 509–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg JT, Casey BJ ( 2005): An integrative theory of attention‐deficit/ hyperactivity disorder based on the cognitive and affective neurosciences. Dev Psychopathol 17: 785–806. [DOI] [PubMed] [Google Scholar]
- Pinter JD, Eliez S, Schmitt JE, Capone GT, Reiss AL ( 2001): Neuroanatomy of Down's syndrome: A high‐resolution MRI study. Am J Psychiatry 158: 1659–1665. [DOI] [PubMed] [Google Scholar]
- Plessen KJ, Bansal R, Zhu H, Whiteman R, Amat J, Quackenbush GA, Martin L, Durkin K, Blair C, Royal J, Hugdahl K, Peterson BS ( 2006): Hippocampus and amygdala morphology in attention‐deficit/hyperactivity disorder. Arch Gen Psychiatry 63: 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport JL, Gogtay N ( 2008): Brain neuroplasticity in healthy, hyperactive and psychotic children: Insights from neuroimaging. Neuropsychopharmacology 33: 181–197. [DOI] [PubMed] [Google Scholar]
- Rapoport J, Chavez A, Greenstein D, Addington A, Gogtay N ( 2009): Autism spectrum disorders and childhood‐onset schizophrenia: Clinical and biological contributions to a relation revisited. J Am Acad Child Adolesc Psychiatry 48: 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redcay E, Courchesne E ( 2005): When is the brain enlarged in autism? A meta‐analysis of all brain size reports. Biol Psychiatry 58: 1–9. [DOI] [PubMed] [Google Scholar]
- Rubia K ( 2002): The dynamic approach to neurodevelopmental psychiatric disorders: Use of fMRI combined with neuropsychology to elucidate the dynamics of psychiatric disorders, exemplified in ADHD and schizophrenia. Behav Brain Res 130( 1/2): 47–56. [DOI] [PubMed] [Google Scholar]
- Schulz KP, Newcorn JH, Fan J, Tang CY, Halperin JM ( 2005): Brain activation gradients in ventrolateral prefrontal cortex related to persistence of symptoms in ADHD in adolescent boys. J Am Acad Child Adolesc Psychiatry 44: 47–54. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese‐Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee Y‐H, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimaki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King M‐C, Skuse D, Geschwind DH, Gilliam TC, Ye K, Wigler M ( 2007): Strong association of de novo copy number mutations with autism. Science 316: 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman LJ, Valera EM, Makris N ( 2005): Structural brain imaging of attention‐deficit/hyperactivity disorder. Biol Psychiatry 57: 1263–1272. [DOI] [PubMed] [Google Scholar]
- Semrud‐Clikeman M, Pliszka SR, Lancaster J, Liotti M ( 2006): Volumetric MRI differences in treatment‐naive vs chronically treated children with ADHD [erratum appears in Neurology. 2006 Dec 12;67:2091]. Neurology 67: 1023–1027. [DOI] [PubMed] [Google Scholar]
- Shaw P, Greenstein D, Lerch J, Clasen L, Lenroot R, Gogtay N, Evans A, Rapoport J, Giedd J ( 2006a) Intellectual ability and cortical development in children and adolescents. Nature 440: 676–679. [DOI] [PubMed] [Google Scholar]
- Shaw P, Lerch J, Greenstein D, Sharp W, Clasen L, Evans A, Giedd J, Castellanos FX, Rapoport J ( 2006b) Longitudinal mapping of cortical thickness and clinical outcome in children and adolescents with attention‐deficit/hyperactivity disorder. Arch Gen Psychiatry 63: 540–549. [DOI] [PubMed] [Google Scholar]
- Shaw P, Eckstrand K, Sharp W, Blumenthal J, Lerch JP, Greenstein D, Clasen L, Evans A, Giedd J, Rapoport JL ( 2007): Attention‐deficit/hyperactivity disorder is characterized by a delay in cortical maturation. Proc Natl Acad Sci USA 104: 19649–19654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw P, Sharp WS, Morrison M, Eckstrand K, Greenstein DK, Clasen LS, Evans AC, Rapoport JL ( 2009): Psychostimulant treatment and the developing cortex in attention deficit hyperactivity disorder. Am J Psychiatry 166: 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonuga‐Barke EJS ( 2002): Psychological heterogeneity in AD/HD—A dual pathway model of behaviour and cognition. Behav Brain Res 130( 1/2): 29–36. [DOI] [PubMed] [Google Scholar]
- Syed Z, Dudbridge F, Kent L ( 2007): An investigation of the neurotrophic factor genes GDNF, NGF and NT3 in susceptibility to ADHD. Am J Med Genet B Neuropsychiatr Genet 144B: 375–378. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Vidal C, Giedd JN, Gochman P, Blumenthal J, Nicolson R, Toga AW, Rapoport JL ( 2001): Mapping adolescent brain change reveals dynamic wave of accelerated gray matter loss in very early‐onset schizophrenia. Proc Natl Acad Sci USA 98: 11650–11655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toplak ME, Dockstader C, Tannock R ( 2006): Temporal information processing in ADHD: Findings to date and new methods. J Neurosci Methods 151: 15–29. [DOI] [PubMed] [Google Scholar]
- Valera EM, Faraone SV, Murray KE, Seidman LJ ( 2007): Meta‐analysis of structural imaging findings in attention‐deficit/hyperactivity disorder. Biol Psychiatry 61: 1361–1369. [DOI] [PubMed] [Google Scholar]
- van der Schot AC, Vonk R, Brans RGH, van Haren NEM, Koolschijn PCMP, Nuboer V, Schnack HG, van Baal GCM, Boomsma DI, Nolen WA, Hulshoff Pol HE, Kahn RS ( 2009): Influence of genes and environment on brain volumes in twin pairs concordant and discordant for bipolar disorder. Arch Gen Psychiatry 66: 142–151. [DOI] [PubMed] [Google Scholar]
- van't Ent D, Lehn H, Derks EM, Hudziak JJ, Van Strien NM, Veltman DJ, De Geus EJC, Todd RD, Boomsma DI ( 2007): A structural MRI study in monozygotic twins concordant or discordant for attention/hyperactivity problems: Evidence for genetic and environmental heterogeneity in the developing brain. Neuroimage 35: 1004–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW, Knopman DS, Petersen RC, Jack CR Jr; Alzheimer's Disease Neuroimaging Initiative ( 2009): MRI and CSF biomarkers in normal, MCI, and AD subjects: Predicting future clinical change. Neurology 73: 294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal CN, Rapoport JL, Hayashi KM, Geaga JA, Sui Y, McLemore LE, Alaghband Y, Giedd JN, Gochman P, Blumenthal J, Gogtay N, Nicolson R, Toga AW, Thompson PM ( 2006): Dynamically spreading frontal and cingulate deficits mapped in adolescents with schizophrenia. Arch Gen Psychiatry 63: 25–34. [DOI] [PubMed] [Google Scholar]
- Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, Stray SM, Rippey CF, Roccanova P, Makarov V, Lakshmi B, Findling RL, Sikich L, Stromberg T, Merriman B, Gogtay N, Butler P, Eckstrand K, Noory L, Gochman P, Long R, Chen Z, Davis S, Baker C, Eichler EE, Meltzer PS, Nelson SF, Singleton AB, Lee MK, Rapoport JL, King M‐C, Sebat J ( 2008): Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320: 539–543. [DOI] [PubMed] [Google Scholar]