

Abstract
Although subtle anatomical anomalies long precede the onset of clinical symptoms in Alzheimer's disease, their impact on the reorganization of brain networks underlying cognitive functions has not been fully explored. A unique window into this reorganization is provided by presymptomatic cases of familial Alzheimer's disease (FAD). Here we studied neural circuitry related to semantic processing in presymptomatic FAD cases by estimating the intracranial sources of the N400 event‐related potential (ERP). ERPs were obtained during a semantic‐matching task from 24 presymptomatic carriers and 25 symptomatic carriers of the E280A presenilin‐1 (PS‐1) mutation, as well as 27 noncarriers (from the same families). As expected, the symptomatic‐carrier group performed worse in the matching task and had lower N400 amplitudes than both asymptomatic groups, which did not differ from each other on these variables. However, N400 topography differed in mutation carrier groups with respect to the noncarriers. Intracranial source analysis evinced that the presymptomatic‐carriers presented a decrease of N400 generator strength in right inferior‐temporal and medial cingulate areas and increased generator strength in the left hippocampus and parahippocampus compared to the controls. This represents alterations in neural function without translation into behavioral impairments. Compared to controls, the symptomatic‐carriers presented a similar anatomical shift in the distribution of N400 generators to that found in presymptomatic‐carriers, albeit with a larger reduction in generator strength. The redistribution of N400 generators in presymptomatic‐carriers indicates that early focal degeneration associated with the mutation induces neural reorganization, possibly contributing to a functional compensation that enables normal performance in the semantic task. Hum Brain Mapp, 2010. © 2009 Wiley‐Liss, Inc.
Keywords: Alzheimer's disease, asymptomatic, N400, generators, mutation
INTRODUCTION
A promising (though infrequently used) research approach on the earlier stages of Alzheimer's disease (AD) is to examine presymptomatic carriers (PresymC) of gene mutations that inevitably cause familial forms of the disease. Familial forms of this kind have been described for mutations in three genes: amyloid protein precursor (APP) gene on chromosome 21, presenilin‐1 (PS‐1) gene on chromosome 14, and presenilin‐2 (PS‐2) gene on chromosome 1 [Hardy, 1997]. All have an autosomal dominant pattern of inheritance. These infrequent mutations virtually have 100% penetrance. Familial Alzheimer's disease (FAD) has an earlier onset than sporadic forms of AD, with the presymptomatic carriers developing a gradual cognitive decline, that at first affects episodic memory, and which afterwards grows into a pervasive dysfunction. Several studies have shown that asymptomatic carriers, although still cognitively intact, exhibit reduction of medial temporal lobe (MTL) volumes [Fox et al., 1996, 2001; Scahill et al., 2002; Schott et al., 2003; Wahlund et al., 1999]. Serial magnetic resonance images (MRI) of nine mutation carriers have shown increased hippocampal atrophy rates, as compared to controls, 5.5 years before disease onset [Ridha et al., 2006], and increased whole‐brain atrophy that is evident 3.5 years before onset. In a diffusion tensor imaging study of 12 asymptomatic mutation carriers, white matter fractional anisotropy was reduced in the fornix and in tracts related to the orbitofrontal region [Ringman et al., 2007].
Several functional resting‐state, neuroimaging studies have also been performed on asymptomatic mutation carriers [Almkvist et al., 2003; Johnson et al., 2001; Mosconi et al., 2006]. An examination of 18 presymptomatic E280A PS‐1 mutation carriers with single photon computerized tomography (SPECT) evidenced reduced perfusion in relation to controls in the hippocampal complex, anterior and posterior cingulate cortex, posterior parietal lobe, and the anterior frontal lobe [Johnson et al., 2001]. Using positron emission tomography (PET), in a sample of seven presymptomatic PS‐1 mutation carriers, reduced glucose metabolism was found bilaterally in the whole brain measures, in inferior parietal lobe, and superior temporal gyrus, as well as in the entorhinal cortex and the hippocampus in the left hemisphere [Mosconi et al., 2006]. The reduced metabolism was widespread and also involved areas without cortical atrophy.
Summarizing, the meager body of evidence related to asymptomatic carriers of FAD mutations is consistent with early abnormalities in the morphometry, anatomical connectivity, and resting‐state functional images of the MTL, especially the hippocampus, which all precede in years the onset of symptoms. This is congruent with follow‐up studies of subjects at risk for sporadic forms of AD, who also display similar pathological and functional alterations years before disease onset [Chetelat and Baron, 2003; Dickerson et al., 2001; Medina et al., 2006]. Given the role of MTL in memory function, it would be important to carry out functional imaging studies of young asymptomatic mutation carriers while they perform tasks challenging different memory functions. Unfortunately, to our knowledge only a single‐case report of this type has been published (although an older, more cognitively impaired, FAD case was also examined in the same work). This study [Mondadori et al., 2006] found increased functional MRI (fMRI) activation in regard to controls in left frontal, temporal, and parietal neocortices and in the left hippocampus during the learning and retrieval of an episodic memory task (pairing of unknown faces with professions). The increased activation related to the memory task is interpreted as the result of compensatory effort to surmount preclinical pathological damage to brain areas underlying memory functions. This interesting finding needs to be replicated in a larger sample, but it is congruent with functional imaging findings in groups of asymptomatic subjects with the apolipoprotein E epsilon4 (APOE4) allelle [Bookheimer et al., 2000; Johnson et al., 2006] and thus at risk for AD [Bassett et al., 2006], and in groups of normally ageing subjects [Reuter‐Lorenz, 2002].
An additional technique, source localization of event related potentials (ERPs), can be applied to study early changes in local brain function, which has excellent temporal resolution (millisecond level) thus complementing the sluggish response of fMRI and PET. By focusing on specific ERP components, different cognitive processes can be explored. In this article, we explore this approach.
Although episodic memory is usually considered to be the first cognitive process impaired in the course of AD, several lines of evidence indicate that semantic memory could also be affected at early stages [Chertkow et al., 2008], and in some cases may appear before the deficits in episodic memory [Dudas et al., 2005; Hodges and Patterson, 1995; Storandt, 2008]. Semantic deficits in AD have been explored with different tasks, including the semantic priming paradigm [Giffard et al., 2005]. Many studies report difficulty with semantic memory in patients with probable AD [Chertkow and Bub, 1990; Grossman et al., 1996]. In addition, deficits in semantic memory can also be found relatively early in AD [Chan et al., 1997; Chertkow and Bub, 1990; Hodges and Patterson, 1995]. This is convenient for our purposes, because a thoroughly studied ERP component, the N400, is related to semantic processing. The N400, an ERP component, is widely used in language research [see review by VanPetten and Luka, 2006]. The N400 refers to a negative voltage deflection that peaks about 400 ms after stimulus presentation that is maximal over centro‐parietal electrode sites. In the original description [Kutas and Hillyard, 1980], participants read sentences that ended with a congruent or an incongruent word. The N400 is larger for incongruent than for congruent words. N400 components are also elicited by nonlinguistic stimuli (pictures, faces), when they do not match the semantic context created by previous stimuli [Barrett et al., 1988; Barrett and Rugg, 1989, 1990; Caldara et al., 2004; Holcomb and McPherson, 1994; Stuss et al., 1992]. Specifically, we propose to examine early deficits in semantic processing with an N400 paradigm, which generates both behavioral and brain activation data.
An additional inducement to study the N400 is that its probable neural generators have been identified by different techniques, including recordings in patients with focal brain damage [Friederici et al., 1999; Hagoort et al., 1996; Swaab et al., 1997], intracranially‐recorded ERPs in patients with implanted electrodes [Allison et al., 1994; Elger et al., 1997; Fernandez et al., 2001; McCarthy et al., 1995; Nobre and McCarthy, 1995; Smith et al., 1986], and source modeling of magnetoencephalography and ERP data [Haan et al., 2000; Halgren et al., 2002; Helenius et al., 1998, 2002; Kwon et al., 2005]. All of these studies agree on the important contribution of the temporal regions including MTL in the generation of N400, although other areas (e.g. in the frontal cortex) would also participate. Given the apparent overlap between brain areas containing the N400 generators and those that exhibit early damage in the temporal course of AD, the N400 is potentially useful for exploring the effects of the neural degeneration associated to AD progression upon semantic function.
Furthermore, several studies have recorded the N400 in patients with AD and have found amplitude reductions of the N400 component in patients as compared to normal controls [Auchterlonie et al., 2002; Castaneda et al., 1997; Ford et al., 1996; Hamberger et al., 1995; Iragui et al., 1996; Olichney et al., 2002, 2006; Olichney and Hillert, 2004; Ostrosky‐Solis et al., 1998; Schwartz et al., 1996]. This suggests an impaired activation of the neural generators of this component in AD. The N400 is also reduced in cases with mild cognitive impairment (MCI) [Olichney et al., 2006]. However, to our knowledge, no study of the N400 in presymptomatic carriers of FAD mutations has been performed.
Summarizing the goal of this study is to examine the feasibility of using ERP source localization to study the local physiopathology of early stages of AD with high temporal resolution. Here we recorded the N400 during a picture semantic matching task in members of a large pedigree from Colombia, some of which carried the E280A PS‐1 mutation for early onset AD [Lopera et al., 1997]. This paradigm was selected because it has been previously applied to Spanish speaking patients with AD showing to be sensitive for discovering semantic deficits by both accuracy of performance and in the ERP data [Castaneda et al., 1997; Ostrosky‐Solis et al., 1998]. Twenty‐four presymptomatic carriers (PresymC), 27 noncarriers (NonC), and 25 symptomatic carriers (SymC) subjects were recruited. Intracranial sources of the N400 are estimated by source reconstruction in each group, as a window into neural circuitry underlying semantic processing. In this case, sources are estimated over the difference waveforms, to isolate components specifically involved in the processing of the task. This is important for source localization, because, as it is the case of fMRI, irrelevant activity must be suppressed for further analysis. We used a Bayesian Model Averaging (BMA) method for source reconstruction, as described by Trujillo‐Barreto et al. [ 2004]. This method has shown significantly less blurring, ghost sources, and underestimation of deep sources than alternative approaches [Trujillo‐Barreto et al., 2004]. The last characteristic is important for studies of damage associated to AD, where regions buried within the MTL are involved. If some of the structures contributing to the N400 generation are damaged in presymptomatic carriers (as suggested by the studies reviewed previously), the neural sources of N400 in these cases would be different to those found in controls. Thus, the comparison between NonC and PresymC could contribute to clarify the role of functional reorganization on semantic processing when early, preclinical, structural degeneration is present.
MATERIALS AND METHODS
Subjects
Participants of this study were 76 subjects from a group of families with a history of FAD reported by Lopera et al. [ 1997]. FAD in this population is caused, with 100% penetrance, by the E280A mutation in the PS‐1 gene in chromosome 14. Each participant gave their informed consent, according to a protocol approved by the Human Subjects Committee of the University of Antioquia. All subjects received a medical, neurological, and neuropsychological examination to determine whether or not they met the National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) and AD, the Related Disorders Association (ADRDA) criteria [McKhann et al., 1984], and the Diagnostic and Statistical Manual of Mental Disorders (DSM IV [American Psychiatric Association, 1994]) criteria for dementia. The medical examination included standard laboratory tests (e.g., serum chemistry and hematology, thyroid and liver function tests, and brain MRI). The neuropsychological testing used the Spanish version of the following tests: a test battery developed by the “Consortium to Establish a Registry for AD” [Karrasch et al., 2005; Morris et al., 1989], the Boston Naming Test [Kaplan et al., 1978], the Boston Diagnostic Aphasia Examination [Goodglass and Kaplan, 1972], and the Rey‐Osterreith Complex Figure test [Rey, 1941]. In addition, blood samples were collected to determine the presence of the PS‐1 mutation. Subjects who were classified as asymptomatic were active and functionally normal, they had no significant cognitive complaints, and no evidence of cognitive impairments on the standardized neuropsychological tests. Subjects with a history of neurological or psychiatric illness were excluded from the study. (For the details of neuropsychological evaluation protocol see [Arango‐Lasprilla et al., 2007]). The presence of E280A mutation in the presenilin 1 gene was previously determined in each subject by genetic testing (E280A, substitution of glutamic acid for alanine in chromosome 14).
This population was divided into three subgroups (Table I). The NonC group consisted of 27 individuals who were cognitively normal and did not carry the E280A PS‐1 mutation. The PresymC group consisted of 24 individuals who were carriers of the E280A PS‐1 mutation but did not present cognitive dysfunction or dementia symptoms. These individuals were expected to subsequently develop AD, but they were in a preclinical stage while the study was in progress. The third group, SympC, consisted of 25 E280A PS‐1 mutation carriers who met the criteria for a clinical research diagnosis of probable AD.
Table I.
Participant descriptive data
| Noncarriers E280A | Presymptomatic carriers E280A | Symptomatic carriers E280A | |
|---|---|---|---|
| Complete sample | |||
| Subset (n) | 27 | 24 | 25 |
| Age | |||
| Mean (min‐max) | 42.8 (23–50) | 39.3 (25–49) | 48.2 (35–53) |
| GDS/FAST Staging systema | 1 | 1 | ≥2 |
| ≤4 | |||
| MMSEb | |||
| Mean (min‐max) | 29 (26–30) | 28 (25–30) | 21 (16–24) |
| Barthel Scalec | 50 | 50 | 50 |
| Subsample for inverse solution analysis | |||
| Subset (n) | 16 | 17 | 14 |
| Age | |||
| Mean (min‐max) | 40 (23–50) | 37 (25–49) | 46 (35–53) |
| GDS/FAST Staging systema | 1 | 1 | ≥2 |
| ≤4 | |||
| MMSEb | |||
| Mean (min‐max) | 29 (26–30) | 29 (25–30) | 21 (16–23) |
| Barthel Scalec | 50 | 50 | 50 |
The three groups were similar in sex and educational level. However, the mean age of the symptomatic carriers group was higher than the other two asymptomatic groups. An analysis of variance (ANOVA) demonstrated a significant age effect (F(2,73) = 11.07, P < 0.00006), and planned comparison showed that age did not differ between NonC and PresymC (P < 0.4), although SympC were different in age from both NonC (P < 0.0004) and PresymC (P < 0.000039). Asymptomatic groups were selected as having a GDS/FAST staging system score of 1 [Auer and Reisberg, 1997] and MiniMental State Examination (MMSE) [Folstein et al., 1975], above 23, which is the normal limit in Colombia. The mean MMSE scores for NonC and PresymC groups were not significantly different from one another. The symptomatic group reached GDS/FAST staging system scores between 2 and 4, and MMSE between 15 and 24, which classified them as very mild dementia The Spanish version of Barthel scale [Aguirre‐Acevedo et al., 2007] in the three groups was of 50, indicating that they functioned at a similar level in everyday activities (Table I).
Stimulation Procedure
The stimuli consisted of 118 pairs of drawings of objects and animals [Snodgrass and Vanderwart, 1980] for which denomination, familiarity, and imaginability had been characterized for Spanish speaking populations [Aveleyra et al., 1996]. These drawings were presented in a computer monitor in an 8 cm2 area. Because subjects sat approximately 1 m away from the monitor, the drawings subtended a vertical and horizontal angle of 4.6°. Pairs of stimuli were selected in which 50% were semantically related (belonging to the same semantic category) whereas the other 50% were not. These were the congruent and incongruent pairs, respectively (see [Bobes et al., 1996] for stimuli examples).
During the experiment, the subjects sat in a comfortable chair in front of the computer monitor and were asked to minimize body and eye movements. The drawings in a pair were sequentially presented, each for 1 s, the first member acting as context, and the second serving to trigger the ERPs recording epoch. The different pairs were presented in pseudorandom order. The task of the subjects was to discriminate between the congruent and incongruent pairs of pictures, by pressing one of two keys during the 2 s after the second stimulus offset. The response was delayed until electroencephalographic (EEG) recording was finished to reduce artifacts, thus reaction time was not measured. The session began with a training phase, using 10 pairs of drawings not employed during the subsequent ERPs recordings.
ERP Recordings
In a subset of the subjects (16 from NonC group, 17 from the PresymC and 14 from the SympC group, Table I), EEG data acquisition was carried out with 120 monopolar derivations, using electrodes mounted in an elastic cap homogeneously distributed over the scalp, as well as two electro‐oculogram (EOG) channels, with a MEDICID‐128 system (Neuronic, SA, Havana). In the remaining subjects (11 from NonC group, 7 from the PresymC and 11 from the SympC group), only 19 derivation were recorded, using Ag‐AgCl disk electrodes placed at the locations of the 10/20 international system on a MEDICID‐III/E system, coinciding with the position of the first 19 electrodes of the 120 channels montage. Data obtained from this last subset of individuals were not considered for current source analysis.
Signals were amplified by a factor of 10,000 and filtered between 0.5 and 30 Hz (3 dB down), and a notch filter with peak at 60 Hz was used. All electrodes were referred to linked earlobes, and the inter‐electrode impedance was always below 10 kΩ. The EEG was digitally recorded at a sampling rate of 200 Hz, with 900 ms epochs, and a 100 ms prestimulus baseline (synchronized with the onset of the stimuli). Each EEG epoch was stored on magnetic disk, and it was visually inspected offline. Those epochs with generalized artifacts or detectable eye‐movement in the EOG were eliminated. In the 120 channels recording, electrodes with excessive noise were eliminated and substituted by an interpolation of the 11 closest neighbors. Evoked responses were obtained for each experimental condition by averaging the congruent and incongruent trials separately. To isolate the N400 effect, difference waveforms were obtained from the subtraction between congruent and incongruent recordings. These ERPs were subjected to low‐pass filtering with a 18 Hz cut‐off (zero phase distortion), and amplitudes were corrected by subtracting the average pre‐stimulus amplitude value. Voltage measures were rereferenced to an average reference for topographic analysis and source resconstruction.
Data Analysis
Behavioral data: Discrimination accuracy was measured using d′ of signal detection theory [Swets, 1964]. Correct classifications of congruent items were considered as Hits, and incorrect classifications of incongruent items were considered as False Alarms. The d' values were submitted to ANOVA analysis using Group as between‐subject factor (NonC, PresymC, and SympC).
Analysis of ERP amplitude effects was carried out for the 19 electrodes of the 10/20 system in the complete sample of 76 subjects (in the cases with 120 channel recordings, other sites were ignored for this analysis). The mean voltage amplitude in predefined time‐windows was submitted to a repeated measures analysis of variance (rmANOVA) using one between‐subject factor: group (NonC, PresymC, and SympC) and two within‐subject factors: congruity (congruent vs. incongruent) and site (with 19 levels corresponding to the electrodes used). When appropriate, the Greenhouse‐Geisser procedure [Keselman and Rogan, 1980] was used to mitigate violations of the sphericity assumptions in the repeated measures ANOVA (the corresponding epsilon values are reported). The following three time‐windows were each used in an rmANOVAs (corresponding to the main ERP components): 230–280, 390–440, and 500–650 ms. The threshold for statistical significance was set at P < 0.01.
The statistical difference between two scalp distributions was tested by applying permutation techniques to the difference waveforms. Note that permutation tests are distribution free, do not assumptions about underlying correlation structure are required, and they provide exact P‐values for any number of subjects, time points, and recording sites. Voltage maps were firstly normalized as suggested by McCarthy and Wood [ 1985] to eliminate amplitude differences between conditions. The variation of ERP topography between groups was assessed for the 390–440 ms time window (in the difference waveforms containing the N400 congruity effect), for which all 10 time points were included. The maximum of the permutated t‐tests (t max) was calculated between two groups for each electrode across all time points. The distribution estimated for max t can then be used to set significance levels that control the experiment wise error for the simultaneous univariate comparisons at each electrode [Blair and Karniski, 1994; Galan et al., 1997].
Source Reconstruction Method
The sources of the N400 component were estimated in the 47 subjects (Table I) with 120 electrode recordings. The sources of the ERP are localized through the solution of the EEG inverse problem (IP). This consists on the estimation of the primary current densities (PCDs) distribution inside the brain that produces a given measured EEG. There are infinite theoretical solutions to this problem, and thus specific additional prior information (or constraints) about the EEG generators must be included to obtain a unique and physiologically valid solution. These constraints may be mathematical (in the LORETA method [Pascual‐Marqui et al., 1994] the smoothest solutions is required) or anatomical in nature. A common procedure is to assume that the possible generators are confined to a given region of the brain (restricting PCDs to different subsets of i.e. gray matter) and to make all statistical inference conditional to that assumption. But different choices of the anatomical constraints lead to completely different current density distributions, which introduces some uncertainty about the model assumptions that must be taken into account. This problem, commonly omitted by traditional inverse solution methods, has been widely treated in the Bayesian literature and it is known as Model Uncertainty [Hoeting et al., 1999]. In this work, we use the Bayesian Model Averaging (BMA) approach firstly described by Trujillo‐Barreto [Penny et al., 2006; Trujillo‐Barreto et al., 2004], which is an application of the Bayesian model inference scheme, under the evidence approximation [MacKay, 1992], to the EEG/MEG inverse problem. This provides a coherent mechanism to account for the type of model uncertainty described earlier.
In short, in this study, solution under each model (anatomical constraint) is obtained using the traditional LORETA method [Pascual‐Marqui et al., 1994]. Different models were defined by constraining the sources to one of 68 anatomical compartments, which were chosen from the “Montreal average brain” (MNI Brain) [Petrides et al., 1993] Probabilistic brain atlas (PBA) [Collins et al., 1994; Mazziotta et al., 1995], the cerebellum (16 areas), and other 12 areas that comprised less than 10 voxels were excluded from consideration. The Bayesian paradigm allows estimating the posterior probability of each model given the data, which represents a measure of “how good” that model is for explaining the data. This measure of goodness expresses a trade‐off between goodness‐of‐fit (data reconstruction error based on that model) and complexity (number of parameters used by the model to explain the data) of the model. Here the number of parameters of the given model is proportional to the total number of voxels contained in the anatomical regions used to constrain the inverse solution in that model. That is, highest probabilities are assigned to the simpler models that explain the data best. Subsequently, model uncertainty is taken into account by averaging the PCDs obtained for each particular anatomical constraint (or model) weighted by their model posterior probabilities. This weighted average provides a PCD map that is unconditional on any model. For a detailed description of the mathematics and properties of the method, see [Trujillo‐Barreto et al., 2004].
The mean voltage amplitude in the time window including the N400 component (311–490 ms poststimulus) were submitted to source reconstruction. Statistical parametric mapping (SPM) was used to make population‐level inference over the calculated sources of the N400 (defined as the difference waveform). The PCD was estimated for each subject's N400 component using the method described earlier. The forward model used in this case consists of three spheres modeling piecewise homogenous compartments: brain, skull, and scalp. The conductivity values selected in our case were 0.33 and 0.022 S m−1 for the brain, scalp, and skull, respectively [Oostendorp et al., 2000; Zhang et al., 2006]. Outside the scalp the surrounding air has zero conductivity. The intracerebral PCDs were estimated over a grid of 20,092 points, constrained to the gray matter. With this information, the physical term (electric lead field) that relates the intracerebral activity to the scalp electric fields was computed. In Trujillo‐Barreto et al. [ 2004], the authors reported the localization error of the BMA method for different EEG and MEG sensors arrays. The localization error for 120 electrodes is below 10%. In terms of distance this means a localization error of less than 8.5 mm.
Note that the ‘Montreal average brain’ (MNI brain) [Petrides et al., 1993] was used as the anatomical model for all subjects. Subsequently, SPMs were computed based on a voxel‐by‐voxel Hotelling T2 test against zero to estimate the statistically significant sources in each group. For localizing the group differences, SPMs were computed for the voxel‐by‐voxel mean difference between groups using the Hotelling T2 test. The resulting probability maps were thresholded at a false discovery rate (FDR) q = 0.05 [Genovese et al., 2002] and were depicted as 3D activation images overlaid on the MNI brain. Clusters of contiguous voxels surviving the threshold were identified, and local maxima were measured.
RESULTS
Both groups of asymptomatic subjects were highly accurate in matching figures. Mean d′ for NonC was 2.98 (SD = 1.76), corresponding to 76% of hits and 9% of false alarms; and for PresymC was 2.89 (SD = 1.2), corresponding to 72% of hits and 6% of false alarms. However, SympC were less accurate (mean d′ = 1.16, SD = 2.07), corresponding to 51% of hits and 29% of false alarms. A one‐way ANOVA showed a significant effect for Group (F(2,70) = 13.63, P < 0.0004). In the planned comparisons d′ was not significantly different between asymptomatic groups (NonC vs. PresymC, P = 0.83), whereas this measure was significantly lower for SympC than for NonC (P < 0.00009) and for PresymC (P < 0.0003).
The grand average ERPs at the 10/20 system sites for the three groups of subjects are shown in Figure 1. The ERPs exhibited three conspicuous components, all prominent at central sites: a first positive peak with latency around 250 ms (P250), followed by a more prominent positivity peaking around 400 ms, and a late positive component (LPC) with latency around 550 ms. No difference between congruent and incongruent recordings were apparent before 280 ms. After this latency, recordings corresponding to incongruent trials were more negative in all groups, producing a peak with a minimum at about 410 ms, and which lasted up to about 525 ms in the three groups of subjects. This corresponds to the N400 component. To observe the N400 more clearly, difference waveforms were obtained by subtracting incongruent from the congruent ones. The grand averages of these difference waveforms were overlaid for the three groups in Figure 2, where the isolated N400 component (the N400 effect) could be observed. At Pz, (where N400 is usually assessed) and other parietal and occipital sites, no difference in amplitude or latency of the N400 was observed between NonC and PresymC, while the N400 amplitude for SymC was reduced. However, at more frontal sites, several amplitude differences can be observed between groups.
Figure 1.
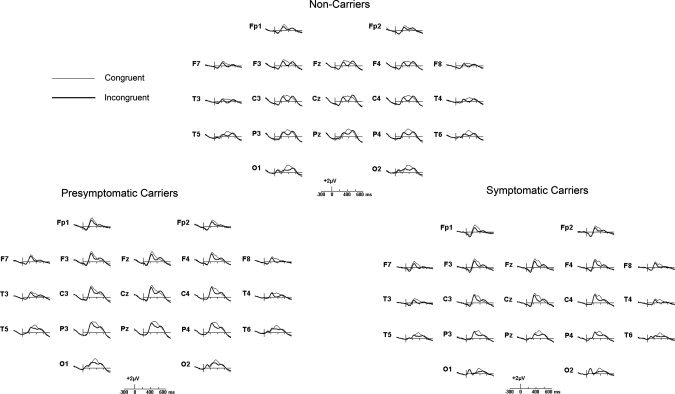
Grand average ERPs obtained during the semantic matching task in the three groups. ERPs associated with congruent figures (thin lines) are overlaid on ERPs elicited by incongruent figures (thick lines). In the upper panel, ERPs obtained in NonC, in the lower panel left the PresymC and in the lower panel right the SympC. Positive deflection point up.
Figure 2.
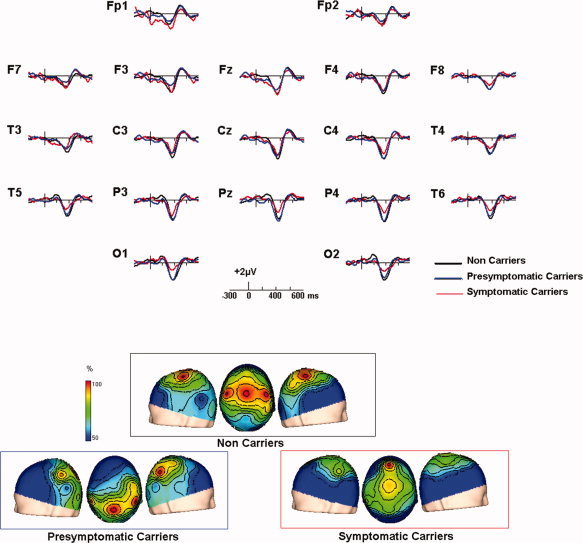
(A) Difference waveforms showing the isolated grand average N400 component in each group. The waveform obtained for NonC (in black), PresymC (in blue) and SympC (in red) are overlaid. Positive deflection point up. (B) Voltage maps representing the scalp distribution of the N400 in each group. In the upper pannel topographic map obtained in NonC, in the lower panel left PresymC and lower panel right SympC. These maps were obtained for the mean amplitude in the time windows including the negative component, expressing amplitudes as a percentage of the maximum. Three different views are depicted: left; center (from above with the nose up) and right. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
A rmANOVA was performed to the ERP amplitudes (congruent and incongruent waveforms) in three different time windows including the main components (Table II). For P250 component (230–280 ms) the main effect of Group was significant. Planned comparisons demonstrated that P250 amplitude was increased for the PresymC (mean = 4.3) compared to the NonC group (mean 2.4, t = 3,77, P < 0.0003) and also compared to the SympC (mean 2.3, t = 3,55, P < 0.0006). No difference was found between NonC and SympC. The effect of Group was not significant in the time window of the N400 component. For the LPC component (500–650 ms), the effect of Group was significant. Planned comparisons revealed that LPC amplitude were similar in NonC (mean = 2.21) and PresymC (mean = 2.09), but there was an amplitude reduction for the LPC in SympC (mean = 0.91) compared to the NonC (t = 3.86, P < 0.0002) and compared to the PresymC (t = 3.43, P < 0.0009).
Table II.
Summary of the results obtained from ERPs repeated measure ANOVA
| P250 (230–280 ms) | P400 (390–440 ms) | P550 (500–650 ms) | |
|---|---|---|---|
| Group | F(2, 72) = 8.8 | F(2, 72) = 3.5 ns | F(2, 72) = 8.99 |
| P < 0.001 | P < 0.0001 | ||
| Congruity | F(1, 72) = 2.02 ns | F(1, 72) = 88.4 | F(1, 72) = 3.77 ns |
| P = 0.00001 | |||
| Congruity × group | F(2, 72) = 0.35 ns | F(2, 72) = 1.47 ns | F(2, 72) = 1.10 ns |
| Site | F(18, 1296) = 30.8 | F(18, 1296) = 38.9 | F(18, 1296) = 53.5 |
| P < 0.001 | P < 0.00001 | P < 0.00001 | |
| ε = 0.15 | ε = 0.24 | ε = 0.26 | |
| Site × group | F(18, 1296) = 2.47 ns | F(18, 1296) = 2.56 | F(18, 1296) = 2.19 ns |
| P < 0.008 | |||
| ε = 0.24 | |||
| Congruity × site | F(18, 1296) = 8.3 | F(18, 1296) = 6.86 | F(18, 1296) = 8.36 |
| P < 0.0001 | P < 0.00001 | P < 0.00001 | |
| ε = 0.31 | ε = 0.21 | ε = 0.26 | |
| Congruity × site × group | F(18, 1296) = 1.38 ns | F(36, 1296) = 2.94 | F(36, 1296) = 0.7 ns |
| P < 0.004 | |||
| ε = 0.21 |
One between subject factors (group), and two within‐subject factors (congruity and site) were included. Test was performed on mean amplitude in three time windows (P < 0.01).
Congruity was only significant in the 390–440 ms time window, consistent with the N400 effect. In this same time window, the triple interaction congruity × site × group was significant, indicating that N400 component differed in either amplitude or topography between groups. Planned comparisons for the amplitude difference between congruent and incongruent trials were carried out in this time window. They showed that significant difference between NonC and SympC (F(1,72) = 4.96, P < 0.029) in central, parietal temporal, and occipital sites (excluding electrodes Fp1, Fp2, F3, F4, F7, F8, Fz, and Cz), whereas no significant differences were evinced between NonC and PresymC (P = 0.55). Planned comparison also showed that differences between NonC and SympC were significant for congruent trials (F(1,72) = 4.04, P < 0.047), but not for incongruent trials (P = 0.75).
The topography of the N400 effect was analysed over the difference waveforms. The normalized scalp distributions of the N400 effect are shown in more detail in Figure 2. Although the NonC group exhibited the maximum at central sites, in PresymC the maximum was observed at parietal sites and in SympC at frontal sites. The permutation test confirmed that these distributions were different. For NonC and PresymC the global test was significant (P < 0.05). Tests of marginal hypotheses showed that the differences were located at F3, F7, and Fz (all P < 0.05). The NonC and the SympC groups also were significantly different (global P < 0.05) with local effects at F3, C3, C4, P3, P4, O1, O2, T3, T4, T5, T6, and Pz electrodes (all P < 0.05). Finally, statistically significant topographic differences also appeared between PresymC and SympC groups (located at C4, P4, T3, and T6 electrodes, global and marginal tests all P < 0.05)
Source analysis was carried out in the N400 time window, which were the only latencies where the main experimental factor Congruity had a significant effect. This allowed us to use the scalp distribution of difference waveform amplitudes mean as input to the source localization analysis. Note that this precluded estimation of current sources in brain areas with nonspecific activity not related to the semantic processing task. The sources were estimated in those subjects with 120 electrode recordings (Table I). The grand mean and SPMs of the source‐solutions for each group are showed in Figure 3. The SPM results were plotted in a “glass brain” display and on selected slices of the MNI average brain (Fig. 3B–D). All clusters of significant voxels, as well as a brief description of their anatomical localization, were summarized in Table III. For NonC group, several sources in the right hemisphere were significantly greater than zero in the Hotelling T2 tests (Fig. 3B). One of these sources extended across the inferior temporal gyrus reaching the anterior fusiform gyrus, whereas other sources were found at the middle temporal gyrus, superior temporal gyrus (STG), medial cingulate (MC), precuneus, and the postcentral sulcus (Table III). In the PresymC group, SPM uncovered sources (Fig. 3C) at the right hippocampus and parahippocampal regions as well as at STG, but in contrast to the NonC group generator activity was not found in the right inferior temporal gyrus neither in the right MC. However, in this group the left hemisphere was also involved in the N400 generation, exhibiting significant sources in the hippocampus and fusiform gyrus. Other clusters of significant voxels are described in Table III. The SympC group showed a generators pattern very similar to the one shown by the PresymC group (Fig. 3D), with sources in the right parahippocampus and STG, but with a less extended generator in the left temporal areas, largely confined to hippocampal and parahippocampal areas (Table III).
Figure 3.
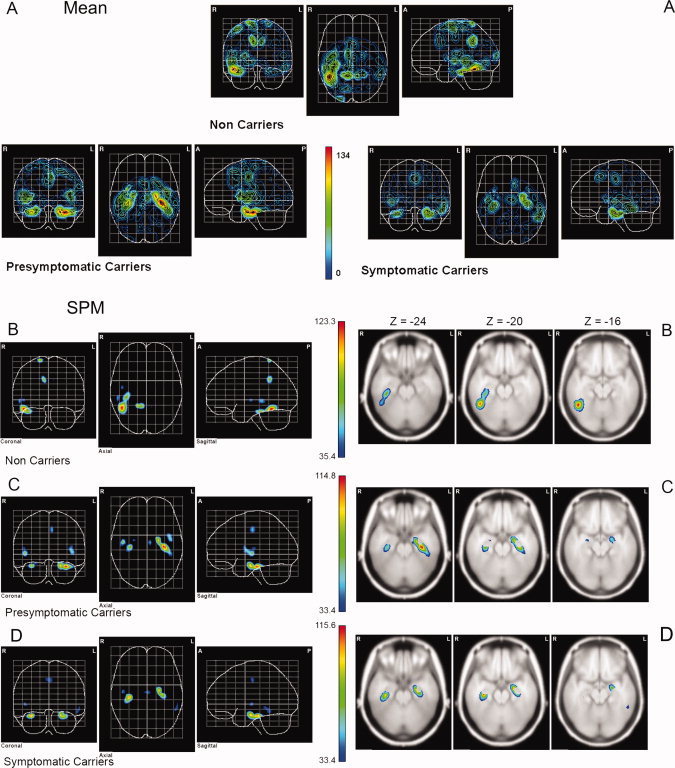
(A) Grand mean of the current source density obtained in each group. (B) Statistical parametric maps of the cortical current density distribution of the N400 component in NonC. Scale represents Hotelling T2 values. Only those voxels where the T value reached significance (q = 0.05, FDR corrected) are displayed. On the left, the maximum intensity projection (onto coronal, axial and sagittal planes) of the BMA solution is displayed on a “glass brain.” On the right, three slices show significant generator sites for the inferior temporal regions. The Z values of the corresponding MNI coordinates are indicated. (C) Statistical parametric maps of the cortical current density distribution of the N400 component in PresymC. (D) Statistical parametric maps of the cortical current density distribution of the N400 component in SympC. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Table III.
Brain regions showing current source densities significantly different from zero (all q < 0.05 FDR corrected for whole brain volume, extent threshold of 10 voxels)
| Cluster | AAL | BA | x | y | z | Vol | T2 | P |
|---|---|---|---|---|---|---|---|---|
| Noncarriers | ||||||||
| 1 | Fusiform_R | 20 | 38 | −30 | −24 | 5280 | 78.9 | 0.000019 |
| 1 | Temporal_Inf_R | 20 | 46 | −50 | −16 | 5280 | 123.3 | 0.000002 |
| 2 | Temporal_Mid_R | 21 | 54 | −34 | −4 | 266 | 51.3 | 0.000175 |
| 2 | Temporal_Mid_R | 22 | 54 | −30 | 0 | 266 | 50.7 | 0.000184 |
| 3 | Temporal_Sup_R | 48 | 46 | −14 | 0 | 52 | 48.1 | 0.000239 |
| 4 | Cingulum_Mid_R | 23 | 6 | −46 | 36 | 843 | 69.1 | 0.000038 |
| 4 | Precuneus_R | 0 | 10 | −46 | 40 | 843 | 75.9 | 0.000023 |
| 5 | Postcentral_R | 1 | 14 | −46 | 76 | 514 | 97.2 | 0.000006 |
| Presymptomatic carriers | ||||||||
| 1 | Fusiform_L | 20 | −34 | −22 | −24 | 4551 | 114.8 | 0.000001 |
| 2 | ParaHippocampal_R | 20 | 34 | −22 | −20 | 1099 | 79.5 | 0.000011 |
| 1 | Hippocampus_L | 35 | −22 | −10 | −20 | 4551 | 96.8 | 0.000003 |
| 3 | Hippocampus_R | 20 | 26 | −10 | −16 | 132 | 46.4 | 0.000202 |
| 4 | Temporal_Sup_R | 48 | 46 | −14 | 0 | 1506 | 70.5 | 0.000021 |
| 4 | Putamen_R | 48 | 35 | −4 | 0 | 1506 | 52.7 | 0.000104 |
| 4 | 48 | 36 | −2 | 0 | 1506 | 51 | 0.000124 | |
| 4 | 48 | 36 | 2 | 0 | 1506 | 39.8 | 0.000434 | |
| 4 | Putamen_R | 48 | 35 | −3 | 2 | 1506 | 47 | 0.000188 |
| 6 | Insula_L | 0 | −46 | −10 | 4 | 1233 | 53.5 | 0.000096 |
| 6 | Rolandic_Oper_L | 48 | −42 | −6 | 4 | 1233 | 57.2 | 0.000067 |
| 4 | 48 | 35 | −4 | 4 | 1506 | 47.4 | 0.000181 | |
| 4 | 48 | 36 | −2 | 4 | 1506 | 39.5 | 0.000449 | |
| 4 | Putamen_R | 48 | 34 | 2 | 4 | 1506 | 39.5 | 0.000447 |
| 4 | Putamen_R | 48 | 32 | 5 | 4 | 1506 | 60.3 | 0.000051 |
| 4 | 48 | 35 | −2 | 7 | 1506 | 36.5 | 0.00066 | |
| 6 | Insula_L | 48 | −42 | −2 | 8 | 1233 | 58.2 | 0.000061 |
| 4 | Putamen_R | 48 | 32 | 2 | 8 | 1506 | 52.1 | 0.000111 |
| 4 | Putamen_R | 48 | 31 | 4 | 8 | 1506 | 50.3 | 0.000133 |
| 4 | Putamen_R | 48 | 32 | 0 | 9 | 1506 | 35.6 | 0.000739 |
| 4 | Putamen_R | 48 | 31 | 1 | 10 | 1506 | 33 | 0.001058 |
| 4 | Rolandic_Oper_R | 48 | 50 | −18 | 12 | 1506 | 37.1 | 0.00061 |
| 4 | Rolandic_Oper_R | 48 | 46 | −14 | 12 | 1506 | 36.2 | 0.000683 |
| 6 | Rolandic_Oper_L | 48 | −42 | 2 | 12 | 1233 | 46 | 0.000211 |
| 7 | Supp_Motor_Area_L | 0 | −6 | −14 | 48 | 638 | 50.8 | 0.000126 |
| Symptomatic carriers | ||||||||
| 1 | ParaHippocampal_L | 36 | −26 | −16 | −24 | 1712 | 98.6 | 0.00002 |
| 1 | Hippocampus_L | 36 | −25 | −14 | −24 | 1712 | 99 | 0.000019 |
| 1 | Hippocampus_L | 35 | −24 | −14 | −21 | 1712 | 99.9 | 0.000018 |
| 2 | ParaHippocampal_R | 20 | 34 | −22 | −20 | 1292 | 115.6 | 0.000009 |
| 1 | Hippocampus_L | 35 | −24 | −11 | −20 | 1712 | 100.7 | 0.000018 |
| 1 | Hippocampus_L | 20 | −26 | −10 | −18 | 1712 | 89.1 | 0.000032 |
| 3 | Temporal_Sup_R | 48 | 46 | −14 | 0 | 20 | 46.5 | 0.000593 |
| 4 | Putamen_R | 48 | 35 | −4 | 0 | 14 | 51.6 | 0.000379 |
| 4 | 48 | 36 | −2 | 0 | 14 | 49.2 | 0.000467 | |
| 4 | Putamen_R | 48 | 35 | −3 | 2 | 14 | 43.6 | 0.000772 |
AAL, anatomical label corresponding to probabilistic brain atlas [Collins et al., 1994; Mazziotta et al., 1995] of the maximum in each cluster. BA, Brodmann area; x, y, z, coordinates from the MNI atlas; Vol, cluster size (in voxels); T2, Hotteling T2 value of peak within significantly activated cluster of voxels; L, left; R, right; P, P value.
The SPMs showing between‐groups differences are shown in Figure 4. The Hotelling T2 values obtained from testing for statistical differences between two groups were multiplied by the difference between module means, to obtain the sign of the difference. Significant differences appeared between NonC and PresymC groups (Fig. 4A, Table IV) in temporal areas of both hemispheres. Some areas showed lower generator‐intensity in PresymC (positive values), all in the right hemisphere (fusiform, inferior temporal, and medial cingulated among other), whereas other areas showed negative values reflecting the recruitment of additional sources at left hippocampus and parahippocampal area (extending to fusiform gyrus), and also at right parahippocampal area and STG. Other clusters of significant voxels are described in Table IV. A very similar pattern appeared for the differences between NonC and SympC (Fig. 4B, Table V). Decreases in generator‐intensity were associated to SympC group in the right hemisphere including inferior temporal, fusiform, parahippocampal, STG, and medial cingulate areas. In the left hemisphere, increased generator‐intensity was observed in the hippocampus (extending to parahippocampal area) and in the inferior temporal gyrus. Other clusters of significant voxels were described in Table V. Despite the similarities in source distributions associated with PresymC and SympC groups, the SPM showed differences in mesial temporal areas bilaterally (Fig. 4C and Table VI). In this case a decrease of generator‐intensity was associated to SympC group in the hippocampus in both hemispheres, and in the right parahippocampus and right superior temporal gyrus.
Figure 4.
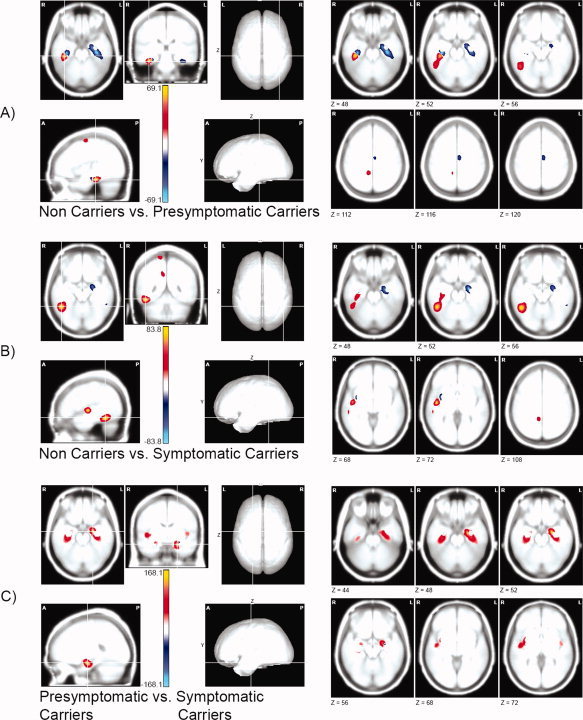
Statistical parametric maps of the difference in cortical current density distribution between groups. The scale represents the Hotelling T2 values (multiplied by the difference between mean modules) for the between groups comparison. Only those voxels where the T2 value reached significance (q = 0.05, FDR corrected) are displayed, with red for positive values and blue for negative values. In the left panel, orthogonal views of the maximum. In the right panel, selected slices displaying principal effects. Z values below each slice indicate the corresponding MNI coordinates. (A) Noncarriers group compared to the presymptomatic carriers group (threshold Hotelling T2 = 21.6, equivalent q = 0.0015). (B) Non‐carriers group compared to the symptomatic carriers group (threshold Hotelling T2 = 21.5, equivalent q = 0.0017). (C) Presymptomatic carriers group compared to the symptomatic carriers group (threshold Hotelling T2 = 20.1, equivalent q = 0.0023). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Table IV.
Brain regions showing significant differences in current source densities between noncarriers and presymptomatic carries groups (all q < 0.05 FDR corrected for whole brain volume, extent threshold of 10 voxels)
| Cluster | AAL | BA | x | y | z | Vol | T2 | P |
|---|---|---|---|---|---|---|---|---|
| Noncarriers > presymptomatic carriers | ||||||||
| 1 | Fusiform_R | 20 | 38 | −30 | −24 | 3618 | 69.1 | 0.000001 |
| 1 | Fusiform_R | 20 | 34 | −30 | −20 | 3618 | 60 | 0.000001 |
| 1 | Temporal_Inf_R | 37 | 46 | −54 | −16 | 3618 | 44.4 | 0.000009 |
| 4 | Cingulum_Mid_R | 23 | 6 | −46 | 36 | 733 | 38.1 | 0.00003 |
| 4 | Precuneus_R | 0 | 10 | −46 | 40 | 733 | 40.1 | 0.00002 |
| 5 | Precentral_R | 6 | 38 | −10 | 64 | 524 | 37.9 | 0.000031 |
| 5 | Frontal_Sup_R | 6 | 34 | −6 | 64 | 524 | 32.9 | 0.00009 |
| 6 | Postcentral_R | 1 | 18 | −46 | 76 | 270 | 42.1 | 0.000014 |
| Presymptomatic carriers > noncarriers | ||||||||
| 1 | ParaHippocampal_L | 20 | −30 | −22 | −24 | 3308 | 64.7 | 0.000001 |
| 1 | Hippocampus_L | 35 | −22 | −10 | −24 | 3308 | 56.6 | 0.000001 |
| 2 | ParaHippocampal_R | 20 | 34 | −26 | −20 | 854 | 69.3 | 0.000001 |
| 3 | Temporal_Sup_R | 48 | 46 | −14 | 0 | 373 | 34.4 | 0.000065 |
| 4 | Putamen_R | 48 | 35 | −4 | 0 | 43 | 31.3 | 0.000129 |
| 4 | 48 | 36 | −2 | 0 | 43 | 30.2 | 0.000165 | |
| 4 | 48 | 36 | 2 | 0 | 43 | 24.1 | 0.00072 | |
| 4 | Putamen_R | 48 | 35 | −3 | 2 | 43 | 27.3 | 0.000326 |
| 6 | Rolandic_Oper_L | 48 | −42 | −6 | 4 | 43 | 23.6 | 0.000817 |
| 4 | 48 | 35 | −4 | 4 | 43 | 26.8 | 0.000373 | |
| 4 | 48 | 36 | −2 | 4 | 43 | 22.3 | 0.001146 | |
| 5 | Putamen_R | 48 | 32 | 5 | 4 | 29 | 34.1 | 0.000069 |
| 6 | Insula_L | 48 | −42 | −2 | 8 | 43 | 26.3 | 0.000416 |
| 5 | Putamen_R | 48 | 32 | 2 | 8 | 29 | 27.2 | 0.000333 |
| 5 | Putamen_R | 48 | 31 | 4 | 8 | 29 | 26.4 | 0.000412 |
| 10 | Cingulum_Mid_L | 0 | −6 | −14 | 44 | 809 | 38.9 | 0.000026 |
AAL, anatomical label corresponding to probabilistic brain atlas [Collins et al., 1994; Mazziotta et al., 1995] of the maximum in each cluster. BA, Brodmann area; x, y, z, coordinates from the MNI atlas; Vol, cluster size (in voxels); T2, Hotteling T2 value of peak within significantly activated cluster of voxels; L, left; R, right; P, P value.
Table V.
Brain regions showing significant differences in current source densities between noncarriers and symptomatic carries groups (all q < 0.05 FDR corrected for whole brain volume, extent threshold of 10 voxels)
| Cluster | AAL | BA | x | y | z | Vol | T2 | P |
|---|---|---|---|---|---|---|---|---|
| Noncarriers > symptomatic carriers | ||||||||
| 1 | Fusiform_R | 20 | 38 | −26 | −24 | 5239 | 34.4 | 0.000095 |
| 1 | ParaHippocampal_R | 20 | 34 | −22 | −20 | 5239 | 47.8 | 0.000008 |
| 1 | Temporal_Inf_R | 20 | 46 | −50 | −16 | 5239 | 83.8 | 0.000001 |
| 3 | Temporal_Mid_R | 21 | 54 | −34 | −4 | 121 | 27.2 | 0.000447 |
| 2 | Temporal_Sup_R | 48 | 46 | −14 | 0 | 1448 | 81.5 | 0.000001 |
| 6 | Cingulum_Mid_R | 23 | 6 | −46 | 36 | 642 | 35.4 | 0.000079 |
| 6 | Precuneus_R | 0 | 10 | −46 | 40 | 642 | 38.4 | 0.000043 |
| 7 | Precentral_R | 6 | 42 | −10 | 60 | 76 | 23.4 | 0.001083 |
| 7 | Precentral_R | 6 | 38 | −10 | 64 | 76 | 25.4 | 0.000671 |
| 8 | Supp_Motor_Area_R | 6 | 10 | −2 | 72 | 77 | 25.9 | 0.000605 |
| 9 | Postcentral_R | 1 | 14 | −46 | 76 | 620 | 63.4 | 0.000001 |
| Symptomatic carriers > noncarriers | ||||||||
| 1 | Hippocampus_L | 35 | −22 | −10 | −20 | 1934 | 78.6 | 0.000001 |
| 3 | Temporal_Inf_L | 20 | −54 | −46 | −16 | 27 | 24 | 0.000946 |
| 5 | Putamen_R | 48 | 36 | −2 | −4 | 363 | 49.2 | 0.000006 |
| 5 | 48 | 36 | 2 | −4 | 363 | 42 | 0.000022 | |
| 5 | 48 | 36 | −6 | −3 | 363 | 37.5 | 0.000052 | |
| 5 | Putamen_R | 48 | 35 | −4 | 0 | 363 | 61.5 | 0.000001 |
| 5 | 48 | 36 | −2 | 0 | 363 | 60.3 | 0.000001 | |
| 5 | 48 | 36 | 2 | 0 | 363 | 55.9 | 0.000002 | |
| 5 | Putamen_R | 48 | 35 | −3 | 2 | 363 | 56.6 | 0.000002 |
| 5 | 48 | 36 | −2 | 4 | 363 | 44.2 | 0.000015 | |
| 5 | Putamen_R | 48 | 34 | 2 | 4 | 363 | 49.8 | 0.000006 |
| 5 | 48 | 34 | −7 | 6 | 363 | 25.6 | 0.000649 | |
| 5 | Putamen_R | 48 | 32 | 2 | 8 | 363 | 51.7 | 0.000004 |
| 5 | Putamen_R | 48 | 31 | 4 | 8 | 363 | 51.8 | 0.000004 |
| 5 | Putamen_R | 48 | 32 | 0 | 9 | 363 | 32.8 | 0.000133 |
| 5 | Putamen_R | 48 | 31 | 1 | 10 | 363 | 31 | 0.000193 |
AAL, anatomical label corresponding to probabilistic brain atlas [Collins et al., 1994; Mazziotta et al., 1995] of the maximum in each cluster. BA, Brodmann area; x, y, z, coordinates from the MNI atlas; Vol, cluster size (in voxels); T2, Hotteling T2 value of peak within significantly activated cluster of voxels; L, left; R, right; P, P value.
Table VI.
Brain regions showing significant differences in current source densities between presymptomatic carries and symptomatic carries groups (all q < 0.05 FDR corrected for whole brain volume, extent threshold of 10 voxels)
| Cluster | AAL | BA | x | y | z | Vol | T2 | P |
|---|---|---|---|---|---|---|---|---|
| Presymptomatic carriers > symptomatic carriers | ||||||||
| 2 | ParaHippocampal_R | 36 | 25 | −14 | −24 | 3130 | 24.5 | 0.000771 |
| 2 | ParaHippocampal_R | 20 | 34 | −22 | −20 | 3130 | 123.2 | 0.000001 |
| 2 | ParaHippocampal_R | 20 | 26 | −20 | −20 | 3130 | 37.8 | 0.000042 |
| 2 | 20 | 25 | −18 | −20 | 3130 | 32.8 | 0.000117 | |
| 1 | Hippocampus_L | 35 | −22 | −10 | −20 | 5914 | 168.8 | 0.000001 |
| 2 | Hippocampus_R | 20 | 26 | −10 | −16 | 3130 | 48.5 | 0.000006 |
| 5 | 48 | 36 | −12 | −4 | 2574 | 24.1 | 0.000851 | |
| 5 | Insula_R | 48 | 37 | −10 | −4 | 2574 | 25.3 | 0.000639 |
| 5 | Putamen_R | 48 | 36 | −2 | −4 | 2574 | 53.1 | 0.000003 |
| 5 | 48 | 36 | 2 | −4 | 2574 | 40.2 | 0.000027 | |
| 5 | 48 | 36 | −6 | −3 | 2574 | 40.3 | 0.000026 | |
| 5 | Temporal_Sup_R | 48 | 46 | −14 | 0 | 2574 | 77.7 | 0.000001 |
| 5 | Putamen_R | 48 | 35 | −4 | 0 | 2574 | 76.4 | 0.000001 |
| 5 | 48 | 36 | −2 | 0 | 2574 | 74.2 | 0.000001 | |
| 5 | 48 | 36 | 2 | 0 | 2574 | 57.3 | 0.000001 | |
| 5 | Putamen_R | 48 | 35 | −3 | 2 | 2574 | 67 | 0.000001 |
| 6 | Rolandic_Oper_L | 48 | −42 | −6 | 4 | 1870 | 49 | 0.000005 |
| 5 | 48 | 35 | −4 | 4 | 2574 | 65.5 | 0.000001 | |
| 5 | 48 | 36 | −2 | 4 | 2574 | 55.8 | 0.000002 | |
| 5 | Putamen_R | 48 | 34 | 2 | 4 | 2574 | 55.4 | 0.000002 |
| 5 | Putamen_R | 48 | 32 | 5 | 4 | 2574 | 76.6 | 0.000001 |
| 5 | 48 | 34 | −7 | 6 | 2574 | 36.2 | 0.000059 | |
| 5 | 48 | 35 | −2 | 7 | 2574 | 49.2 | 0.000005 | |
| 6 | Insula_L | 48 | −42 | −2 | 8 | 1870 | 50.9 | 0.000004 |
| 5 | Putamen_R | 48 | 32 | 2 | 8 | 2574 | 64.8 | 0.000001 |
| 5 | Putamen_R | 48 | 31 | 4 | 8 | 2574 | 63 | 0.000001 |
| 5 | Putamen_R | 48 | 32 | 0 | 9 | 2574 | 45.1 | 0.000011 |
| 5 | Putamen_R | 48 | 31 | 1 | 10 | 2574 | 41.3 | 0.000022 |
| 6 | Rolandic_Oper_L | 48 | −42 | 2 | 12 | 1870 | 38.6 | 0.000036 |
| Symptomatic carriers > presymptomatic carriers | ||||||||
| 1 | Hippocampus_L | 20 | −26 | −10 | −16 | 34 | 94.5 | 0.000001 |
| 1 | Amygdala_L | 34 | −26 | −6 | −12 | 34 | 31.7 | 0.000149 |
AAL, anatomical label corresponding to probabilistic brain atlas [Collins et al., 1994; Mazziotta et al., 1995] of the maximum in each cluster. BA, Brodmann area; x, y, z, coordinates from the MNI atlas; Vol, cluster size (in voxels); T2, Hotteling T2 value of peak within significantly activated cluster of voxels; L, left; R, right; P, P value.
DISCUSSION
ERPs evoked during the semantic matching task elicited the P250, N400, and LPC components in the three groups of subjects. The P250 and LPC were not modulated by congruency, but showed differences in amplitude between groups. Presymptomatic carriers of the E280A PS‐1 mutation showed increased in P250 amplitude in respect to the non carriers but they showed normal amplitude of the LPC component. The symptomatic group showed amplitude values of the P250 similar to the non carriers group, while LPC component was reduced. In the three groups, incongruent pictures elicited the N400 component, which is the main focus of this work. Compared to normal controls, symptomatic (demented) carriers presented poorer performance in the semantic matching task, as well as a change in the anatomical distribution of the generators of the N400. In contrast, the presymptomatic carriers of the E280A PS‐1 mutation presented normal task performance and normal N400 amplitude and latency. However, they also reveal, in relation to noncarrier members of the same families, different N400 topography, and a significant change in the anatomical distribution of the estimated N400 generators, with a similar pattern to that of the demented subjects, but with larger current densities than the later. These shifts with respect to controls consisted in a decrease of generator strength in right inferior‐temporal and medial cingulate areas, and the recruitment of generators in the left mesial temporal region.
N400 in Alzheimer Disease
Some of our findings in mildly demented subjects are consistent with previous studies in patients with AD. Prior reports have found that in patients with AD the coexistence of normal amplitude of the earlier ERP components, like P250, with amplitude reduction of the LPC [Revonsuo et al., 1998]. These authors suggested that this probably reflect that degenerative process associate with AD affected differently the high‐level integration processes than the earlier sensory‐perceptual processes. Other late positive components like P600 in word repetition tasks or the P300 in oddball paradigms were consistenly reduced in patients with AD [Olichney et al., 2006; Olichney and Hillert, 2004; Polich and Corey‐Bloom, 2005]. The increase in P250 amplitude found in PresymC in respect to controls was unexpected; it has not been reported for AD or MCI cases and needs to be replicated.
Our most interesting findings related to semantic processing and the N400 are consistent with previous studies using similar paradigms in patients with AD. Here, the symptomatic group exhibited striking deterioration of their semantic matching performance, as well as lower N400 amplitudes, suggesting deficits in semantic processing. These behavioral and ERP deficits have also been found in other studies of patients with AD, using both verbal material [Ford et al., 1996; Iragui et al., 1996; Revonsuo et al., 1998] and pictures [Auchterlonie et al., 2002; Castaneda et al., 1997; Ford et al., 2001; Ostrosky‐Solis et al., 1998]. Changes in N400 topographic distribution have also been reported in patients with mild AD [Olichney et al., 2006]. These impairments may be related to deficits in other semantic memory tasks in patients with probable AD [Chertkow and Bub, 1990; Grossman et al., 1996], which can be found relatively early in the disease course [Chertkow and Bub, 1990; Hodges and Patterson, 1995]. However, in our task, participants had to discriminate between congruent and incongruent picture pairs, but delayed their responses until after the second stimulus was removed from the screen. Therefore, contributions from short‐term memory problems must also be considered as an explanation of the inaccurate of the symptomatic carriers' performance. On the other hand, presymptomatic mutation carriers performed the semantic matching task as accurately as non‐carriers (NonC) (which also concurs with previous reports). Note however that reaction‐time was not measured, so we could not ascertain if the accurate performance was associated with a time cost. In neuropsychological studies of the Antioquia families, presymptomatic mutation carriers of the E280A PS‐1 mutation were also found to have intact processing of semantic associations, although they performed worse than noncarriers on linguistic tasks that required processing of the meaning of words [Arango‐Lasprilla et al., 2003, 2007]. Importantly, here the presymptomatic carriers did not differ in N400 amplitude or latency from controls, which at first glance suggests an intact neural basis for the processing of semantic associations.
N400 Neural Sources
The most original aspect of this study was the examination of deficits in neural activity as indexed by source modeling of the N400. We expected this approach uncovered more subtle alterations of this component which were suggested by the changes in scalp topography that was found between groups (indicative of variations in N400 neural sources). To our knowledge, no previous study has explored the N400 or its intracranial sources in presymptomatic carriers of FAD genes. Several studies have examined the sources of N400 in normal controls, which we briefly review now. As mentioned in the introduction, the EEG inverse problem is ill posed and therefore the sources estimated depend critically on the prior assumptions adopted to formulate an inversion scheme. This probably contributed to variability of previous reports on the sources of the N400 in normal subjects, which in each case have adopted different models. One study, using a small number of current dipoles, located the N400 sources bilaterally in the temporal lobes, though predominantly in the left superior temporal sulcus [Simos et al., 1997]. Another study using LORETA (a distributed source solution, with current source strength constrained to change smoothly over space) reported generators in bilateral frontal and temporal areas [Hamm et al., 2002]. Finally, an additional study using VARETA (which allows the degree of smoothness of the solution to vary across space) yields more extended distribution of N400 generators including bilateral parahippocampal, occipitotemporal, anterior and posterior temporal lobes, and frontal regions [Silva‐Pereyra et al., 2003]. However, despite their discrepancies, all these studies found N400 generators in the temporal lobes, which is consistent with local field potential recordings from intracranial electrodes in MTL of epileptic patients [Allison et al., 1994; Elger et al., 1997; Fernandez et al., 2001; McCarthy et al., 1995; Nobre and McCarthy, 1995; Smith et al., 1986].
Here we attempted to circumvent the problem of model selection using the BMA approach mentioned in the introduction [Trujillo‐Barreto et al., 2004]. With BMA all possible anatomical configurations of sources were averaged each weighted by the evidence that supports them (i.e. the probability of the anatomical configuration given the data). Furthermore, to improve the reliability of the results, instead of reporting mean current density at each voxel (as in many previous reports) we used a SPM approach [Henson et al., 2007] and only considered results where the mean current density for the subject groups (or the density differences between groups) was significantly deviant from zero. Using these methods we found that the intracranial sources for the N400 in our control group (NonC) were located principally within the temporal lobe (inferior temporal gyrus, anterior fusiform gyrus, superior temporal gyrus), in concordance with the previously reviewed studies carried out in normal subjects, although other locations were also involved. However, in contrast to previous researches, here larger sources were found in the right hemisphere than in the left one. Note that the earlier studies employed linguistic materials. Our distinct hemispheric lateralization could be due to the use of pictures instead of written words, which is congruent with the differential lateralization of scalp topography when the N400 elicited by pictures and words are compared [Ganis et al., 1996; West and Holcomb, 2002; Willems et al., 2008].
Neural Reorganization in Presymptomatic Carriers
We first considered N400 generators in the presymptomatic carriers as compared to noncarriers. The anatomical distribution of N400 sources was significantly different between the two groups. Some generators were diminished in intensity, in particular, those located at anterior‐fusiform, inferior‐temporal, and medial‐cingulate areas of the right hemisphere. These sites overlapped in part with the regions initially affected by neuropathological changes in AD [Arnold et al., 1991; Braak and Braak, 1991]. However, the reduction in generator intensity at these sites was accompanied by significant increases of generator intensity in the left temporal lobe (hippocampal, parahippocampal and fusiform areas). Note that current density was not significant in these areas in the noncarriers subjects. The coexistence of brain areas which respectively presented decreased and increased activation has been described for the blood oxygenation level dependent (BOLD) responses during episodic memory processing in a young asymptomatic FAD mutation carrier [Mondadori et al., 2006], as well as in members of families at risk for late‐onset AD [Bassett et al., 2006], in carriers of the APOE4 allele [Bondi et al., 2005; Bookheimer et al., 2000; Fleisher et al., 2005] and in nonselected samples of individual from FAD pedigrees [Johnson et al., 2006; Mosconi et al., 2006].
The enhanced activity in some brain areas in asymptomatic subjects at risk for AD have been explained as a compensatory recruitment of well functioning neural populations to balance dysfunction in other populations already affected by the neuropathological changes [Mondadori et al., 2006]. We did not expect the precise anatomical location of decreased/increased activations in these previous studies to completely coincide with our results due to important differences in the tasks and memory domains involved. Nevertheless, an interesting coincidence was found between the increased BOLD response found at left temporo‐mesial areas in previous studies [Bondi et al., 2005; Bookheimer et al., 2000; Fleisher et al., 2005; Mondadori et al., 2006], and the increased PCDs in this study. Therefore, functional reorganization (analogous to that suggested by previous studies) could be operating in our sample of PresymC, who presented normal N400 latency and amplitude, as well as normal accuracy in the matching task, despite the changes in N400 generators.
Some neuroimaging studies of normal cognitive aging should be mentioned here. These studies have shown greater activation in memory‐related brain areas (among other sites) in older adults than in the younger adults, even when performance is age‐equivalent. This age‐related over activation has also been interpreted as a sign of an enduring neural capacity for functional reorganization or redistribution of resources in response to metabolic and neurobiological declines related to aging [see Reuter‐ Lorenz and Cappell, 2008; for a review about the compensation‐related utilization of neural circuits hypothesis, CRUNCH].
Finally, it is important to emphasize that despite a similar shift in anatomical distribution of N400 generators for both asymptomatic carriers and demented cases, significant differences appeared between the two groups respect to current source intensity. The decrease of generator intensity in the right infero‐temporal area, relative to controls, was larger for symptomatic carriers. Furthermore, the increase of generator strength in the left temporal mesial areas was smaller than in asymptomatic carriers. This result is congruent with reports of decreased task‐ related BOLD activity in patients in the early stages of AD [Small et al., 1999] and MCI [Dickerson et al., 2005], as compared to age matched controls, and with the finding of decreased of BOLD responses in middle age mutation carriers who fulfilled criteria for amnesic MCI [Mondadori et al., 2006]. The reduced intensity of N400 generators suggested that in the course of the degenerative process this reorganized network was less functional, and semantic deficits (as well as generator intensity decreases) appeared in the symptomatic group.
In conclusion, functional alterations in neural circuitry related to semantic processing (as indexed by the sources of the N400) accompany the neuropathologic changes in AD and are detectable before onset of dementia symptoms. These electrophysiological indicators apparently monitor the functional reorganization induced by early brain damage in AD. Therefore, ERP source localization in conjunction with other neuroimaging modalities may contribuited to the understanding of the earliest stages of this disease.
Acknowledgements
We thank Dr. Eduardo Martinez for providing statistical advice and all the family members of the Colombian kindred for their support to this project.
REFERENCES
- Aguirre‐Acevedo DC, Gomez RD, Moreno S, Henao‐Arboleda E, Motta M, Muñoz C, Arana A, Pineda DA, Lopera F ( 2007): Validity and reliability of the CERAD‐Col neuropsychological battery. Rev Neurol 45: 655–660. [PubMed] [Google Scholar]
- Allison T, Ginter H, McCarthy G, Nobre AC, Puce A, Luby M, Spencer DD ( 1994): Face recognition in human extrastriate cortex. J Neurophysiol 71: 821–825. [DOI] [PubMed] [Google Scholar]
- Almkvist O, Axelman K, Basun H, Jensen M, Viitanen M, Wahlund LO, Lannfelt L ( 2003): Clinical findings in nondemented mutation carriers predisposed to Alzheimer's disease: A model of mild cognitive impairment. Acta Neurol Scand Suppl 179: 77–82. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association ( 1994): Diagnostic and Statistical Manual of Mental Disorders, 4th ed. Washington D.C: American Psychiatric Association. [Google Scholar]
- Arango‐Lasprilla JC, Iglesias J, Lopera F ( 2003): Neuropsychological study of familial Alzheimer's disease caused by mutation E280A in the presenilin 1 gene. Am J Alzheimers Dis Other Demen 18: 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arango‐Lasprilla JC, Cuetos F, Valencia C, Uribe C, Lopera F ( 2007): Cognitive changes in the preclinical phase of familial Alzheimer's disease. J Clin Exp Neuropsychol 29: 892–900. [DOI] [PubMed] [Google Scholar]
- Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW ( 1991): The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer's disease. Cereb Cortex 1: 103–116. [DOI] [PubMed] [Google Scholar]
- Auchterlonie S, Phillips NA, Chertkow H ( 2002): Behavioral and electrical brain measures of semantic priming in patients with Alzheimer's disease: Implications for access failure versus deterioration hypotheses. Brain Cogn 48: 264–267. [PubMed] [Google Scholar]
- Auer S, Reisberg B ( 1997): The GDS/FAST staging system. Int Psychogeriatr 9 ( Suppl 1): 167–171. [DOI] [PubMed] [Google Scholar]
- Aveleyra E, Gómez C, Ostrosky‐Solis F, Rigalt C, Cruz F ( 1996): Adaptación de los estímulos no verbales de Snodgrass y Vanderwart en población hispanohablante: Criterios para la denominación, concordancia de la imagen, familiaridad y complejidad visual. Rev Mex Psicol 13: 5–19. [Google Scholar]
- Barrett SE, Rugg MD ( 1989): Event‐related potentials and the semantic matching of faces. Neuropsychologia 27: 913–922. [DOI] [PubMed] [Google Scholar]
- Barrett SE, Rugg MD ( 1990): Event‐related potentials and the semantic matching of pictures. Brain Cogn 14: 201–212. [DOI] [PubMed] [Google Scholar]
- Barrett SE, Rugg MD, Perrett DI ( 1988): Event‐related potentials and the matching of familiar and unfamiliar faces. Neuropsychologia 26: 105–117. [DOI] [PubMed] [Google Scholar]
- Bassett SS, Yousem DM, Cristinzio C, Kusevic I, Yassa MA, Caffo BS, Zeger SL ( 2006): Familial risk for Alzheimer's disease alters fMRI activation patterns. Brain 129: 1229–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair R, Karniski W ( 1994): Distribution‐free statistical analyses of surface and volumetric maps In: Thatcher RW, Hallett M, Zeffiro T, John ER, Huerta M, editors. Functional Neuroimaging. New York: Academic Press; pp 19–28. [Google Scholar]
- Bobes MA, Lei ZX, Ibanez S, Yi H, Valdes‐Sosa M ( 1996): Semantic matching of pictures in schizophrenia: A cross‐cultural ERP study. Biol Psychiatry 40: 189–202. [DOI] [PubMed] [Google Scholar]
- Bondi MW, Houston WS, Eyler LT, Brown GG ( 2005): FMRI evidence of compensatory mechanisms in older adults at genetic risk for Alzheimer disease. Neurology 64: 501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak‐Vance MA, Mazziotta JC, Small GW ( 2000): Patterns of brain activation in people at risk for Alzheimer's disease. N Engl J Med 343: 450–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E ( 1991): Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol (Berl) 82: 239–259. [DOI] [PubMed] [Google Scholar]
- Caldara R, Jermann F, Arango GL, Van der Linden M ( 2004): Is the N400 category‐specific? A face and language processing study. Neuroreport 15: 2589–2593. [DOI] [PubMed] [Google Scholar]
- Castaneda M, Ostrosky‐Solis F, Perez M, Bobes MA, Rangel LE ( 1997): ERP assessment of semantic memory in Alzheimer's disease. Int J Psychophysiol 27: 201–214. [DOI] [PubMed] [Google Scholar]
- Chan AS, Butters N, Salmon DP ( 1997): The deterioration of semantic networks in patients with Alzheimer's disease: A cross‐sectional study. Neuropsychologia 35: 241–248. [DOI] [PubMed] [Google Scholar]
- Chertkow H, Bub D ( 1990): Semantic memory loss in dementia of Alzheimer's type. What do various measures measure? Brain 113: 397–417. [DOI] [PubMed] [Google Scholar]
- Chertkow H, Whatmough C, Saumier D, Duong A ( 2008): Cognitive neuroscience studies of semantic memory in Alzheimer's disease. Prog Brain Res 169: 393–407. [DOI] [PubMed] [Google Scholar]
- Chetelat G, Baron JC ( 2003): Early diagnosis of Alzheimer's disease: Contribution of structural neuroimaging. Neuroimage 18: 525–541. [DOI] [PubMed] [Google Scholar]
- Collins DL, Neelin P, Peters TM, Evans AC ( 1994): Automatic 3D intersubject registration of MR volumetric data in standardized Talairach space. J Comput Assist Tomogr 18: 192–205. [PubMed] [Google Scholar]
- Dickerson BC, Goncharova I, Sullivan MP, Forchetti C, Wilson RS, Bennett DA, Beckett LA, Toledo‐Morrell L ( 2001): MRI‐derived entorhinal and hippocampal atrophy in incipient and very mild Alzheimer's disease. Neurobiol Aging 22: 747–754. [DOI] [PubMed] [Google Scholar]
- Dickerson BC, Salat DH, Greve DN, Chua EF, Rand‐Giovannetti E, Rentz DM, Bertram L, Mullin K, Tanzi RE, Blacker D, Albert MS, Sperling RA ( 2005): Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology 65: 404–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas RB, Clague F, Thompson SA, Graham KS, Hodges JR ( 2005): Episodic and semantic memory in mild cognitive impairment. Neuropsychologia 43: 1266–1276. [DOI] [PubMed] [Google Scholar]
- Elger CE, Grunwald T, Lehnertz K, Kutas M, Helmstaedter C, Brockhaus A, Van RD, Heinze HJ ( 1997): Human temporal lobe potentials in verbal learning and memory processes. Neuropsychologia 35: 657–667. [DOI] [PubMed] [Google Scholar]
- Fernandez G, Heitkemper P, Grunwald T, Van RD, Urbach H, Pezer N, Lehnertz K, Elger CE ( 2001): Inferior temporal stream for word processing with integrated mnemonic function. Hum Brain Mapp 14: 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher AS, Houston WS, Eyler LT, Frye S, Jenkins C, Thal LJ, Bondi MW ( 2005): Identification of Alzheimer disease risk by functional magnetic resonance imaging. Arch Neurol 62: 1881–1888. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR ( 1975): Mini‐mental state. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12: 189–198. [DOI] [PubMed] [Google Scholar]
- Ford JM, Woodward SH, Sullivan EV, Isaacks BG, Tinklenberg JR, Yesavage JA, Roth WT ( 1996): N400 evidence of abnormal responses to speech in Alzheimer's disease. Electroencephalogr Clin Neurophysiol 99: 235–246. [DOI] [PubMed] [Google Scholar]
- Ford JM, Askari N, Mathalon DH, Menon V, Gabrieli JD, Tinklenberg JR, Yesavage J ( 2001): Event‐related brain potential evidence of spared knowledge in Alzheimer's disease. Psychol Aging 16: 161–176. [DOI] [PubMed] [Google Scholar]
- Fox NC, Warrington EK, Freeborough PA, Hartikainen P, Kennedy AM, Stevens JM, Rossor MN ( 1996): Presymptomatic hippocampal atrophy in Alzheimer's disease. A longitudinal MRI study. Brain 119: 2001–2007. [DOI] [PubMed] [Google Scholar]
- Fox NC, Crum WR, Scahill RI, Stevens JM, Janssen JC, Rossor MN ( 2001): Imaging of onset and progression of Alzheimer's disease with voxel‐compression mapping of serial magnetic resonance images. Lancet 358: 201–205. [DOI] [PubMed] [Google Scholar]
- Friederici AD, von Cramon DY, Kotz SA ( 1999): Language related brain potentials in patients with cortical and subcortical left hemisphere lesions. Brain 122: 1033–1047. [DOI] [PubMed] [Google Scholar]
- Galan L, Biscay R, Rodriguez JL, Perez‐Abalo MC, Rodriguez R ( 1997): Testing topographic differences between event related brain potentials by using non‐parametric combinations of permutation tests. Electroencephalogr. Clin Neurophysiol 102: 240–247. [DOI] [PubMed] [Google Scholar]
- Ganis G, Kutas M, Sereno MI ( 1996): The search for “common sense”: An electrophysiological study of the comprehension of words and pictures in reading. J Cogn Neurosci 8: 89–106. [DOI] [PubMed] [Google Scholar]
- Genovese CR, Lazar NA, Nichols T ( 2002): Thresholding of statistical maps in functional neuroimaging using the false discovery rate. NeuroImage 15: 870–878. [DOI] [PubMed] [Google Scholar]
- Giffard B, Desgranges B, Eustache F ( 2005): Semantic memory disorders in Alzheimer's disease: Clues from semantic priming effects. Curr Alzheimer Res 2: 425–434. [DOI] [PubMed] [Google Scholar]
- Goodglass H, Kaplan E ( 1972): Assessment of Aphasia and Related Disorders. Philadelphia: Lea & Febiger. [Google Scholar]
- Grossman M, D'Esposito M, Hughes E, Onishi K, Biassou N, White‐Devine T, Robinson KM ( 1996): Language comprehension profiles in Alzheimer's disease, multi‐infarct dementia, and frontotemporal degeneration. Neurology 47: 183–189. [DOI] [PubMed] [Google Scholar]
- Haan H, Streb J, Bien S, Rösler F ( 2000): Individual cortical current density reconstructions of the semantic N400 effct: Using a generalized minimum norm model with diferent constraints (L1 and L2 norm). Hum Brain Mapp 11: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagoort P, Brown CM, Swaab TY ( 1996): Lexical‐semantic event‐related potential effects in patients with left hemisphere lesions and aphasia, and patients with right hemisphere lesions without aphasia. Brain 119: 627–649. [DOI] [PubMed] [Google Scholar]
- Halgren E, Dhond RP, Christensen N, Van PC, Marinkovic K, Lewine JD, Dale AM ( 2002): N400‐like magnetoencephalography responses modulated by semantic context, word frequency, and lexical class in sentences. NeuroImage 17: 1101–1116. [DOI] [PubMed] [Google Scholar]
- Hamberger MJ, Friedman D, Ritter W, Rosen J ( 1995): Event‐related potential and behavioral correlates of semantic processing in Alzheimer's patients and normal controls. Brain Lang 48: 33–68. [DOI] [PubMed] [Google Scholar]
- Hamm JP, Johnson BW, Kirk IJ ( 2002): Comparison of the N300 and N400 ERPs to picture stimuli in congruent and incongruent contexts. Clin Neurophysiol 113: 1339–1350. [DOI] [PubMed] [Google Scholar]
- Hardy J ( 1997): Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci 20: 154–159. [DOI] [PubMed] [Google Scholar]
- Helenius P, Salmelin R, Service E, Connolly JF ( 1998): Distinct time courses of word and context comprehension in the left temporal cortex. Brain 121: 1133–1142. [DOI] [PubMed] [Google Scholar]
- Helenius P, Salmelin R, Service E, Connolly JF, Leinonen S, Lyytinen H ( 2002): Cortical activation during spoken‐word segmentation in nonreading‐impaired and dyslexic adults. J Neurosci 22: 2936–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson RN, Mattout J, Singh KD, Barnes GR, Hillebrand A, Friston K ( 2007): Population‐level inferences for distributed MEG source localization under multiple constraints: Application to face‐evoked fields. NeuroImage 38: 422–438. [DOI] [PubMed] [Google Scholar]
- Hodges JR, Patterson K ( 1995): Is semantic memory consistently impaired early in the course of Alzheimer's disease? Neuroanatomical and diagnostic implications. Neuropsychologia 33: 441–459. [DOI] [PubMed] [Google Scholar]
- Hoeting JA, Madigan D, Raftery AE, Volinsky CT ( 1999): Bayesian model averaging: A tutorial. Statistical Science 14: 382–417. [Google Scholar]
- Holcomb PJ, McPherson WB ( 1994): Event‐related brain potentials reflect semantic priming in an object decision task. Brain Cogn 24: 259–276. [DOI] [PubMed] [Google Scholar]
- Iragui V, Kutas M, Salmon DP ( 1996): Event‐related brain potentials during semantic categorization in normal aging and senile dementia of the Alzheimer's type. Electroencephalogr Clin Neurophysiol 100: 392–406. [PubMed] [Google Scholar]
- Johnson KA, Lopera F, Jones K, Becker A, Sperling R, Hilson J, Londono J, Siegert I, Arcos M, Moreno S, Madrigal L, Ossa J, Pineda N, Ardila A, Roselli M, Albert MS, Kosik KS, Rios A ( 2001): Presenilin‐1‐associated abnormalities in regional cerebral perfusion. Neurology 56: 1545–1551. [DOI] [PubMed] [Google Scholar]
- Johnson SC, Schmitz TW, Trivedi MA, Ries ML, Torgerson BM, Carlsson CM, Asthana S, Hermann BP, Sager MA ( 2006): The influence of Alzheimer disease family history and apolipoprotein E epsilon4 on mesial temporal lobe activation. J Neurosci 26: 6069–6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan E, Goodglass H, Weintraub S ( 1978): Boston Naming Test. Philadelphia: Lea & Febiger. [Google Scholar]
- Karrasch M, Sinerva E, Gronholm P, Rinne J, Laine M ( 2005): CERAD test performances in amnestic mild cognitive impairment and Alzheimer's disease. Acta Neurol Scand 111: 172–179. [DOI] [PubMed] [Google Scholar]
- Kutas M, Hillyard SA ( 1980): Reading senseless sentences: Brain potentials reflect semantic incongruity. Science 207: 203–205. [DOI] [PubMed] [Google Scholar]
- Kwon H, Kuriki S, Kim JM, Lee YH, Kim K, Nam K ( 2005): MEG study on neural activities associated with syntactic and semantic violations in spoken Korean sentences. Neurosci Res 51: 349–357. [DOI] [PubMed] [Google Scholar]
- Lopera F, Ardilla A, Martinez A, Madrigal L, Arango‐Viana JC, Lemere CA, Arango‐Lasprilla JC, Hincapié L, Arcos‐Burgos M, Ossa JE, Behrens IM, Norton J, Lendon C, Goate AM, Ruiz‐Linares A, Rosselli M, Kosik KS ( 1997): Clinical features of early‐onset Alzheimer disease in a large kindred with an E280A presenilin‐1 mutation. JAMA 277: 793–799. [PubMed] [Google Scholar]
- MacKay DJC ( 1992): Bayesian interpolation. Neural Comput 4: 415–447. [Google Scholar]
- Mazziotta JC, Toga AW, Evans A, Fox P, Lancaster J ( 1995): A probabilistic atlas of the human brain: Theory and rationale for its development. The International Consortium for Brain Mapping (ICBM). NeuroImage 2: 89–101. [DOI] [PubMed] [Google Scholar]
- McCarthy G, Wood CC ( 1985): Scalp distributions of event‐related potentials: An ambiguity associated with analysis of variance models. Electroencephalogr Clin Neurophysiol 62: 203–208. [DOI] [PubMed] [Google Scholar]
- McCarthy G, Nobre AC, Bentin S, Spencer DD ( 1995): Language‐related field potentials in the anterior‐medial temporal lobe. I. Intracranial distribution and neural generators. J Neurosci 15: 1080–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM ( 1984): Clinical diagnosis of Alzheimer's disease: Report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 34: 939–944. [DOI] [PubMed] [Google Scholar]
- Medina D, Toledo‐Morrell L, Urresta F, Gabrieli JD, Moseley M, Fleischman D, Bennett DA, Leurgans S, Turner DA, Stebbins GT ( 2006): White matter changes in mild cognitive impairment and AD: A diffusion tensor imaging study. Neurobiol Aging 27: 663–672. [DOI] [PubMed] [Google Scholar]
- Mondadori CR, Buchmann A, Mustovic H, Schmidt CF, Boesiger P, Nitsch RM, Hock C, Streffer J, Henke K ( 2006): Enhanced brain activity may precede the diagnosis of Alzheimer's disease by 30 years. Brain 129: 2908–2922. [DOI] [PubMed] [Google Scholar]
- Morris JC, Heyman A, Mohs RC, Hughes JP, van BG, Fillenbaum G, Mellits ED, Clark C ( 1989): The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). I. Clinical and neuropsychological assessment of Alzheimer's disease. Neurology 39: 1159–1165. [DOI] [PubMed] [Google Scholar]
- Mosconi L, Sorbi S, de Leon MJ, Li Y, Nacmias B, Myoung PS, Tsui W, Ginestroni A, Bessi V, Fayyazz M, Caffarra P, Pupi A ( 2006): Hypometabolism exceeds atrophy in presymptomatic early‐onset familial Alzheimer's disease. J Nucl Med 47: 1778–1786. [PubMed] [Google Scholar]
- Nobre AC, McCarthy G ( 1995): Language‐related field potentials in the anterior‐medial temporal lobe. II. Effects of word type and semantic priming. J Neurosci 15: 1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olichney JM, Hillert DG ( 2004): Clinical applications of cognitive event‐related potentials in Alzheimer's disease. Phys Med Rehabil Clin N Am 15: 205–233. [DOI] [PubMed] [Google Scholar]
- Olichney JM, Morris SK, Ochoa C, Salmon DP, Thal LJ, Kutas M, Iragui VJ ( 2002): Abnormal verbal event related potentials in mild cognitive impairment and incipient Alzheimer's disease. J Neurol Neurosurg Psychiatry 73: 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olichney JM, Iragui VJ, Salmon DP, Riggins BR, Morris SK, Kutas M ( 2006): Absent event‐related potential (ERP) word repetition effects in mild Alzheimer's disease. Clin Neurophysiol 117: 1319–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostendorp TF, Delbeke J, Stegeman DF ( 2000): The conductivity of the human skull: Results of in vivo and in vitro measurements. IEEE Trans Biomed Eng 47: 1487–1492. [DOI] [PubMed] [Google Scholar]
- Ostrosky‐Solis F, Castaneda M, Perez M, Castillo G, Bobes MA ( 1998): Cognitive brain activity in Alzheimer's disease: Electrophysiological response during picture semantic categorization. J Int Neuropsychol Soc 4: 415–425. [DOI] [PubMed] [Google Scholar]
- Pascual‐Marqui RD, Michel CM, Lehmann D ( 1994): Low resolution electromagnetic tomography: A new method for localizing electrical activity in the brain. Int J Psychophysiol 18: 49–65. [DOI] [PubMed] [Google Scholar]
- Penny WD, Mattout J, Trujillo‐Barreto NJ ( 2006): Bayesian model selection and averaging In: Friston KJ, et al., editors. Statistical Parametric Mapping: The Analysis of Functional Brain Images. Oxford: Academic Press; pp 454–467. [Google Scholar]
- Petrides M, Alivisatos B, Evans AC, Meyer E ( 1993): Dissociation of human mid‐dorsolateral from posterior dorsolateral frontal cortex in memory processing. Proc Natl Acad Sci USA 90: 873–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polich J, Corey‐Bloom J ( 2005): Alzheimer's disease and P300: Review and evaluation of task and modality. Curr Alzheimer Res 2: 515–525. [DOI] [PubMed] [Google Scholar]
- Reuter‐Lorenz P ( 2002): New visions of the aging mind and brain. Trends Cogn Sci 6: 394. [DOI] [PubMed] [Google Scholar]
- Reuter‐Lorenz PA, Cappell KA ( 2008): Neurocognitive aging and the compensation hypothesis. Curr Directions Psychol Sci 17: 177–182. [Google Scholar]
- Revonsuo A, Portin R, Juottonen K, Rinne JO ( 1998): Semantic processing of spoken words in Alzheimer's disease: An electrophysiological study. J Cogn Neurosci 10: 408–420. [DOI] [PubMed] [Google Scholar]
- Rey A ( 1941): L'examen psychologique dans les cas d'encephalopathie traumatique. Arch Psychol 28: 286–340. [Google Scholar]
- Ridha BH, Barnes J, Bartlett JW, Godbolt A, Pepple T, Rossor MN, Fox NC ( 2006): Tracking atrophy progression in familial Alzheimer's disease: A serial MRI study. Lancet Neurol 5: 828–834. [DOI] [PubMed] [Google Scholar]
- Ringman JM, O'Neill J, Geschwind D, Medina L, Apostolova LG, Rodriguez Y, Schaffer B, Varpetian A, Tseng B, Ortiz F, Fitten J, Cummings JL, Bartzokis G ( 2007): Diffusion tensor imaging in preclinical and presymptomatic carriers of familial Alzheimer's disease mutations. Brain 130: 1767–1776. [DOI] [PubMed] [Google Scholar]
- Scahill RI, Schott JM, Stevens JM, Rossor MN, Fox NC ( 2002): Mapping the evolution of regional atrophy in Alzheimer's disease: Unbiased analysis of fluid‐registered serial MRI. Proc Natl Acad Sci USA 99: 4703–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott JM, Fox NC, Frost C, Scahill RI, Janssen JC, Chan D, Jenkins R, Rossor MN ( 2003): Assessing the onset of structural change in familial Alzheimer's disease. Ann Neurol 53: 181–188. [DOI] [PubMed] [Google Scholar]
- Schwartz TJ, Kutas M, Butters N, Paulsen JS, Salmon DP ( 1996): Electrophysiological insights into the nature of the semantic deficit in Alzheimer's disease. Neuropsychologia 34: 827–841. [DOI] [PubMed] [Google Scholar]
- Silva‐Pereyra J, Rivera‐Gaxiola M, Aubert E, Bosch J, Galan L, Salazar A ( 2003): N400 during lexical decision tasks: A current source localization study. Clin Neurophysiol 114: 2469–2486. [DOI] [PubMed] [Google Scholar]
- Simos PG, Basile LF, Papanicolaou AC ( 1997): Source localization of the N400 response in a sentence‐reading paradigm using evoked magnetic fields and magnetic resonance imaging. Brain Res 762: 29–39. [DOI] [PubMed] [Google Scholar]
- Small SA, Perera GM, DeLaPaz R, Mayeux R, Stern Y ( 1999): Differential regional dysfunction of the hippocampal formation among elderly with memory decline and Alzheimer's disease. Ann Neurol 45: 466–472. [DOI] [PubMed] [Google Scholar]
- Smith ME, Stapleton JM, Halgren E ( 1986): Human medial temporal lobe potentials evoked in memory and language tasks. Electroencephalogr Clin Neurophysiol 63: 145–159. [DOI] [PubMed] [Google Scholar]
- Snodgrass JG, Vanderwart M ( 1980): A standardized set of 260 pictures: Norms for name agreement, image agreement, familiarity, and visual complexity. J Exp Psychol [Hum Learn] 6: 174–215. [DOI] [PubMed] [Google Scholar]
- Storandt M ( 2008): Cognitive deficits in the early stages of Alzheimer's disease. Curr Directions Psychol Sci 17: 198–202. [Google Scholar]
- Stuss DT, Picton TW, Cerri AM, Leech EE, Stethem LL ( 1992): Perceptual closure and object identification: Electrophysiological responses to incomplete pictures. Brain Cogn 19: 253–266. [DOI] [PubMed] [Google Scholar]
- Swaab T, Brown C, Hagoort P ( 1997): Spoken sentence comprehension in aphasia: Event‐related potential evidence for a lexical integration deficit. J Cogn Neurosci 9: 39–66. [DOI] [PubMed] [Google Scholar]
- Trujillo‐Barreto NJ, ubert‐Vazquez E, Valdes‐Sosa PA ( 2004): Bayesian model averaging in EEG/MEG imaging. NeuroImage 21: 1300–1319. [DOI] [PubMed] [Google Scholar]
- VanPetten C, Luka BJ ( 2006): Neural localization of semantic context effects in electromagnetic and hemodynamic studies. Brain Lang 97: 279–293. [DOI] [PubMed] [Google Scholar]
- Wahlund LO, Basun H, Almkvist O, Julin P, Axelman K, Shigeta M, Jelic V, Nordberg A, Lannfelt L ( 1999): A follow‐up study of the family with the Swedish APP 670/671 Alzheimer's disease mutation. Dement Geriatr Cogn Disord 10: 526–533. [DOI] [PubMed] [Google Scholar]
- West WC, Holcomb PJ ( 2002): Event‐related potentials during discourse‐level semantic integration of complex pictures. Brain Res Cogn Brain Res 13: 363–375. [DOI] [PubMed] [Google Scholar]
- Willems RM, Ozyurek A, Hagoort P ( 2008): Seeing and hearing meaning: ERP and fMRI evidence of word versus picture integration into a sentence context. J Cogn Neurosci 20: 1235–1249. [DOI] [PubMed] [Google Scholar]
- Zhang Y, van Drongelen W, He B ( 2006): Estimation of in vivo brain‐to‐skull conductivity ratio in humans. Appl Phys Lett 89: 223903–2239033. [DOI] [PMC free article] [PubMed] [Google Scholar]