

Abstract
Sphingolipidoses are monogenic lipid storage diseases caused by variants in enzymes of lipid synthesis and metabolism. We describe an autosomal recessive complex neurological disorder affecting consanguineous kindred. All four affected individuals, born at term following normal pregnancies, had mild to severe intellectual disability, spastic quadriplegia, scoliosis and epilepsy in most, with no dysmorphic features. Brain MRI findings were suggestive of leukodystrophy, with abnormal hyperintense signal in the periventricular perioccipital region and thinning of the body of corpus callosum. Notably, all affected individuals were asymptomatic at early infancy and developed normally until the age of 8–18 months, when deterioration ensued. Homozygosity mapping identified a single 8.7 Mb disease-associated locus on chromosome 1q41–1q42.13 between rs1511695 and rs537250 (two-point LOD score 2.1). Whole exome sequencing, validated through Sanger sequencing, identified within this locus a single disease-associated homozygous variant in DEGS1, encoding C4-dihydroceramide desaturase, an enzyme of the ceramide synthesis pathway. The missense variant, segregating within the family as expected for recessive heredity, affects an evolutionary-conserved amino acid of all isoforms of DEGS1 (c.656A>G, c.764A>G; p.(N219S), p.(N255S)) and was not found in a homozygous state in ExAC and gnomAD databases or in 300 ethnically matched individuals. Lipidomcs analysis of whole blood of affected individuals demonstrated augmented levels of dihydroceramides, dihydrosphingosine, dihydrosphingosine-1-phosphate and dihydrosphingomyelins with reduced levels of ceramide, sphingosine, sphingosine-1-phosphate and monohexosylceramides, as expected in malfunction of C4-dihydroceramide desaturase. Thus, we describe a sphingolipidosis causing a severe regressive neurological disease.
Subject terms: Disease genetics, Genetics research
Introduction
Sphingomyelins, a class of complex sphingolipids, are major lipid constituents of the plasma membrane. In the nervous system, they are essential in formation of myelin by glial cells, enabling efficiency and speed of action potentials [1–4]. Sphingomyelins are synthesized from ceramides and phosphatidylcholine by the action of sphingomyelin synthases (SMS). Three SMS genes have been found in humans, namely sphingomyelin synthase 1 (SMS1), sphingomyelin synthase 2 (SMS2), and SMS-related protein (SMSr). Of the three pathways that contribute to sphingomyelin synthesis, the most common synthetic pathway involves the condensation of ceramide and CDP-choline by SMS1. Sphingomyelin is degraded to ceramide and phosphocholine by the activity of sphingomyelinases. Thus, sphingolipids are also important precursors of ceramides, bioactive lipids implicated in various biological responses ranging from cell growth and differentiation to apoptosis [1–4].
Sphingolipidoses are monogenic inborn errors of metabolism, in which malfunction of enzymes of sphingolipid metabolism culminates in lower levels of downstream sphingolipids, as well as accumulation of nondegradable lipids in the lysosomes, causing severe storage diseases. The resulting sphingolipidoses are mostly regressive in their clinical course, with phenotypic deterioration becoming clinically evident as lipid accumulation reaches critical levels. This is combined in some of the diseases with defective generation of myelin [1–4].
At least 14 disorders of phospholipids, sphingolipids and fatty acids biosynthesis have been described [1–4]. Of those, the most notable sphingolipidoses are diseases of functional deficiencies in Hexosaminidase A (Tay Sachs disease), α-galactosidase (Fabry’s disease), Arylsulfatase A (metachromatic leukodystrophy; MLD), β-galactosidase (Krabbe disease), β-glucosidase (Gaucher disease), sphingomyelinase (Niemann-pick disease), and ceramidase (Farber’s disease). As the substrates accumulating and those that fail to form in each disease are different, the clinical phenotypes are different: from blindness and muscular weakness in Tay Sachs to skin rash and kidney failure in Fabry’s disease, to enlarged liver and spleen in Gaucher, to dermatitis and skeletal deformation in Farber disease. However, varying degrees of intellectual disability and absence of myelin exist in most diseases [1–4]. Fabry disease is unusual both in its x-linked recessive heredity (while other sphingolipidoses are autosomal recessive) and in the absence of intellectual disability [1–4].
We now describe a novel autosomal recessive complex neurological regressive disorder with varying degrees of intellectual disability, spastic paraplegia and epilepsy, caused by a variant in DEGS1, encoding C4-dihydroceramide desaturase, the last enzymatic step in ceramide synthesis.
Materials and methods
The study was approved by the Schneider Medical Center and Soroka Medical Center institutional review board (approval #5071G). DNA and whole blood samples were obtained following written informed consent.
Clinical phenotyping
Affected individuals and their siblings and parents were studied following informed consent. Phenotyping of affected individuals was determined by senior pediatric neurologist and geneticist.
Linkage and sequence analysis
Linkage analysis and whole exome sequencing (WES) were performed as previously described [5, 6]. In some detail: DNA samples of the four affected individuals, their parents and 1 healthy sibling of each of the two families studied were assayed using Illumina Omni Express arrays (GeneByGene, Houston, TX 77008, USA). Candidate regions were identified using homozygosity mapping, assuming a single founder for the disease haplotype in the kindred. Multipoint LOD score was calculated using SUPERLINK_ONLINE_SNP1.1 (http://cbl-hap.cs.technion.ac.il/superlink-snp/), assuming autosomal recessive heredity with 0.99 penetrance and susceptibility allele frequency of 0.0001. WES was done using paired-end (2 × 101) protocol at a mean coverage of 30-fold (HiSeq2000, Illumina, Theragene Etex, republic of Korea). Of all exonic nucleotides, 85–90% were covered by >10 reads. For exome enrichment, we used SureSelect XT V5 Library Prep Kit targeting 51 Mb regions. Sequencing, read alignment, variant calling, and annotation were performed by Theragen Etex Bioinformatics Team. WES data were then analyzed through using QIAGEN’s Ingenuity® Variant Analysis™ software (http://www.qiagenbioinformatics.com/ingenuity-variant-analysis; QIAGEN Redwood City), excluding variants observed with an allele frequency ≥1% in the 1000 Genomes Project (http://www.internationalgenome.org/) or the Allele Frequency Community (http://www.allelefrequencycommunity.org/), or variants appearing in a homozygous state in our in-house WES database of 300 controls. Variants were then further sifted based on their being predicted deleterious (as listed in HGMD® or ClinVar), previously classified as disease-associated (pathogenic or likely pathogenic) according to computed ACMG guidelines classification, or being associated with loss of function by causing frameshift, in-frame indel, start/stop codon change, missense or splice site loss up to 5 bases into intron (as predicted by MaxEntScan). Of remaining variants, we selected only those within the chromosome 1 disease-associated locus and segregating within the family as expected for autosomal recessive heredity (assayed through Sanger sequencing). The disease-causing variant has been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar), accession number SCV000897760.
Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry
Analysis was done as previously described [7]. In brief: UPLC-Q-TOF-MS/MS analysis of sphingolipids. Sphingolipids were analyzed on an AB Seriex 5600+ quadrupole Time-of-Flight (Q-TOF) Premier mass spectrometer coupled with a Water Acquity Ultra Performance Liquid Chromatography (UPLC). A reversed-phase analytical column Agilent zorbax sb-C8 (2.1 × 150 mm, 3.5 µm) was used for the separation of sphingomyelins and ceramides. Solvent A was water with 0.1% FA. The gradient was comprised of an increase of solvent B (methanol) from 80 to 99% over 20 min, from 99 to 100% in 15 min and reducing to 80% in 1 min, then holding at 80% for the last 9 min. Constant flow at a rate of 0.3 ml/min was running in a Waters UPLC (Waters Corp, Milford, MA, USA) coupled with an AB TripleTOF 5600 plus System (AB SCIEX, Framingham, USA). The optimal Mass condition (scan range) was 100–1500 m/z. The source voltage was +5.5 kV and the source temperature was 600 ℃ to get the positive ions. The pressure of Gas 1 (Air) or Gas 2 (Air) was set to 50 psi. The pressure of Curtain Gas (N2) was set to 30 psi. Injection volume was set at 10 µl with the ultra-violet (UV) detector set at 254 nm. Maximum allowed error was set to ±5 ppm. Declustering potential (DP) was 100 V, and collision energy (CE) was 10 V. For MS/MS acquisition mode, the parameters were the same except for the collision energy being set at 40 ± 20 V, ion release delay (IRD) at 67, ion release width (IRW) at 25. The mass calibration was performed before each analysis employing the Automated Calibration Delivery System. The analysis of sphingolipids was performed in triplicates.
Identification of sphingolipid species
Different sphingomyelins and ceramides were identified by comparing MS/MS ions of analysts with those of sphingolipid standards in ChemSpider base (http://www.chemspider.com/) through the software Peakview (https://sciex.com.cn/products/software/peakview-software). Ceramide and sphingomyelin standards were purchased from Avanti Polar Lipids (https://avantilipids.com/). All standards were dissolved by LC-grade methanol in a concentration of 100 μg/ml. Product [M+H]+ ions at m/z 208.2, 210.3, 236.2, 238.3, 264.4, and 266.4 corresponding to doubly dehydrated product ion fragments were respectively selected for the detection of ceramides and sphingomyelins with d14:1, d14:0, d16:1, d16:0, d18:1, and d18:0 sphingoid base backbones. Neutral loss research was set to allow passage of m/z 184.07 and 141.2 corresponding to the loss of phosphocholine and phosphorylethanolamine moiety combining with the selection of different precursor ions to identify different sphingomyelins and CPEs, respectively. Maximum allowed error for the reliability was set to ±10 ppm. The correctness of sphingomyelin identification was also rectified by the retention time of standards and the content of sphingolipids were referred to the intensity of analytes.
Results
Delineation of the disease phenotype
Four individuals of a large inbred consanguineous Arab Israeli family presented with a complex neurological disorder. Segregation of the phenotype within the family was highly suggestive of autosomal recessive heredity (Fig. 1a). All affected individuals had mild to severe intellectual disability, spastic quadriplegia, sustained clonus in both Achilles tendons, and three had also epilepsy, with no further findings on physical examination or dysmorphic features. Notably, all affected individuals were born at term following normal pregnancies, with borderline high birthweights (3500, 3700, 3800, 3900 g), and were asymptomatic and developed normally until the age of 8–18 months, when deterioration ensued.
Fig. 1.
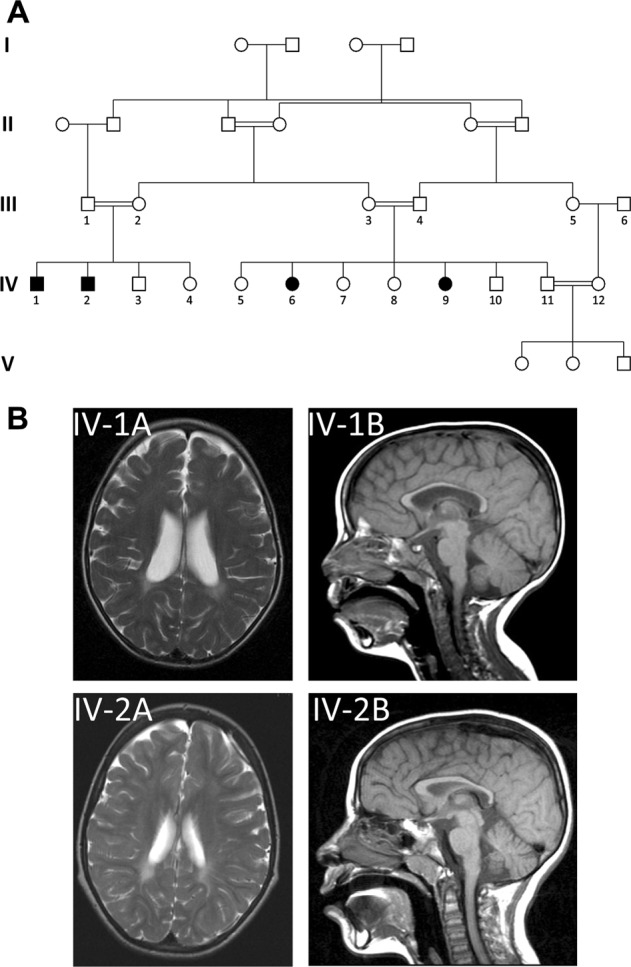
The studied kindred and disease phenotype: a The studied kindred. b Brain MRI of IV-1 at age 3.5 years and of IV-2 at age 7 years demonstrated moderate dilatation of the lateral ventricles with abnormal hyperintense signal in the periventricular perioccipital region, as well as thinning of the body of the corpus callosum
In detail: IV-1 is a 24-year old male. Except for starting to walk on tiptoes at age 18 months, he achieved normal developmental milestones up to age 3 years, when regression in motor skills ensued. On loss of independent ambulation, the patient could walk using a posterior walker, until being confined to a wheelchair at the age of 18 years following complications of scoliosis corrective surgery. At the most recent follow-up, findings on neurological evaluation included proximal upper limb and distal weakness, lower limb weakness (1/5), general hyperreflexia with sustained clonus in both Achilles tendons, and bilateral extensor Babinski sign. Head circumference was 58 cm (95th percentile) and there were no dysmorphic features. Cranial nerves, muscle tone, sensation, and finger-to-nose test were normal. There was no intention tremor or dysmetria, but dysarthric speech was noted. Intelligence was within normal range—graduated from a regular high school, could read, write, speak two languages, and was fully continent, receiving no medications.
IV-2, younger sibling of IV-1, is a 21-year old male. By age 18 months, he was able to walk independently (while leaning on walls) and spoke three-word sentences. Thereafter, regression, significantly more rapid than in his brother, ensued: he lost ambulation completely at 3 years and is currently confined to bed with no ability to sit, stand or walk; cannot actively move his limbs and is incontinent. Verbal ability was lost at age 5–6 years and his vocabulary is currently limited to “mother”. However, he is communicative non-verbally and understands basic social events (visiting relatives). Until 17 years of age he was fed a regular diet, but since then is fed only minced food. Generalized tonic-clonic seizures, effectively controlled with valproic acid, appeared at age 20 years. Neurological examination at the last follow-up demonstrated extreme spasticity with contractures in the wrists, elbows, and knees, scissoring of the lower limbs, and clenched fists. Very hyper-reactive deep tendon reflexes were noted, as well as clonus and bilateral extensor Babinski sign. Non-operable severe scoliosis was evident. The patient maintains eye contact, visually following and fixating, and has a social smile, but cannot communicate verbally. Cranial nerves were normal and head circumference was 55 cm (40th percentile).
IV-6 is a 24-year old female, first cousin to IV-1 and IV2. At age 1 year could sit and clap hands and say mom and dad, but did not yet crawl or stand. Thereafter, she began to regress until she could no longer sit or talk. She was diagnosed with cerebral palsy and underwent three tendon release operations. Tonic-clonic seizures appeared at age 3 years and effectively controlled with prysoline. At present, she is bedridden and cannot speak, but understands simple situations and is communicative. She is fed minced food and is incontinent. At the last follow-up, neurological evaluation showed head circumference 51 cm (<2nd percentile), general spasticity with hyperreactive deep tendon reflexes, positive Babinski sign, and clonus bilaterally. Scoliosis was evident. There was no nystagmus. Generalized contractures in the wrists, elbows, knees, and ankles were noted. The patient was able to visually follow and fixate, and had a social smile, yet was unable to speak.
IV-9 is a 17-year-old-female, sister to IV-6. Examination by a child neurologist at age 1 year yielded no abnormalities and development was normal until age 18 months: she crawled, stood independently, was able to walk a few steps unsupported and spoke two-word sentences. Thereafter, she began to regress and lost all motor milestones. She underwent several tendon release operations and is scheduled for surgery for severe scoliosis. Epilepsy (successfully controlled with clobazam) appeared at age 4 years, yet was milder than in IV-6, involving only focal eye deviation and facial spasms. She is currently confined to a wheelchair and speaks only few words. At the last follow-up, neurological examination revealed findings similar to those in IV-6. Head circumference was 53 cm (5th percentile).
In the affected individuals, complete blood count as well as routine blood chemistry (including liver function tests) were within normal limits. Brain MRI of IV-1 at age 3.5 years and of IV-2 at age 7 years (Fig. 1b) demonstrated moderate dilatation of the lateral ventricles with abnormal hyperintense signal in the periventricular perioccipital region, as well as thinning of the body of the corpus callosum. Due to low compliance, MRI of individuals IV-6 and IV-9, follow-up MRI of IV-1 and IV-2, as well as nerve conduction velocity testing could not be performed.
Genetic studies
Linkage analysis, testing all four affected individuals, their parents and an unaffected sibling for each of the two affected families within the kindred, identified a single homozygosity locus (Fig. 2a) shared by and unique to the affected individuals, segregating in the kindred as expected for autosomal recessive heredity of the disease: 8.7 Mbp segment on chromosome q41–1q42.13 between dbSNPs rs1511695 and rs537250 (positioned at 220,448,330 and 229,159,643 accordingly on assembly GRCh37 / Hg19). Two-point LOD score was 2.1. WES data of individuals IV-1 and IV-6 were filtered for normal variants as aforementioned. Following exclusion of common variants (with allele frequency >0.5% in any of the public databases of 1000 Genomes project, ExAC, NHLBI ESP exomes, CGI or other datasets represented in Allele Frequency Community), only two homozygous variants (and no compound heterozygous variants), were identified within the chromosome 1 locus: a missense variant in DNAH14 (NM_001373.1, c.11789C>T; p.(P3930L); data not shown) and a missense variant in DEGS1 (known also as DES1), converting Asparagine to Serine in all of its three known isoforms: NM_003676.4, c.764A>G, p.(N255S); NM_001321541.1, c.764A>G, p.(N255S); NM_001321542.1, c.656A>G, p.(N219S), (Fig. 2b). In line with the homozygosity mapping data, both co-segregated within the kindred, as was validated through Sanger sequencing (data not shown). Both variants were not found at all in the Exome Aggregation Consortium (ExAC) and gnomAD databases. However, the DNAH14 variant (dbSNP ID 533472746) was in an amino acid that is not conserved throughout evolution (Sup Fig. 1), was predicted to be benign by both SIFT and PolyPhen-2 software, and had Allele Frequency Community (AFC) summarized frequency of 0.401. Therefore, we concluded that this variant is highly unlikely to be causative for the disease phenotype. In contrast, the DEGS1 missense variant (dbSNP ID 768180196), within the putative cytosolic domain of the encoded protein, affected an amino acid extremely conserved throughout evolution (Fig. 2c, d), was predicted to be damaging by both SIFT and PolyPhen-2 (CADD score 24) and had an AFC summarized frequency of 0.058. Moreover, only 2 DEGS1 loss-of-function (LoF) variants have been reported in ExAC and only 14 in gnomAD, none of which in a homozygous state. We thus concluded that the likely causative variant of this monogenic autosomal recessive disease is the missense variant in DEGS1, encoding delta 4-desaturase, sphingolipid 1 (known also as C4-dihydroceramide desaturase).
Fig. 2.

Identification of the DEGS1 variant: a Homozygosity mapping: SNPs colored red, blue and dashed red represent homozygosity for major allele, heterozygosity and homozygosity for the minor allele, respectively; arrow marks a single chromosome 1 homozygous locus shared by affected individuals (Homozygosity-Mapper). b The c.764A>G DEGS1 variant (Sanger sequencing). c evolutionary conservation of the p.(N255S) variant. d Location of the variant within the cytosolic domain of DEGS1
Determination of the biochemical effects of the DEGS1 variant
C4-dihydroceramide desaturase, encoded by DEGS, is a membrane bound desaturase localized to the endoplasmic reticulum (ER) membrane, whose protein structure has not yet been resolved; hence, we could not reliably predict the structural effect of the DEGS1 variant and its putative functional effect. However, we could determine the actual functional effects of this variant through UPLC-TOF-MS/MS analysis of whole blood of three affected individuals (Fig. 3): in the de-novo synthesis pathway of ceramide, the last step in the conversion of dihydroceramide into ceramide is C4-dihydroceramide desaturase, crucial in the regulation of the balance between sphingolipids and dihydrosphingolipids. In line with the expected effects of a loss of function variant in DEGS1, compared to healthy control (individual IV-5), affected individuals (IV-1, IV-6, and IV-9) had significantly lower blood levels of ceramide and higher levels of dihydroceramides. Other findings were also in line with malfunction of C4-dihydroceramide desaturase: affected individuals had higher levels of dihydrosphingosine but lower levels of sphingosine, higher levels of diydrosphingosine-1-phosphate but lower levels of sphingosine-1-phosphate, higher levels of dihydrosphingomyelins and lower levels of monohexosylceramides. Notably, blood levels of ceramide-1-phosphates and of sphingomyelin were not significantly altered.
Fig. 3.

Biochemical consequences of the DEGS1 variant in patients’ whole blood: a–f Compared to healthy control (WT, IV-5), affected individuals (Mut-1, 2, 3 are IV-1, IV-6, IV-9, respectively) had higher levels of dihydroceramides, dihydrosphingosine, dihydrosphingosine-1-phosphate, and dihydrosphingomyelins, and reduced levels of ceramides, sphingosine, sphingosine-1-phosphate and monohexosylceramides. g, h Levels of sphingomyelins and ceramide-1-phosphates were similar between the healthy control and the patients. (i) Schema demonstrating that these results are consistent with inhibition of conversion of dihydroceramide to ceramide due to the DEGS1 variant
Discussion
We delineate an early-onset autosomal recessive disease presenting with progressive spastic paraplegia, scoliosis and variable degrees of intellectual disability and epilepsy. Through combined linkage analysis and whole exome sequencing we narrowed down the possible disease-causing variants to two adjacent co-segregating missense variants in DNAH14 and in DEGS1. Based on lack of evolutionary conservation, three different prediction software and high prevalence in ExAC database, we ruled out the DNAH14 as being disease-causing. In contrast, the DEGS1 variant was in an evolutionary conserved residue of a highly conserved gene that plays an essential role in a pathway of ceramide/phospholipids synthesis, that is essential from plants to primates and humans. Segregation of the DEGS1 variant within the affected kindred, its absence in a homozygous state and low frequency in a heterozygous state in known databases as well as in 300 ethnically matched controls, were in line with the DEGS1 variant being pathogenic. This was further substantiated through lipidomics analysis of whole blood samples of affected individuals, demonstrating findings as expected in malfunction of C4-dihydroceramide desaturase (encoded by DEGS1), affecting the de-novo synthesis pathway of ceramide: the patients had higher levels of dihydroceramides, dihydrosphingosine, dihydrosphingosine-1-phosphate and dihydrosphingomyelins. The opposite was true with ceramides, sphingosine, sphingosine-1-phosphate, and monohexosylceramides. These results were consistent with inhibition of conversion of dihydroceramides to ceramides due to the dihydroceramide desaturase variant [1–4, 8, 9]. Of note, the levels of sphingomyelins and ceramide-1-phosphates were similar between the healthy control and the patients. This is an unexpected finding because the levels of ceramides, the precursors of both sphingomyelins and ceramide-1-phosphates, were reduced in the patients. One possible explanation is that there is compensatory downregulation of sphingomyelinases and ceramide-1-phosphatases in the patients [1–4, 8, 9].
The double bond that distinguishes dihydroceramides from ceramides markedly alters the biophysical properties of the molecules, modifying their elastic properties and packing behavior [3, 9, 10]. Dihydroceramide desaturase, crucial in the regulation of the balance between sphingolipids and dihydrosphingolipids, spans the ER membrane twice and contains three His-containing consensus motifs that are characteristic of a group of membrane fatty acid desaturases [11]. Two distinct dihydroceramide desaturases, DEGS1 and DEGS2, catalyze the desaturation reaction, with DEGS1 accounting for ceramide synthesis in most tissues [9–11]. DEGS1 expression is upregulated during the 4th–9th week of human embryogenesis [12], implicating a pivotal role of ceramide/phosphosphingolipids synthesis in neural system development. In mice, homozygous Degs1 null variant causes accumulation of dihydroceramide (DhCer) with relatively low ceramide (Cer) levels, resulting in incompletely penetrant lethality. Surviving mice had failure to thrive, scaly skin, sparse hair, and tremors, as well as abnormal liver function, and numerous blood chemistry and hematologic abnormalities, culminating in death by the age of 8–10 weeks [13]. Notably, the human homozygous variant had similar effects in terms of DhCer and Cer levels, causing a clear neurological disease, yet was not lethal and did not cause apparent skin and hair phenotypes.
The findings suggestive of hypomyelinating leukodystrophy evident in patients IV-1 and IV-2 are in line with the known roles of sphingolipids in myelin formation, as well as with the central and peripheral demyelination seen in other disorders of sphingolipid metabolism, such as MLD and Krabbe disease [1–4, 8, 14]. While sphingolipids are essential in formation of myelin [1–4], defective myelination can be mediated not only by defective synthesis of sphingomyelins but also through accumulation of DhCer, as seen in the affected individuals [1–4, 8, 11]. In fact, DhCer accumulation has been shown to affect reactive oxygen species (ROS) generation and apoptosis as well as cell cycle regulation and induction of autophagy [2, 4, 11], processes shown to be specifically affected by suppression of DEGS1 in cell culture [2, 4, 11, 15]. Experiments in model membranes and in Drosophila suggest that accumulation of DhCer (with enhanced DhCer/Cer ratio) is detrimental by destabilizing membranes and increasing their permeability, suggesting that the accumulation of DhCer plays a major role in the disease pathophysiology [16]. A substantial role in DhCer accumulation in disease causation can also explain the progressive nature of the disease phenotype and the onset only 8–18 months after birth, as is seen in other storage diseases [2, 3].
Sphingolipids are particularly abundant in the nervous system. Being amphiphilic, they localize to cellular membranes and many of their roles in health and disease result from membrane reorganization and from lipid interaction with proteins within cellular membranes [1, 10, 11]. Sphingolipid metabolism is an intricate network in which modulation of a single step affects many other components [1, 10, 11, 16]. Neuronal sphingolipid synthesis takes place across multiple cellular compartments, beginning at the cytosolic leaflet of the ER, where palmitoyl CoA and serine are converted to ceramide. Ceramide is then transported either to the luminal leaflet of the ER, where it is converted by ceramide galactosyltransferase to the essential neuronal sphingolipid galactosylceramide; or to the Golgi complex, where it is converted to either glucosylceramide on the cytosolic side of the Golgi via glucosylceramide synthase, or to sphingomyelin on the luminal side by sphingomyelin synthase. Transport of ceramide from the ER to the Golgi complex is facilitated by either ceramide transfer protein (CERT) or vesicular transport [1, 10, 11]. Thus, the disease pathophysiology is likely complex, and is likely not limited to the effects of the altered levels of the molecules directly upstream and downstream of DEGS1 that we demonstrate, or specifically to the ER.
Significant variability in the severity of the clinical phenotype, from practically normal IQ with motor impairment and relatively large head (IV-1) to severe delay and small head (IV-6), was evident in the patients sharing homozygosity of the same DEGS1 variant. The biochemical data, with similar results for the different affected individuals, suggest that this is not directly due to variations in DEGS1 activity. Thus, the phenotypic variation of the disease is likely due to a combination of environmental and genetic modifiers that are yet to be determined, that were not identified through careful analysis of the WES data of the phenotypically variant individuals IV-1 and IV6. Follow-up MRI and nerve conduction velocity studies, not available at this point, might shed light on the phenotypic variability in disease progression. As suggested in two recent studies done parallel to and independent of ours [17, 18], further detailed studies of the human patients and in model systems will be needed to fully understand the disease mechanism, possibly enabling therapeutic modalities through complementation of components of the pathway whose production is reduced, as well as attempts at removal or abrogation of formation of components whose accumulation is destructive.
Supplementary information
Acknowledgements
The study was supported by the Legacy Heritage Bio-Medical program of the Israel Science Foundation grant No. 1798/16 (to OSB); by the National Knowledge Center for Rare/Orphan Diseases sponsored by the Israel ministry of Science, Technology and Space; and by the US National Institutes of Health Grant P01 CA097132 (to CM). We thank the Lipidomics Core Facility at Stony Brook University, Stony Brook, USA for analyzing sphingolipids.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Vadim Dolgin, Rachel Straussberg
Supplementary information
The online version of this article (10.1038/s41431-019-0444-z) contains supplementary material, which is available to authorized users.
References
- 1.Olsen ASB, Faergeman NJ. Sphingolipids: membrane microdomains in brain development, function and neurological diseases. Open Biol. 2017;7:1–17. doi: 10.1098/rsob.170069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grassi S, Chiricozzi E, Mauri L, Sonnino S, Prinetti A. Sphingolipids and neuronal degeneration in lysosomal storage disorders. J Neurochem. 2019;148:600–11. doi: 10.1111/jnc.14540. [DOI] [PubMed] [Google Scholar]
- 3.Sandhoff K. Neuronal sphingolipidoses: Membrane lipids and sphingolipid activator proteins regulate lysosomal sphingolipid catabolism. Biochimie. 2016;130:146–51. doi: 10.1016/j.biochi.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 4.Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol. 2018;19:175–91. doi: 10.1038/nrm.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perez Y, Menascu S, Cohen I, Kadir R, Basha O, Shorer Z, et al. RSRC1 mutation affects intellect and behavior through aberrant splicing and transcription, downregulating IGFBP3. Brain. 2018;141:961–70. doi: 10.1093/brain/awy045. [DOI] [PubMed] [Google Scholar]
- 6.Kadir R, Harel T, Markus B, Perez Y, Bakhrat A, Cohen I, et al. ALFY-controlled DVL3 autophagy regulates Wnt signaling, determining human brain size. PLoS Genet. 2016;12:e1005919. doi: 10.1371/journal.pgen.1005919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li F, Xu R, Low BE, Lin CL, Garcia-Barros M, Schrandt J, et al. Alkaline ceramidase 2 is essential for the homeostasis of plasma sphingoid bases and their phosphates. FASEB J. 2018;32:3058–69. doi: 10.1096/fj.201700445RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Magaye RR, Savira F, Hua Y, Kelly DJ, Reid C, Flynn B, et al. The role of dihydrosphingolipids in disease. Cell Mol Life Sci. 2019;76:1107–34. doi: 10.1007/s00018-018-2984-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wattenberg BW. The long and the short of ceramides. J Biol Chem. 2018;293:9922–3. doi: 10.1074/jbc.H118.003522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tidhar R, Futerman AH. The complexity of sphingolipid biosynthesis in the endoplasmic reticulum. Biochim Biophys Acta. 2013;1833:2511–8. doi: 10.1016/j.bbamcr.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 11.Fabrias G, Muñoz-Olaya J, Cingolani F, Signorelli P, Casas J, Gagliostro V, et al. Dihydroceramide desaturase and dihydrosphingolipids: debutant players in the sphingolipid arena. Prog Lipid Res. 2012;51:82–94. doi: 10.1016/j.plipres.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Yi H, Xue L, Guo MX, Ma J, Zeng Y, Wang W, et al. Gene expression atlas for human embryogenesis. FASEB J. 2010;24:3341–50. doi: 10.1096/fj.10-158782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–9. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Parikh S, Bernard G, Leventer RJ, van der Knaap MS, van Hove J, Pizzino A, et al. A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephalopathies. Mol Genet Metab. 2015;114:501–15. doi: 10.1016/j.ymgme.2014.12.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kraveka JM, Li L, Szulc ZM, Bielawski J, Ogretmen B, Hannun YA, et al. Involvement of dihydroceramide desaturase in cell cycle progression in human neuroblastoma cells. J Biol Chem. 2007;282:16718–28. doi: 10.1074/jbc.M700647200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siddique MM, Li Y, Chaurasia B, Kaddai VA, Summers SA. Dihydroceramides: from bit players to lead actors. J Biol Chem. 2015;290:15371–9. doi: 10.1074/jbc.R115.653204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pant DC, Dorboz I, Schlüter A, Fourcade S, Launay N, Joya J, et al. Loss of the sphingolipid desaturase DEGS1 causes hypomyelinating leukodystrophy. J Clin Invest. 2019. 10.1172/JCI123959. [DOI] [PMC free article] [PubMed]
- 18.Karsai G, Kraft F, Haag N, Korenke GC, Hanisch B, Othman A, et al. DEGS1-associated aberrant sphingolipid metabolism impairs nervous system function in humans. J Clin Invest. 2019. 10.1172/JCI124159. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.