

Abstract
It is vitally important to identify the genetic determinants of complex brain‐related disorders such as autism, dementia, mood disorders, and schizophrenia. However, the search for genes predisposing individuals to these illnesses has been hampered by their genetic and phenotypic complexity and by reliance upon phenomenologically based qualitative diagnostic systems. Neuroimaging endophenotypes are quantitative indicators of brain structure or function that index genetic liability for an illness. These indices will significantly improve gene discovery and help us to understand the functional consequences of specific genes at the level of systems neuroscience. Here, we review the feasibility of using neuroanatomic and neuropsychological measures as endophenotypes for brain‐related disorders. Specifically, we examine specific indices of brain structure or function that are genetically influenced and associated with neurological and psychiatric illness. In addition, we review genetic approaches that capitalize on the use of quantitative traits, including those derived from brain images. Hum Brain Mapp, 2007. © 2007 Wiley‐Liss, Inc.
Keywords: genetics, neuroimaging, endophenotype, MRI, PET, anatomy, functional MRI, fMRI
INTRODUCTION
Identification of the genetic determinants of complex brain‐related disorders such as autism, dementia, mood disorders, and schizophrenia is of paramount importance. These neurological and psychiatric illnesses represent a significant economic burden [Uhl and Grow, 2004] and are associated with substantial morbidity and mortality, but their underlying pathologies are poorly understood. Identifying the specific genes that contribute to risk for these illnesses should provide critical information on the causes of these diseases that may lead to the development of novel diagnostic and therapeutic strategies. Genetic studies of brain‐related mental diseases have made some recent progress, but the field as a whole lags behind other complex diseases in the identification of disease‐related genes and the subsequent biological inferences that result from discovering a causal component of the pathological chain. There are many possible reasons for this, including a historical overdependence on underpowered genetic study designs. However, a major reason for the slower progress in the genetics of mental illness is the relative lack of emphasis on quantitative endophenotypes that index disease risk [Gershon and Goldin, 1986; Gottesman and Gould, 2003]. Endophenotypes are markers that are genetically correlated with disease liability, and can be measured in all individuals (both affected and unaffected). They may also provide much greater power to localize and identify disease‐related quantitative trait loci (QTLs) than does affection status alone [Blangero et al., 2003]. Biological endophenotypes are measurable intermediate phenotypes that are generally closer to the action of the gene and thus exhibit higher genetic signal‐to‐noise ratios [Gottesman and Gould, 2003].
Imaging methods are being successfully applied in a wide range of neurological and psychiatric disorders, providing important insights in the pathophysiology of these illnesses and clues for the development of novel treatments. Indeed, there is growing evidence that a host of common brain disorders, including addictions [Thompson et al., 2004], attention deficit hyperactivity disorder [Sowell et al., 2003b], dementia [Thompson et al., 2006], anxiety disorders, mood disorders [Bearden et al., in press], seizure disorders [Lin et al., in press], and schizophrenia [Cannon et al., 2002], are associated with alterations in brain structure and function. Although early large‐scale brain‐imaging research focused young, healthy, normal adult subjects [Mazziotta et al., 2001], in the past decade, normative studies of brain structure and function have been extended to the entire human lifespan, from childhood through extreme old age [Gogtay et al., 2004; Sowell et al., 2003a; Thompson et al., 2006]. Together these streams of research, which delineate the normal variation in brain structure and function as well as the pathological extremes of this variation, provide a backdrop for understanding genetic influences on neruoanatomy and neurophysiology. Since in vivo brain imaging methods are typically repeatable, provide quantitative data, and are often far more sensitive than behavioral observations to subtle brain changes, such markers have the potential to be endophenotypic markers [Hariri and Weinberger, 2003]. In this manuscript, we review current data suggesting that neuroanatomic and functional neuroimaging measures can be valid endophenotypes for a host of neurological and psychiatric illnesses. In addition, we will outline genetic methods that should capitalize on the complexity of imaging data and facilitate novel gene discovery, as well as test hypotheses about the action of specific, known genes.
DEFINING AN ENDOPHENOTYPE
Although twin, family and adoption studies have demonstrated that most neurological and psychiatric illnesses are substantially heritable (Table II gives heritability estimates for various illnesses), with a few notable exceptions, the molecular genetic origins of these illnesses remain elusive. Undoubtedly, the search for genetic loci involved in these disorders has been hampered by the genetic complexity of these illnesses, heterogeneity of disease expression (e.g., variable symptom presentation), and comorbidity with other disorders that may distort clinical presentation (e.g., substance abuse). Because genes predisposing to neurological or psychiatric illnesses may be transmitted without expression of the clinical phenotype (formal diagnosis), interest has arisen in developing endophenotypes, indicators of processes mediating between genotype and phenotype. Using endophenotypic markers may be advantageous, since they are generally less complex than their associated phenotype and thus may be more readily linked to a specific genetic locus [Gershon and Goldin, 1986; Glahn et al., 2004; Gottesman and Gould, 2003; Leboyer et al., 1998; Lenox et al., 2002]. In addition, endophenotypes for complex human psychiatric and neurological disorders could potentially be extended to animal models [Gottesman and Gould, 2003], advancing our understanding of the neurobiology of psychiatric disorders, and furthering the development of novel medications [Nestler et al., 2002]. Furthermore, endophenotypes may be necessary to resolve the status of family members in genetic studies of neurological and psychiatric disorders.
Table II.
Structural brain abnormalities in neurological and psychiatric illnesses
| Complex genetic brain disorder | Illness heritability | Frontal lobe | Temporal lobe | Cerebellum | Hippocampus/limbic system | White‐matter hyperintensity |
|---|---|---|---|---|---|---|
| Addictions | 0.4–0.6 [Begleiter and Porjesz, 1999] | ↓ [Lingford‐Hughes et al., 2003] | ↓ [Lingford‐Hughes et al., 2003] | ↓ [Lingford‐Hughes et al., 2003] | ↓ [Geuze et al., 2005] | ↑ [Sullivan and Pfefferbaum, 2001] |
| Alzheimer's & dementia | 0.5–0.7 [Bergem et al., 1997] | ↓ [Scheltens et al., 2002] | ↓ [Scheltens et al., 2002] | ↓ [Karas et al., 2003] | ↓ [Geuze et al., 2005] | ↑ [Hanyu et al., 1999] |
| ADHD | 0.60–0.90 [Rhee et al., 1999] | ↓ [Castellanos et al., 1996, 2002; Mostofsky et al., 2002] | ↓ [Castellanos et al., 2002] | ↓ [Castellanos et al., 2002] | ↓ [Castellanos et al., 2002; Geuze et al., 2005] | |
| Autism | ↓ [Carper and Courchesne, 2005; McAlonan et al., 2005] | ↓ [McAlonan et al., 2005] | ↓ [McAlonan et al., 2005; Pierce and Courchesne, 2001] | ↑ [Geuze et al., 2005; Rojas et al., 2004; Schumann et al., 2004] | ||
| Epilepsy & seizure disorders | 0.60–0.88 [Berkovic et al., 1998] | ↓ [Geuze et al., 2005] | ||||
| Mood disorders | 0.37–0.59 [Merikangas et al., 2002] | ↓ [Monkul et al., 2005] | ↓ [Monkul et al., 2005] | ↓ [Mills et al., 2005] | ↓ [Geuze et al., 2005; Monkul et al., 2005] | ↑ [Monkul et al., 2005] |
| PTSD & anxiety disorders | 0.20–0.66 [Stein et al., 2002] | ↓ [Fennema‐Notestine et al., 2002] | ↓ [Geuze et al., 2005; Hull, 2002; Wignall et al., 2004] | ↑ [Canive et al., 1997] | ||
| Schizophrenia | 0.70–0.83 [Cannon et al., 1998] | ↓ [Wright et al., 2000] | ↓ [Wright et al., 2000] | ↓ [Wright et al., 2000] | ↓ [Geuze et al., 2005; Wright et al., 2000] | ↔ [Brown et al., 1992] |
Decrease (↓), increase (↑), or evidence for no change (↔) in overall or gray matter volume.
For a marker to be considered an endophenotype, it must be shown to (1) be highly heritable, (2) be associated with the phenotype (formal diagnosis), (3) be independent of clinical state, and (4) impairment must cosegregate with the illness within a family, with nonaffected family members showing impairment relative to the general population [Gershon and Goldin, 1986; Glahn et al., 2004; Gottesman and Gould, 2003; Leboyer et al., 1998; Lenox et al., 2002]. In the current review, we focus on the heritability of neuroanatomic and functional neuroimaging measurements. We also briefly describe how brain anatomy and physiology are altered in individuals with neurological and psychiatric illnesses and their relatives.
HERITABILITY OF NEUROANATOMY
The size, shape, and complexity of the primate brain vary considerably between individuals and a significant portion of this variability is influenced by genetic factors. While very early stages of primate brain development are predominantly mediated by genetic programs [Rubenstein and Rakic, 1999; Rubenstein et al., 1999], later stages of development, organization, and brain maturation result from a complex interaction of genetic and environmental influences [Rakic, 1988]. Genetic influence is typically expressed as a heritability statistic (denoted h 2), which reflects the percentage of the variation in a trait (e.g., brain volume) attributable to genetic factors. In contrast, “environmental” factors refer to trait variation not included in the heritability estimate, including nutrition, education and experience or experimental errors such as lack of reproducibility in the test. Studies in nonhuman primates have provided heritability estimates for brain weight ranging between 42% and 75% [Cheverud et al., 1990a, b; Mahaney et al., 1993]. Human magnetic resonance imaging studies have expanded upon these initial findings (see [Thompson et al., 2002] for a review), reporting high heritabilities for whole brain volumes, with somewhat lower heritabilities for lobar volumes (Table I). Reduced heritability estimates for lobar structures might be associated with the reliability of delineating lobar regions rather than an intrinsic reduction in the genetic influences of these regions. In contrast, ventricular volume seems to be mediated almost entirely by environmental factors [Baare et al., 2001; Wright et al., 2002] (see Styner et al. [2005] for contrary evidence). Estimated heritability for sulcal shape or length seems to vary considerably based on the sulcus in question, though these measurements traditionally suffer from poor reliability.
Table I.
Heritability of human brain phenotypes
| Brain region | Sample sizea | Heritabilityb |
|---|---|---|
| Intracranial volume | 471 [254–1330] | 0.87 [0.72–0.92] [Atwood et al., 2004; Baare et al., 2001; Hulshoff Pol et al., 2004; Posthuma et al., 2000, 2002] |
| Whole brain volume | 194 [38–278] | 0.78 [0.56–1.00] [Baare et al., 2001; Bartley et al., 1997; Geschwind et al., 2002; Hulshoff Pol et al., 2004; Narr et al., 2002; Posthuma et al., 2000; Wright et al., 2002] |
| Total gray matter volume | 256 [256–258] | 0.88 [0.82–1.00] [Baare et al., 2001; Hulshoff Pol et al., 2004; Posthuma et al., 2000] |
| Total white matter volume | 256 [256–258] | 0.85 [0.82–0.87] [Baare et al., 2001; Hulshoff Pol et al., 2004; Posthuma et al., 2000] |
| Frontal volume | 278 | 0.65 [0.64–0.66] [Geschwind et al., 2002] |
| Temporal volume | 278 | 0.58 [0.56–0.60] [Geschwind et al., 2002] |
| Parietal volume | 278 | 0.52 [0.50–0.53] [Geschwind et al. 2002] |
| Occipital volume | 278 | 0.33 [Geschwind et al., 2002] |
| Cerebellum volume | 149 [40–258] | 0.74 [0.66–0.87] [Posthuma et al., 2000; Wright et al., 2002] |
| Calosal volume | 125 [80–170] | 0.44–1.0 [Narr et al., 2002; Pfefferbaum et al., 2004] |
| Sulcus shape or length | 58 [38–78] | 0.22 [0.10–0.77] [Bartley et al., 1997; Eckert et al., 2002] |
| White matter hyperintensity | 810 [290–1330] | 0.76 [0.73–0.78] [Atwood et al., 2004; Carmelli et al., 1998] |
Mean n [range].
Heritability = mean h 2 [range].
Bartley et al. [1997] reported 94% heritability for brain volume, based on structural equation modeling in 10 monozygotic (MZ) and nine dizygotic (DZ) twin pairs. In elderly twins, Sullivan et al. [2001] reported that the volume of the hippocampus was less heritable (h 2 = 0.4) than that of the adjacent temporal horns (h 2 = 0.6), corpus callosum (h 2 = 0.8), and intracranial volume (h 2 = 0.8). They suggested that environmental differences, perhaps interacting with genetic differences, may exert especially strong or prolonged influences on hippocampal size. A lower heritability figure for hippocampal size is consistent with its role in memory encoding, its vulnerability to plasma cortisol levels, and its plasticity in later life [Maguire et al., 2000; see also Lyons et al., 2001 for a related MRI study in monkeys]. In a similar vein, Baaré et al. [2001] found that individual differences in lateral ventricle volume were best explained by a structural equation model containing common (58%) and unique (42%) environmental factors, indicating genes to be of little or no influence. The same authors found that genetic factors almost entirely accounted for individual differences in whole brain (90%), gray (82%) and white (88%) matter volume, in a study based on a sizeable sample of 54 MZ and 58 DZ twin pairs, and 34 of their full siblings [Baare et al., 2001]. In their multivariate analysis of body height, and volumes of gray matter, white matter and the intracranial space, Baaré et al. noted that a large part of the genetic influences were common to the three brain measures, and a smaller part was shared with height. Some genes may therefore have a general effect on the brain, while other genes may affect specific volumes. More recently, Pfefferbaum et al. [2001] used diffusion imaging, which is sensitive to myelination levels and fiber orientation, to quantify the microstructure of the corpus callosum in 15 MZ and 18 DZ pairs. They found that anterior interhemispheric connecting pathways, in the callosal genu, were more susceptible than splenial pathways to environmental influences, perhaps reflecting the prolonged maturation of the frontal cortex well into adulthood [Sowell et al., 1999]. Using bivariate genetic modeling, these authors also noted that intracranial volume and corpus callosum area were tightly correlated, a correlation due entirely to shared genetic effects between these two brain structures.
Most current reports of lobular or region specific heritability are based on region of interest analyses where specific brain areas are manually traced. Yet, this approach may be less sensitive to small variations in brain structure or tissue type distribution than more modern modeling methods [Good et al., 2002]. For example, Thompson et al. [2001] used an elastic deformation procedure to model the genetic influences on neuroanatomic variation between healthy mono‐ and dizygotic twin pairs at each voxel on the surface of the cortex. This analysis indicated that genetic factors significantly influenced cortical gray matter density in subregions of prefrontal and temporal lobes, particularly Broca's and Wernicke's language areas. However, other areas within these structures were less heritable, suggesting that heritability estimates based on gross lobular regions of interest blur more subtle local differences (Fig. 1; see Rogers et al., this issue, for similar work in nonhuman primates). A rather provocative implication of this study was that cognitive performance (specifically full‐scale IQ) was correlated with brain structure volumes in the very regions where structure is under greatest genetic control [Thompson et al., 2001]. Although a wide body of work (reviewed in Gray and Thompson [2004]) supports the hypothesis that genetic variations contribute profoundly to IQ, and that gray matter volume is correlated with IQ, the high heritability of frontal gray matter volume suggests that it may serve as a possible endophenotype for IQ. Taking this idea further, Posthuma et al. [2000] also found high heritability for gray matter volumes. However, using a cross‐twin cross‐trait (bivariate genetic) analysis to compute genetic correlations, they demonstrated that the linkage between gray matter and IQ is highly genetic, in other words it is strongly mediated by common genetic differences [Posthuma et al., 2000]. Genetic factors may therefore contribute to structural differences in the brain that are linked with cognitive differences. The direction of causation is less clear. Logically, genetic factors may influence brain volume, which in turn influences cognition. In practice, each factor is likely to influence the others. Genes with pleiotropic effects may influence both brain volume and cognition, without there necessarily being a direct effect of brain volume on cognition. In addition, individuals with higher IQ may also seek more intellectual stimulation, develop more synapses, and seek environments correlated with their genotypes (see Gray and Thompson [2004] for an analysis of alternate explanations).
Figure 1.
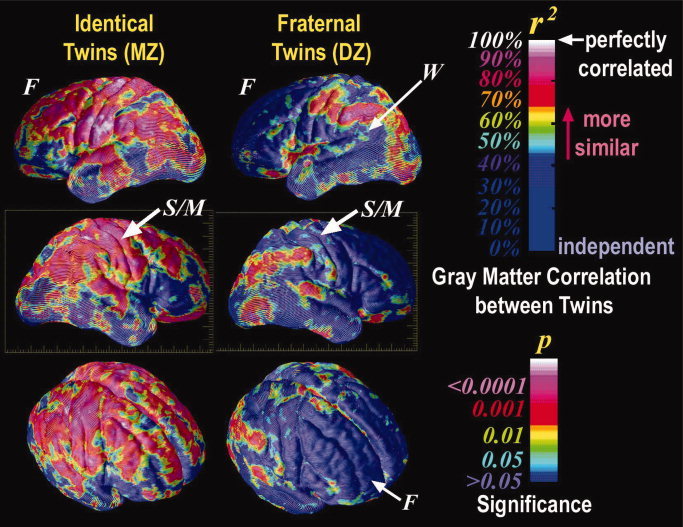
Genetic influences on human neuroanatomy. Ten monozygotic twin pairs (n = 20 subjects) are almost perfectly correlated in their gray matter distribution, with near identity in frontal (F), sensorimotor (S/M), and perisylvian language cortices. In contrast, 10 dizygotic twin pairs (n = 20) are significantly less alike in frontal cortices, but are 90–100% correlated for gray matter in perisylvian language‐related cortex, including supramarginal and angular territories and Wernicke's language area (W). The significance of these increased similarities, visualized in color, is related to the local intraclass correlation coefficents (r) (From Thompson et al., Nat Neurosci, 2001, 4(12), 1253–1258, © Nature Publishing Group, reproduced by permission). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Region of interest methods tends to focus on a relatively small number of brain areas and potentially miss important covariation between structures. Such covariation could suggest that different brain structures are being influenced by the same gene or sets of genes. In contrast, modern morphometric methods can simultaneously model even minor neuroanatomic variations throughout the brain [Ashburner et al., 2003], particularly given advances in MRI technology which provide exquisite soft‐tissue contrast and very high resolution (e.g. less than 1 mm3). Modern image analysis methods and high‐resolution images should significantly improve estimates of the heritability of neuroanatomic regions and, more importantly, localize genes influencing brain structure. For example, Wright et al. [1999] parcellated 92 regional gray matter volumes in 10 MZ and 9 DZ twin pairs, scanned with MRI. Inter‐regional relationships were summarized by principal component analysis of the resulting genetic correlation matrix [Wright et al., 1999]. This identified shared genetic effects on the frontal‐parietal cortices and bilateral temporal cortex and insula. As the size and scope of these studies increase, decomposition of the genetic correlation matrix is likely to be a key exploratory tool to identify supraregional brain systems [Wright et al., 1999], which share common genetic influences, systems which may cut across conventional anatomic boundaries. Such a factor‐analytic approach would be of interest to apply to voxel‐based studies as well, given the interdependencies in structure volumes and the likely overlap in the sets of genes that influence the volumes of different substructures.
NEUROANATOMY OF BRAIN DISORDERS
Although neurological and psychiatric illnesses are typically identified through behavioral observation, these illnesses are closely associated with anomalous brain structure. Advances in neuroimaging have left little doubt that particular diseases preferentially disturb specific neuroanatomic structures. Furthermore, the distribution of structural anomalies seemingly reflects the underlying pathology, often dissociating illnesses; see Table II for a targeted summary of comparative findings. For example, early and disproportionate hippocampal atrophy in individuals with memory complaints typically points to a diagnosis of Alzheimer's disease. In contrast, focal atrophy of the temporal lobe, frontal lobe, or both, makes Alzheimer's disorder less probable and the potential for other dementias such as Pick's disease more likely [Scheltens et al., 2002]. Similar, though less robust, dissociations have been reported for psychiatric illnesses. For instance, reduced hippocampal volumes are consistently reported in both major depression [Videbech and Ravnkilde, 2004] and schizophrenia [Honea et al., 2005; Wright et al., 2000]. However, gray‐matter reductions observed in affective disorders appear to be limited to the limbic system [McDonald et al., 2004b]. In contrast, schizophrenia is associated with widespread volumetric diminution [Cannon et al., 2002; Wright et al., 2000].
The neuroanatomic anomalies seen in individuals affected with neurological or psychiatric illnesses often predate overt symptom expression [Fleisher et al., 2005; Harris et al., 2004; Moffat et al., 2000], though typically in a less severe form. In addition, many of the neuroanatomic variations associated with illness have been observed in individuals at risk for these same disorders. For example, women with mild cognitive impairment (MCI) and the apolipoprotein E epsilon4 (ApoE4) genotype have smaller hippocampal volumes and are more likely to develop Alzheimer's disease than MCI women without the ApoE4 variant [Fleisher et al., 2005]. Similar, though less significant trends were observed in males [Fleisher et al., 2005]. Adolescents and young adults from families with more than one individual who abuses alcohol have significantly smaller amygdala volumes than comparison subjects without ancestral history for alcoholism [Hill et al., 2001]. Unaffected family members of individuals with schizophrenia or individuals with bipolar disorder have discrete gray matter volume deficits but comparable white matter anomalies, suggesting both distinct and common abnormalities associated with genetic liability for these major psychiatric illnesses [McDonald et al., 2004a]. While unaffected cotwins of patients with schizophrenia have widespread gray matter reductions, healthy monozygotic cotwins have significantly lower gray matter volumes in polar and dorsolateral prefrontal cortex than healthy dizygotic cotwins, suggesting that these regions are sensitive to genetic liability for schizophrenia [Cannon et al., 2002]. Taken together, findings of anomalous neuroanatomy in individuals affected by neurological and psychiatric illnesses and those at increased genetic risk for these disorders suggest that at least a portion of these indices may be appropriate endophenotypic markers [McDonald et al., 2004a].
HERITABILITY OF FUNCTIONAL NEUROIMAGING
Functional neuroimaging involves the assessment of physiologic or metabolic states in a living organism. Most human functional neuroimaging experiments use radioactive ligands (e.g., SPECT or PET) or less invasive magnetic resonance procedures (e.g., fMRI). Functional neuroimaging experiments may be grouped into those that include a cognitive or psychological challenge and those that index resting metabolic state. Those experiments that include a challenge typically focus on an a priori brain region or a system putatively linked to the behavioral, cognitive, or physiological demands placed upon the subject (e.g., prefrontal activity during a working memory task). Since PET [Ragland et al., 1997] and fMRI [Callicott et al., 1999] signals are impacted by cognitive or behavioral performance, neuroactivation studies should consider performance level when examining individual differences or familial variability. Given these concerns, the complexity of functional neuroimaging analyses and the relatively high cost of collecting these data, few studies have examined genetic influences on functional neuroimaging signals. Indeed, to our knowledge, there are no published heritability estimates for functional neuroimaging measures. However, there is a good deal of circumstantial evidence supporting the hypothesis that functional neuroimaging measures are influenced by genetics. First, conceptually similar electrophysiological measures (e.g. ERP, EEG) have repeatedly been shown to be highly heritable (van Beijsterveldt and van Baal, 2002; van Beijsterveldt et al., 1996] and have even been successfully applied in large‐scale linkage analysis [Edenberg et al., 2004]. Second, a number of recent manuscripts have reported associations between specific genes and functional activation patterns [Hariri et al., 2002, 2003; Meyer‐Lindenberg et al., 2005; Pezawas et al., 2004], suggesting genetic underpinnings for functional neuroimaging markers. Third, several investigators have demonstrated that healthy siblings or cotwins of individuals with psychiatric disorders have activation patterns similar to their affected family member [Hirvonen et al., 2005; Karlsgodt et al., 2007; Macdonald et al., 2006], suggesting that at least a portion of the aberrant signal detected in the proband may be genetically mediated. Finally, many of the cognitive and behavioral challenges applied in neuroimaging experiments have high levels of heritability [Bouchard and McGue, 1981; Glahn et al., 2007], implying that the activation pattern associated with this challenge may also be heritable. Thus, while there is currently little published data on the heritability of functional neuroimaging measures, much of brain function is genetically mediated, so that, assuming adequate reliability of the measurements, functional neuroimaging data is likely heritable.
FUNCTIONAL NEUROIMAGING OF BRAIN DISORDERS
Functional neuroimaging methods provide information on the integration and interaction of brain regions in large‐scale spatially distinct neural networks during cognitive or behavioral challenges or in response to physiological stimuli. Such data are critical for understanding the neuropsychological response to information processing demands at the systems level and, thus, may be particularly sensitive to neuropathologic changes found in individuals with brain‐related disorders [Gothelf et al., 2005; Hirvonen et al., 2006; Liguori et al., 2006; Seidman et al., 2006]. Furthermore, there is growing evidence that functional neuroimaging methods can distinguish between behaviorally asymptomatic individuals at high and low genetic risk for a neurological or psychiatric illness [Small, 2006; Whalley et al., 2006] For example, Bookheimer et al. [2000] studied 30 neurologically healthy individuals who performed a declarative memory task during a functional MRI experiment. Sixteen of these subjects had at least one copy of the apolipoprotein (APOE) ε4 allele, a gene strongly linked to Alzheimer's dementia [Corder et al., 1993; Saunders et al., 1993]. Although the mean age and level of education were similar in the two groups, those individuals with an ε4 allele had significantly stronger imaging responses in regions affected by Alzheimer's disease, including the left hippocampal, parietal, and prefrontal regions, than persons who were ε3 homozygous [Bookheimer et al., 2000]. In addition, the degree of baseline brain activation correlated with the degree of decline in memory 2 years after the scan, suggesting that patterns of brain activation are sensitive to genetic risk of Alzheimer's disease and predicted subsequent memory impairment. More recently, a number of studies have demonstrated that functional neuroimaging methods are sensitive to polymorphisms in genes associated with neurotransmitter systems [Egan et al., 2001; Mattay et al., 2003; Pezawas et al., 2005; Smolka et al., 2005] or neurodevelopment [Hariri et al., 2003]. Together, these data suggest that markers of brain function may be more sensitive to subtle genetic variations than overt behavior [Hariri and Weinberger, 2003]. This claim is supported by Hariri et al. [2002], who found that a functional polymorphism in the promoter region of the human serotonin transporter gene (SLC6A4) predicted amygdala response to fearful or angry faces, while behavioral measures of normal fear or pathological anxiety did not statistically differ between groups. However, questions remain about the generalizability or reproducibility of genetic association studies employing functional neuroimaging markers and the utility of this approach for genes lacking a clear functional link to brain processing. Nonetheless, the application of functional neuroimaging methods to genetic association studies represents a significant proof of concept for the integration of imaging and genetic approaches to understanding neurological and psychiatric illnesses.
DETECTION, LOCALIZATION AND IDENTIFICATION OF DISEASE‐RELATED QUANTITATIVE TRAIT LOCI
In this postgenomic era, one approach to understanding the genetic architecture of a complex phenotype follows a specific route often described as positional cloning. Initially, an underlying QTL is localized by a genomic scan to a potentially large chromosomal region. This localization is usually accomplished by linkage analysis using data on the cosegregation of phenotypes and genetic markers in families. Whole genome scanning using an association approach in unrelated samples is an alternative design, although it is still in its infancy as huge numbers of single nucleotide polymorphisms (SNP) are typically required to adequately cover the genome and there is a lack of statistical methods or power to adequately control Type I and II error. After a QTL is localized, its chromosomal location should be refined through fine mapping methods, typically including the saturation of the positional candidate region with additional genetic markers. It is possible to simultaneously exploit information on both linkage and linkage disequilibrium (whose effective signal spans a much smaller region than a linkage signal does).
The genetic analysis of brain structure is one of the most critically important areas of biomedical science. However, the genetic architecture of the brain is complex, involving multiple genetic and environmental components and their interactions. The specific QTLs that are involved in the developmental pathways producing adult brain structure, and their individual effects on the general population, are still largely unknown. Recent studies have pointed to specific genes that may have significant influence on variation in human brain structure [Evans et al., 2005; Mekel‐Bobrov et al., 2005] but the significance of these genes has not yet been quantified, and there are certainly many other genes that affect variation in brain morphology. The complexity of the genotype–phenotype relationships relevant to such complex phenotypes requires that statistical inference play a prominent role in the dissection of the underlying genetic architecture. Statistical genetic methods suitable for this immense task are still in their early development.
In recent years, substantial advances have been made in the techniques for finding QTLs influencing disease‐related traits. High‐throughput genotyping methods have revolutionized the search for complex disease loci and the resulting emphasis on linkage studies utilizing total genome scans represents the current state‐of‐the‐science. Additional molecular advances in high‐throughput resequencing and SNP typing are also of considerable value for identifying the functional variants in positional candidate loci identified by linkage‐based genome scans. Given an adequate sampling design, it will be possible to localize important genes involved in both normal and pathological variation using a genomic scan strategy coupled with follow‐up fine mapping using linkage disequilibrium/association‐based methods. It is now clear, with the advent of high throughput resequencing, that the next phase of genetic research requires new methods to identify functionally relevant polymorphisms in positional candidate genes.
Evidence is accumulating rapidly that it is feasible to identify the underlying genes influencing quantitative variation by combining molecular and statistical approaches. A number of specific functional genes that influence complex phenotypes in humans have been successfully identified, including genes underlying QTLs for taste sensitivity to PTC [Kim et al., 2003], asthma [Zhang et al., 2003], IgE serum levels [Zhang et al., 2003], stroke [Gretarsdottir et al., 2003], and osteoporosis [Styrkarsdottir et al., 2003]. Each of these genes was found through linkage‐based localization of QTLs. Similar methods will eventually identify genes influencing brain structure.
MAPPING GENES INFLUENCING QUANTITATIVE RISK FACTORS
Much of the recent success in the initial localization of QTLs influencing quantitative disease risk factors in extended human pedigrees is attributable to the development of the variance component method of linkage analysis. Variance component linkage/disequilibrium methods have several advantages over classical penetrance‐model‐based quantitative trait linkage methods. The simpler parameterization of variance component models leads to a more parsimonious and better‐estimated set of focal parameters. In variance component‐based linkage analysis, the focal parameter is the QTL‐specific additive genetic variance or QTL‐specific heritability. For a simple one‐locus model with a diallelic QTL, the QTL‐specific additive genetic variance can be written as σ = 2p q(1 − p q)α2, where p q is the allele frequency of the QTL polymorphic variant and α is half the displacement between the two homozygous genotypes. Thus, at minimum, a penetrance‐based linkage analysis would need to specify or estimate p q and α, while in variance component‐based linkage analysis, these two parameters are absorbed into σ. We have recently shown that little power is gained over the simpler variance component parameterization even when the penetrance model can be specified [Goring et al., 2001]. Moreover, the power for variance component‐based linkage analysis can greatly exceed that of penetrance‐model based analysis when the penetrance model is misspecified, as is always the case when there are multiple QTLs or many functional variants at a QTL. The variance component approach can accommodate pedigrees of any size and complexity, while penetrance‐based models rapidly become computationally intractable as pedigree size/complexity increases. Since it is now clear that large complex pedigrees have substantially more power per sampled individual than do smaller families [Blangero et al., 2000a, b, 2001, 2003], the advantage of using variance component methods for localizing QTLs is considerable. Hence, the optimal design for identifying genes that influence neuroanatomic variation is large extended pedigrees.
DETECTING GENETIC ASSOCIATION
In addition to linkage‐based approaches to QTL localization and identification, it is possible to examine the potential effect of known polymorphic variation on a given trait using an association‐based paradigm. Association studies either involve family‐based sampling or involve a sampling strategy including unrelated individuals. Genetic association tests are dependent on the sequence variant in question having a true functional effect on the phenotype, or possibly being in linkage disequilibrium with a functional variant. Linkage disequilibrium simply represents the statistical correlation between two sequence variations due to a shared history and, thus, is a purely stochastic phenomenon. The extent of linkage disequilibrium is highly circumscribed (usually less than 200–300 kb) and thus can be exploited to limit the continued search for the responsible gene(s) to a much smaller genomic region than is possible with linkage (linkage signals can span more than 20 Mb). However, linkage represents a much more predictable force than disequilibrium, since it is ruled by a biophysical phenomenon with a well‐known and precise mathematical form.
Association studies of quantitative endophenotypes, like neuroimaging measures, usually involve simple tests for differences in means between marker genotypes, typically a single nucleotide polymorphism or SNP. The total relative signal of a genetic marker is given by the marker‐specific genetic variance [σ = 2p m(1 − p m)α2] or heritability [h = σ/(1 + σ)]. In the simple case of a single variant underlying a QTL, the marker‐specific heritability is related to the QTL‐specific heritability by the equation σ = ρ2σ, where ρ is the correlation between the marker variant and the true functional variant [Blangero et al., 2005]. Thus, ρ is a direct measure of linkage disequilibrium and the higher ρ values reflect greater relative signal due to the marker. If ρ is 1, then the marker is itself the functional variant or is in such strong statistical correlation with the true variant that the two could not be separated using statistical inference.
LEVELS OF INFERENCE IN GENETIC ASSOCIATION
Association studies can have different levels of focus. In the extreme case, it is possible to scan the whole genome for association using chip‐based genotyping. In such cases, more than 500,000 SNPs may be analyzed [Lawrence et al., 2005]. At the other extreme, a study may be interested in testing a point hypothesis about a specific genetic marker in a known candidate gene. This latter situation should only be undertaken in the presence of extremely strong prior evidence for a marker. Testing a single marker may be appropriate in replication studies or in studies done subsequent to the definitive identification of the molecular functionality of a variant. All other studies of candidate genes should generally focus on exhaustive (or nearly exhaustive) examination of the sequence variation within the gene. A HapMap‐based approach that employs common marker variants chosen to cover major blocks of linkage disequilibrium within a gene may or may not prove useful. If rare variants are important in quantitative variation, such disequilibrium‐based approaches will not work, since rare variants will not be in high correlation with common marker variants. Given the increasing evidence for the importance of rare variation, the future of human genetic analysis is certain to lay in the area of large‐scale comprehensive resequencing in which all variants are identified and tested for statistical association [Blangero, 2004]. Such an approach requires new statistical techniques for the prioritization of the most likely function.
There are several critical issues in the design of a valid association study, including the overall power of a study to detect a true association is a function of both the sample size and the number of variants to be tested. The multiple testing problem can be potentially enormous and dramatically limit the power of association studies. Figure 2 shows the power function for a hypothetical study involving the analysis of a single quantitative endophenotype studied in 1,000 unrelated subjects. The three lines represent the level of variant examination being undertaken running the gamut from the analysis of a single SNP (blue line) to the comprehensive analysis of a single gene (red line, 50 total sequence variants) to the genome‐wide analysis of 5,000,000 markers (black line). Each power function is based on an experiment‐wide significance level of 0.05 as calculated by a standard Bonferroni correction. As is clearly shown in Figure 2, the reduction in power that is observed in multiple testing can be appreciable. If we are studying only a single SNP, we have 80% power to detect an association that accounts for as little as 0.8% of the total phenotypic variance of the endophenotype. For a study in which a gene is comprehensively analyzed with 50 variants, we have 80% power to detect an association that accounts for as little as 1.7% of the total variation, while for a genome‐wide association study, we require a large genetic signal that accounts for at least 3.7% of the total phenotypic variation before we achieve 80% power of detection. This latter marker‐specific heritability would be on the high side for a single functional variant. The Bonferroni correction is conservative but the overall pattern of these results is retained for other potentially more powerful multiple test correction methods such as utilization of a false discovery rate [Benjamini and Hochberg, 1995].
Figure 2.
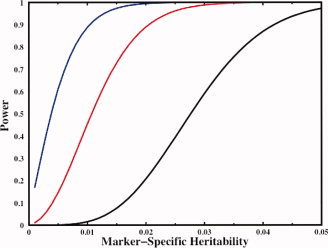
Power to detect genetic association. A power function for a hypothetical study involving the analysis of a single quantitative endophenotype studied in 1,000 unrelated subjects. The three lines represent the level of variant examination being undertaken running the gamut from the analysis of a single SNP (blue line) to the comprehensive analysis of a single gene (red line, 50 sequence variants) to the genome‐wide analysis of 5,000,000 markers (black line). Each power function is based on an experiment‐wide significance level of 0.05 as calculated by a standard Bonferroni correction. For a single SNP, we have 80% power to detect an association that accounts for as little as 0.8% of the total phenotypic variance of the endophenotype. For a gene with 50 variants, we have 80% power to detect an association that accounts for 1.7% of total variation, while for a genome‐wide association study, we require a large genetic signal that accounts for at least 3.7% of the total phenotypic variation before we achieve 80% power of detection. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
This simple example also suggests the great importance of providing accurate experiment‐wide P‐values for results when publishing. A nominal significance level of 0.05 has little place in modern complex disease genetics, since the likelihood of a study typing only a single variant is very small. Thus, the nominal P‐values published so widely in the incipient brain endophenotype association literature have little validity for inference in the presence of multiple testing.
Another important consideration in the design of a valid association study is the avoidance of genetic stratification in which individuals from diverse genetic backgrounds are mixed within a single study. It is well known that such hidden stratification leads to excessive Type 1 error due to the confounding of population‐level genetic variation with phenotypic variation. A number of methods have been proposed for detecting and correcting for such hidden heterogeneity. However, the best study design actively avoids such stratification by focusing on more homogenous samples or by the utilization of family‐based data, which is not influenced by this issue. In fact, family‐based studies are probably the most justifiable design for the genetic analysis of quantitative brain structure and function, since both linkage and linkage disequilibrium information can be jointly exploited to localize and identify underlying QTLs.
CONCLUSIONS
Genetically complex brain‐related disorders cost the United States as much as $1.2 trillion annually [Uhl and Grow, 2004], in addition to the enormous personal cost to patients and their caregivers. Any novel insights into biological mechanisms that predispose individuals to these illnesses hold the promise of potential new therapies and significant reduction of this considerable burden and improve the lives of individuals affected by these illnesses. The use of neuroimaging measures as endophenotypes for neurological and psychiatric illnesses could lead to new gene discoveries, and a better understanding of the biological mechanisms in these disorders. Thus the application of imaging measures in molecular genetics investigations (and vice versa) could significantly improve our understanding of brain function and structure. In this manuscript, we reviewed evidence that many neurological and psychiatric illnesses are associated with identifiable neuroanatomic and neurophysiologic changes and that many of these measures are heritable. Additional information may be needed when studying a specific illness, but nonetheless, we strongly advocate the use of neuroimaging measures in genetic studies.
Despite our enthusiasm for integrating genetic and neuroimaging investigations, we must discuss several caveats. First, both imaging and genetic data are complex. Each has millions of possible variables and the potential for spurious findings is tremendous. To minimize the computational requirements and to limit the number of potential statistical tests, many groups reduce the imaging data to a small number of scalar values per subject (e.g., prefrontal gray matter density or amygdala activation). Alternately, one could use only a small subset of the available genetic information by focusing on a single SNP or haplotype. These data reduction procedures are currently necessary because the mathematical procedures needed to control Type I error are not yet available. However, by significantly reducing one or both data types, important relationships will be missed. Thus, the development of high‐dimensional procedures to allow whole genome—full brain analyses is necessary to fully explore these rich sources of data.
Independent of the complexity of these two data types, one should be concerned with measurement reliability. Large‐scale genetic studies routinely apply methods to determine the reliability and validity of each sample being processed (e.g., SNP consistency, gender matching, relationship within pedigree) and procedures to minimize potential genotyping error. Similar methods are not ubiquitously applied in neuroimaging studies, though quality assurance procedures typically reduce error related to data acquisition. Most neuroimaging studies require significant postprocessing to render raw data into behaviorally/genetically meaningful information (e.g., statistical activation maps for fMRI studies or hippocampal demarcation for neuroanatomic studies). However, the reliability of these measures for specific experiments is often not provided. Although many neuroimaging measurements are quite reliable [Specht et al., 2003], the relative reliability across individuals or over time of specific dependent measures (e.g. the exact locus of activation vs. the mean activation vs. the maximum statistical value in a region for a functional neuroimaging experiment) should be considered when choosing measures to apply in genetic analyses.
Another caveat is related to sample size. Genetic studies typically require large sample sizes (between 1,000 and 3,000) to be powered to detect medium to small gene effects. In contrast, most functional neuroimaging studies include 10–20 subjects. Such small studies require strong prior hypotheses (which are generally lacking in complex disease genetics) and extreme ascertainment strategies to have any chance of detecting a true relationship. This disparity in typical sample sizes is a potential point of friction between the traditional geneticist and those originally trained as neuroimagers. Hariri and Weinberger [2003] recently proposed that functional neuroimaging data might represent a final common pathway for neuronal activity before it is transformed into a myriad of possible behaviors. As such, functional neuroimaging data could be significantly more sensitive to gene action than overt behavior and thus far smaller sample sizes may be needed to detect activity using neuroimaging data than behavioral data. To date, this hypothesis has not been formally tested and the number of subjects needed for imaging genomics studies is an open debate.
Despite possible limitations, we believe that the integration of neuroimaging and genetics information will significantly advance our understanding of systems‐level neuroscience and provide novel insights into brain‐related disorders.
REFERENCES
- Ashburner J, Csernansky JG, Davatzikos C, Fox NC, Frisoni GB, Thompson PM ( 2003): Computer‐assisted imaging to assess brain structure in healthy and diseased brains. Lancet Neurol 2: 79–88. [DOI] [PubMed] [Google Scholar]
- Atwood LD, Wolf PA, Heard‐Costa NL, Massaro JM, Beiser A, D'Agostino RB, DeCarli C ( 2004): Genetic variation in white matter hyperintensity volume in the Framingham Study. Stroke 35: 1609–1613. [DOI] [PubMed] [Google Scholar]
- Baare WF, Hulshoff Pol HE, Boomsma DI, Posthuma D, de Geus EJ, Schnack HG, van Haren NE, van Oel CJ, Kahn RS ( 2001): Quantitative genetic modeling of variation in human brain morphology. Cereb Cortex 11: 816–824. [DOI] [PubMed] [Google Scholar]
- Bartley AJ, Jones DW, Weinberger DR ( 1997): Genetic variability of human brain size and cortical gyral patterns. Brain 120 (Part 2): 257–269. [DOI] [PubMed] [Google Scholar]
- Bearden CE, Thompson PM, Dalwani M, Hayashi KM, Lee AD, Nicoletti M, Trakhenbroit M, Glahn DC, Brambilla P, Sassi RB, Mallinger AG, Frank E, Kupfer DJ, Soares JC: Greater cortical gray matter density in lithium‐treated patients with bipolar disorder. Biol Psychiatry (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begleiter H, Porjesz B ( 1999): What is inherited in the predisposition toward alcoholism? A proposed model. Alcohol Clin Exp Res 23: 1125–1135. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y ( 1995): Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc B 57: 289–300. [Google Scholar]
- Bergem AL, Engedal K, Kringlen E ( 1997): The role of heredity in late‐onset Alzheimer disease and vascular dementia. A twin study. Arch Gen Psychiatry 54: 264–270. [DOI] [PubMed] [Google Scholar]
- Berkovic SF, Howell RA, Hay DA, Hopper JL ( 1998): Epilepsies in twins: Genetics of the major epilepsy syndromes. Ann Neurol 43: 435–445. [DOI] [PubMed] [Google Scholar]
- Blangero J ( 2004): Localization and identification of human quantitative trait loci: King harvest has surely come. Curr Opin Genet Dev 14: 233–240. [DOI] [PubMed] [Google Scholar]
- Blangero J, Williams JT, Almasy L ( 2000a): Quantitative trait locus mapping using human pedigrees. Hum Biol 72: 35–62. [PubMed] [Google Scholar]
- Blangero J, Williams JT, Almasy L ( 2000b): Robust LOD scores for variance component‐based linkage analysis. Genet Epidemiol 19 ( Suppl 1): S8–S14. [DOI] [PubMed] [Google Scholar]
- Blangero J, Williams JT, Almasy L ( 2001): Variance component methods for detecting complex trait loci. Adv Genet 42: 151–181. [DOI] [PubMed] [Google Scholar]
- Blangero J, Williams JT, Almasy L ( 2003): Novel family‐based approaches to genetic risk in thrombosis. J Thromb Haemost 1: 1391–1397. [DOI] [PubMed] [Google Scholar]
- Blangero J, Goring HHH, Kent JWJ, Williams JT, Peterson C, Almasy L, Dyer TD ( 2005): Quantitative trait nucleotide analysis using Bayesian model selection. Hum Biol 77: 541–559. [DOI] [PubMed] [Google Scholar]
- Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak‐Vance MA, Mazziotta JC, Small GW ( 2000): Patterns of brain activation in people at risk for Alzheimer's disease. N Engl J Med 343: 450–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard TJ Jr, McGue M ( 1981): Familial studies of intelligence: A review. Science 212: 1055–1059. [DOI] [PubMed] [Google Scholar]
- Brown FW, Lewine RJ, Hudgins PA, Risch SC ( 1992): White matter hyperintensity signals in psychiatric and nonpsychiatric subjects. Am J Psychiatry 149: 620–625. [DOI] [PubMed] [Google Scholar]
- Callicott JH, Mattay VS, Bertolino A, Finn K, Coppola R, Frank JA, Goldberg TE, Weinberger DR ( 1999): Physiological characteristics of capacity constraints in working memory as revealed by functional MRI. Cerebral Cortex 9: 20–26. [DOI] [PubMed] [Google Scholar]
- Canive JM, Lewine JD, Orrison WW Jr, Edgar CJ, Provencal SL, Davis JT, Paulson K, Graeber D, Roberts B, Escalona PR, Calais L ( 1997): M.R.I. reveals gross structural abnormalities in PTSD. Ann NY Acad Sci 821: 512–515. [DOI] [PubMed] [Google Scholar]
- Cannon TD, Kaprio J, Lonnqvist J, Huttunen M, Koskenvuo M ( 1998): The genetic epidemiology of schizophrenia in a Finnish twin cohort. A population‐based modeling study. Arch Gen Psychiatry 55: 67–74. [DOI] [PubMed] [Google Scholar]
- Cannon TD, Thompson PM, van Erp TG, Toga AW, Poutanen VP, Huttunen M, Lonnqvist J, Standerskjold‐Nordenstam CG, Narr KL, Khaledy M, Zoumalan CI, Dail R, Kaprio J ( 2002): Cortex mapping reveals regionally specific patterns of genetic and disease‐specific gray‐matter deficits in twins discordant for schizophrenia. Proc Natl Acad Sci USA 99: 3228–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmelli D, DeCarli C, Swan GE, Jack LM, Reed T, Wolf PA, Miller BL ( 1998): Evidence for genetic variance in white matter hyperintensity volume in normal elderly male twins. Stroke 29: 1177–1181. [DOI] [PubMed] [Google Scholar]
- Carper RA, Courchesne E ( 2005): Localized enlargement of the frontal cortex in early autism. Biol Psychiatry 57: 126–133. [DOI] [PubMed] [Google Scholar]
- Castellanos FX, Giedd JN, Marsh WL, Hamburger SD, Vaituzis AC, Dickstein DP, Sarfatti SE, Vauss YC, Snell JW, Lange N, Kaysen D, Krain AL, Ritchie GF, Rajapakse JC, Rapoport JL ( 1996): Quantitative brain magnetic resonance imaging in attention‐deficit hyperactivity disorder. Arch Gen Psychiatry 53: 607–616. [DOI] [PubMed] [Google Scholar]
- Castellanos FX, Lee PP, Sharp W, Jeffries NO, Greenstein DK, Clasen LS, Blumenthal JD, James RS, Ebens CL, Walter JM, Zijdenbos A, Evans AC, Giedd JN, Rapoport JL ( 2002): Developmental trajectories of brain volume abnormalities in children and adolescents with attention‐deficit/hyperactivity disorder. JAMA 288: 1740–1748. [DOI] [PubMed] [Google Scholar]
- Cheverud JM, Falk D, Hildebolt C, Moore AJ, Helmkamp RC, Vannier M ( 1990a): Heritability and association of cortical petalias in rhesus macaques (Macaca mulatta). Brain Behav Evol 35: 368–372. [DOI] [PubMed] [Google Scholar]
- Cheverud JM, Falk D, Vannier M, Konigsberg L, Helmkamp RC, Hildebolt C ( 1990b): Heritability of brain size and surface features in rhesus macaques (Macaca mulatta). J Hered 81: 51–57. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak‐Vance MA ( 1993): Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261: 921–923. [DOI] [PubMed] [Google Scholar]
- Eckert MA, Leonard CM, Molloy EA, Blumenthal J, Zijdenbos A, Giedd JN ( 2002): The epigenesis of planum temporale asymmetry in twins. Cereb Cortex 12: 749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg HJ, Dick DM, Xuei X, Tian H, Almasy L, Bauer LO, Crowe RR, Goate A, Hesselbrock V, Jones K, Kwon J, Li TK, Nurnberger JI Jr, O'Connor SJ, Reich T, Rice J, Schuckit MA, Porjesz B, Foroud T, Begleiter H ( 2004): Variations in GABRA2, encoding the α 2 subunit of the GABA(A) receptor, are associated with alcohol dependence and with brain oscillations. Am J Hum Genet 74: 705–714. Epub Mar 12, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE, Goldman D, Weinberger DR ( 2001): Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci USA 98: 6917–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PD, Gilbert SL, Mekel‐Bobrov N, Vallender EJ, Anderson JR, Vaez‐Azizi LM, Tishkoff SA, Hudson RR, Lahn BT ( 2005): Microcephalin, a gene regulating brain size, continues to evolve adaptively in humans. Science 309: 1717–1720. [DOI] [PubMed] [Google Scholar]
- Fennema‐Notestine C, Stein MB, Kennedy CM, Archibald SL, Jernigan TL ( 2002): Brain morphometry in female victims of intimate partner violence with and without posttraumatic stress disorder. Biol Psychiatry 52: 1089–1101. [DOI] [PubMed] [Google Scholar]
- Fleisher A, Grundman M, Jack CR Jr, Petersen RC, Taylor C, Kim HT, Schiller DH, Bagwell V, Sencakova D, Weiner MF, DeCarli C, DeKosky ST, van Dyck CH, Thal LJ; Alzheimer's Disease Cooperative Study ( 2005): Sex, apolipoprotein E epsilon 4 status, and hippocampal volume in mild cognitive impairment. Arch Neurol 62: 953–957. [DOI] [PubMed] [Google Scholar]
- Gershon E, Goldin L ( 1986): Clinical methods in psychiatric genetics. I. Robustness of genetic marker investigative strategies. Acta Psychiatr Scand 74: 113–118. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Miller BL, DeCarli C, Carmelli D ( 2002): Heritability of lobar brain volumes in twins supports genetic models of cerebral laterality and handedness. Proc Natl Acad Sci USA 99: 3176–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuze E, Vermetten E, Bremner JD ( 2005): MR‐based in vivo hippocampal volumetrics: 2. Findings in neuropsychiatric disorders. Mol Psychiatry 10: 160–184. [DOI] [PubMed] [Google Scholar]
- Glahn DC, Bearden CE, Niendam TA, Escamilla MA ( 2004): The feasibility of neuropsychological endophenotypes in the search for genes associated with bipolar affective disorder. Bipolar Disord 6: 171–182. [DOI] [PubMed] [Google Scholar]
- Glahn DC, Almasy L, Blangero J, Burk GM, Estrada J, Peralta JM, Meyenberg N, Castro MP, Barrett J, Nicolini H, Raventos H, Escamilla MA ( 2007): Adjudicating neurocognitive endophenotypes for schizophrenia. Am J Med Genet B Neuropsychiatr Genet 144: 242–249. [DOI] [PubMed] [Google Scholar]
- Gogtay N, Giedd JN, Lusk L, Hayashi KM, Greenstein D, Vaitiuzis C, Nugent TF, Herman DH, Classen L, Toga AW, Rapoport JL, Thompson PM ( 2004): Dynamic mapping of human cortical development during childhood and adolescence. Proc Natl Acad Sci USA 101: 8174–8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good CD, Scahill RI, Fox NC, Ashburner J, Friston KJ, Chan D, Crum WR, Rossor MN, Frackowiak RS ( 2002): Automatic differentiation of anatomical patterns in the human brain: Validation with studies of degenerative dementias. Neuroimage 17: 29–46. [DOI] [PubMed] [Google Scholar]
- Goring HH, Williams JT, Blangero J ( 2001): Linkage analysis of quantitative traits in randomly ascertained pedigrees: Comparison of penetrance‐based and variance component analysis. Genet Epidemiol 21 ( Suppl 1): S783–S788. [DOI] [PubMed] [Google Scholar]
- Gothelf D, Eliez S, Thompson T, Hinard C, Penniman L, Feinstein C, Kwon H, Jin S, Jo B, Antonarakis SE, Morris MA, Reiss AL ( 2005): COMT genotype predicts longitudinal cognitive decline and psychosis in 22q11.2 deletion syndrome. Nat Neurosci 8: 1500–1502. [DOI] [PubMed] [Google Scholar]
- Gottesman II, Gould TD ( 2003): The endophenotype concept in psychiatry: Etymology and strategic intentions. Am J Psychiatry 160: 636–645. [DOI] [PubMed] [Google Scholar]
- Gray JR, Thompson PM ( 2004): Neurobiology of intelligence: science and ethics. Nat Rev Neurosci 5: 471–482. [DOI] [PubMed] [Google Scholar]
- Gretarsdottir S, Thorleifsson G, Reynisdottir ST, Manolescu A, Jonsdottir S, Jonsdottir T, Gudmundsdottir T, Bjarnadottir SM, Einarsson OB, Gudjonsdottir HM, Hawkins M, Gudmundsson G, Gudmundsdottir H, Andrason H, Gudmundsdottir AS, Sigurdardottir M, Chou TT, Nahmias J, Goss S, Sveinbjornsdottir S, Valdimarsson EM, Jakobsson F, Agnarsson U, Gudnason V, Thorgeirsson G, Fingerle J, Gurney M, Gudbjartsson D, Frigge ML, Kong A, Stefansson K, Gulcher JR ( 2003): The gene encoding phosphodiesterase 4D confers risk of ischemic stroke. Nat Genet 35: 131–138. [DOI] [PubMed] [Google Scholar]
- Hanyu H, Imon Y, Sakurai H, Iwamoto T, Takasaki M, Shindo H, Kakizaki D, Abe K ( 1999): Regional differences in diffusion abnormality in cerebral white matter lesions in patients with vascular dementia of the Binswanger type and Alzheimer's disease. Eur J Neurol 6: 195–203. [DOI] [PubMed] [Google Scholar]
- Hariri AR, Weinberger DR ( 2003): Imaging genomics. Br Med Bull 65: 259–270. [DOI] [PubMed] [Google Scholar]
- Hariri AR, Mattay VS, Tessitore A, Kolachana B, Fera F, Goldman D, Egan MF, Weinberger DR ( 2002): Serotonin transporter genetic variation and the response of the human amygdala. Science 297: 400–403. [DOI] [PubMed] [Google Scholar]
- Hariri AR, Goldberg TE, Mattay VS, Kolachana BS, Callicott JH, Egan MF, Weinberger DR ( 2003): Brain‐derived neurotrophic factor val66met polymorphism affects human memory‐related hippocampal activity and predicts memory performance. J Neurosci 23: 6690–6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JM, Whalley H, Yates S, Miller P, Johnstone EC, Lawrie SM ( 2004): Abnormal cortical folding in high‐risk individuals: A predictor of the development of schizophrenia? Biol Psychiatry 56: 182–189. [DOI] [PubMed] [Google Scholar]
- Hill SY, De Bellis MD, Keshavan MS, Lowers L, Shen S, Hall J, Pitts T ( 2001): Right amygdala volume in adolescent and young adult offspring from families at high risk for developing alcoholism. Biol Psychiatry 49: 894–905. [DOI] [PubMed] [Google Scholar]
- Hirvonen J, van Erp TG, Huttunen J, Aalto S, Nagren K, Huttunen M, Lonnqvist J, Kaprio J, Hietala J, Cannon TD ( 2005): Increased caudate dopamine D2 receptor availability as a genetic marker for schizophrenia. Arch Gen Psychiatry 62: 371–378. [DOI] [PubMed] [Google Scholar]
- Hirvonen J, van Erp TG, Huttunen J, Aalto S, Nagren K, Huttunen M, Lonnqvist J, Kaprio J, Cannon TD, Hietala J ( 2006): Brain dopamine d1 receptors in twins discordant for schizophrenia. Am J Psychiatry 163: 1747–1753. [DOI] [PubMed] [Google Scholar]
- Honea R, Crow TJ, Passingham D, Mackay CE ( 2005): Regional deficits in brain volume in schizophrenia: a meta‐analysis of voxel‐based morphometry studies. Am J Psychiatry 162: 2233–2245. [DOI] [PubMed] [Google Scholar]
- Hull AM ( 2002): Neuroimaging findings in post‐traumatic stress disorder. Systematic review. Br J Psychiatry 181: 102–110. [PubMed] [Google Scholar]
- Hulshoff Pol HE, Brans RG, van Haren NE, Schnack HG, Langen M, Baare WF, van Oel CJ, Kahn RS ( 2004): Gray and white matter volume abnormalities in monozygotic and same‐gender dizygotic twins discordant for schizophrenia. Biol Psychiatry 55: 126–130. [DOI] [PubMed] [Google Scholar]
- Karas GB, Burton EJ, Rombouts SA, van Schijndel RA, O'Brien JT, Scheltens P, McKeith IG, Williams D, Ballard C, Barkhof F ( 2003): A comprehensive study of gray matter loss in patients with Alzheimer's disease using optimized voxel‐based morphometry. Neuroimage 18: 895–907. [DOI] [PubMed] [Google Scholar]
- Karlsgodt KH, Glahn DC, van Erp TG, Therman S, Huttunen M, Manninen M, Kaprio J, Cohen MS, Lonnqvist J, Cannon TD ( 2007): The relationship between performance and fMRI signal during working memory in patients with schizophrenia, unaffected co‐twins, and control subjects. Functional magnetic resonance imaging study of cognitive control in the healthy relatives of schizophrenia patients. Increased caudate dopamine D2 receptor availability as a genetic marker for schizophrenia. Schizophr Res 89: 191–197. Epub Oct 5, 2006. [DOI] [PubMed] [Google Scholar]
- Kim UK, Jorgenson E, Coon H, Leppert M, Risch N, Drayna D ( 2003): Positional cloning of the human quantitative trait locus underlying taste sensitivity to phenylthiocarbamide. Science 299: 1221–1225. [DOI] [PubMed] [Google Scholar]
- Lawrence RW, Evans DM, Cardon LR ( 2005): Prospects and pitfalls in whole genome association studies. Philos Trans R Soc Lond B Biol Sci 360: 1589–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leboyer M, Bellivier F, Nosten‐Bertrand M, Jouvent R, Pauls D, Mallet J ( 1998): Psychiatric genetics: Search for phenotypes. Trends Neurosci 21: 102–105. [DOI] [PubMed] [Google Scholar]
- Lenox RH, Gould TD, Manji HK ( 2002): Endophenotypes in bipolar disorder. Am J Med Genet 114: 391–406. [DOI] [PubMed] [Google Scholar]
- Liguori M, Fera F, Gioia MC, Valentino P, Manna I, Condino F, Cerasa A, La Russa A, Clodomiro A, Paolillo A, Nistico R, Vercillo L, Cittadella R, Quattrone A ( 2006): Investigating the role of brain‐derived neurotrophic factor in relapsing‐remitting multiple sclerosis. Genes Brain Behav 6: 177–183. [DOI] [PubMed] [Google Scholar]
- Lin JJ, Salamon N, Lee AD, Dutton RA, Geaga JA, Hayashi KM, London ED, Luders E, Toga AW, Engel J Jr, Thompson PM: Reduced Neocortical Thickness & Complexity Mapped in Mesial Temporal Lobe Epilepsy with Hippocampal Sclerosis. Cereb Cortex (in press). [DOI] [PubMed] [Google Scholar]
- Lingford‐Hughes AR, Davies SJ, McIver S, Williams TM, Daglish MR, Nutt DJ ( 2003): Addiction. Br Med Bull 65: 209–222. [DOI] [PubMed] [Google Scholar]
- Lyons DM, Yang C, Sawyer‐Glover AM, Moseley ME, Schatzberg AF ( 2001): Early life stress and inherited variation in monkey hippocampal volumes. Arch Gen Psychiatry 58: 1145– 1151. [DOI] [PubMed] [Google Scholar]
- Macdonald AW III, Becker TM, Carter CS ( 2006): Functional magnetic resonance imaging study of cognitive control in the healthy relatives of schizophrenia patients. Biol Psychiatry 60: 1241–1249. Epub Aug 30, 2006. [DOI] [PubMed] [Google Scholar]
- Maguire EA, Gadian DG, Johnsrude IS, Good CD, Ashburner J, Frackowiak RS, Frith CD ( 2000): Navigation‐related structural change in the hippocampi of taxi drivers. Proc Natl Acad Sci USA 97: 4398–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahaney MC, Williams‐Blangero S, Blangero J, Leland MM ( 1993): Quantitative genetics of relative organ weight variation in captive baboons. Hum Biol 65: 991–1003. [PubMed] [Google Scholar]
- Mattay VS, Goldberg TE, Fera F, Hariri AR, Tessitore A, Egan MF, Kolachana B, Callicott JH, Weinberger DR ( 2003): Catechol O‐methyltransferase val158‐met genotype and individual variation in the brain response to amphetamine. Proc Natl Acad Sci USA 100: 6186–61191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazziotta J, Toga A, Evans A, Fox P, Lancaster J, Zilles K, Woods R, Paus T, Simpson G, Pike B, Holmes C, Collins L, Thompson P, MacDonald D, Iacoboni M, Schormann T, Amunts K, Palomero‐Gallagher N, Geyer S, Parsons L, Narr K, Kabani N, Le Goualher G, Boomsma D, Cannon T, Kawashima R, Mazoyer B ( 2001): A probabilistic atlas and reference system for the human brain: International consortium for brain mapping (ICBM). Philos Trans R Soc Lond Ser B Biol Sci 356: 1293–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlonan GM, Cheung V, Cheung C, Suckling J, Lam GY, Tai KS, Yip L, Murphy DG, Chua SE. ( 2005): Mapping the brain in autism. A voxel‐based MRI study of volumetric differences and intercorrelations in autism. Brain 128 (Part 2): 268–276. [DOI] [PubMed] [Google Scholar]
- McDonald C, Bullmore ET, Sham PC, Chitnis X, Wickham H, Bramon E, Murray RM ( 2004a): Association of genetic risks for schizophrenia and bipolar disorder with specific and generic brain structural endophenotypes. Arch Gen Psychiatry 61: 974–984. [DOI] [PubMed] [Google Scholar]
- McDonald C, Zanelli J, Rabe‐Hesketh S, Ellison‐Wright I, Sham P, Kalidindi S, Murray RM, Kennedy N ( 2004b): Meta‐analysis of magnetic resonance imaging brain morphometry studies in bipolar disorder. Biol Psychiatry 56: 411–417. [DOI] [PubMed] [Google Scholar]
- Mekel‐Bobrov N, Gilbert SL, Evans PD, Vallender EJ, Anderson JR, Hudson RR, Tishkoff SA, Lahn BT ( 2005): Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens . Science 309: 1720–1722. [DOI] [PubMed] [Google Scholar]
- Merikangas KR, Chakravarti A, Moldin SO, Araj H, Blangero JC, Burmeister M, Crabbe J Jr, Depaulo JR Jr, Foulks E, Freimer NB, Koretz DS, Lichtenstein W, Mignot E, Reiss AL, Risch NJ, Takahashi JS ( 2002): Future of genetics of mood disorders research. Biol Psychiatry 52: 457–477. [DOI] [PubMed] [Google Scholar]
- Meyer‐Lindenberg A, Mervis CB, Sarpal D, Koch P, Steele S, Kohn P, Marenco S, Morris CA, Das S, Kippenhan S, Mattay VS, Weinberger DR, Berman KF ( 2005): Functional, structural, and metabolic abnormalities of the hippocampal formation in Williams syndrome. J Clin Invest 115: 1888–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills NP, Delbello MP, Adler CM, Strakowski SM ( 2005): MRI analysis of cerebellar vermal abnormalities in bipolar disorder. Am J Psychiatry 162: 1530–1532. [DOI] [PubMed] [Google Scholar]
- Moffat SD, Szekely CA, Zonderman AB, Kabani NJ, Resnick SM ( 2000): Longitudinal change in hippocampal volume as a function of apolipoprotein E genotype. Neurology 55: 134–136. [DOI] [PubMed] [Google Scholar]
- Monkul ES, Malhi GS, Soares JC ( 2005): Anatomical MRI abnormalities in bipolar disorder: Do they exist and do they progress? Aust N Z J Psychiatry 39: 222–226. [DOI] [PubMed] [Google Scholar]
- Mostofsky SH, Cooper KL, Kates WR, Denckla MB, Kaufmann WE ( 2002): Smaller prefrontal and premotor volumes in boys with attention‐deficit/hyperactivity disorder. Biol Psychiatry 52: 785–794. [DOI] [PubMed] [Google Scholar]
- Narr KL, Cannon TD, Woods RP, Thompson PM, Kim S, Asunction D, van Erp TG, Poutanen VP, Huttunen M, Lonnqvist J, Standerksjold‐Nordenstam CG, Kaprio J, Mazziotta JC, Toga AW ( 2002): Genetic contributions to altered callosal morphology in schizophrenia. J Neurosci 22: 3720–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler E, Gould E, Manji H, Buncan M, Duman R, Greshenfeld H, Hen R, Koester S, Lederhendler I, Meaney M, Robbins T, Winsky L, Zalcman S ( 2002): Preclinical models: Status of basic research in depression. Biol Psychiatry 52: 503–528. [DOI] [PubMed] [Google Scholar]
- Pezawas L, Verchinski BA, Mattay VS, Callicott JH, Kolachana BS, Straub RE, Egan MF, Meyer‐Lindenberg A, Weinberger DR ( 2004): The brain‐derived neurotrophic factor val66met polymorphism and variation in human cortical morphology. J Neurosci 24: 10099–10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezawas L, Meyer‐Lindenberg A, Drabant EM, Verchinski BA, Munoz KE, Kolachana BS, Egan MF, Mattay VS, Hariri AR, Weinberger DR ( 2005): 5‐HTTLPR polymorphism impacts human cingulate‐amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci 8: 828–834. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, Carmelli D ( 2001): Genetic regulation of regional microstructure of the corpus callosum in late life. Neuroreport 12: 1677–1681. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, Carmelli D ( 2004): Morphological changes in aging brain structures are differentially affected by time‐linked environmental influences despite strong genetic stability. Neurobiol Aging 25: 175–183. [DOI] [PubMed] [Google Scholar]
- Pierce K, Courchesne E ( 2001): Evidence for a cerebellar role in reduced exploration and stereotyped behavior in autism. Biol Psychiatry 49: 655–664. [DOI] [PubMed] [Google Scholar]
- Posthuma D, de Geus EJ, Neale MC, Hulshoff Pol HE, Baare WEC, Kahn RS, Boomsma D ( 2000): Multivariate genetic analysis of brain structure in an extended twin design. Behav Genet 30: 311–319. [DOI] [PubMed] [Google Scholar]
- Posthuma D, De Geus EJ, Baare WF, Hulshoff Pol HE, Kahn RS, Boomsma DI ( 2002): The association between brain volume and intelligence is of genetic origin. Nat Neurosci 5: 83–84. [DOI] [PubMed] [Google Scholar]
- Ragland JD, Glahn DC, Gur RC, Censits DM, Smith RJ, Mozley PD, Alavi A, Gur RE ( 1997): PET regional cerebral blood flow change during working and declarative memory: Relationship with task performance. Neuropsychology 11: 222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P ( 1988): Specification of cerebral cortical areas. Science 241: 170–176. [DOI] [PubMed] [Google Scholar]
- Rhee H, Waldman ID, Hay DA, Levy F ( 1999): Sex differences in genetic and environmental influences on DSM‐III‐R attention‐deficit/hyperactivity disorder. J Abnorm Psychol 108: 24–41. [DOI] [PubMed] [Google Scholar]
- Rojas DC, Smith JA, Benkers TL, Camou SL, Reite ML, Rogers SJ ( 2004): Hippocampus and amygdala volumes in parents of children with autistic disorder. Am J Psychiatry 161: 2038–2044. [DOI] [PubMed] [Google Scholar]
- Rubenstein JL, Rakic P ( 1999): Genetic control of cortical development. Cereb Cortex 9: 521–523. [DOI] [PubMed] [Google Scholar]
- Rubenstein JL, Anderson S, Shi L, Miyashita‐Lin E, Bulfone A, Hevner R ( 1999): Genetic control of cortical regionalization and connectivity. Cereb Cortex 9: 524–532. [DOI] [PubMed] [Google Scholar]
- Saunders AM, Strittmatter WJ, Schmechel D, George‐Hyslop PH, Pericak‐Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper‐MacLachlan DR, Alberts MJ, Hulette C, Crain B, Goldgaber D, Roses AD ( 1993): Association of apolipoprotein E allele epsilon 4 with late‐onset familial and sporadic Alzheimer's disease. Neurology 43: 1467–1472. [DOI] [PubMed] [Google Scholar]
- Scheltens P, Fox NC, Barkhof F, De Carli C ( 2002): Structural magnetic resonance imaging in the practical assessment of dementia: Beyond exclusion. Lancet Neurol 1: 13–21. [DOI] [PubMed] [Google Scholar]
- Schumann CM, Hamstra J, Goodlin‐Jones BL, Lotspeich LJ, Kwon H, Buonocore MH, Lammers CR, Reiss AL, Amaral DG ( 2004): The amygdala is enlarged in children but not adolescents with autism; the hippocampus is enlarged at all ages. J Neurosci 24: 6392–6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman LJ, Thermenos HW, Poldrack RA, Peace NK, Koch JK, Faraone SV, Tsuang MT ( 2006): Altered brain activation in dorsolateral prefrontal cortex in adolescents and young adults at genetic risk for schizophrenia: An fMRI study of working memory. Schizophr Res 85: 58–72. [DOI] [PubMed] [Google Scholar]
- Small GW ( 2006): Diagnostic issues in dementia: Neuroimaging as a surrogate marker of disease. J Geriatr Psychiatry Neurol 19: 180–185. [DOI] [PubMed] [Google Scholar]
- Smolka MN, Schumann G, Wrase J, Grusser SM, Flor H, Mann K, Braus DF, Goldman D, Buchel C, Heinz A ( 2005): Catechol‐O‐methyltransferase val158met genotype affects processing of emotional stimuli in the amygdala and prefrontal cortex. J Neurosci 25: 836–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowell ER, Thompson PM, Holmes CJ, Jernigan TL, Toga AW ( 1999): In vivo evidence for post‐adolescent brain maturation in frontal and striatal regions. Nat Neurosci 2: 859–861. [DOI] [PubMed] [Google Scholar]
- Sowell ER, Peterson BS, Thompson PM, Welcome SE, Henkenius AL, Toga AW ( 2003a): Mapping cortical change across the human lifespan. Nat Neurosci 6: 309–315. [DOI] [PubMed] [Google Scholar]
- Sowell ER, Thompson PM, Welcome SE, Henkenius AL, Toga AW, Peterson BS ( 2003b): Cortical abnormalities in children and adolescents with attention‐deficit hyperactivity disorder. Lancet 362: 1699–1707. [DOI] [PubMed] [Google Scholar]
- Specht K, Willmes K, Shah NJ, Jancke L ( 2003): Assessment of reliability in functional imaging studies. J Magn Reson Imaging 17: 463–471. [DOI] [PubMed] [Google Scholar]
- Stein MB, Jang KL, Taylor S, Vernon PA, Livesley WJ ( 2002): Genetic and environmental influences on trauma exposure and posttraumatic stress disorder symptoms: A twin study. Am J Psychiatry 159: 1675–1681. [DOI] [PubMed] [Google Scholar]
- Styner M, Lieberman JA, McClure RK, Weinberger DR, Jones DW, Gerig G ( 2005): Morphometric analysis of lateral ventricles in schizophrenia and healthy controls regarding genetic and disease‐specific factors. Proc Natl Acad Sci USA 102: 4872–4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styrkarsdottir U, Cazier JB, Kong A, Rolfsson O, Larsen H, Bjarnadottir E, Johannsdottir VD, Sigurdardottir MS, Bagger Y, Christiansen C, Reynisdottir I, Grant SF, Jonasson K, Frigge ML, Gulcher JR, Sigurdsson G, Stefansson K ( 2003): Linkage of osteoporosis to chromosome 20p12 and association to BMP2. PLoS Biol 1: E69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan EV, Pfefferbaum A ( 2001): Magnetic resonance relaxometry reveals central pontine abnormalities in clinically asymptomatic alcoholic men. Alcohol Clin Exp Res 25: 1206–1212. [PubMed] [Google Scholar]
- Sullivan EV, Pfefferbaum A, Swan GE, Carmelli D ( 2001): Heritability of hippocampal size in elderly twin men: Equivalent influence from genes and environment. Hippocampus 11: 754–762. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Cannon TD, Narr KL, van Erp T, Poutanen VP, Huttunen M, Lonnqvist J, Standertskjold‐Nordenstam CG, Kaprio J, Khaledy M, Dail R, Zoumalan CI, Toga AW ( 2001): Genetic influences on brain structure. Nat Neurosci 4: 1253–1258. [DOI] [PubMed] [Google Scholar]
- Thompson P, Cannon TD, Toga AW ( 2002): Mapping genetic influences on human brain structure. Ann Med 34: 523–536. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Hayashi KM, Simon S, Geaga J, Hong MS, Sui Y, Lee JY, Toga AW, Ling WL, London ED ( 2004): Structural abnormalities in the brains of human subjects who use methamphetamine. J Neurosci 24: 6028–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PM, Hayashi KM, Dutton RA, Chiang MC, Leow AD, Sowell ER, de Zubicaray GI, Becker JT, Lopez OL, Aizenstein HJ, et al. 2006. Tracking Alzheimer's disease. In: De Leon MJ, Federoff H, Hirsch J, Martin GM, Morrison J, Snider A, editors. Proceedings of the New York Academy of Sciences, New York. [DOI] [PMC free article] [PubMed]
- Uhl GR, Grow RW ( 2004): The burden of complex genetics in brain disorders. Arch Gen Psychiatry 61: 223–229. [DOI] [PubMed] [Google Scholar]
- van Beijsterveldt CE, Molenaar PC, de Geus EJ, Boomsma DI ( 1996): Heritability of human brain functioning as assessed by electroencephalography. Am J Hum Genet 58: 562–573. [PMC free article] [PubMed] [Google Scholar]
- van Beijsterveldt CE, van Baal GC ( 2002): Twin and family studies of the human electroencephalogram: A review and a meta‐analysis. Biol Psychol 61: 111–138. [DOI] [PubMed] [Google Scholar]
- Videbech P, Ravnkilde B ( 2004): Hippocampal volume and depression: A meta‐analysis of MRI studies. Am J Psychiatry 161: 1957–1966. [DOI] [PubMed] [Google Scholar]
- Whalley HC, Simonotto E, Moorhead W, McIntosh A, Marshall I, Ebmeier KP, Owens DG, Goddard NH, Johnstone EC, Lawrie SM ( 2006): Functional imaging as a predictor of schizophrenia. Biol Psychiatry 60: 454–462. [DOI] [PubMed] [Google Scholar]
- Wignall EL, Dickson JM, Vaughan P, Farrow TF, Wilkinson ID, Hunter MD, Woodruff PW ( 2004): Smaller hippocampal volume in patients with recent‐onset posttraumatic stress disorder. Biol Psychiatry 56: 832–836. [DOI] [PubMed] [Google Scholar]
- Wright IC, Sharma T, Ellison ZR, McGuire PK, Friston KJ, Brammer MJ, Murray RM, Bullmore ET ( 1999): Supra‐regional brain systems and the neuropathology of schizophrenia. Cereb Cortex 9: 366–378. [DOI] [PubMed] [Google Scholar]
- Wright IC, Rabe‐Hesketh S, Woodruff PW, David AS, Murray RM, Bullmore ET ( 2000): Meta‐analysis of regional brain volumes in schizophrenia. Am J Psychiatry 157: 16–25. [DOI] [PubMed] [Google Scholar]
- Wright IC, Sham P, Murray RM, Weinberger DR, Bullmore ET ( 2002): Genetic contributions to regional variability in human brain structure: Methods and preliminary results. Neuroimage 17: 256–271. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Leaves NI, Anderson GG, Ponting CP, Broxholme J, Holt R, Edser P, Bhattacharyya S, Dunham A, Adcock IM, Pulleyn L, Barnes PJ, Harper JI, Abecasis G, Cardon L, White M, Burton J, Matthews L, Mott R, Ross M, Cox R, Moffatt MF, Cookson WO ( 2003): Positional cloning of a quantitative trait locus on chromosome 13q14 that influences immunoglobulin E levels and asthma. Nat Genet 34: 181–186. [DOI] [PubMed] [Google Scholar]