

Abstract
This article describes a suite of computational approaches suitable for deriving various quantitative phenotypes from structural magnetic resonance (MR) images obtained in rodents and used subsequently in genetic studies of complex traits. We begin by introducing the basic principles of genetic studies of complex traits in experimental models. We then illustrate the use of MR‐based computational anatomy in vivo and ex vivo, and in combination with histology. This work was carried out in two inbred strains of rats, namely spontaneously hypertensive rats and Brown Norway rats; these are parental strains of the only existing panel of recombinant inbred strains of rats. The rats were scanned in vivo at two time points (at 8 and 12 weeks of age) and ex vivo (at 12 weeks of age). We describe between‐strain differences and across‐time changes in brain and kidney volumes, as well as regional variations in brain structure using surface‐ and deformation‐based approaches. We conclude by discussing the power of the population‐based computational analysis of MR images, and their fusion with histology, in studies of complex traits. Hum Brain Mapp, 2007. © 2007 Wiley‐Liss, Inc.
Keywords: phenotype, complex traits, recombinant inbred strain, rat, magnetic resonance imaging, histology, neuroanatomy
INTRODUCTION
Most common human diseases, such as hypertension or depression, are complex genetic traits; their pathogenesis is multifactorial involving multiple genes and environments. The action of these two groups of factors is intricate and includes various types of gene–gene and gene–environment interactions [Botstein and Risch, 2003; Hoh and Ott, 2004; Pausova et al., 1999]. The latter type of interactions may also involve long‐term epigenetic modifications of the genome, such as DNA methylation and histone acetylation [Richards, 2006]. Complex genetic traits are also characterized by genetic heterogeneity: distinct sets of genes may cause the development of the same trait in different individuals [Lander and Schork, 1994].
Genetic dissection of complex traits (or phenotypes) is carried out by measuring genotypes and phenotypes and examining, statistically, their relationships [Hoh and Ott, 2003; Lander and Kruglyak, 1995; Mashimo et al., 2006; Morton, 2005; Twigger et al., 2005]. This can be performed at either genome‐wide level or single‐gene level. A genome‐wide scan is conducted without any a priori hypothesis by investigating genotype–phenotype relationships using about 500 variable number of tandem repeats markers or 500,000 single nucleotide polymorphisms evenly distributed throughout the genome. Candidate‐gene studies involve examination of genotype–phenotype relationships at a single‐gene locus; a candidate gene is selected on the basis of its known physiological role in the phenotype under study.
The phenotypes under study can be either qualitative (e.g. presence [affected] or absence [not affected] of hypertension or depression) or quantitative (e.g. blood pressure or number/duration of depressive episodes). Complex genetic traits develop due to an overlapping subset of several pathogenetic mechanisms. Highly heritable traits, such as the degree and distribution of adiposity or volume of the anterior cingulate cortex, segregate in families with hypertension and depression, respectively [Pausova et al., 2001]. These traits represent intermediate phenotypes (or endophenotypes) in the chain of pathophysiological events leading from specific gene variants to the development of a complex genetic trait [Williams et al., 1996]. Because genetic architecture of these intermediate phenotypes is expected to be less complex than that of hypertension and depression themselves, the likelihood of identifying their genetic determinants is probably higher.
The multifaceted genetic nature of common human diseases has made it rather difficult to identify the responsible genes. The use of experimental animal models represents an attractive complementary approach to genetic studies of common diseases in humans [Darvasi, 1998; Flint et al., 2005; Nadeau et al., 2000]. Experimental models allow investigators to control both the genes and environment (e.g. diet, social interactions), and to acquire detailed in vivo and ex vivo phenotypes at different levels of analysis. Several experimental models exist that were created from genetically defined rats and mice. These models are generated by specific breeding schemes, starting most often from two genetically diverse, inbred strains (so‐called progenitor strains). Inbred strains are produced by selective brother–sister mating, which leads to the development of homozygozity at all chromosomal loci and, hence, genotype stability over time. In other words, breeding within an inbred strain produces genetically identical animals. One such experimental model are recombinant inbred strains; they are a set of inbred strains produced by crossing two progenitor strains followed by brother–sister breeding of their progeny for at least 20 generations. The panel consists of strains in which various random combinations of parental genomes are fixed (Fig. 1). As the strains are genetically reproducible, genotyping for a genome‐wide scan does not have to be repeated in each new generation of animals. In fact, most panels of these strains have already been genotyped with genome‐wide sets of DNA markers and these genotypes can be readily used in all subsequent studies carried out in these strains [Reuveni et al., 2007; Twigger et al., in press]. Furthermore, because all animals of a given recombinant inbred strain are genetically identical, various types of phenotypes obtained in different individuals can be considered in the same way as if they were obtained in the same individual. Under carefully controlled conditions, for example, variations in blood pressure recorded with radiotelemetry in one animal and adiposity measured with magnetic resonance imaging (MRI) in another can be correlated as if obtained from the same animal. Likewise, gene–environment interactions can be investigated in a more direct way, as genetically “identical” animals can be exposed to both experimental and control environments. The relative disadvantage of recombinant inbred strains is the fact that most existing panels consist of a relatively limited number of strains (n = 20–100) and, as such, they do not have sufficient power to map “small” gene effects. To improve mapping power and resolution, and to keep the advantages of studying inbred strains, it has been proposed recently to create The Collaborative Cross, a panel of 1,000 recombinant inbred strains of rats originating from eight genetically diverse progenitor strains. Although costly, this panel is hoped to be effective for studying complex polygenic networks and interactions between genes, environments and other factors [The Complex Trait Consortium, 2004].
Figure 1.
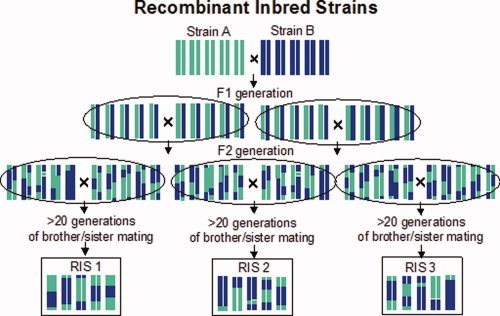
Recombinant Inbred Strains (RIS) are a set of inbred strains that are produced by intercrossing two progenitor strains followed by intercross inbreeding of their progeny for at least 20 generations. The panel consists of strains in which various random combinations of parental genomes are fixed. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Magnetic resonance imaging (MRI) provides multiple opportunities for the acquisition of three‐dimensional quantitative phenotypes in the various genetically well‐defined experimental animal models and, as such, it is likely to facilitate genetic dissection of complex traits [Badea et al., 2006]. Here, we will focus on anatomical MR imaging. Anatomical MRI can capture a wealth of information over large areas in a relatively small amount of time, as compared with traditional histological sectioning, while preserving the morphology of the individual organs and structures of interest. It allows for the longitudinal imaging of living organs and structures with a submillimetric resolution (∼200 μm isotropic), as well as for high‐resolution (∼50 μm isotropic) explorations of finer substructures in fixed specimens (e.g. brains or kidneys). Its versatility enables the visualization, detection, and quantification of global and local differences between time points and across strains. These range from volume differences, such as the volumes of brain ventricles, to complex changes in local shape, such as the branching of cerebellar folia.
By comparing two populations of experimental animals belonging, for example, to two distinct strains, as opposed to two strain representatives, we can factor out inherent inter‐individual variability and focus on actual strain differences. Given the large number of comparisons required for a systematic analysis across time points, organs/structures or strains, and of the sheer size of each dataset to be analyzed (a 50 μm isotropic resolution scan of a fixed brain typically consists of more than 10 million voxels), an automated computational approach is necessary for a thorough anatomical characterization.
Automating this generalized comparison process usually requires a common coordinate space. We use automated image registration as a means for establishing anatomical correspondences for every location in every pair of images. The resulting deformation fields, that is the set of three‐dimensional vectors representing the displacements between each pair of corresponding locations in two images, capture the morphological differences between specimens obtained at different time points and in different strains. Together with the volume comparison of semiautomatically delineated structures, the statistical analysis of these fields, the so‐called deformation‐based morphometry, provides us with an automated way to detect and characterize anatomical differences between populations in a quantitative manner [Ali et al., 2005; Chung et al., 2001; Davatzikos et al., 1996; Gaser et al., 1999; Nieman et al., 2006; Thompson et al., 1997].
Yet, in spite of the remarkable boost to signal‐to‐noise ratio (SNR) provided by high‐field MR scanners, the spatial resolution, contrast, and specificity of the acquired images still fall short of the level of anatomical or functional details afforded by classical histology or immunohistochemistry. At present, MR imaging cannot detect changes at the molecular or cellular level unless such changes translate directly into variations in volume or MR intensity. By registering post mortem histological slices or gene expression maps with in vivo and ex vivo MR volumes, we can establish a one‐to‐one spatial correspondence between the various modalities, with the in vivo MR volume acting as an adequate anatomical framework. This enables the extraction of 3D, geometrically correct, phenotypes from histology. It also makes it possible to analyze structures extending across more than one 2D slice, and to compute the volume of regions of interest that can only be defined on the histological slices [MacKenzie‐Graham et al., 2004].
In this article, we aim to demonstrate the remarkable efficiency of MR imaging and joint MR/histological fusion for longitudinal in vivo and high‐resolution ex vivo anatomical phenotyping in two inbred strains of rats, namely Brown Norway (BN) and spontaneously hypertensive rats (SHR). The BN and SHR rats are the parental strains of the only existing panel of recombinant inbred strains in the rat [Pravenec et al. 1989].
MATERIALS AND METHODS
Animals
Male BN rats (n = 4) and SHR rats (n = 4) from Charles River UK Ltd (Margate, Kent, UK) underwent in vivo MRI of the left kidney and the brain at 7–8 weeks (late adolescence; body weight: 234 ± 13 g [BN] and 215 ± 8 g [SHR]) and 12–13 weeks (early adulthood; body weight: 307 ± 18 g [BN] and 317 ± 7 g [SHR]) of age. Each of these animals also underwent ex vivo imaging and histology. Animal care was upheld in accordance with the UK Animals (Scientific Procedures) Act of 1986 and within approved project and personal license guidelines.
MR Imaging
All MRI data were acquired on a 7T Bruker Biospec Avance 70/20 USR system (Bruker Biospin MRI GmbH, Ettlingen, Germany) equipped with a 20‐cm inner‐bore diameter gradient‐set for in vivo imaging of the kidneys and a 12‐cm inner‐bore diameter gradient‐set for in vivo imaging of the brain and all ex vivo imaging.
In Vivo MR Acquisition
Each animal was anaesthetised with isoflurane and a mixture N2O with 5%CO2/95%O2 in a ratio of 1/3 to 2/3, respectively. Anaesthesia was induced with 3% isoflurane and then maintained with 1.5% isoflurane throughout the imaging period. At the end of the second scanning session (at 12‐weeks), the anaesthetic was increased to 4% isoflurane to induce deep anesthesia and the animals were perfused intracardially (peristaltic pump, MasterFlex, Cole‐Parmer, UK) with 300 ml of cold 0.9% saline, followed by 300 ml of cold 4% paraformaldehyde. Following perfusion, the head and the kidneys were removed, placed in 4% paraformaldehyde and stored at 4°C.
Images of the kidneys were acquired using a fast spin echo sequence with respiratory gating. Data were collected with TE of 10.18 ms, an effective TE (TEeff) of 32.46 ms, a TR of 5 s and eight echoes per excitation. The field of view (FOV) was 60 × 60 mm2 and the image resolution was 0.234 × 0.234 mm2 in‐plane with a 1 mm slice thickness and there were 50 slices per volume. We collected a total of 10 image volumes divided into two series of five 50‐slice volumes with a 0.2 mm z‐shift between each volume to allow for super‐resolution postprocessing. The total imaging time per kidney was 26 min.
Images of the brain were collected using a fast spin echo sequence with a TE of 8.61 ms, a TEeff of 36.88 ms, a TR of 39.5 s, and eight echoes per excitation. Image resolution was 0.25 × 0.25 mm2 in‐plane with a 0.25 mm slice thickness. Each volume consisted of 108 slices with a FOV of 32 × 32 mm2. Sixteen volumes (one signal average per volume) were collected for each brain, for an acquisition time of 84 min. The volumes were then linearly aligned and averaged to improve SNR.
Ex Vivo MR Acquisition
All ex vivo MR imaging of the brain and kidneys was carried out between 19 and 22 weeks after sacrifice. The soft tissue on the outside of the skull and the lower jaw bone were carefully removed prior to imaging. Perfused fixed tissues were imaged in 25‐mm diameter tubes having been wrapped in gauze to prevent movement. The tubes were filled with perfluoropolyether (Fomblin LC08; Performance Fluids, Nelson, UK) to prevent the tissue drying out during data acquisition.
Kidneys were imaged using a fast spin echo sequence with a TE of 8.00 ms, a TEeff of 25.5 ms, a TR of 5 s. We used eight echoes per excitation and two signal averages per kidney. Data were acquired as a 3D dataset with a FOV of 26.37 × 19.81 × 19.81 mm3, an isotropic resolution of 103 μm and an acquisition time of 12.8 h.
Brains were imaged within the skull using a fast spin echo sequence with a TE of 8.24 ms, TEeff of 19.06 ms, a TR of 5 s, eight echoes per excitation, and two signal averages. Data were acquired as a 3D dataset with a FOV of 32.96 × 19.81 × 19.81 mm3, an isotropic resolution of 103 μm and an acquisition time of 12.8 h.
Histology
Each brain and kidney were dehydrated in ascending grades of alcohol (70%, 80%, 96% for 90 min each, then 100%, 100%, 100% for 60 min each); immersed into two different xylene solutions (90 min each); and finally immersed into two different paraffin solutions (at 60–65°C for 120 min each) (Leica TP1020). They were then embedded into paraffin moulds orientated so that they were cut coronally. Each embedded block was then mounted on a microtome (Rotary Microtome Cut 4060). Sections were cut into 20 μm thickness. All sections were floated in a warm deionized water bath (∼40°C). The sections were then mounted on (2%) gel‐coated slides and air dried overnight on a hot plate (at 40°C), Consecutive brain sections were alternatively stained with standard protocol Nissl (0.5% cresyl violet) and Haematoxylin and Eosin (H&E) to reveal cell bodies and highlight nuclear structures [Wootton et al., 1995]. Kidney slices were stained with H&E. All sections were then mounted with DPX under a glass coverslip and imaged on a histology slide scanner (Pathscan Enabler II, Electron Microscopy Sciences, US) at 4,000 dpi.
Super‐Resolution of Kidney Images
Kidney MR images were postprocessed using Irani and Peleg [1990] super‐resolution approach. This technique takes as an input a sequence of lower resolution images of the same object acquired with different (known) relative displacements and iteratively reconstructs a higher‐resolution estimate of that object. We postprocessed both series of five images for each kidney and averaged the two higher‐resolution reconstructed images to increase the SNR (Fig. 3).
Figure 3.
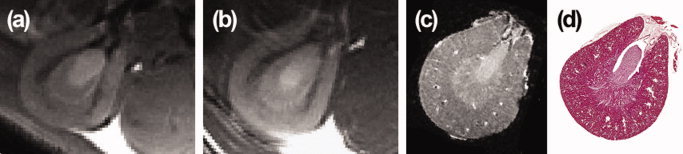
Four coronal slices through the left kidney of a BN rat: in vivo MRI at 8‐weeks (a), in vivo MRI at 12‐weeks (b), ex vivo MRI at 12‐weeks (c), corresponding H&E stained histological slide (d). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Image Registration and Population Averaging of Brain Scans
As mentioned in the Introduction, image registration greatly facilitates anatomical phenotyping by modeling shape differences between all pairs of input images. To characterize anatomical differences across strains and between time points, we combined the images of all individuals belonging to a chosen population and a time point (for instance, in vivo SHR brains acquired at 8‐weeks) into an image average, which provided representation of the average anatomy of that population. Our averaging strategy followed the atlas building approach of Guimond et al. [2000], which ensures that the obtained average does not depend on the choice of a particular reference; the resulting average is the true barycenter (geometric center) of the population.
All brain registrations were performed with the rigidity adaptable technique described previously [Pitiot and Guimond, 2006]. In this multi‐step approach, the input images are first registered with a classical block‐matching algorithm. The obtained raw deformation field is then regularized by estimating a rigid (or affine) transformation over a sliding spherical neighborhood whose radius (the rigidity radius) decreases with each step. A large radius globally matches individual structures (an infinitely large radius corresponds to a global rigid or affine registration) while a small radius allows for the recovery of small local distortions. This is close in spirit to the use of a multi‐resolution approach [see Maintz and Viergever, 1998] for an overview) but gives us finer control over the flexibility of the deformation field (see [Pitiot and Guimond, 2006] for details).
To remove extrinsic deformations unrelated to actual anatomical differences (such as variations of the position of the head in the acquisition FOV, different overall brain sizes, MR biases, etc.), the input images were first corrected for radio‐frequency field inhomogeneities and globally registered with respect to position, orientation and scale. For the correction of field inhomogeneities, we used the N3 nonparametric nonuniform intensity‐normalization method, which eliminates the dependence of the field estimate on anatomy [Sled et al., 1998]. For the global registration, we used only an infinitely large rigidity radius and estimated a global affine transformation. The intensity of the input images were matched by fitting a line to the joint histogram of the input image pairs, using linear regression and outlier rejection [Guimond et al., 2000]. These globally normalized images were then registered to the selected reference image, which was previously positioned so as to match the orientation of standard histological atlases (for the convenience of visual inspection). The registered images were then averaged voxelwise to produce a first mean‐intensity image. Finally, the deformation fields were averaged vectorwise and the resulting average field was applied to the first mean‐intensity image to produce the first average‐image estimate. This process was repeated iteratively until convergence.
We computed the average brain image for each population to produce six averages: in vivo SHR brain at 8‐weeks (inSHR‐8) and 12‐weeks (inSHR‐12), same for BN (inBN‐8 and inBN‐12), ex vivo SHR brain (exSHR‐12) and ex vivo BN brain (exBN‐12). We also registered each brain scan to each average: for instance, all BN brain images at 12‐weeks were registered to the BN average at 8‐weeks, all SHR brain images at 8‐weeks were registered to BN average at 8‐weeks, etc.
Volumetric Analysis
Since the population average adequately summarizes a population's anatomy and presents a higher SNR than any individual scan, it is the ideal image onto which to delineate structures of interest. As an illustration, we manually painted the brain on the ex vivo BN average (exBN‐12). For maximal accuracy, we delineated the structures of interest on the 2D view orthogonal to their long axis while checking for consistency (and occasionally touching up) in the other perpendicular views, using custom‐made in‐house software. Using the same protocol, we also delineated the kidneys obtained in vivo at both time points on the postprocessed super‐resolution in vivo images.
The labels defined on the ex vivo BN brain average were then projected onto the ex vivo SHR average using the average deformation field we previously computed between them; this projection was validated by visual inspection and manually corrected for small registration inaccuracies. Finally, the ex vivo BN and SHR labels were projected onto every in vivo and ex vivo BN and SHR brain scan respectively, also using the previously computed deformation fields. This registration approach enables us to limit the tedious manual delineation process to a single brain average, chosen for its maximal resolution and SNR.
The volumes of the structures of interest were estimated by counting the number of labeled voxels for each brain and kidney scan. The significance of volume differences between strains and between time points was assessed using, respectively, a two‐sample and a paired Student's t‐test, as well as a 2‐way Analysis of Variance (alpha level set at 0.05).
Additionally, we estimated the shape of the average kidney for each strain and time point. To this end, we established first correspondences across the set of kidney delineations for every location on the kidney surface. We used a surface‐deformation strategy that iteratively adapts the shape of a deformable model (a triangular mesh in our case) to the delineated surface [Fleuté et al., 1999]. This deformation process follows that previously described in Pitiot et al. [2004]. The final deformed models were then rigidly registered to each other to remove arbitrary nonanatomical deformations and averaged per strain and per time point.
Deformation‐Based Morphometry
In as much as a population average represents the inherent anatomy of that population free from individual biological variations, the deformation field computed between a population member and the corresponding average models the extent to which the anatomy of that member differs from that of the population. Consequently, the set of all deformation fields models the overall population variability. By comparing interpopulation to intrapopulation deformations, we can identify and quantify anatomical differences in shape between populations. The now standard Hotelling T 2 test, originally employed in morphometric studies of the human brain [Cao and Worsley, 1999; Davatzikos et al., 1996; Thompson and Toga, 1999], can be also applied to the rodent brain [Niemann et al., 2006] to detect areas of statistically significant variations in shape. These correspond to locations at which the differences between the mean displacement vectors of two populations, computed between every scan in a population and a given target average brain, are statistically significant. In other words, voxels with high values of Hotteling T 2 highlight locations at which inter‐population deformations are significantly larger than intrapopulation deformations; the higher the T 2, the more difference in shape at that particular location. Note that as opposed to the volumetric approach outlined above, deformation‐based morphometry highlights areas that do not necessarily coincide with manually defined structures. It is therefore free from a priori considerations about anatomical nomenclature and arbitrary atlas granularity. In particular, it can detect changes at the substructure level or across groups of structures.
Another standard voxelwise morphological information, namely differences in the local volume, can be estimated from the determinant of the Jacobian of the deformation field computed between the population members and the population average [Chung et al., 2001; Thompson et al., 2000]. We used a standard two‐sample t‐test of the log‐Jacobian values to identify areas of significant local difference in volume between two populations.
In our study, both maps were thresholded following the false discovery rate (FDR) methodology [Genovese et al., 2006]; we set the threshold at FDR of 5%.
Between‐strain differences were obtained from the analysis of the deformation fields computed from the registration of the SHR volumes to the BN averages. We performed the analysis on the in vivo scans at 8‐weeks and 12‐weeks, and the ex vivo scans at 12‐weeks. Same‐strain, cross‐time point changes were computed from the analysis of the fields computed from the registration of the in vivo strain volumes at 12‐weeks to the same strain volumes at 8‐weeks.
MRI/Histology Fusion
The first step to the fusion of 2D histological information with MRI is the reconstruction of a 3D volume from successive pairs of 2D sections. Typically, the many processing steps that a slab of tissue undergoes during preparation induce a number of mis‐alignments that need to be carefully corrected during the reconstruction. In particular, the cutting and glass‐mounting steps yield arbitrary translations and rotations between successive slices, as well as nonlinear shrinkages and distortions due to chemical reactions. Our rigidity adaptable approach [Pitiot and Guimond, 2006] was designed to deal with these specific issues of biomedical image registration. In addition to a user‐specifiable rigidity radius, the characteristics of the deformation field are also controlled by the geometry and topology of the images to be registered. In particular, unrelated image components (e.g., those on either side of a gap) are automatically treated independently. This results in an optimum correspondence across sections while keeping tissue distortions to a minimum.
In addition to reconstructing a geometrically coherent volume by spatially realigning 2D slices, we also need to correct the substantial intensity inhomogeneities induced by the histological processes, such as small changes in slice thickness or staining artifacts. In this study, we used Malandain et al. [2004]'s histogram affine matching technique.
The second step in the fusion of MR and histology consists of the registration of the reconstructed 3D histological volume with the MR volume. The challenge here is to find an MR section that corresponds precisely to each histological section. This process not only corrects the non‐linear distortions induced in each slice but it also helps recover the general 3D orientation lost during the cutting of the slices. We used the hybrid fusion technique developed by Malandain et al. [2004]. It consists of an iterative 2‐phase approach where the initial histological volume is globally affinely registered to the MR volume, and its 2D sections are realigned alternatively. By constraining the registration process, in particular by keeping constant the thickness of the slices, this hybrid technique yields more trustworthy results than standard fluid or elastic approaches [Maintz and Viergever, 1998] without sacrificing the overall accuracy.
RESULTS
Average Images and Volumes
Figure 2a,b,e,f,m,n show the brain averages obtained for each population (inBN‐8, inBN‐12, inSHR‐8, inSHR‐12, exBN‐12, and exSHR‐12 respectively). To facilitate visual comparison, we show corresponding slices obtained by affinely registering all in vivo and ex vivo BN and SHR average images to the BN average of images obtained at 8‐weeks of age (inBN‐8), and we superimpose an identical regular grid over each image. In vivo volumes were up‐sampled 2.5 times to match the size of the ex vivo volumes using nearest neighbor interpolation so as not to smooth them artificially (hence their pixelized aspect). Not surprisingly, we measured an average 1.7‐fold improvement in SNR between individual scans and the average (n = 4) brains, where SNR was calculated as the ratio of the mean intensity in an homogeneous area over the intensity variance in this same area. Overall, the average images have better contrast and less noise than the individual scans, a clear indicator of both the adequacy of the registration process and the biological (anatomical) homogeneity of each population (in terms of those features that can be captured by MRI).
Figure 2.
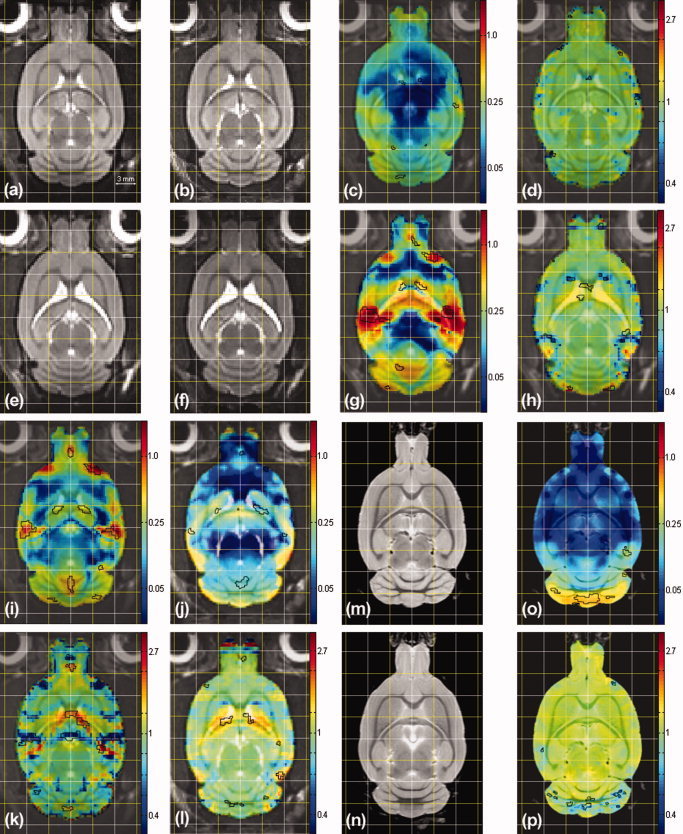
A selection of deformation‐based morphometry results highlighting morphological differences between strain and between time points (see text for details). All measures are in mm.
Table I gives the brain and kidney volumes for each strain at the two time points. Body weight did not differ between BN and SHR rats and increased significantly in both strains between the first and second scan (time: P < 0.0001). Kidney volume showed a similar (but more modest) trend (time: P < 0.04). When corrected for body size, however, kidney volume demonstrated a decrease that was significantly greater in SHR and than in BN rats (strain × time interaction: P < 0.04). Brain volume increased (by about 7%) over time (P < 0.0001); when corrected for body size, there was a relative decrease in brain volume (time: P < 0.0001) that was more pronounced in SHR rats (strain × time interaction: P = 0.03). This simply reflects strain differences in the disproportionate growth of the body and the brain over the 4‐week period, respectively. As for strain differences, absolute brain size was higher (by about 5%) in BN than SHR rats (strain: P = 0.0004); this difference disappeared after correcting for body size. Finally, we observed a significant reduction (by about 4%) of absolute brain size assessed from ex vivo, as compared with in vivo scans obtained at 12 weeks of age.
Table I.
Brain and kidney volumes of BN and SHR rats estimated by the registration‐based projection approach
| Strain | Organ | 8‐week in vivo | 12‐week in vivo | 12‐week ex vivo |
|---|---|---|---|---|
| BN | Brain (mm3) | 2341 ± 54 | 2514 ± 53† | 2414 ± 62 |
| Brain/body weight (mm3/g) | 10.3 ± 0.4 | 8.3 ± 0.4 | 7.9 ± 0.3 | |
| Kidney (mm3) | 915 ± 142 | 1128 ± 129 | N/A | |
| Kidney/body weight (mm3/g) | 4.0 ± 0.4 | 3.8 ± 0.4 | N/A | |
| Body weight (g) | 229 ± 15 | 307 ± 18 | ||
| SHR | Brain (mm3) | 2234 ± 27* | 2393 ± 4*† | 2297 ± 27* |
| Brain/body weight (mm3/g) | 10.4 ± 0.3 | 7.5 ± 0.1 | 7.2 ± 0.2* | |
| Kidney (mm3) | 948 ± 54 | 1021 ± 123 | N/A | |
| Kidney/body weight (mm3/g) | 4.4 ± 0.3 | 3.2 ± 0.4† | N/A | |
| Body weight (g) | 215 ± 8 | 317 ± 7 |
Values are given as mean ± standard deviation. Statistically significant differences (P < 0.05, ANOVA analysis with strain and time as the main factors) between BN and SHR and the two time points are denoted by *and †, respectively.
Deformation Based Morphometry
Figure 2 also illustrates within‐strain morphological changes across the two time points (horizontal axis) and between‐strain differences at each time point (vertical axis). The first row compares the BN population scanned in vivo at 12‐weeks relative to 8‐weeks. In addition to the average images (a,b), we show in (c) a map of the mean norm of the deformation fields superimposed in color (we use a logarithmic color map, the values are in mm) on top of the reference population average (inBN‐8) and, in (d), we show the Jacobian map of the field (again, we use a logarithmic color map, the values are also in mm). The areas of significant (FDR of 5%) changes are delineated in black on both maps. The second row similarly compares SHR rats. The between‐strain differences are illustrated as follows. The first column compares the SHR population relative to the BN one at 8‐weeks, and the second column at 12‐weeks. In the black box, we show in (o) the map of the mean norm of the deformation fields calculated between BN and SHR rats scanned ex vivo, and the associated Jacobian maps in (p) with, again, the areas of significant differences in black.
We first focus on the comparison of the BN and SHR strains at both time points (first and second columns). The most striking strain differences evidenced by the thresholded mean deformation map in Figure 2i,j are localized in four regions: the olfactory bulb, junction of the olfactory bulb and the forebrain, the ventricles and the cerebellum. Analysis of the Jacobian maps highlights the differences in ventricular volume between the two strains, at both 8‐weeks and 12‐weeks. The intricate pattern that the Jacobian map exhibits in the cerebellum embodies dramatic variations in the structure of white matter in the cerebellar folia. While the main folia seem to be the same in the two strains, we observe a variety of secondary white‐matter structures in the cerebellar folia of the BN, which have no counterparts in SHR. Those are more easily noticed when comparing a sagittal view of two representative rat brains (one BN rat: BN001, and one SHR rat: SHR001) side by side in Figure 4 (see in particular the black arrows). Here we also show the sagittal slice extracted from the histological volumes reconstructed from consecutive axial sections of BN001 registered to its ex vivo MRI in (e), and the sagittal section which best matches the sagittal image (d) from a stack of sagittal histological sections cut in the cerebellum of SHR001. Note that the rugged appearance of Figure 4e is due to the inferior resolution along the sagittal axis (20 μm) of the reconstructed histological volume and artefacts (tears and holes) in the axial histological sections. We observe that comparisons based on the ex vivo data elicited more robust statistics (in the cerebellum in particular where a larger number of changes in the structure of the folia were detected, (Fig 4o,p) and better defined (due to the increased resolution) statistical maps than those based on the in vivo deformation fields. This was not the case for the ventricles, however; in both strains, they appeared to collapse after fixation.
Figure 4.
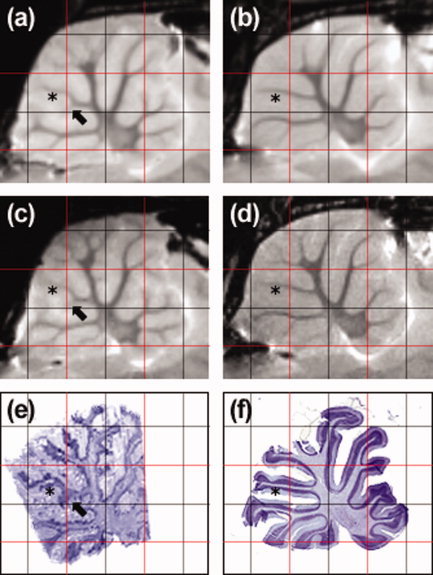
Comparison of the cerebellar folia of BN and SHR rats on corresponding (affinely registered) MR and histological sagittal slices: ex vivo SHR average at 12‐weeks (a); ex vivo BN average at 12‐weeks (b); ex vivo SHR rat representative (SHR001) at 12‐weeks (c); ex vivo BN rat representative (BN001) at 12‐weeks (d); slice through volume reconstructed from axial histological sections of BN001 (e); histological section sliced mid‐sagitally in cerebellum of SHR001 and registered to ex vivo SHR average (f). Black arrows highlight one of the secondary white‐matter structures in the cerebellar folia of the BN, which have no counterparts in SHR. Black stars indicate the branch shared between the two strains, to which this secondary structure is attached. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Comparison of the first and second row of Figure 2 highlights very clear developmental differences between BN and SHR over the 4‐week period. Both the mean deformation and the Jacobian maps of the SHR population (g,h) show a dramatic increase in the size of ventricles between 8 and 12 weeks. We did not observe this change in BN rats. We also note changes in the cerebellum of SHR rats at 12‐weeks relative to 8‐weeks.
DISCUSSION
In this article, we defined a computational strategy for the qualitative and quantitative phenotyping of rodents′ brains and kidneys by MRI and histology. We described the set of semi‐ and fully‐automated techniques that form the backbone of our image analysis infrastructure and illustrated their applications on the study of the anatomical characteristics of two rat strains, BN and SHR, which are the parental strains of the only existing panel of recombinant inbred strains in the rat [Pravenec et al. 1989].
Our volumetric and deformation‐based morphometry approaches successfully detected statistically significant morphological differences, at a global and local scale, respectively, both between the two strains and during the transition from late adolescence (8‐weeks) to early adulthood (12‐weeks). The difficulty of analyzing the many morphological differences captured by coregistering the various rat populations is alleviated by condensing them into a small number of statistical maps: two average images, a mean deformation field, a Hotelling's T 2 and a log‐Jacobian t‐test field per pair of population.
Several factors determine the quality of the population‐based statistical approach presented here.
First, successful MR phenotyping requires a delicate balance between acquisition time and image quality. For instance, our ex vivo brain scans took nine times longer to acquire than their in vivo counterparts but their analysis revealed more robust morphological differences. Furthermore, since their resolution was more than twice that of the in vivo scans, the areas of strain differences were twice more precisely defined. The issue of time becomes even more pressing when considering histology, as serial sectioning and staining is so time‐consuming that often only a part of the brain can be processed. Yet, as illustrated in Figure 4, histology is the modality of choice to reveal the cytoarchitecture underlying cortical structures and the fine cellular details that are still out of the reach of MR imaging. Our phenotyping strategy combines in vivo and ex vivo MRI with histology in an attempt to maximize the amount of anatomical information collected while minimizing the overall acquisition time. In this framework, in vivo MR imaging becomes a fast noninvasive screening tool, which is ideal for identifying promising regions of interest. Ex vivo MR imaging confirms or disproves such in vivo findings, refines the geometry of the regions and potentially detects finer anatomical phenotypes that were not captured by the coarser and noisier in vivo scans. The identified areas are then processed histologically to complement the MR phenotypes with information from the cellular level. Note that, obviously, this multi‐step approach is not amenable to longitudinal (e.g. developmental) analysis. But the use of inbred strains overcomes this limitation in that genetically identical individuals can be studied at different ages. Statistical results obtained from ex vivo volumes must also be considered in the light of potential artefacts introduced during the perfusion and fixation processes [e.g. Goto and Goto, 2006]. The very clear strain differences in the size of ventricles as observed in vivo were not present ex vivo. Furthermore, the brain size derived from ex vivo scans was underestimated (by about 4%) compared with in vivo measurements. We suspect that this is due in part to the collapsed ventricles. These observations clearly highlight the effects of fixation and/or the mode of delivery of the fixative.
Second, in as much as the resolution of the acquired images limits the spatial resolution of the detected phenotypes, population size determines to a large extent the threshold above which a phenotype will become detectable, given the population's overall anatomical variability. Therefore, for a given population (e.g. strain and environment), it is desirable to assess the extent of the population's variability with respect to the phenotype of interest to estimate the minimum size given the desired detection threshold. Let us consider two MR‐based anatomical phenotypes studied here, namely the brain size and the local shape in the cerebellum. When expressed as a coefficient of variation (SD/Mean), the variance is about 2% for the brain size; it is very similar in both strains. Given this degree of variation, we can estimate a sample size necessary for detecting strain differences in the above phenotypes. If we were to set both the confidence interval and statistical power at 95%, we would need only three animals in each group to reject the null hypothesis of no strain differences in brain size. Similarly, we would need four animals to reject the null hypothesis of no strain differences in the local shape in the cerebellum. Clearly, given the fidelity of MR‐derived phenotypes and their relatively low within‐strain (biological) variability, only a small number of animals is necessary for detecting strain differences in brain anatomy here. In the same vein, as few as three animals may be necessary to characterize each of the 32 strains constituting the panel of recombinant inbred strains of rats. But the variance across all 32 strains cannot be known a priori; this will determine statistical power needed for genome‐wide scans using the panel.
Finally, as a prerequisite for population averaging and standard deformation based morphometry, registration also largely influences the accuracy and robustness of the anatomical analysis. Clearly, deformation based morphometry will only be able to detect morphological differences that were successfully captured by the registration process. In this respect, the control that our rigidity adaptable technique gives us over the registration process (in the form of a user‐specifiable regularization radius) enables us to select the scale of the phenotypes to be detected. Our geometry‐ and topology‐dependent regularization technique also insures that the registration takes into account some elements about the anatomy of the input images. This achieves a good compromise between a region‐of‐interest (ROI) based approach (e.g. volumetric morphometry), which can only detect phenotypes at the level of the delineated ROI and a voxelwise approach (e.g. deformation based morphometry), which detect changes at the substructure level independently from their complex anatomical interactions (typically, this can result in arbitrary, nonbiologically plausible deformations). In addition, there are several ways to choose the registration target for deformation‐based morphometry that will influence subsequent statistical analysis of phenotype–genotype relationships. For example, if we use as a target one of the two strain averages, than the overall magnitude of the displacement field will be minimal for the individuals belonging to the “target” strain. This is unlikely to be the case if the registration target is an average of both strains.
We believe that the approach described here demonstrates the high value of computational MRI and MRI/histology strategies for the qualitative and quantitative anatomical phenotyping of rodents′ organs and tissues for genetic studies of complex traits. Future advancements in this area are likely to take place both on the data acquisition and computational sides. In the former area, the use of MR‐based molecular imaging [e.g. with Mn2+, Koretsky and Silva, 2004] will enhance further the phenotyping potential of MR by increasing its specificity and spatial resolution. In the latter area, implementation of fast and computationally effective approaches will be necessary to meet the demands of walking through the genome, marker by marker, in numerous (up to 100) lines of inbred strains; the use of 500 markers, for example, will require the registration and subsequent deformation‐based morphometry analysis of brain images obtained in each of the 500 distinct subpopulations as defined by their genotype. Overall, we believe that the combination of MR‐based “organ‐wide” and genome‐wide scans opens up new avenues of studies of complex traits underlying disorders such as depression and hypertension.
Acknowledgements
The authors thank Dr. Anderson and Miss. Christie in preparing the histological sections.
Contributor Information
Alain Pitiot, Email: alain.pitiot@nottingham.ac.uk.
Zdenka Pausova, Email: zdenka.pausova@nottingham.ac.uk.
REFERENCES
- Ali AA, Dale AM, Badea A, Johnson GA ( 2005): Automated segmentation of neuroanatomical structures in multispectral MR microscopy of the mouse brain. Neuroimage 27: 425–435. [DOI] [PubMed] [Google Scholar]
- Badea CT, Bucholz E, Hedlund LW, Rockman HA, Johnson GA ( 2006): Related imaging methods for morphological and functional phenotyping of the rodent heart. Toxicol Pathol 34: 111–117. [DOI] [PubMed] [Google Scholar]
- Botstein D, Risch N ( 2003): Discovering genotypes underlying human phenotypes: Past successes for Mendelian disease, future approaches for complex disease. Nat Genet 33: 228–237. [DOI] [PubMed] [Google Scholar]
- Cao J, Worsley KJ ( 1999): The dection of local shape changes via the geometry of Hotelling's T‐2 fields. Ann Stat 27: 925–942. [Google Scholar]
- Chung MK, Worsley KJ, Paus T, Cherif C, Collins DL, Giedd JN, Rapoport JL, Evans AC ( 2001): A unified statisticla approach to deformation‐based morphometry. NeuroImage 14: 595–606. [DOI] [PubMed] [Google Scholar]
- Darvasi A ( 1998): Experimental strategies for the genetic dissection of complex traits in animal models. Nat Genet 18: 19–24. [DOI] [PubMed] [Google Scholar]
- Davatzikos C, Vaillant M, Resnick S, Prince JL, Letovsky S, Bryan RN ( 1996): A computerized method for morphological analysis of the corpus callosum. J Comput Assist Tomo 20: 88–97. [DOI] [PubMed] [Google Scholar]
- Fleute M, Lavallee S, Julliard R ( 1999): Incorporating a statistically based shape model into a system for computer‐assisted anterior cruciate ligament surgery. Med Image Anal 3: 209–222. [DOI] [PubMed] [Google Scholar]
- Flint J, Valdar W, Shifman S, Mott R ( 2005): Strategies for mapping and cloning quantitative trait genes in rodents. Nat Rev Genet 6: 271–286. [DOI] [PubMed] [Google Scholar]
- Gaser C, Volz H‐P, Kiebel S, Riehemann S, Sauer H ( 1999): Detecting structural changes in whole brain based on nonlinear deformations—Application to schizophrenia research. NeuroImage 10: 107–113. [DOI] [PubMed] [Google Scholar]
- Genovese CR, Roeder K, Wasserman L ( 2006): False discovery control with p‐value weighting. Biometrika 93: 509–524. [Google Scholar]
- Goto N, Goto J. Morphometric evaluations of the human nervous system. Hum Cell 19: 49–64. [DOI] [PubMed] [Google Scholar]
- Guimond A, Meunier J, Thirion J‐P ( 2000): Average brain models: A convergence study. Comput Vis Image Understand 77: 192–210. [Google Scholar]
- Hoh J, Ott J ( 2003): Mathematical multi‐locus approaches to localizing complex human trait genes. Nat Rev Genet 4: 701–709. [DOI] [PubMed] [Google Scholar]
- Hoh J, Ott J ( 2004): Genetic dissection of diseases: design and methods. Curr Opin Genet Dev 14: 229–232. [DOI] [PubMed] [Google Scholar]
- Hubner N, Wallace CA, Zimdahl H, Petretto E, Schulz H, maciver F, Mueller M, Hummel O, Monti J, Zidek V, Musilova A, Kren V, Causton H, Game L, Born G, Schmidt S, Muller A, Cook SA, Kurtz TW, Whittaker J, Pravenec M, Aitman TJ ( 2005): Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nat Genet 37: 243–253. [DOI] [PubMed] [Google Scholar]
- Irani M, Peleg S ( 1990): Super Resolution From Image Sequences, ICPR 2, 115–120.
- Koretsky AP, Silva AC ( 2004): Manganese‐enhanced magnetic resonance imaging (MEMRI). NMR Biomed 17: 527–531. [DOI] [PubMed] [Google Scholar]
- Lander E, Kruglyak L ( 1995): Genetic dissection of complex traits: Guidelines for interpreting and reporting linkage results. Nat Genet 11: 241–247. [DOI] [PubMed] [Google Scholar]
- Lander ES, Schork NJ ( 1994): Genetic dissection of complex traits. Science 265: 2037–2048. [DOI] [PubMed] [Google Scholar]
- MacKenzie‐Graham A, Lee EF, Dinov ID, Bota M, Shattuck DW, Ruffins S, Yuan H, Konstantinidis F, Pitiot A, Ding Y, Hu G, Jacobs RE and Toga AW ( 2004): A multimodal, multidimensional atlas of the C57BL/6J mouse brain. J Anat 204: 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maintz JBA, Viergever MA ( 1998): A survey of medical image registration. Med Image Anal 2: 1–36. [DOI] [PubMed] [Google Scholar]
- Malandain G, Bardinet E, Nelissen K, Vanduffel W ( 2004): Fusion of autoradiographs with an MR volume using 2‐D and 3‐D linear transformations. NeuroImage 23: 111–127. [DOI] [PubMed] [Google Scholar]
- Mashimo T, Voigt B, Tsurumi T, Naoi K, Nakanishi S, Yamasaki K, Kuramoto T, Serikawa T ( 2006): A set of highly informative rat simple sequence length polymorphism (SSLP) markers and genetically defined rat strains. BMC Genet 4: 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton NE ( 2005): Linkage disequilibrium maps and association mapping. J Clin Invest 115: 1425–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau JH, Singer JB, Matin A, Lander ES ( 2000): Analysing complex genetic traist with chromosome substitution strains. Nat Genet 24: 221–225. [DOI] [PubMed] [Google Scholar]
- Nieman BJ, Flenniken AM, Adamson SL, Henkelman RM, Sled JG ( 2006): Anatomical phenotyping in the brain and skull of a mutant mouse by magnetic resonance imaging and computer tomography. Physiol Genom 24: 154–162. [DOI] [PubMed] [Google Scholar]
- Pausova Z, Tremblay J, Hamet P ( 1999): Gene‐environment interactions in hypertension. Curr Hypertens Rep 1: 42–50. [DOI] [PubMed] [Google Scholar]
- Pausova Z, Gossard F, Gaudet D, Tremblay J, Kotchen T, Cowley AW, Hamet P ( 2001): Heritability estimates of obesity measures in siblings with and without hypertension. Hypertension 38: 41–47. [DOI] [PubMed] [Google Scholar]
- Pitiot A, Guimond A ( 2006): Geometrical Regularization of Displacement Fields with Application to Biological Image Registration. WBIR'06, 101–109.
- Pitiot A, Delingette H, Thompson PM, Ayache N ( 2004): Expert knowledge guided segmentation system for brain MRI. NeuroImage 23: S85–S96. [DOI] [PubMed] [Google Scholar]
- Pravenec M, Klir P, Kren V, Zicha J, Kunes J ( 1989): An analysis of spontaneous hypertension in spontaneously hypertensive rats by means of new recombinant inbred strains. J Hypertens 7: 217–222. [PubMed] [Google Scholar]
- Reuveni E, Ramensky VE, Gross C ( 2007): Mouse SNP Miner: An annotated database of mouse functional single nucleotide polymorphisms. BMC Genom 8: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards EJ ( 2006): Inherited epigenetic variation—revisiting soft inheritance. Nat Rev Genet 7: 395–401. [DOI] [PubMed] [Google Scholar]
- Sled JG, Zijdenbos AP, Evans AC ( 1998): A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans Med Imag 17: 87–97. [DOI] [PubMed] [Google Scholar]
- The Complex Trait Consortium ( 2004): The collaborative cross, a community resource for the genetic analysis of complex traits. Nat Genet 36: 1133–1137. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Toga AW ( 1999): Brain Warping. San Deigo: Academic Press. [Google Scholar]
- Thompson PM, MacDonald D, Mega MS, Holmes CJ, Evans AC, Toga AW ( 1997): Detection and mapping of abnormal brain structure with a probabilistic atlas of cortical surfaces. J Comput Assist Tomo 21: 567–581. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Giedd JN, Woods RP, MacDonald D, Evans AC, Toga AW ( 2000): Growth patterns in the developing human brain detected using continuum‐mechanical tensor mapping. Nature 404: 190–193. [DOI] [PubMed] [Google Scholar]
- Twigger SN, Pasko D, Nie J, Shimoyama M, Bromberg S, Campbell D, Chen J, dela Cruz N, Fan C, Foote C, Harris G, Hickmann B, Ji Y, Jin W, Li D, Mathis J, Nenasheva N, Nigam R, Petri V, Reilly D, Ruotti V, Schauberger E, Seiler K, Slyper R, Smith J, Wang W, Wu W, Zhao L, Zuniga‐Meyer A, Tonellato PJ, Kwitek AE, Jacob HJ ( 2005) Tools and strategies for physiological genomics: The Rat Genome Database. Physiol Genom 23: 246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigger SN, Shimoyama M, Bromberg S, Kwitek AE, Jacob HJ; RGD Team: The Rat Genome Database, update 2007—easing the path from disease to data and back again. Nucleic Acids Res (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GH, Hollenberg NK, Hopkins PM, Jeunemaitre X ( 1996): The role of intermediate phenotypes in essential hypertension: Non‐modulation as a model. Endocr Res 22: 675–680. [DOI] [PubMed] [Google Scholar]
- Wootton R, Springall D, Polak J. editor ( 1995): Image Analysis in Histology (Postgraduate Medical Science), Cambridge University Press. [Google Scholar]