

Abstract
Treatment modalities for cancer radiation therapy have become increasingly diversified given the growing number of facilities providing proton and carbon-ion therapy in addition to the more historically accepted photon therapy. An understanding of high-LET radiobiology is critical for optimization of charged particle radiation therapy and potential DNA damage response. In this review, we present a comprehensive summary and comparison of these types of therapy monitored primarily by using DNA damage biomarkers. We focus on their relative profiles of dose distribution and mechanisms of action from the level of nucleic acid to tumor cell death.
Keywords: proton therapy, DNA damage, carbon-ion radiation therapy
Introduction
In addition to the more historically accepted photon therapy, a growing number of facilities are providing proton and carbon-ion (C-ion) therapy. Consequently, treatment modalities for cancer radiation therapy have become increasingly diversified. An understanding of high–linear energy transfer (LET) radiobiology is critical for optimization of charged particle radiation therapy. Although the physical aspects of charged particle radiobiology are reasonably well understood, the differences in the DNA damage response from these various radiation qualities should be further studied to fully benefit from these new therapies. In this review, we present a comprehensive summary and comparison of these 3 types of therapy monitored primarily by using DNA damage biomarkers.
Important Facts about Photon Therapy
The absorption of energy from radiation in biologic material may lead to excitation or ionization. The raising of an electron in an atom or molecule to a higher energy level without actual ejection of the electron is called excitation. If the radiation has sufficient energy to eject 1 or more orbital electrons from the atom or molecule, the process is called ionization, and that radiation is said to be ionizing radiation. The important characteristic of ionizing radiation is the localized release of large amounts of energy. The energy dissipated per ionizing event is about 33 eV, which is more than enough to break a strong chemical bond, such as a C=C bond (4.9 eV) [1].
Despite the fact that modern photon radiation therapy is delivered with much safer techniques, with computed tomography scanners and more accurate delivery with intensity-modulated radiation therapy, the relative depth dose distributions of photons are still much higher than those of protons [2]. Photons deposit their peak dose close to their entrance into the tissue, and thereafter, there is an exponential decrease of deposited dose with increasing depth [3] (see Figure 1 for illustration).
Figure 1.
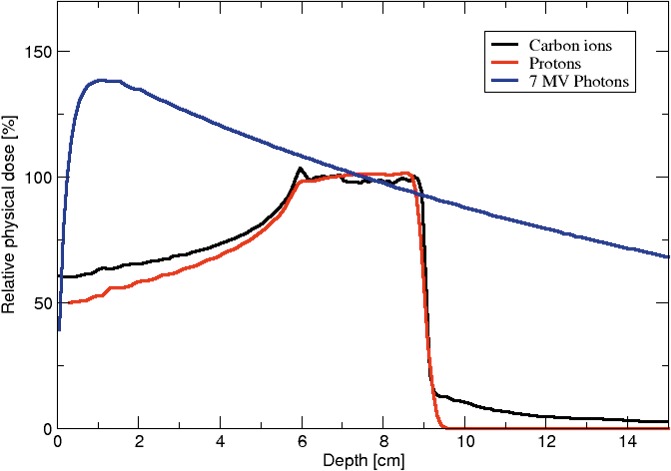
Energy deposition profile as a function of depth into tissue for various radiation therapy modalities.
The Superior Energy Deposition and Tissue Relative Biological Efficiency of Charged Particles and Protons
In tissues and cells, heavy charged particles have been studied to deposit energy along their traversed path, forming a characteristic track structure unlike low-LET radiation that deposits its energy homogenously over targets [4]. The tracks consist of a cylindrical central core where most of the energy is deposited and an outer penumbra that contains energy from sporadically ionized electrons or δ-rays originating from ionization events in the track core.
The specific distribution of energy deposition in tissue by a proton beam allows precise targeting of tumors, while requiring equally precise coordinates to avoid damage to healthy tissue. As previously discussed, low-energy (ie, keV) photons deposit their energy primarily at the entry of the tissue, whereas for high-energy photons (ie, MeV), their energy deposition peaks a few centimeters inside tissue (see Figure 1). In contrast, as long as charged particles move relatively fast (>100 MeV/n), energy deposited is relatively low, until the velocity of the particle gradually decreases with depth, allowing more energy deposition along their linear track. The unit used to measure the energy deposited by charged particles is called linear energy transfer (LET) with the unit of keV/μm. Therefore, charged particles start at lower LET at the entry of the tissue but have a rapid LET increase at the end of the track, where most of the energy is deposited in short and large bursts of energy in a region called the Bragg peak, where most of the particle energy is deposited in a thin slice. The initial energy of the particle beam determines the position of the Bragg peak, and coincides with the tumor location, leading to a much lower damage to the tissue in front of as well as behind the tumor as compared to photon therapy [5, 6]. Figure 1 illustrates the difference of energy deposition as a function of depth into tissue for both MeV photons, protons, and carbons. In these profiles, the strong advantage of charged particles with much localized energy deposition deep in the tissue, with little build-up before the tumor, can be seen. On the other hand, the superiority of carbon therapy is not obvious from simple energy deposition and one must bring up the concept of relative biological effectiveness (RBE) and how heavier ions have higher RBE for DNA damage and cell death than proton radiation, as discussed below.
Owing to a complex 3D structure, tumors typically require targeting by multiple particle beams that have partially overlapping Bragg peaks, combining to form a spread-out Bragg peak (SOBP) that encompasses the entire tumor [7] as illustrated in Figure 1. Naturally, the strict requirement to match the SOBP with a tumor location poses a danger of imprecise targeting of the proton beam, which can damage the neighboring healthy tissue: for example, injuring the gastrointestinal tract while treating prostate cancer. The most common method to improve tumor targeting is intensity-modulated proton therapy, which combines a narrow beam of varied intensity with a precise adjustment of location by using a magnetic field [8].
Finally, the fact that the damage to healthy tissue is significantly lower than in photon therapy permits dose escalation and hypofractionation (fewer, but higher-dose irradiations for the same total dose received by the tumor), which may lead to a greater therapeutic advantage [7] and potentially tumor reoxygenation for better cell killing by increasing blood vessel leakage [9]. However, using higher individual doses may also change the long-term risk of increased damage to healthy tissue [5].
The Rise of Proton and Charged Particle Therapy
The limitations of energy deposition profiles of photon, combined with the growing technologic advances in delivering particle beams precisely, have led the way for the growing adoption of particle-based therapy for cancer treatment over the last 3 decades [10, 11]. This adoption started in 1975 with the pioneering work of scientists at the Lawrence Berkeley National Laboratory using the Bevalac, a weak-focusing proton synchrotron combined with the SuperHILAC linear accelerator as an injector for heavy ions, to deliver charged-particle radiation therapy including proton and C-ions to patients. Even though the Bevalac is now decommissioned, this work has led to the opening of 11 centers in the world at the moment, treating patients with C-ions (https://www.ptcog.ch/index.php/facilities-in-operation) [12, 13]. In Japan, National Institute of Radiological Sciences began conducting clinical trials on carbon-ion radiation therapy (CIRT) as early as 1994. Globally, to date more than 96 000 patients have been treated with particle beams, and about 15 000 patients have undergone CIRT [14–16].
Understanding the Link Between DNA Damage and Cell Death in the Context of Radiation
To compare and adjust the treatment profiles of different types of radiation therapy, the impact of radiation therapy on the tumor is represented by its RBE against photon therapy.
Given the complexity of response of tissues to charged particles, biological effect must be included when preparing the treatment plan of heavy ion radiation therapy, to minimize the radiosensitivity of normal tissue while maximizing death of cancerous cells. One promising computer model is the Local Energy Model developed at GSI, Darmstadt, Germany [17, 18]. The Local Effect Model assumes that DNA damage and cell death can be solely predicted by the amount of energy deposited in a small subnuclear volume, independently of the radiation quality [17]. With such an approach, one can predict survival curves for any particles, based solely on biological properties derived from x-ray data alone. It is therefore a much more attractive model than using empirical survival curves to predict the response of both tumor and healthy tissue. For proton therapy, an RBE value of 1.1 is typically used in clinical situations to calculate the equivalent biologic dose for proton therapy relative to photon therapy [19]. However, the LET increases drastically in the last few microns of the particle track [20], which has been shown to translate into a higher RBE both in vitro and in vivo [21–26] and there is an increased concern that the RBE of 1.1 is an oversimplification, which may have clinical implications [26–28].
RBE greater than unity is thought to reflect the fact that DNA lesions induced by charged particles are more complex [29]. However, a unified formalism able to model DNA complexity in a way that can predict cell survival for various radiation qualities remains to be accepted [30]. Therefore, RBE is often measured for each cell, particle, and energy of interest. Survival curves are typically fitted with a decreasing exponential with a linear quadratic dose dependence, introduced by Douglas and Fowler [31] for both of the radiation reference (x-ray) and the particle of interest. The alpha-beta ratio is typically thought to represent the intrinsic radiosensitivity of the cell type, with alpha being the linear term and beta being the quadratic term. RBE is then calculated as the ratio of the reference dose to the dose of the test radiation necessary to produce the same level of cell survival, often 10% survival. A recent review suggests that tissues with high alpha-beta ratio, such as the brain, may be disproportionally sensitive to proton therapy and require an RBE adjustment along the particle track as a function of LET and alpha-beta tissue property to limit the damage done to healthy tissue [32–34]. Finally, most RBE studies have been done in vitro, since they require a relatively high-throughput process, and the RBE in vivo may be further confounded by off-target systemic effects [35].
The accuracy of the Local Effect Model mentioned above [17] suggests that the spatial distribution of radiation-induced DNA double-strand breaks (DSBs) in the nucleus is the critical factor for predicting cell death. Therefore, characterizing such spatial distribution in tumor cell becomes a critical endeavor to feed computer model for better treatment planning. The level of DSBs can be experimentally quantified by detecting small nuclear domain referred to as radiation-induced foci (RIF), which are formed at the site of DSBs by proteins involved in DNA repair. For example, RIF formed by either 53BP1 (tumor suppressor p53 binding protein 1) or by the phosphorylation of H2AX (γH2AX) have been studied after proton radiation [36–38]. Figure 2A through 2C illustrates the detection of Bragg peak using γH2AX as a marker of DSBs, showing its usefulness for characterizing radiation therapy. The ensuing damages from δ-rays generated by charged particles are simpler and thought to be repaired efficiently, while the more complex damages in the core need activation of several repair pathways working in concert [13]. Oike et al [39] computed LET distribution in the treatment of tumor cells and compared it to 53BP1 foci size distribution in fresh biopsy samples of uterine cervical cancer, with RIF showing bimodal peaks at the center of the SOBP just like predicted for LET. Presumably, the low-LET peak led to small foci similar in size to the ones induced by x-ray (ie, simple DSBs). In contrast, the higher-LET peaks presumably induced larger RIF, only visible in irradiated tumor as the larger peak. These data are evidence that C-ion–induced complex lesions can be detected in clinical settings by using 53BP1 RIF size distribution as a surrogate marker of DSB complexity and probably cell death [39].
Figure 2.
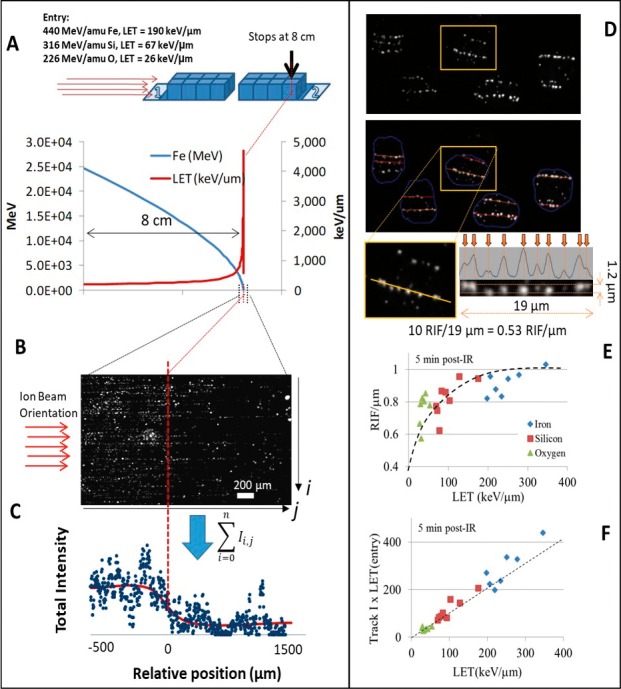
Characterizing Bragg peak using DNA damage biomarker (reprint from Vadhavkar et al41). (A) Schematic showing the slide arrangement for ions to traverse 8 cm of MCF10A grown as confluent monolayer. Stage position allows the theoretical computation of the energy and linear energy transfer (LET) as illustrated in the graph for Fe ion. (B) Visualization of 8-cm point in the slides where the particle is expected to stop, using γH2AX labeling. (C) By summing the amount of signal along the i axis in (B), one can visualize the drop of DNA damage, once the particle has stopped at position 0 (0 represents the expected stopping point of the ion). (D) Illustration of image algorithm used to detect individual tracks. Software automatically identifies tracks (colored lines overlapping 53BP1 radiation-induced foci [RIF] in gray image). Intensity profiles are determined by summing the signal perpendicular to tracks with a 1.2-μm diameter so that local maxima can be more easily detected. This allows the computation of the number of RIF/μm. (E) Experimental RIF/μm measured for the 3 ions of interest here (Fe with Z = 26, Si with Z = 14, and O with Z = 8). Dashed line helps visualize saturation of response with LET. (F) The normalized total intensity of 53BP1 detected in all RIF along tracks per unit length (41) is proportional to LET as indicated by the dashed line. Abbreviations: 53BP1, tumor suppressor p53 binding protein 1; γH2AX, phosphorylation of H2AX.
To better understand the role of DNA damage in cell death, our group characterized the spatial property of RIF induced by particles of increasing LET and we modified the Local Effect model to introduce the concept of RIF coalescence as a new mechanism to predict cell death for any LET [40]. In the RIF coalescence model, DSBs move into single repair unit characterized by large RIF before the repair machinery kicks in. This model is contrary to the classic “contact-first” model where DSBs are assumed as immobile and repaired at the lesion site [41, 42]. The RIF coalescence model, first suggested by Aten et al [43], and recently confirmed by our laboratory by using time-lapse fluorescent microscopy of 53BP1 fused to GFP in response to high doses of x-ray [44], has the advantage of being much more efficient molecularly. In this model, the spatial distribution of DSBs becomes a critical factor influencing DNA repair efficiency. For example, high-LET radiation generates several close-by DSBs along its linear track, increasing significantly the probability of its coalescing into the same RIF, which can lead to increased chromosomal rearrangement [45] or DNA misrepair in general. Evidence of RIF coalescence is shown in Figure 2D and 2E where one can see that the number of RIF/μm reaches a plateau at about 200 keV/μm. Above such LET, the linear density of DSBs keeps increasing with LET (as indicated by the linear increase in the intensity of 53bp1, Figure 2F), while the number of RIF only increases modestly and thus the average number of DSBs per RIF increases [46, 47]. Meanwhile, low doses of low-LET radiation, such as x-rays, generate DSBs randomly in the nucleus and do not cause RIF coalescence. We thus propose that RIF coalescence is an alternative explanation of DSB complexity, which can lead to hypersensitivity to high-LET radiation for patient or cell type with strong clustering properties. With this model, one can explain why mutation, cell death, and cancer is much more prevalent following exposure to charged particles, as the damaged cells are being overwhelmed by the number of DSBs to repair within single repair units. This model is a paradigm shift where increased spatial proximity of DSBs is an alternate explanation for hypersensitivity to high LET in contrast to the classical DNA damage complexity as the primary mechanism [48–50], which we will describe next.
DNA Repair Pathways and Radiation Quality
In contrast to RIF coalescence occurring over several micrometers in the nucleus, at the DNA scale, damages induced by high-LET irradiation are also much different from x-rays [51, 52] and are characterized by multiple bases being affected within a few turns of DNA helix. These are referred to as clustered DNA damage or multiple damage sites [53–55] and are considered much more complex to repair than simple DNA lesions. These complex lesions can be observed in DSBs, single strand breaks (SSBs) and oxidative clustered DNA lesions, such as abasic sites, and/or base damages (oxidized purines or pyrimidines) [56, 57] and occur within 20- to 30-bp regions measuring 10 to 20 nm [58, 59]. Immunofluorescent imaging using DNA repair markers such as γ-H2AX and 53BP1, XRCC1, and hOGG1 foci, surrogate markers for DSBs, SSBs, and base damages, respectively, have been used to detect tracks and the sites of clustered DNA damage [49].
The impact of these complex lesions has been proposed as another explanation for higher biological effects of charged particles. For example, the biological impacts of C-ions and x-rays were compared by Zhao et al [60] whereby three human tumor cell lines showed increased survival for x-rays compared to C-ions, while neutral comet assays showed substantially higher amount of residual DSBs, 0.5 and 4 hours after C-ion irradiation than with x-rays. Strong induction of γH2AX after 30 minutes was observed in all cell lines and persisted for 24 hours after C-ion exposure [60]. For human pancreatic cancer stemlike cells, colony and spheroid formation and tumorigenicity were higher than for noncancer stemlike cells, and a higher decrease in surviving fractions was observed with C-ions than with x-rays, with increasing doses [61]. At the genetic level, decondensation of heterochromatin, along with twisting of the tracks proximal to the heterochromatic regions, has been observed after C-ion irradiation [62]. These clustered double-stranded lesions introduce substantial amount of challenge in the DNA repair processes [63]. To prevent genomic instability by unrepaired DSBs or cell death (by mitotic catastrophe, apoptosis, or senescence), multiple DNA repair pathways are activated in response to clustered DNA damage. Three critical pathways, namely, homologous recombination (HR), nonhomologous end joining (NHEJ), and alternative end joining, are usually used to repair DSBs [64]. Studies suggest that DSBs induced by high-LET irradiation might be preferentially repaired by HR [58, 65], by recruiting DNA strand invasion proteins and Rad51 [67]. One interpretation of these results is the inability of the Ku heterodimer to bind to the DNA owing to short DNA fragments induced by clustered damage prohibiting an essential step in the NHEJ pathway [59]. There are also reports suggesting HR might also be the preferential repair pathway for proton irradiation of human cancer cells, even with low-LET proton irradiation [37, 67]. On the other hand, data from Gerelchuluun et al [65] point at the NHEJ pathway playing an important role in repairing DSBs induced by both clinical proton and C-ion beams. Recent reports have also indicated that NHEJ might influence DNA repair following C-ion damage [66, 68]. Shibata et al [69] provide an interesting report that examined the factors that favored the speed of DNA repair using NHEJ and HR in cells in G2 phase. With chromatin complexity as one of the factors, the group demonstrated that DNA lesion complexity was the second factor favoring the choice of NHEJ to repair complex DSBs induced by high-LET radiation. Their results show that, while the first attempt to repair DSBs in G2 is made by NHEJ, there is a competing backup NHEJ pathway that occurs in the absence of Ku but requires DNA-dependent protein kinase catalytic subunit (DNA-PKcs). Their model depicts that, following a lesion, quick binding of DNA-PK recruits NHEJ as the first repair pathway. If the next steps can progress without hindrance, NHEJ rapidly repairs the DSBs. If on the other hand, rejoining cannot occur owing to the lesion or chromatin complexity, DSB end resection and HR pathway is chosen. Again if resection cannot occur, NHEJ takes over again and efficiently repairs the DSBs, provided this happens in an earlier stage before committing to HR.
Response to clustered DNA damage repair is affected by a transition of ataxia-telangiectasia mutated (ATM) and Rad3-related transition at lesion sites and switch from NHEJ to HR [70, 71]. Yet another mechanism of DNA damage induced by CIRT is mitotic catastrophe, ensuing from atypical mitosis activated by lack of G2 endpoint [49, 72]. Cellular senescence as indicated by high expression of senescence-associated beta-galactosidase SA-β-Gal and low levels of Ki-67, was also found to be induced by high-LET radiation such as 80 keV/μm C-ions, as compared to X-rays, on human melanoma cells. Five days post irradiation, persistent levels of ATM kinase activation and 53BP1 expression were noted with C-ions as compared to X-rays. 53BP1 was found to colocalize with XRCC1, for both X-rays and C-ions, signifying formation of SSB and DSB complexes [73].
The initial RIF number per Gy has been found to be comparable between photon and proton irradiation, and independent of LET in cells irradiated with protons in therapeutic beams with relative low LET [36, 37], whereas studies focusing on higher LET values, both with protons and C-ions, have demonstrated increasing RIF number with higher LET [74, 75]. The residual number of DNA damage after 24 to 26 hours, which is presumably reflecting unrepaired DSBs, has been shown to be LET dependent in more studies, also in the clinical relevant LET range for proton irradiation [36, 38, 75, 76] (Figure 3). In the studies with protons in therapeutic beams, this is pointing towards the repair rather than the induction of DNA damage, which is different from photon irradiation. This increased number of residual foci could be due to preference for HR as repair mechanism, as HR compared to NHEJ is a slower repair mechanism [37].
Figure 3.
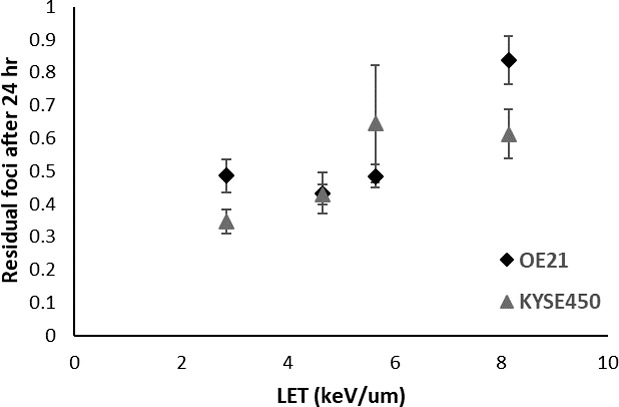
Residual numbers of γH2AX foci formed per cell (24 hours relative to initial number of foci at 0.5 hours after irradiation), in OE21 and KYSE450 cells irradiated with 8-Gy proton beam irradiation (235 MeV). Data are extracted from the study of Hojo et al.39 Error bars represent the combined relative standard error of the mean of the original data. Abbreviations: γH2AX, phosphorylation of H2AX; LET, linear energy transfer.
Other Caveats about Proton and Particle Therapy
In addition to direct DNA damage, exposure to protons generates reactive oxygen species (ROS), which further damage the DNA and alter normal functions of the cell directly in the proton path as well the neighboring cells [6]. However, ROS production is stimulated by tissue oxygen content, which is frequently reduced in tumor environment, suggesting tumor oxygenation as a method to improve the effectiveness of proton therapy. On the other hand, excessive ROS production is also likely to cause bystander damage to cells that are not directly targeted by the protons. Thus, while proton therapy is particularly efficient at causing tumor cell apoptosis [77], it may also be the mechanism behind increased apoptosis as well as necrosis of surrounding tissue [78].
Other tissue-level effects of proton radiation therapy include altered angiogenesis and inflammation. Interestingly, while photon therapy has been shown to increase angiogenesis, proton therapy has the opposite effect of inhibiting it [79]. Lower angiogenesis is therapeutically beneficial: it limits tumor survival by reducing nutrient availability. In contrast, the research on inflammatory changes due to proton therapy is comparatively scarce and conflicting: protons have been indicated to increase both traditionally inflammatory and anti-inflammatory cytokines. Ultimately, the relative importance of inflammation in therapeutic outcomes may be determined on the tissue that is receiving the irradiation and the possibility of it causing systemic effects, which could lead to impaired responses to infection or cognitive damage [79–81].
Future Considerations
In spite of the aforementioned benefits and the excellent outcome presented in clinical data, CIRT is still limited to about 11 centers globally. The primary hindrance in establishing CIRT centers is the estimated high costs of initial investment, maintenance, and treatment to patients. To better compare the experimental, preclinical, and clinical data from CIRT to conventional photon and proton therapy, thorough investigations should be conducted in beam line and SOBP characterization, dose dynamics including understanding of absorbed dose, cellular damage, and repair kinetics and well-designed clinical studies. A special emphasis on studies characterizing better DNA repair following high LET, including the coalescence of DSBs into single unit of repair, will be essential for a future model to predict accurate biological response of the tumor, while preserving healthy tissue from potentially long-term effect currently unseen with photon therapy. The increased availability of CIRT to the patient population and clinicians in North America will highly depend on such studies.
ADDITIONAL INFORMATION AND DECLARATIONS
Conflicts of Interest: The authors have no conflicts of interest to disclose.
Acknowledgments: This review was written under the support by National Aeronautics and Space Administration NNL15AA08I to S.V.C. (principal investigator). We would like to thank Niels Bassler, PhD, Medical Radiation Physics, Department of Physics at Stockholm, Sweden, for providing us with Figure 1.
References
- 1.Hall E, Hall EJ, Giaccia AJ. Radiobiology for Radiologists. Philadelphia: Lippincott Williams & Wilkins;; 2012. [Google Scholar]
- 2.Mitin T, Zietman AL. Promise and pitfalls of heavy-particle therapy. J Clin Oncol. 2014;32:2855–63. doi: 10.1200/JCO.2014.55.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goitein M. Radiation Oncology: A Physicist's Eye View. New York, NY: Springer Science & Business Media;; 2007. [Google Scholar]
- 4.Cashman J, Dykstra B, Clark-Lewis I, Eaves A, Eaves C. Changes in the proliferative activity of human hematopoietic stem cells in NOD/SCID mice and enhancement of their transplantability after in vivo treatment with cell cycle inhibitors. J Exp Med. 2002;196:1141–50. doi: 10.1084/jem.20010916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blanchard P, Gunn GB, Lin A, Foote RL, Lee NY, Frank SJ. Proton therapy for head and neck cancers. Sem Radiat Oncol. 2018;28:53–63. doi: 10.1016/j.semradonc.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 6.Held KD, Kawamura H, Kaminuma T, Paz AES, Yoshida Y, Liu Q, Willers H, Takahashi A. Effects of charged particles on human tumor cells. Front Oncol. 2016;6:23. doi: 10.3389/fonc.2016.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patyal B. Dosimetry aspects of proton therapy. Technol Cancer Res Treat. 2007;6:17–23. doi: 10.1177/15330346070060S403. [DOI] [PubMed] [Google Scholar]
- 8.Wilkens JJ, Oelfke U. Optimization of radiobiological effects in intensity modulated proton therapy. Med Phys. 2005;32:455–65. doi: 10.1118/1.1851925. [DOI] [PubMed] [Google Scholar]
- 9.Paul-Gilloteaux P, Potiron V, Delpon G, Supiot S, Chiavassa S, Paris F, Costes SV. Optimizing radiotherapy protocols using computer automata to model tumour cell death as a function of oxygen diffusion processes. Sci Rep. 2017;7:2280. doi: 10.1038/s41598-017-01757-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noda K, Furukawa T, Fujisawa T, Iwata Y, Kanai T, Kanazawa M, Kitagawa A, Komori M, Minohara S, Murakami T, Muramatsu M, Sato S, Takei Y, Tashiro M, Torikoshi M, Yamada S, Yusa K. New accelerator facility for carbon-ion cancer-therapy. J Radiat Res. 2007;48:A43–54. doi: 10.1269/jrr.48.a43. [DOI] [PubMed] [Google Scholar]
- 11.Combs SE, Jakel O, Haberer T, Debus J. Particle therapy at the Heidelberg Ion Therapy Center (HIT): integrated research-driven university-hospital-based radiation oncology service in Heidelberg, Germany. Radiother Oncol. 2010;95:41–4. doi: 10.1016/j.radonc.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 12.Castro JR, Saunders WM, Tobias CA, Chen GT, Curtis S, Lyman JT, Collier JM, Pitluck S, Woodruff KA, Blakely EA, Tenforde T, Char D, Phillips TL, Alpen EL. Treatment of cancer with heavy charged particles. Int J Radiat Oncol Biol Phys. 1982;8:2191–8. doi: 10.1016/0360-3016(82)90569-7. [DOI] [PubMed] [Google Scholar]
- 13.Mohamad O, Sishc BJ, Saha J, Pompos A, Rahimi A, Davis AJ, Kim DWM. Carbon ion radiotherapy: a review of clinical experiences and preclinical research, with an emphasis on DNA damage/repair. Cancers (Basel) 2017;9:E66. doi: 10.3390/cancers9060066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohno T. Particle radiotherapy with carbon ion beams. EPMA J. 2013;4:9. doi: 10.1186/1878-5085-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ebner DK, Kamada T. The emerging role of carbon-ion radiotherapy. Front Oncol. 2016;6:140. doi: 10.3389/fonc.2016.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohno T, Kanai T, Yamada S, Yusa K, Tashiro M, Shimada H, Torikai K, Yoshida Y, Kitada Y, Katoh H, Ishii T, Nakano T. Carbon ion radiotherapy at the Gunma University Heavy Ion Medical Center: new facility set-up. Cancers (Basel) 2011;3:4046–60. doi: 10.3390/cancers3044046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scholz M, Kellerer A, Kraft-Weyrather W, Kraft G. Computation of cell survival in heavy ion beams for therapy: the model and its approximation. Radiat Environ Biophys. 1997;36:59–66. doi: 10.1007/s004110050055. [DOI] [PubMed] [Google Scholar]
- 18.Elsässer T, Weyrather WK, Friedrich T, Durante M, Iancu G, Krämer M, Kragl G, Brons S, Winter M, Weber KJ, Scholz M. Quantification of the relative biological effectiveness for ion beam radiotherapy: direct experimental comparison of proton and carbon ion beams and a novel approach for treatment planning. Int J Radiat Oncol Biol Phys. 2010;78:1177–83. doi: 10.1016/j.ijrobp.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 19.Mahajan A. Normal tissue complications from low-dose proton therapy. Health Phys. 2012;103:586–9. doi: 10.1097/HP.0b013e3182611114. [DOI] [PubMed] [Google Scholar]
- 20.Mohan R, Mahajan A, Minsky BD. New strategies in radiation therapy: exploiting the full potential of protons. Clin Cancer Res. 2013;19:6338–43. doi: 10.1158/1078-0432.CCR-13-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saager M, Peschke P, Brons S, Debus J, Karger CP. Determination of the proton RBE in the rat spinal cord: is there an increase towards the end of the spread-out Bragg peak? Radiother Oncol. 2018. pii:S0167-8140(18)30134-8. [DOI] [PubMed]
- 22.Bettega D, Calzolari P, Chauvel P, Courdi A, Herault J, Iborra N, Marchesini R, Massariello P, Poli GL, Tallone L. Radiobiological studies on the 65MeV therapeutic proton beam at Nice using human tumour cells. Int J Radiat Biol. 2000;76:1297–303. doi: 10.1080/09553000050151565. [DOI] [PubMed] [Google Scholar]
- 23.Sørensen BS, Overgaard J, Bassler N. In vitro RBE-LET dependence for multiple particle types. Acta Oncol. 2011;50:757–62. doi: 10.3109/0284186X.2011.582518. [DOI] [PubMed] [Google Scholar]
- 24.Britten RA, Nazaryan V, Davis LK, Klein SB, Nichiporov D, Mendonca MS, Wolanski M, Nie X, George J, Keppel C. Variations in the RBE for cell killing along the depth-dose profile of a modulated proton therapy beam. Radiat Res. 2012;179:21–8. doi: 10.1667/RR2737.1. [DOI] [PubMed] [Google Scholar]
- 25.Wouters BG, Skarsgard LD, Gerweck LE, Carabe-Fernandez A, Wong M, Durand RE, Nielson D, Bussiere MR, Wagner M, Biggs P, Paganetti H, Suit HD. Radiobiological intercomparison of the 160 MeV and 230 MeV proton therapy beams at the Harvard Cyclotron Laboratory and at Massachusetts General Hospital. Radiat Res. 2015;183:174–87. doi: 10.1667/RR13795.1. [DOI] [PubMed] [Google Scholar]
- 26.Lühr A, von Neubeck C, Pawelke J, Seidlitz A, Peitzsch C, Bentzen SM, Bortfeld T, Debus J, Deutsch E, Langendijk JA, Loeffler JS, Mohan R, Scholz M, Sørensen BS, Weber DC, Baumann M, Krause M. “Radiobiology of Proton Therapy”: results of an international expert workshop. Radiother Oncol. 2018. pii:S0167-8140(18)30277-9. [DOI] [PubMed]
- 27.Peeler CR, Mirkovic D, Titt U, Blanchard P, Gunther JR, Mahajan A, Mohan R, Grosshans DR. Clinical evidence of variable proton biological effectiveness in pediatric patients treated for ependymoma. Radiother Oncol. 2016;121:395–401. doi: 10.1016/j.radonc.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buchsbaum JC. Are treatment toxicity issues in particle therapy a clarion call for biologic treatment planning overall? Int J Radiat Oncol Biol Phys. 2017;97:1085–6. doi: 10.1016/j.ijrobp.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 29.Goodhead DT. Mechanisms for the biological effectiveness of high-LET radiations. J Radiat Res. 1999;40:1–13. doi: 10.1269/jrr.40.s1. [DOI] [PubMed] [Google Scholar]
- 30.Cucinotta FA, Durante M. Cancer risk from exposure to galactic cosmic rays: implications for space exploration by human beings. Lancet Oncol. 2006;7:431–5. doi: 10.1016/S1470-2045(06)70695-7. [DOI] [PubMed] [Google Scholar]
- 31.Douglas BG, Fowler JF. The effect of multiple small doses of x rays on skin reactions in the mouse and a basic interpretation. Radiat Res. 1976;66:401–26. [PubMed] [Google Scholar]
- 32.Girdhani S, Sachs R, Hlatky L. Biological effects of proton radiation: what we know and don't know. Radiat Res. 2013;179:257–72. doi: 10.1667/RR2839.1. [DOI] [PubMed] [Google Scholar]
- 33.Paganetti H. Relative biological effectiveness (RBE) values for proton beam therapy: variations as a function of biological endpoint, dose, and linear energy transfer. Phys Med Biol. 2014;59:R419–72. doi: 10.1088/0031-9155/59/22/R419. [DOI] [PubMed] [Google Scholar]
- 34.Jones B, McMahon S, Prise K. The radiobiology of proton therapy: challenges and opportunities around relative biological effectiveness. Clin Oncol (R Coll Radiol) 2018;30:285–92. doi: 10.1016/j.clon.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 35.Newhauser WD, Durante M. Assessing the risk of second malignancies after modern radiotherapy. Nat Rev Cancer. 2011;11:438–48. doi: 10.1038/nrc3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaudhary P, Marshall TI, Currell FJ, Kacperek A, Schettino G, Prise KM. Variations in the processing of DNA double-strand breaks along 60-MeV therapeutic proton beams. Int J Radiat Oncol Biol Phys. 2016;95:86–94. doi: 10.1016/j.ijrobp.2015.07.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grosse N, Fontana AO, Hug EB, Lomax A, Coray A, Augsburger M, Paganetti H, Sartori AA, Pruschy M. Deficiency in homologous recombination renders Mammalian cells more sensitive to proton versus photon irradiation. Int J Radiat Oncol Biol Phys. 2014;88:175–81. doi: 10.1016/j.ijrobp.2013.09.041. [DOI] [PubMed] [Google Scholar]
- 38.Hojo H, Dohmae T, Hotta K, Kohno R, Motegi A, Yagishita A, Makinoshima H, Tsuchihara K, Akimoto T. Difference in the relative biological effectiveness and DNA damage repair processes in response to proton beam therapy according to the positions of the spread out Bragg peak. Radiat Oncol. 2017;12:111. doi: 10.1186/s13014-017-0849-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oike T, Niimi A, Okonogi N, Murata K, Matsumura A, Noda S-E, Kobayashi D, Iwanaga M, Tsuchida K, Kanai T, Ohno T, Shibata A, Nakano T. Visualization of complex DNA double-strand breaks in a tumor treated with carbon ion radiotherapy. Sci Rep. 2016;6:22275. doi: 10.1038/srep22275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vadhavkar N, Pham C, Georgescu W, Deschamps T, Heuskin AC, Tang J, Costes SV. Combinatorial DNA damage pairing model based on X-ray-induced foci predicts the dose and LET dependence of cell death in human breast cells. Radiat Res. 2014;182:273–81. doi: 10.1667/RR13792.1. [DOI] [PubMed] [Google Scholar]
- 41.Nikiforova MN, Stringer JR, Blough R, Medvedovic M, Fagin JA, Nikiforov YE. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science. 2000;290:138–41. doi: 10.1126/science.290.5489.138. [DOI] [PubMed] [Google Scholar]
- 42.Soutoglou E, Dorn JF, Sengupta K, Jasin M, Nussenzweig A, Ried T, Danuser G, Misteli T. Positional stability of single double-strand breaks in mammalian cells. Nat Cell Biol. 2007;9:675–82. doi: 10.1038/ncb1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aten JA, Stap J, Krawczyk PM, van Oven CH, Hoebe RA, Essers J, Kanaar R. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science. 2004;303:92–5. doi: 10.1126/science.1088845. [DOI] [PubMed] [Google Scholar]
- 44.Georgescu W, Osseiran A, Rojec M, Liu Y, Bombrun M, Tang J, Costes SV. Characterizing the DNA damage response by cell tracking algorithms and cell features classification using high-content time-lapse analysis. PLoS One. 2015;10:e0129438. doi: 10.1371/journal.pone.0129438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anderson RM, Stevens DL, Goodhead DT. M-FISH analysis shows that complex chromosome aberrations induced by alpha-particle tracks are cumulative products of localized rearrangements. Proc Natl Acad Sci U S A. 2002;99:12167–72. doi: 10.1073/pnas.182426799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Costes SV, Ponomarev A, Chen JL, Nguyen D, Cucinotta FA, Barcellos-Hoff MH. Image-based modeling reveals dynamic redistribution of DNA damage into nuclear sub-domains. PLoS Comput Biol. 2007;3:e155. doi: 10.1371/journal.pcbi.0030155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ponomarev AL, Costes SV, Cucinotta FA. Stochastic properties of radiation-induced DSB: DSB distributions in large scale chromatin loops, the HPRT gene and within the visible volumes of DNA repair foci. Int J Radiat Biol. 2008;84:916–29. doi: 10.1080/09553000802499212. [DOI] [PubMed] [Google Scholar]
- 48.Rydberg B, Cooper B, Cooper PK, Holley WR, Chatterjee A. Dose-dependent misrejoining of radiation-induced DNA double-strand breaks in human fibroblasts: experimental and theoretical study for high- and low-LET radiation. Radiat Res. 2005;163:526–34. doi: 10.1667/rr3346. [DOI] [PubMed] [Google Scholar]
- 49.Asaithamby A, Hu B, Chen DJ. Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc Natl Acad Sci U S A. 2011;108:8293–8. doi: 10.1073/pnas.1016045108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nikitaki Z, Nikolov V, Mavragani IV, Mladenov E, Mangelis A, Laskaratou DA, Fragkoulis GI, Hellweg CE, Martin OA, Emfietzoglou D, Hatzi VI, Terzoudi GI, Iliakis G, Georgakilas AG. Measurement of complex DNA damage induction and repair in human cellular systems after exposure to ionizing radiations of varying linear energy transfer (LET) Free Radic Res. 2016;50:S64–78. doi: 10.1080/10715762.2016.1232484. [DOI] [PubMed] [Google Scholar]
- 51.Amornwichet N, Oike T, Shibata A, Nirodi CS, Ogiwara H, Makino H, Kimura Y, Hirota Y, Isono M, Yoshida Y, Ohno T, Kohno T, Nakano T. The EGFR mutation status affects the relative biological effectiveness of carbon-ion beams in non-small cell lung carcinoma cells. Sci Rep. 2015;5:11305. doi: 10.1038/srep11305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amornwichet N, Oike T, Shibata A, Ogiwara H, Tsuchiya N, Yamauchi M5, Saitoh Y, Sekine R, Isono M, Yoshida Y, Ohno T, Kohno T, Nakano T. Carbon-ion beam irradiation kills X-ray-resistant p53-null cancer cells by inducing mitotic catastrophe. PLoS One. 2014;9:e115121. doi: 10.1371/journal.pone.0115121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chatterjee A, Holley WR. Biochemical mechanisms and clusters of damage for high-LET radiation. Adv Space Res. 1992;12:33–43. doi: 10.1016/0273-1177(92)90087-e. [DOI] [PubMed] [Google Scholar]
- 54.Ward J. Biochemistry of DNA lesions. Radiat Res. 1985;104:S103–11. [PubMed] [Google Scholar]
- 55.Ward J. The complexity of DNA damage: relevance to biological consequences. Int J Radiat Biol. 1994;66:427–32. doi: 10.1080/09553009414551401. [DOI] [PubMed] [Google Scholar]
- 56.Hada M, Georgakilas AG. Formation of clustered DNA damage after high-LET irradiation: a review. J Radiat Res. 2008;49:203–10. doi: 10.1269/jrr.07123. [DOI] [PubMed] [Google Scholar]
- 57.Sutherland B, Bennett P, Schenk H, Sidorkina O, Laval J, Trunk J, Monteleone D, Sutherland J. Clustered DNA damages induced by high and low LET radiation, including heavy ions. Phys Med. 2001;17:202–4. [PubMed] [Google Scholar]
- 58.Wang H, Wang X, Zhang P, Wang Y. The Ku-dependent non-homologous end-joining but not other repair pathway is inhibited by high linear energy transfer ionizing radiation. DNA Repair (Amst) 2008;7:725–33. doi: 10.1016/j.dnarep.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 59.Watanabe R, Wada S, Funayama T, Kobayashi Y, Saito K, Furusawa Y. Monte Carlo simulation of radial distribution of DNA strand breaks along the C and Ne ion paths. Radiat Prot Dosim. 2011;143:186–90. doi: 10.1093/rpd/ncq539. [DOI] [PubMed] [Google Scholar]
- 60.Zhao J, Guo Z, Zhang H, Wang Z, Song L, Ma J, Pei S, Wang C. The potential value of the neutral comet assay and γH2AX foci assay in assessing the radiosensitivity of carbon beam in human tumor cell lines. Radiol Oncol. 2013;47:247–57. doi: 10.2478/raon-2013-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oonishi K, Cui X, Hirakawa H, Fujimori A, Kamijo T, Yamada S, Yokosuka O, Kamada T. Different effects of carbon ion beams and X-rays on clonogenic survival and DNA repair in human pancreatic cancer stem-like cells. Radiother Oncol. 2012;105:258–65. doi: 10.1016/j.radonc.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 62.Jakob B, Splinter J, Conrad S, Voss KO, Zink D, Durante M, Löbrich M, Taucher-Scholz G. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. 2011;39:6489–99. doi: 10.1093/nar/gkr230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lorat Y, Timm S, Jakob B, Taucher-Scholz G, Rübe CE. Clustered double-strand breaks in heterochromatin perturb DNA repair after high linear energy transfer irradiation. Radiother Oncol. 2016;121:154–61. doi: 10.1016/j.radonc.2016.08.028. [DOI] [PubMed] [Google Scholar]
- 64.Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med. 2015;66:129–43. doi: 10.1146/annurev-med-081313-121208. [DOI] [PubMed] [Google Scholar]
- 65.Gerelchuluun A, Manabe E, Ishikawa T, Sun L, Itoh K, Sakae T, Suzuki K, Hirayama R, Asaithamby A, Chen DJ, Tsuboi K. The major DNA repair pathway after both proton and carbon-ion radiation is NHEJ, but the HR pathway is more relevant in carbon ions. Radiat Res. 2015;183:345–56. doi: 10.1667/RR13904.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saha J, Wilson P, Thieberger P, Lowenstein D, Wang M, Cucinotta FA. Biological characterization of low-energy ions with high-energy deposition on human cells. Radiat Res. 2014;182:282–91. doi: 10.1667/RR13747.1. [DOI] [PubMed] [Google Scholar]
- 67.Fontana AO, Augsburger MA, Grosse N, Guckenberger M, Lomax AJ, Sartori AA, Pruschy MN. Differential DNA repair pathway choice in cancer cells after proton- and photon-irradiation. Radiother Oncol. 2015;116:374–80. doi: 10.1016/j.radonc.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 68.Zhou X, Zhang X, Xie Y, Tanaka K, Wang B, Zhang H. DNA-PKcs inhibition sensitizes cancer cells to carbon-ion irradiation via telomere capping disruption. PLoS One. 2013;8:e72641. doi: 10.1371/journal.pone.0072641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shibata A, Conrad S, Birraux J, Geuting V, Barton O, Ismail A, Kakarougkas A, Meek K, Taucher-Scholz G, Löbrich M, Jeggo PA. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011;30:1079–92. doi: 10.1038/emboj.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saha J, Wang M, Cucinotta FA. Investigation of switch from ATM to ATR signaling at the sites of DNA damage induced by low and high LET radiation. DNA Repair. 2013;12:1143–51. doi: 10.1016/j.dnarep.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 71.Whalen MK, Gurai SK, Zahed-Kargaran H, Pluth JM. Specific ATM-mediated phosphorylation dependent on radiation quality. Radiat Res. 2008;170:353–64. doi: 10.1667/RR1354.1. [DOI] [PubMed] [Google Scholar]
- 72.Kobayashi D, Oike T, Shibata A, Niimi A, Kubota Y, Sakai M, Amornwhichet N, Yoshimoto Y, Hagiwara Y, Kimura Y, Hirota Y, Sato H, Isono M, Yoshida Y, Kohno T, Ohno T, Nakano T. Mitotic catastrophe is a putative mechanism underlying the weak correlation between sensitivity to carbon ions and cisplatin. Sci Rep. 2017;7:40588. doi: 10.1038/srep40588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang X, Ye C, Sun F, Wei W, Hu B, Wang J. Both complexity and location of DNA damage contribute to cellular senescence induced by ionizing radiation. PLoS One. 2016;11:e0155725. doi: 10.1371/journal.pone.0155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Suetens A, Konings K, Moreels M, Quintens R, Verslegers M, Soors E, Tabury K, Grégoire V, Baatout S. Higher initial DNA damage and persistent cell cycle arrest after carbon ion irradiation compared to X-irradiation in prostate and colon cancer cells. Front Oncol. 2016;6:87. doi: 10.3389/fonc.2016.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Antonelli F, Campa A, Esposito G, Giardullo P, Belli M, Dini V, Meschini S, Simone G, Sorrentino E, Gerardi S, Cirrone GA, Tabocchini MA. Induction and repair of DNA DSB as revealed by H2AX phosphorylation foci in human fibroblasts exposed to low- and high-LET radiation: relationship with early and delayed reproductive cell death. Radiat Res. 2015;183:417–31. doi: 10.1667/RR13855.1. [DOI] [PubMed] [Google Scholar]
- 76.Kavanagh JN, Currell FJ, Timson DJ, Savage KI, Richard DJ, McMahon SJ, Hartley O, Cirrone GA, Romano F, Prise KM, Bassler N, Holzscheiter MH, Schettino G. Antiproton induced DNA damage: proton like in flight, carbon-ion like near rest. Sci Rep. 2013;3:1770. doi: 10.1038/srep01770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Finnberg N, Wambi C, Ware JH, Kennedy AR. Gamma-radiation (GR) triggers a unique gene expression profile associated with cell death compared to proton radiation (PR) in mice in vivo. Cancer Biol Ther. 2008;7:2023–33. doi: 10.4161/cbt.7.12.7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Merchant TE. Clinical controversies: proton therapy for pediatric tumors. Semin Radiat Oncol. 2013;23:97–108. doi: 10.1016/j.semradonc.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Girdhani S, Lamont C, Hahnfeldt P, Abdollahi A, Hlatky L. Proton irradiation suppresses angiogenic genes and impairs cell invasion and tumor growth. Radiat Res. 2012;178:33–45. doi: 10.1667/rr2724.1. [DOI] [PubMed] [Google Scholar]
- 80.Giedzinski E, Rola R, Fike JR, Limoli CL. Efficient production of reactive oxygen species in neural precursor cells after exposure to 250 MeV protons. Radiat Res. 2005;164:540–4. doi: 10.1667/rr3369.1. [DOI] [PubMed] [Google Scholar]
- 81.Nielsen S, Bassler N, Grzanka L, Swakon J, Olko P, Andreassen CN, Overgaard J, Alsner J, Sørensen BS. Differential gene expression in primary fibroblasts induced by proton and cobalt-60 beam irradiation. Acta Oncol. 2017;56:1406–12. doi: 10.1080/0284186X.2017.1351623. [DOI] [PubMed] [Google Scholar]