

SUMMARY
People with Down syndrome (DS; trisomy 21) display a different disease spectrum relative to the general population, including lower rates of solid malignancies and higher incidence of neurological and autoimmune conditions. However, the mechanisms driving this unique clinical profile await elucidation. We completed a deep mapping of the immune system in adults with DS using mass cytometry to evaluate 100 immune cell types, which revealed global immune dysregulation consistent with chronic inflammation, including key changes in the myeloid and lymphoid cell compartments. Furthermore, measurement of interferon-inducible phosphorylation events revealed widespread hypersensitivity to interferon-α in DS, with cell-type-specific variations in downstream intracellular signaling. Mechanistically, this could be explained by overexpression of the interferon receptors encoded on chromosome 21, as demonstrated by increased IFNAR1 surface expression in all immune lineages tested. These results point to interferon-driven immune dysregulation as a likely contributor to the developmental and clinical hallmarks of DS.
Graphical Abstract
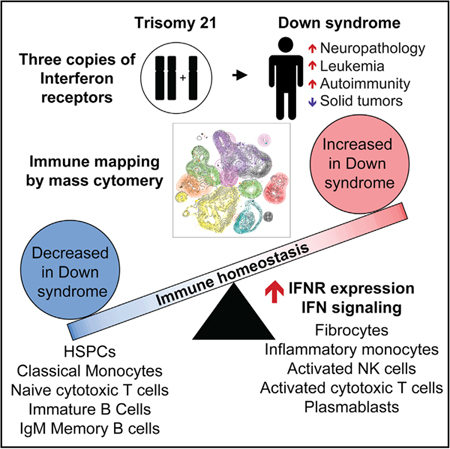
In Brief
Waugh et al. undertook deep mapping of the immune system in adults with trisomy 21, revealing global immune dysregulation reminiscent of inflammatory states, concurrent with widespread hypersensitivity to IFN-α. These data highlight immune dysregulation and IFN hyperactivity as contributors to the comorbidities more common in people with Down syndrome.
INTRODUCTION
Down syndrome (DS) is caused by trisomy of chromosome 21 (trisomy 21 [T21]), the most common human chromosomal abnormality affecting ~1 in 700 live births (de Graaf et al., 2017). In addition to the well-recognized developmental features of DS, individuals with T21 experience a different disease spectrum relative to the typical population, showing lower rates of certain medical conditions, such as most solid malignancies (Hasle et al., 2016), while being strongly predisposed to others, such as leukemias (Maloney et al., 2015), various autoimmune disorders (Mårild et al., 2013; Madan et al., 2006; Anwar et al., 1998; Karlsson et al., 1998), pulmonary hypertension (King and Tulloh, 2011), and Alzheimer’s disease (AD) (Hartley et al., 2014). With the exception of AD, the high prevalence of which has been linked to the presence of the amyloid precursor protein gene (APP) on chromosome 21 (chr21) (Hartley et al., 2014), the processes driving this differential disease spectrum await elucidation. Therefore, identification of these mechanisms may illuminate strategies for better diagnostics and therapeutics, both in people with DS and in the typical population affected by the conditions modulated by T21.
It is well documented that people with DS display alterations within the immune compartment, yet available literature does not agree on the extent and etiology of this phenomenon (reviewed in Ram and Chinen, 2011). Furthermore, few studies have investigated the immune system of adults with DS. Given the remarkable increase in life expectancy among people with DS (de Graaf et al., 2017), there is a clear need to understand how immune alterations may contribute to the unique disease spectrum observed across their lifespan. There are multiple genes encoded on chr21 with roles in immune control, including four of the six interferon (IFN) receptors (IFNRs): the two subunits for the type I ligands, IFNAR1 and IFNAR2; the type II IFNR subunit IFNGR2; and IL10RB, which serves as a subunit of the receptor for the type III IFN ligands (IFNL1/IL29, IFNL2/IL28A, and IFNL3/IL28B) (de Weerd and Nguyen, 2012). Recently, we revealed that T21 causes consistent activation of the IFN transcriptional response, as demonstrated by overexpression of IFN-stimulated genes (ISGs) in multiple cell types (Sullivan et al., 2016). A follow-up plasma proteomics study revealed changes in the circulating proteome indicative of chronic autoinflammation in DS, including elevated levels of potent inflammatory cytokines linked to IFN signaling, such as interleukin-6 (IL-6), tumor necrosis factor α (TNF-α), and MCP-1 (Sullivan et al., 2017). However, the contribution of hyperactive IFN signaling, inflammatory cytokines, and specific immune cell types to the different disease spectrum in DS remains to be defined. Within this framework, we set out to identify alterations in the immune system of adults with DS via mass cytometry (Bandura et al., 2009), which enables the concurrent measurement of dozens of immune cell types, as well as a wealth of phenotypic markers and signaling epitopes (Spitzer and Nolan, 2016). First, we assayed for shifts in immune homeostasis by simultaneous quantification of 100 immune cell types in adults with DS relative to typical controls. These efforts revealed global immune dysregulation in people with DS, with changes in key myeloid and lymphoid cells types reminiscent of that observed in inflammatory and autoimmune states in the typical population. Second, we measured expression of the type I IFNR subunit IFNAR1, which revealed surface protein overexpression across the entire immune system. Finally, using a panel of antibodies recognizing eight phospho-epitopes known to be induced by IFN, we revealed extensive hyper-reactivity to IFN-α in primary immune cells of adults with DS. Altogether, these results support the notion of immune dysregulation and IFN hyperactivity as potential drivers of the different disease spectrum in DS.
RESULTS
Adults with T21 Display Global Immune Dysregulation
In order to define the impacts of T21 on the immune system, we employed mass cytometry to characterize white blood cells from a cohort of 36 adults, half with T21, the other half being age- and gender-matched euploid controls (D21) (Figure S1A; Table S1A). Importantly, none of the participants reported signs of active infection for at least 2 weeks prior to blood draw. As expected from an unbiased selection of individuals with T21, this group displayed a wide range of comorbidities (Table S2). We employed a panel of 28 antibodies that would enable resolution of 100 distinct lymphoid and myeloid cell types (Table S3). After blood processing, staining, and cytometry, we used a variety of analytical tools to investigate this rich dataset. We used clustering algorithms to identify bulk cell communities impacted by T21, followed by further exploration via manual gating, and, in select cases, cross-platform validation using flow cytometry.
First, we normalized data between batches and used the visualization tool for stochastic neighbor embedding (viSNE) (Amir et al., 2013) to reduce dimensionality and visualize the global structure of these data (Figure S1B; STAR Methods). To quantitate similarities in the overall shape of viSNE plots, we calculated Jenson-Shannon (JS) divergence scores, a statistical method used to measure similarities between two probability distributions (Amir et al., 2013), for every pairwise comparison of viSNE plots corresponding to each of the 36 individuals in this cohort. Hierarchical clustering did not illuminate gross overall differences by cohort or batch, although there was a trend for T21 samples to aggregate together (Figure 1A). Next, we employed the clustering algorithm PhenoGraph (Levine et al., 2015) to delineate communities of events with similar marker expression within viSNE plots, which identified 31 clusters, only some of which mapped precisely to major viSNE communities (e.g., cluster 8) (Figure 1B). Notably, several PhenoGraph clusters were significantly depleted (e.g., clusters 2, 7, and 8) or enriched (e.g., clusters 13, 25, and 28) in T21 samples (Figures 1C and 1D; Table S4A). A shortcoming of PhenoGraph is that the clusters identified cannot be easily mapped to recognized immune subtypes. Thus, we used surface marker expression to reveal the composition of bulk immune subtypes overlaid on viSNE plots (Figures 1E, S1C, and S1D). This exercise revealed that clusters 2, 7, and 8 are subsets of B cells (CD27hi, plasmablasts, and CD27low, respectively, all depleted among bulk CD45+ in T21), cluster 13 is non-classical monocytes (elevated in T21), cluster 25 is T cells expressing high levels of granzyme B (elevated in T21), and cluster 28 is an unidentified cell type negative for CD3, CD19, CD14, CD16, and CD56 but highly expressing CD38 and CD123. Therefore, the impact of T21 on the immune system could not be easily interpreted in the context of current literature using a clustering algorithm, such as PhenoGraph.
Figure 1. Mass Cytometry Reveals Global Immune Dysregulation in Adults with T21.

Intact single events enriched for non-granulocytes of hematopoietic origin (CD45+CD66−) were run through viSNE and then compared between individuals with T21 (n = 18) and D21 controls (n = 18).
(A) Similarities between overall shape of individual viSNE plots were quantified through pairwise comparisons by Jenson-Shannon (JS) scores, followed by hierarchical clustering.
(B) PhenoGraph was used to resolve 31 clusters within viSNE plots.
(C) PhenoGraph clusters were uniformly colored according to increased (red) or decreased (blue) frequency among total events in T21 viSNE plots compared to those of D21 controls. Significantly different clusters are labeled with numbers.
(D) Box and whisker plots displaying the data for PhenoGraph Clusters 8 (a subset of B cells) and 25 (a subset of T cells expressing high levels of granzyme B).
(E) Overlay of immune cell subsets defined by surface marker expression on events represented in viSNE plots.
(F) A topographical analysis, kernel density estimate (KDE), was applied to viSNE plots to visualize areas with more (red) or less (blue) event density in samples with T21. Color is overlaid on a viSNE plot showing concatenated events from all T21 samples.
(G) Manual gating of areas within viSNE plots highlighted by KDE. An area of CD4+ T cells (black) depleted of CD45RA+CD27+ events (blue) were interpreted as a subset of naive CD4+ T cells whereas an area enriched for CD45RA—CD27+ events (red) were interpreted as a subset of CD27+ memory CD4+ T cells.
In all cases, statistical significance was determined by a Student’s t test (*p ≤ 0.05 and **p ≤ 0.01). See also Figure S1. All box and whisker plots denote the median within a box extending from the 25th to 75th percentiles and error bars span minimum to maximum values within the datasets.
To further investigate differences among aggregated T21 and D21 viSNE plots without restricting events by cluster identity, we utilized topographical analysis by kernel density estimate (KDE). Interestingly, KDE indicated that PhenoGraph clusters contained distinct areas of both depletion and enrichment of events within the T21 viSNE plots (Figure 1F). For example, the area encompassing CD4+ T cells showed subareas of both depletion and enrichment in T21 samples (Figures 1E and 1F). To test if differences highlighted by KDE within the CD4+ T population are statistically significant and correspond to established CD4+ T cell subsets, we manually gated based on expression of CD45RA and CD27, two markers used to discriminate naive, effector, and memory CD4+ T cells (Figure 1G). Indeed, gated populations largely overlap, albeit not precisely, with these subsets of CD4+ T cells. Thus, although abundance of total CD4+ T cells is not significantly different in DS, a subset of naive CD4+ T cells is significantly depleted, while a subset of CD27+ memory CD4+ T cells is significantly enriched (Figure 1G).
Altogether, these results justified a detailed characterization of 100 immune cell types using manual gating (Figure S2; Table S5), which identified many key immune cell types that are either significantly depleted or enriched in adults with DS (Figure S3; Table S4B). Salient results selected for further analyses are (1) a significant decrease in circulating human stem and progenitor cells (HSPCs); (2) a shift in the myeloid compartment toward subsets associated with inflammation; (3) hyper-activation of the natural killer (NK) and cytotoxic T cell compartments; and (4) a clear dysregulation of the B cell lineage (Figure S3). Of note, an extensive analysis of the impact of co-occurring conditions within the T21 group failed to identify comorbidities that could consistently explain the immune alterations in DS (Table S6 and accompanying online portal described in STAR Methods). Therefore, the most parsimonious interpretation is that T21 causes these immune changes, which could in turn contribute to the development of co-morbidities more frequent in DS.
People with DS Show Fewer Circulating HSPCs along with a Marked Increase in Fibrocytes
A key result is the significant depletion of circulating HSPCs in adults with T21 (Figures 2A and 2B). Although most HSPCs reside in the bone marrow, a small fraction reach the periphery through a regulated process known as ‘‘mobilization’’ (Hoggatt and Pelus, 2011). Interestingly, the few circulating HSPCs in individuals with T21 are shifted toward the CD38− hematopoietic stem cell (HSCs) and multipotent progenitor (MPPs) states, concomitant with a significant decrease in the less pluripotent oligopotent progenitor (OPP) state (Figures 2C and 2D).
Figure 2. Hematopoietic Stem Cells, Progenitors, and Fibrocytes Are Significantly Dysregulated in Individuals with T21.

(A and B) Hematopoietic stem and progenitor cells (HSPCs) were detected by CD34 and CD45 staining in the peripheral blood as depicted in Figures S2A and S2D then compared between individuals with T21 (n = 18) and D21 controls (n = 18).
(B) Representative dot plots denoting frequency among the parent gate.
(C and D) Among HSPCs, CD38 staining was used to delineate hematopoietic stem cells (HSCs) and multipotent progenitors (MPPs) from oligopotent progenitors (OPPs) in individuals with T21 (n = 18) and D21 controls (n = 18).
(D) Representative dot plots denoting frequency among the parent gate.
(E and F) Fibrocytes were detected by expression of COL1 among circulating HSPCs in individuals with T21 (n = 8) and D21 controls (n = 9), as depicted in Figure S2A, S2D, and S2F. See Figure S2G for COL1 metal minus one control.
(F) Representative dot plots denoting frequency among the parent gate.
(G–M) Among fibrocytes, the expression levels of CD45RO (G), COL1 (H and I), and HLA-DR (J–M) were compared by geometric mean metal intensity (gMMI). In (I) and (M), representative histograms are shown with numbers denoting gMMIs among the parent gate indicated in titles. Green represents an individual with T21 and gray a D21 control.
(N) On a flow cytometer, positive staining of pro-collagen resolved fibrocytes among all hematopoietic (CD45+) events from white blood cells of children with T21 (n = 26) versus D21 controls (n = 7), as depicted in Figure S4A.
(O) Lineage tree depicts subsets of HSPCs within the context of hematopoiesis and bone-marrow-derived fibrocytes. Color denotes fold-change where red is increased and blue is decreased frequency of subsets among HSPCs in individuals with T21 compared to D21 controls.
In all cases, statistical significance was determined by a Student’s t test (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001). All box and whisker plots denote the median within a box extending from the 25th to 75th percentiles and error bars span minimum to maximum values within the indicated datasets.
Fibrocytes, a subset of circulating CD34+CD45+CD38− pro-genitor cells that express collagen, have been associated with chronic inflammation, autoimmunity, and fibrotic diseases, particularly of the lung (Galligan and Fish, 2013). To investigate possible dysregulation of the fibrocyte subset, we determined intracellular collagen 1 (COL1) expression among HSPCs directly ex vivo in a subcohort of 8 adults with T21 and 9 D21 controls (Table S1A). Remarkably, fibrocytes comprised a significantly larger fraction of circulating HSPCs in people with DS (Figures 2E, 2F, and S2). Although fibrocytes with T21 did not show differences in expression of the marker CD45RO often used to identify this subset (Figure 2G), they expressed significantly more COL1 on a per-cell basis (Figures 2H and 2I). Significantly fewer circulating fibrocytes in adults with T21 expressed the major histocompatibility complex class II HLA-DR (Figures 2J–2M). Interestingly, fibrocytes expressing HLA-DR can present antigen to T cells (Chesney et al., 1997), and reduced HLA-DR expression on antigen-presenting cells has been associated with poor outcomes from sepsis (Cheadle, 1993; Lekkou et al., 2004).
The observed increase in fibrocytes could have a number of clinical implications, as fibrocytes have been involved in the etiology of comorbidities common in DS, such as pulmonary hypertension and autoimmune conditions (Galligan and Fish, 2013). Furthermore, children with DS have elevated risk of mortality from sepsis (Garrison et al., 2005). Therefore, to deepen our characterization of fibrocytes, we completed a cross-antibody and cross-platform validation by measuring pro-collagen 1 expression among CD45+ cells by flow cytometry on a cohort of 33 children, 26 with DS (Table S1B). Indeed, children with T21 display markedly increased frequency of fibrocytes among all CD45+ white blood cells (Figures 2N, S4A, and S4B).
Altogether, these results show that people with DS display marked dysregulation of HSPC homeostasis, with fewer total mobilized cells that are shifted to a less differentiated state and highly enriched for fibrocytes (Figure 2O).
The Myeloid Compartment of People with T21 Shows Changes Associated with Inflammation and Cytotoxicity
Another interesting result is the obvious shift among functionally distinct monocyte subsets from the classical toward intermediate and nonclassical states (Figures 3A and 3B). To validate this finding, we employed standard flow cytometry on a much larger cohort (82 total adults, 37 with T21; Figure S4C; Table S1B). Indeed, we observed a significant decrease in classical monocytes, along with an increase in intermediate monocytes (Figures 3C and 3D). An increase in intermediate or ‘‘inflammatory’’ monocytes is in line with a previous study of children with T21 (Bloemers et al., 2010) and is commonly associated with various inflammatory and autoimmune conditions (Karlmark et al., 2012). We did not observe alterations in abundance of circulating myeloid dendritic cells (mDCs) (Figure S3; Table S4B), in contrast to a previous report of lower numbers of mDCs in children with DS (Bloemers et al., 2010). The only other myeloid subtype that was significantly altered in abundance in adults with T21 is the CD1c+ conventional dendritic cell (cDC; Figures 3E and 3F), which has been proposed to promote autoimmunity through T cell activation (Cooles et al., 2018; Leal Rojas et al., 2017).
Figure 3. Monocyte and Dendritic Cell Homeostasis Are Disrupted in the Peripheral Blood of Individuals with T21.

(A and B) Within mass cytometry data, monocyte subsets were delineated by expression of canonical markers as depicted in Figure S2 and then compared between individuals with T21 (n = 18) and D21 controls (n = 18).
(B) Representative dot plots are shown with numbers denoting frequency among monocytes.
(C and D) Flow cytometry was used to evaluate the frequency (C) and number (D) of classical and intermediate monocytes within the peripheral blood of individuals with T21 (n = 37 for frequency and n = 31 for relative cell number) and D21 controls (n = 45 for frequency and n = 41 for relative cell number), as depicted in Figure S4C.
(E–K) Phenotypic characterization of myeloid subsets by mass cytometry. In (E) and (F), CD1c expression was used to resolve a subset of conventional dendritic cells (cDCs) among myeloid DCs (mDCs) in individuals with T21 (n = 18) and D21 controls (n = 18). In (G) and (I), the levels of CD16 or CD56 protein expression were compared between individuals with T21 (n = 18) and D21 controls (n = 18) by geometric mean metal intensity (gMMI). In (H) and (J), representative histograms are shown with numbers denoting gMMIs among the parent gate indicated in titles. Green represents an individual with T21 and gray a D21 control. Plasmacytoid DCs are denoted as pDCs.
(K) Lineage tree depicts subsets of the myeloid lineage within the monocyte and mDC compartments of individuals with T21 compared to D21 controls. Color denotes fold change, where red is increased and blue is decreased frequency of subsets among monocyte or mDC compartments in T21 compared to D21 controls.
In all cases, statistical significance was determined by a Student’s t test (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001). All box and whisker plots denote the median within a box extending from the 25th to 75th percentiles and error bars span minimum to maximum values within the indicated datasets.
In order to further investigate markers used to distinguish myeloid subsets, we defined levels of CD16 surface expression in CD16+ monocyte populations. CD16 is expressed on the surface of various immune cell types, where it binds to the Fc region of immunoglobulin Gs (IgGs) to mediate antibody-dependent cell-mediated cytotoxicity (ADCC), also acting as a lysis receptor during immune attack of cancerous and virally infected cells (Mandelboim et al., 1999; Yeap et al., 2016). Surface expression of CD16 was significantly higher on both the intermediate and non-classical CD16+ monocyte subsets in individuals with T21 (Figures 3G and 3H). Furthermore, expression of CD56, a recognized marker of cytotoxic potential among myeloid cells (Van Acker et al., 2017), was significantly higher among diverse dendritic cell (DC) subsets in people with T21 (Figures 3I and 3J).
Overall, alterations within the myeloid compartment of adults with DS are consistent with a heightened state of inflammation and increased cytotoxic potential (Figure 3K).
Natural Killer and Cytotoxic T Cells with T21 Are Hyperactivated
Our antibody panel enabled a deep analysis of cellular immunity, including subsets of innate lymphoid cells (ILCs) and T cells (Figure S2; Table S5). Among the NK cell subset of ILCs, the CD56++CD16− and CD56+CD16− subsets represent the immature precursors and cytokine-producing pools of NK cells that express low amounts of cytotoxic molecules, while the CD56+CD16+ NK subset is considered the most cytotoxic, and the CD56− NK subset is considered terminally differentiated and dysfunctional (Nolte et al., 2009). Adults with T21 have similar frequencies of both bulk NK cells and the five subsets defined by CD56 and CD16 expression among circulating non-granulocytes (Figures 4A–4C and S2; Table S5). However, the slight depletion of CD56+CD16− NK cells and trend toward enrichment of CD56+CD16+ NK cells among circulating non-granulocytes from individuals with T21 (Figure 4C) are in line with a previous report of increased frequency of bulk CD56+CD16+ cells among peripheral blood mononuclear cells (PBMCs) (Cossarizza et al., 1991). To probe deeper into this issue, we analyzed a larger cohort (89 total adults, 32 with T21; Table S1B) using flow cytometry, thus increasing our power to resolve differences among the two major NK cell subsets, CD56++CD16− and CD56+CD16+ (Figure S4C). These efforts revealed no difference in the precursor CD56++CD16− subset but a significant increase in the frequency (but not number) of cytotoxic CD56+CD16+ NK cells in adults with T21 (Figure 4D). We next measured expression of various markers associated with increased activation and enhanced cytotoxicity of NK cells (i.e., CD16, CD38, CD8, and CD11c) (Addison et al., 2005). Most strikingly, all of these markers were significantly upregulated on the CD56+CD16+ cytotoxic subset of NK cells in T21 samples (Figures 4E and 4F). Thus, the most cytotoxic subset of NK cells is not only more abundant but also shows a heightened state of activation.
Figure 4. NK and CD8+ T Cells in Adults with T21 Display Signs of Heightened Activation and Increased Cytotoxic Potential.

(A–C) Mass cytometry was used to identify (A) bulk NK cells and (B and C) NK cell subsets, as described in Figures S2 and Table S5, within the peripheral blood of individuals with T21 (n = 18) and D21 controls (n = 18).
(D) Flow cytometry was used to determine the frequency of CD56++CD16— and CD56+CD16+ NK cell subsets among single events within the peripheral blood of individuals with T21 (n = 38) and D21 controls (n = 51), as depicted in Figure S4C.
(E) Within mass cytometry data, the level of protein expression indicated in heatmap titles was compared between individuals with T21 (n = 18) and D21 controls (n = 18) by geometric mean metal intensity (gMMI) for the indicated NK cell populations. Heatmaps display average gMMIs (left) or Arcsinh ratios (T21 gMMIs — D21 gMMIs, right) for the listed cell types.
(F) A representative histogram, with numbers denoting gMMIs among the parent gate indicated in the title. Green represents an individual with T21 and gray a D21 control.
(G) Mass cytometry was used to discriminate different subsets of T cells within the peripheral blood of individuals with T21 (n = 18) and D21 controls (n = 18) as described in Figure S2 and Table S5. Heatmaps indicate the log2 fold change (FC; T21 over D21) in frequency within all T cells for each of the indicated subsets.
(H) Box and whisker plot showing the depletion of naive CD8+ T cells in individuals with T21.
(I) Representative example of CD8+ T cell subset distribution in D21 versus T21 samples.
(J) Mass cytometry was used as in (G) to delineate subsets of CD8+ T cells that were negative or positive for granzyme B (GZMB) and/or PD1. Heatmaps indicate the log2 fold change (FC, T21 over D21) in frequency within all T cells for each of the indicated subsets.
(K) Box and whisker plots showing data for naive CD8+ T cells divided by GZMB and PD1 expression status.
(L) Representative example of GZMB and PD1 expression among CD8+ T cells in D21 versus T21 samples.
All statistical significance was determined by a Student’s t test (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001). All box and whisker plots denote the median within a box extending from the 25th to 75th percentiles and error bars span minimum to maximum values within the indicated datasets.
Regarding the T cell compartment, current literature is contradictory with respect to abundance of even bulk T cells in DS (reviewed in Ram and Chinen, 2011). We observed no difference in abundance of bulk T cells among circulating non-granulocytes (Figures S3 and S5A) and confirmed this result in a larger cohort (89 total adults, 39 with T21; Table S1B) using flow cytometry (Figures S5B and S4C). However, our antibody panel also enabled the dissection of 57 T cell subsets, including subsets of CD8+ T cells, CD4+ T cells, and regulatory T cells (Tregs). In agreement with previous reports (Pellegrini et al., 2012; Schoch et al., 2017), we observed a significant increase in Treg frequency among circulating non-granulocytes of hematopoietic lineage (Figures S3, S5C, and S5D). Next, we tested for differences in relative abundance within the T cell lineage. Of the 57 subsets analyzed, 11 were statistically different in people with T21 (Figure S5E; Table S4C). CD8+ T cells display a significant depletion of the naive compartment, concurrent with enrichment of the more differentiated memory and effector subsets (Figures 4G–4I). This phenomenon was accompanied by signs of hyperactivation of CD8+ T cells in the T21 samples, as evidenced by higher expression of the cytotoxic effector protein granzyme B (GZMB) and the inhibitory receptor PD1 (Figure 4J). This effect was particularly evident for naive CD8+ T cells (Figures 4J–4L), an unusual observation given that these proteins are normally expressed in activated CD8+ T cells. Interestingly, type I IFN has been shown to license naive CD8+ T cells to upregulate GZMB in the absence of cognate antigen to mediate cytotoxicity (Urban et al., 2016).
Altogether, these results indicate increased activation of the cytotoxic NK and CD8+ T cell compartments in DS.
Adults with DS Show Changes in B Cell Homeostasis Consistent with Inflammation, Autoimmunity, and Aging
The significant depletion of total B cells from the circulation of adults with T21 (Figures S3, 5A, and 5B) is in line with a previous study in children (de Hingh et al., 2005), but disagrees with the postulated normalization with age in DS (Ram and Chinen, 2011). Because this sustained B cell depletion well into adulthood could have important implications for the differential disease spectrum of people with DS, we validated this result using flow cytometry on a much larger cohort (Figure 5C; 83 total adults, 40 with T21; Table S1B).
Figure 5. The B Cell Compartment of Adults with T21 Is Skewed toward More Differentiated Subsets Associated with Inflammatory and Autoimmune States.

(A, B, and D–M) B cells were delineated among non-granulocytes by mass cytometry and manual gating as depicted in Figure S2 and then compared between individuals with T21 (n = 18) and D21 controls (n = 18).
(B, E, F, H, and J) Representative dot plots of the indicated cell type labeled in green with numbers denoting frequency among the parent gate indicated in titles. BMEM, memory B cells; BNA, naive B cells; PBs, plasmablasts.
(C) Flow cytometry was used to evaluate the number and frequency of circulating B cells among single events in individuals with T21 (n = 40 for frequency and n = 32 for relative cell number) and D21 controls (n = 53 for frequency and n = 45 for relative cell number), as depicted in Figure S4C.
(D, G, and I) Within mass cytometry data, B cell subsets were resolved among B cells of individuals with T21 (n = 18) and D21 controls (n = 18), as depicted in Figure S2.
(J and K) An alternative CD19HI gating strategy was used to identify CD11c+ B cells with numbers denoting frequency among the parent gate, B cells (left), or atypical/IgM IgD double-negative memory B cells (DN aBMEM).
(J) Representative dot plots on both sides of the arrow correspond to the same individual with T21 or D21 control.
(L) Representative histograms from an individual with T21 with numbers denoting geometric mean metal intensities (gMMIs) of PD1 expression among the indicated parent gate.
(M) Lineage tree depicts B cell subsets of individuals with T21 compared to D21 controls. Color denotes fold change, where red is increased and blue is decreased frequency of subsets among the B cells in T21 compared to D21 controls.
In all cases, statistical significance was determined by a Student’s t test (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001). All box and whisker plots denote the median within a box extending from the 25th to 75th percentiles and error bars span minimum to maximum values within the indicated datasets.
To investigate this further, we completed a deep analysis of B cell homeostasis by analyzing the markers of differentiation CD27, IgD, IgM, and CD38. Interestingly, we observed a depletion of the less differentiated immature naive (immature BNA), pre-switched activated, and IgM memory B cells (IgM BMEM) (Figures 5D–5F), concomitant with an increase in terminally differentiated plasmablasts (PBs) in DS (Figures 5G and 5H). These data are in line with previous studies of children with T21 showing a depletion of circulating naive B cells and an increased ability to differentiate into PBs in vitro (Verstegen et al., 2010; Carsetti et al., 2015).
We next sought to determine if there were shifts within the B cell compartment for subsets commonly associated with various autoimmune and inflammatory states: anergic B cells, CD11c+ B cells, class-delta switched memory B cells (c-delta BMEM), and IgM IgD double-negative atypical memory B cells (DN aBMEM). We did not observe a significant difference in anergic B cells, a subset that is strongly tied to self-reactive B cells in autoimmunity (Rosenspire and Chen, 2015). Instead, we observed a significant increase in c-delta BMEM (Figures 5F and 5I), which have been described as a likely ‘‘sink’’ for autoreactive B cells (Zheng et al., 2004). We also observed a significant increase in B cells that express CD11c (Figure 5I),a signature phenotype of age-associated B cells (ABCs) that are increased in the circulation of aged individuals with various autoimmune disorders (Rubtsov et al., 2011; Hao et al., 2011; Rubtsova et al., 2015; Karnell et al., 2017). Although CD11c+ B cells have been heavily studied in mouse models of autoimmunity, less is known about the CD11c+ ABCs, aBMEMs, and exhausted PD1+ B cells in humans (Rubtsova et al., 2015; Karnell et al., 2017; Portugal et al., 2017). Therefore, to determine if the CD11c+ fraction of B cells circulating among individuals with T21 were enriched among the subset of aBMEMs detected by our antibody panel, we applied the same gating of DN aBMEMs previously applied to bulk B cells (Figure S2; Table S5) but restricted this exercise to CD11c+ B cells that highly express the canonical CD19 marker of B cells (Figure 5J). We did this because a collective hallmark of these intriguing CD11c+ B cells is low expression of classical B cell markers, except CD19, and expression of receptors not usually expressed by B cells, including CD11c itself and PD1 (Portugal et al., 2017). This exercise revealed an enrichment of CD11c+CD19HI B cells in the DN aBMEM compartment, with significantly increased abundance in people with DS (Figures 5J and 5K). Notably, no B cell subset (either CD11c+ or CD11c−) expressed PD1 (Figure 5L).
Altogether, our analysis of the circulating B cell compartment revealed depletion of less differentiated states concurrent with increased levels of terminally differentiated subsets associated with inflammation and autoimmunity (Figure 5M).
Significant IFNR Overexpression in the Immune System of People with DS
We previously showed that T21 causes constitutive activation of the IFN transcriptional response in multiple cell types (Sullivan et al., 2016), along with changes in the circulating proteome indicative of autoinflammation (Sullivan et al., 2017), all of which could potentially be explained by the fact that four of the six IFNRs are encoded on chr21 (de Weerd and Nguyen, 2012). Therefore, we analyzed surface protein expression of the IFNAR1 subunit on 100 immune cell types identified by our panel and gating strategy. Remarkably, IFNAR1 overexpression was obvious and global (Figures 6A–6C and S6; Table S4D). Next, we analyzed RNA sequencing (RNA-seq) data from bulk white blood cells (WBCs) from a cohort of 19 individuals, 10 with T21 and the rest being age-and gender-matched D21 controls, to test for mRNA overexpression of all four IFNRs encoded on chr21 (Tables S1C and S4E). Indeed, the mRNAs for all four subunits were significantly overexpressed in circulating WBCs of people with DS (Figure 6D).
Figure 6. Global Upregulation of IFNAR1 Surface Protein Expression on Circulating Immune Cells of Individuals with T21.

(A–C) A total of 100 immune cell types were resolved by manual gating on expression of canonical markers as depicted in Figure S2, and then levels of IFNAR1 surface protein expression were compared between individuals with T21 (n = 18) and D21 controls (n = 18) by geometric mean metal intensity (gMMI).
(A) Each dot represents the mean gMMI for IFNAR1 among a gated cell type for all samples of a given karyotype.
(B) Volcano plot showing statistical analysis of IFNAR1 expression. Horizontal dashed line indicates the statistical cut-off of *p ≤ 0.05 as defined by Student’s t test. See Table S4D for full statistical analysis.
(C) Representative histograms of IFNAR1 protein expression with numbers denoting gMMIs. pDCs, plasmacytoid dendritic cells; mDCs, myeloid DCs; ILCs, innate lymphoid cells.
(D) Scatterplots displaying IFNR mRNA expression as defined by RNA-seq of bulk white blood cells of individuals with T21 (n = 10) versus D21 controls (n = 9). Statistical significance was calculated using DESeq2. mRNA expression values are displayed in reads per kilobase per million (RPKM).
See Table S4E for full statistical analysis.
Diverse Immune Cell Types with T21 Are Hyper-responsive to IFN-α Stimulation
Next, we embarked on a deep characterization of signaling events downstream of stimulation with the type I IFN ligand IFN-α−2a. To this end, we designed an antibody panel detecting cell surface markers and intracellular signaling epitopes that would enable single-cell resolution of the impact of both IFN-α and karyotype on eight recognized IFN-inducible signaling events (i.e., phosphorylation of STAT1–STAT6, ERK1/2, and 4E-BP1), in nine major immune subtypes (Table S3; Figure S2). We then adapted our blood processing protocol to include a 30-min stimulation with IFN-α−2a for 8 adults with T21 and 8 age- and gender-matched controls (Table S1A).
First, we analyzed the impact of IFN-α stimulation on the various phospho-epitopes in D21 samples. Expectedly, most signaling events were significantly elevated upon stimulation (Figure 7A; Table S4F). Interestingly, we observed different configurations of IFN-induced signaling among cell types. For example, although all cell types displayed a significant increase in pSTAT1, pSTAT3, and pSTAT5, the fold change was greatest for pSTAT1 in plasmacytoid dendritic cells (pDCs), pSTAT3 in monocytes, and pSTAT5 in HSPCs (Figures 7A–7C, S7A, and S7B).
Figure 7. Global IFN-α Hypersensitivity among Circulating Immune Cells from Individuals with T21.

Whole blood was incubated directly ex vivo for 30 min with (+IFN) or without (basal) IFN-α−2a before processing for mass cytometry. Phospho-epitopes among bulk immune subtypes were resolved by manual gating on expression of canonical markers, as depicted in Figure S2, and then compared between individuals with T21 (n = 8) and D21 controls (n = 8).
(A–G) Arcsinh ratios shown in volcano plots and heatmaps were generated from geometric mean metal intensity (gMMIs) of each of the 8 phospho-epitopes among 9 immune subtypes.
(H) Radar plots scaled to the minimum and maximum gMMIs for the indicated phospho-epitope, where each spoke represents a distinct immune subset, with cohort and stimulation status denoted by line and color. Dot plots represent fold changes in the indicated phospho-epitopes within the indicated immune subset from each biological replicate, with lines connecting samples from the same individual with and without IFN stimulation.
In all cases, statistical significance was determined by a Student’s t test (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001).
We then investigated the impact of T21 on basal levels of phosphorylation prior to IFN-α stimulation, which identified many phospho-epitopes that were significantly elevated at baseline (Figure 7D; Table S4G). Most prominent among these was phosphorylation of the mTOR effector 4E-BP1 (Figure 7E), consistent with previous findings reporting activation of the mTOR pathway in DS (Iyer et al., 2014). CD7+ ILCs were the most impacted cell type in unstimulated conditions, showing significantly elevated levels of 4 of the 8 epitopes measured (Figure S7C).
Finally, we defined the impact of T21 on IFN-stimulated signaling events, which revealed that immune cells from people with T21 show significant hyper-activation of many phospho-epitopes (Figure 7F; Table S4H). For example, pSTAT1 and p4E-BP1 were significantly hyper-induced in multiple cell types tested (Figures S7D and S7E). Of note, there is clear cell-type specificity in the impact of T21 on IFN-inducible signaling. For instance, pSTAT1 and p4E-BP1 were hyper-activated most strongly in the myeloid lineage (i.e., monocytes and DCs), while pSTAT4 was hyper-activated in lineages involved in cellular immunity (i.e., T cells and CD7+ ILCs) (Figures 7G, 7H, and S7F). This agrees with the fact that whereas pSTAT1 is the canonical transcription factor downstream of type I IFNs, STAT4 plays more restricted roles, such as directly inducing expression of GZMB in T cells upon IFN stimulation (Newby et al., 2017). Interestingly, CD8+ T cells from individuals with T21 display increased expression of GZMB (Figures 4J–4L). Phosphorylation of ERK1/2 is another example of the cell-type-specific effects of T21, as it is only significantly upregulated under basal and/or induced conditions in monocytes and DCs from individuals with T21 (Figures 7H and S7F; Tables S4G and S4H). Finally, we noted that fibrocytes showed significantly elevated levels of IFN-inducible signaling relative to COL1-negative HSPCs, both under baseline and IFN-inducible conditions, supporting the notion that differentiation of COL1-positive fibrocytes is driven by JAK/STAT signaling during inflammation (Figure S7G) (Galligan and Fish, 2013).
Altogether, these results demonstrate that T21 increases the response to IFN stimulation, leading to hyper-activation of cell-type-specific downstream signaling cascades. Furthermore, key negative regulators of IFN signaling (e.g., SOCS1 and USP18) were not found to be significantly upregulated at the mRNA level in individuals with T21 (Table S4E). Altogether, these data suggest the absence of desensitization to IFN signaling despite what is likely to be a lifelong overexpression of the IFNRs throughout the immune system, pointing instead to chronic IFN hyperactivity in DS.
DISCUSSION
Using mass cytometry, we uncovered drastic alterations in immune homeostasis in adults with DS across all detected branches of the immune system. Repeatedly, these changes were similar to those associated with the chronic state of low-grade inflammation observed during typical aging and in various inflammatory and autoimmune states in the general population. Consistently, these changes could be tied to increased IFN signaling.
Many immune alterations associated with typical aging are thought to originate from HSPCs (Akunuru and Geiger, 2016). Mobilization of HSPCs to the peripheral blood increases with age (Rundberg Nilsson et al., 2016) and is promoted by various pro-inflammatory cytokines (Hoggatt and Pelus, 2011). Therefore, we were surprised to find that adults with DS show a significant decrease in circulating HSPCs. This paradox could be explained by chronic hyperactivity in type I IFN signaling, which has been shown to induce a quiescent state in the HSPC pool (Pietras et al., 2014). The decrease in mobilized HSPCs was accompanied by a significant increase in circulating fibrocytes, which are known to be elevated in several autoimmune and chronic inflammatory states, where they are considered amplifiers of the immune response (Galligan and Fish, 2013). Of note, the massive increase in fibrocytes observed during myelofibrosis is known to be driven by hyperactive JAK/STAT signaling, and myelofibrosis is effectively treated with JAK inhibitors (Harrison et al., 2012). Thus, the observed disruption in HSPC homeostasis in people with DS could potentially be explained by hyperactive IFN signaling.
Phenotypic alterations within the DC, NK, and T cell compartments of adults with T21 suggest heightened states of activation and cytotoxicity, which could be linked to the development of autoimmunity in DS. For instance, DCs from adults with DS express significantly higher levels of CD56, a hallmark of cytotoxic myeloid cells (Van Acker et al., 2017). Additionally, the mDC compartment is enriched for CD1c+ cDCs in DS. CD1c is a molecule that presents lipid antigens to T cells (Roy et al., 2014) and directly activates intrathyroidal T cells during autoimmune thyroiditis (Roura-Mir et al., 2005), a condition that is highly prevalent in DS (Karlsson et al., 1998). Furthermore, there are clear signs of increased activation of NK and cytotoxic T cells in DS, including significant upregulation of GZMB on even the naive CD8+ T cells. Dysregulated cytotoxic responses by immune cells destroy target organs in various autoimmune disorders that are more prevalent in people with DS but also protect from solid tumors, all downstream of IFN signaling (Xing et al., 2014; Byrne and Turk, 2011; Gogas et al., 2006; Parker et al., 2016). Even the humoral B cell compartment showed changes associated with autoimmunity and inflammation, such as the significant increase in frequency of PBs among circulating B cells, as these antibody-secreting cells increase in circulation following vaccination, during viral infection, or upon bouts of autoimmune activity (Buckner et al., 2013; Odendahl et al., 2005; Lee et al., 2011; Kunkel and Butcher, 2003; Dörner and Lipsky, 2004). The increased frequency of CD11c+ B cells is another hallmark of chronic inflammatory and autoimmune states, particularly in older typical people (Rubtsov et al., 2011; Hao et al., 2011; Rubtsova et al., 2015; Karnell et al., 2017; Portugal et al., 2017). Therefore, multiple changes in cell types involved in cellular and humoral immunity observed in DS could be tied to their elevated risk of autoimmunity.
Many of the immune alterations observed represent potential therapeutic nodes to improve health outcomes in DS, most prominent among them the obvious IFN hyperactivity. We observed that IFNAR1 is significantly increased on the surface of most primary immune cell types circulating in adults with DS, in contrast to a recent publication reporting equal surface expression on primary monocytes, T cells, and B cells using standard flow cytometry (Martínez et al., 2016). Our results agree with an earlier publication that indirectly determined increased surface levels of IFNARs on PBMCs with T21 using radiolabeled type I IFN ligands (Gerdes et al., 1993). Although somewhat expected given the increased gene dosage, this global overexpression is still remarkable, because surface levels of IFNAR protein have been shown to be controlled by negative feedback loops (reviewed in Ivashkiv and Donlin, 2014). Furthermore, signaling downstream of IFNARs is also controlled by negative feedback loops via induction of negative regulators such as ISG15 (Ivashkiv and Donlin, 2014). However, our results discard the notion of ‘‘IFN desensitization’’ in DS. Although the adults in our study have likely experienced a life-long increase in IFN signaling, they remain hyper-responsive to IFN stimulation. When we assayed for multiple IFN-induced signaling events, we observed both increased basal signaling and hyper-activation. Of note, increased basal phosphorylation of 4E-BP1 agrees with reports showing activation of the mTOR pathway in DS (Iyer et al., 2014) and is supported by the fact that mTOR is activated by type I and II IFNs (Kaur et al., 2007). This suggests that mTOR inhibitors, such as rapamycin, could potentially have therapeutic benefits in DS, which is supported by a recent study of rapamycin in a mouse model of DS (Duval et al., 2018). Additionally, these results point to JAK/STAT signaling as an obvious node of intervention in DS. JAK inhibitors are either US Food and Drug Administration (FDA) approved or in clinical trials to treat various autoimmune disorders that are more prevalent in DS, including many autoinflammatory and autoimmune skin conditions (Banerjee et al., 2017; Welsch et al., 2017). Recently, we reported the first two cases of JAK inhibition in DS, with remarkable therapeutic benefit for alopecia areata (Rachubinski et al., 2019), which encourages the development of clinical trials for JAK inhibitors in DS. Lastly, our results reveal many parallels between systemic lupus erythematosus (SLE), a condition associated with increased type I IFN signaling, and DS. For example, our plasma proteomics analysis found many cytokines commonly elevated in DS (Sullivan et al., 2017) and SLE (Idborg et al., 2018), such as IL-10, CXCL10, IL-6, TNF-a, MCP-1, CRP, VEGFA, IL-15, IL-8, and eotaxin. Although there are a few case reports of SLE co-occurring with DS (Bakkaloglu et al., 1994; Feingold and Schneller, 1995; Suwa et al., 2000), an exhaustive investigation of SLE-like pathology in DS is missing.
Although increased IFN signaling is a likely driver of immune dysregulation in DS, additional studies will be necessary to determine definitive cause-effect relationships between IFN hyperactivity and DS phenotypes and define whether anti-IFN strategies will have therapeutic benefits in DS. This report establishes the needed foundation for these studies, with clear potential to also advance our understanding of the impact of immune dysregulation in the typical population.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joaquin Espinosa (joaquin.espinosa@cuanschutz.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All participants were enrolled under study protocols approved by the Colorado Multiple Institutional Review Board (COMIRB #14–2300 for children and COMIRB #15–2170 for adults). Written informed consent was obtained from parents or guardians of each participant, and assent was obtained from participants over the age of 7 years who were cognitively able to assent. All procedures were performed in accordance with COMIRB guidelines and regulations. Demographics and other data about research participants can be found in Table S1.
METHOD DETAILS
Study design
The objective of this study was to determine baseline changes in immune homeostasis within the myeloid and lymphoid lineages of adults with DS in comparison to typical controls. To achieve this goal, we utilized mass cytometry to compare white blood cells enriched for non-granulocytes from 36 adults (18 with DS). This sample size was determined a priori based on previous studies of the transcriptome and proteome of individuals with DS. Key results from mass cytometry were pursued in larger cohorts by cross-platform validation using flow cytometry. None of the participants reported signs of active infection for at least 2 weeks prior to blood draw. All samples were processed directly ex vivo. All data that passed quality control metrics from the mass cytometry or flow cytometry instruments were included. The number of biological replicates for each experiment are detailed in Table S1. All immune and clinical variables collected are provided in Table S2, and also made accessible to readers through the following online portal: https://trisome.shinyapps.io/Waugh_et_al_Cell_Reports_Online_Portal/.
Sample processing for mass cytometry
Whole blood was collected into BD Vacutainer® K2 EDTA tubes (BD, 366643) and processed for mass cytometry analyses directly ex vivo using a protocol adapted from previous studies (O’Gorman et al., 2015). For baseline characterization, 0.5 mL of whole blood underwent red blood cell (RBC) lysis and fixation (BD Biosciences #558049), was washed in 1x PBS (Rockland #MB-008) and Cell Staining Buffer (CSB, Fluidigm #201068), then stored at −80°C in CSB. For IFNa stimulations, 0.5 mL of whole blood was treated with 10,000 U/mL of IFNα−2a (PBL #11100–1) for 30 min at 37°C in 5% CO2, then processed and stored as described above. For staining, samples were thawed at room temperature, washed in CSB, barcoded according to Fluidigm’s Cell-ID 20-Plex Pd Barcoding Kit (#PRD023), then combined prior to staining. Extracellular staining with antibodies was done in CSB, then simultaneous detection of intracellular transcription factors and phospho-epitopes was accomplished using BD PharMingen’s Transcription Factor Phospho Buffer Set (#563239). All antibodies used for staining were either bought pre-conjugated to metal isotopes or were conjugated using the Maxpar Antibody Labeling Kit (Fluidigm 201160B). Barcoded and stained cells were labeled with Cell-ID Intercalator-Ir (Fluidigm, #201192A), then analyzed on a Helios instrument (Fluidigm) with the antibody-metal conjugates listed in Table S3.
Sample processing for flow cytometry
Complete blood counts (CBCs) were acquired on an AcT 10 (Beckman Coulter), then blood was centrifuged at 700 g for 15 min to separate plasma, buffy coat, and RBCs. White blood cells (WBCs) were enriched from the buffy coat fraction by RBC lysis according to manufacturer’s instructions (BD, #555899). WBCs were then stained in FACS Buffer (1x PBS, 1 mM EDTA, 25 mM HEPES pH 7.0, 1% FBS) with the following fluorochrome-conjugated Abs to identify indicated cell populations: CD3 (OKT3; BioLegend), CD19 (HIB19; BioLegend), CD56 (5.1H11; BioLegend), CD16 (B73.1; BioLegend), and CD14 (63D3; BioLegend). Events were acquired on a MoFlo Astrios EQ (Beckman Coulter) then analyzed using FlowJo software (Tree Star v10.4.0). CBCs were used to back-calculate relative cell number per uL of blood for indicated cell types among single events.
For analysis of fibrocytes by flow cytometry, fresh blood (5 mL) was prepared as previously described (Moeller et al., 2009). Briefly, buffy coat cells were removed from blood centrifuged for 10 min at 450 g at room temperature. Following RBC lysis by addition of ammonium chloride (0.15 M, pH 7.3), leukocytes were washed, verified for viability by trypan blue exclusion, and prepared to a concentration of 1×106 cells/mL. Cells were stained with the fluorochrome-conjugated anti-CD45 (PB986; BD Biosciences), fixed and permeabilized according to the manufacturer’s protocol (Fix/Perm Solution 554714; BD Biosciences), and then labeled with rabbit anti-human pro-collagen I (MAB1913; Millipore). Secondary antibody (FITC-conjugated goat anti-rabbit IgG; AP132F; Millipore) was applied according to the manufacturer’s protocols. The negative thresholds were set using fluorescence minus one (FMO) labeled cells from both typical children and children with DS. Events were acquired using an FC500 flow cytometer then processed using CXP software (Beckman Coulter Inc., Brea, CA, USA).
Pre-processing of mass cytometry data
Pre-processing of mass cytometry data included normalization within and between batches via polystyrene beads embedded with lanthanides as previously described (Finck et al., 2013), transformation using hyperbolic arcsinh, then normalization again between batches prior to viSNE analysis using the empirical Bayes approach, Combat (‘sva’ bioconductor package v3.26.0) in R-3.4.3.
Analysis of mass cytometry data using viSNE
To visualize the global structure of myeloid and lymphoid lineages, non-granulocytes (CD66−) of the hematopoietic lineage (CD45+) were enriched by manual gating among single events (Figure S2A), equally subsampled to 2,600–6,000 events, then run through a Barnes-Hut implementation of the t-SNE algorithm, viSNE (Amir et al., 2013), in the R package ‘Rtsne’ (v0.13), using optimized parameters (Figure S1B). All markers listed in Table S3 to characterize myeloid and lymphoid lineages were selected for viSNE, excluding CD34, due to low abundance of CD34+ HSPCs (< 2% of CD45+CD66-events; Amir et al., 2013), as well as CD66, CD45, and IFNAR1. JS divergence scores were calculated for all pairs of viSNE plots as previously described (Amir et al., 2013) to quantify overall similarity among samples, then organized by hierarchical clustering to visualize overall structure within the dataset.
Comparison of distinct regions within viSNE plots
The high-dimensional clustering algorithm PhenoGraph (Levine et al., 2015) was applied by the ‘mass cytometry_cluster’ function in the mass cytometry kit bioconductor package (v1.12.0, Chen et al., 2016) of R-3.5.1 to partition CD45+CD66-single events into subpopulations by expression of all markers listed above for viSNE. To apply topographical analysis to viSNE plots, the ‘kde2d’ function in the MASS library (v7.3.50) of R-3.4.3 was used to get kernel density estimates (KDEs) for a 300×300 grid overlaid onto viSNE plots. Differences in cohort KDEs were visualized by subtraction of concatenated T21 viSNE plots from concatenated D21 viSNE plots (ggplot2 v3.0.0). All manual gating to resolve immune cell types was done in FlowJo, CytoBank, or resultant GatingML files in R that were then applied to indicated analyses.
RNA-seq analysis
WBCs were cryopreserved in CryoStor CS10 Freezing Media (#210102) at ~10×106 cells/mL. WBCs were thawed quickly at 37°C and immediately spiked into 9 mL of complete RPMI media then centrifuged at 500 g for 5 min and media removed. Cells were resuspended in 600 μL RLT Plus (QIAGEN) and Beta-mercaptoethanol (BME) lysis buffer (10 μL BME:1 mL RLT Plus). RNA was extracted using the AllPrep DNA/RNA/Protein Mini Kit according to manufacturer’s instructions (QIAGEN, 80004). RNA quality was determined by Agilent 2200 TapeStation and quantified by Qubit (Life Technologies). Samples with RINs of R 6.8 and a minimum of 1 μg were used to prepare next-generation sequencing libraries with the Illumina TruSeq Stranded mRNA Library Prep Kit and sequenced with an Illumina NovaSeq 6000 instrument. Analysis of library complexity and high per-base sequence quality across all reads was performed using FastQC software. Other bioinformatic steps included trimming of low-quality bases, short reads, and adaptor sequences, with the fastqc-mcf. tool; removal of mycoplasma, mitochondria, and rRNA contaminant sequences with FASTQ Screen; read alignment to GRCh37/hg19 using TopHat2; filtering of high-quality mapped reads with SAMtools; and final quality performed using RSeQC. Gene level counts were obtained using HTSeq (Anders et al., 2015).
Heatmaps, box and whisker, volcano, and radar plots
Generation of heatmaps and volcano plots was done using ggplot2 (v3.1.0) and radar plots using the ‘radarchart’ function of the fmbs package (v0.6.3) in R-3.5.1. All box and whisker plots denote the median within a box extending from the 25th to 75th percentiles and error bars span minimum to maximum values within the indicated datasets (Prism vs7 and 8; GraphPad).
QUANTIFICATION AND STATISTICAL ANALYSIS
The number of biological replicates, or ‘‘n,’’ is specified for each experiment in Table S1. All statistical significance was analyzed using a paired (Figure 7A and Table S4F) or unpaired (all other figures and tables) two-tailed Student’s t test in Prism (vs7 and 8; GraphPad), Microsoft Excel (v16.16.5), or R-3.5.1. A p value ≤ 0.05 was considered statistically significant and is denoted by an asterisk, in which *p ≤ 0.05, **p ≤ 0.01, **p ≤ 0.001, and ***p ≤ 0.0001. Correction for multiple hypotheses testing was performed by the Benjamini-Hochberg method (Klipper-Aurbach et al., 1995). All Benjamini-Hochberg adjusted p values can be found in Tables S4 and S6.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-human CD45 (clone HI30) | Fluidigm | Cat#3089003; RRID: AB_2661851 |
| Mouse monoclonal anti-human CD66a/c/d/e (clone B1.1/CD66) | BD Biosciences | Cat# 551354; RRID: AB_394166 |
| Mouse monoclonal anti-human IgM (clone MHM-88) | BioLegend | Cat#314502; RRID: AB_493003 |
| Mouse monoclonal anti-human CD19 (clone HIP19) | Fluidigm | Cat#3142001; RRID: AB_2651155 |
| Mouse monoclonal anti-human CD123 (clone 6H6) | Fluidigm | Cat#3143014B, RRID: AB_2811081 |
| Mouse monoclonal anti-human CD4 (clone RPA-T4) | Fluidigm | Cat#3145001; RRID: AB_2661789 |
| Mouse monoclonal anti-human IgD (clone IA6–2) | Fluidigm | Cat#3146005B; RRID:AB_2811082 |
| Mouse monoclonal anti-human CD11c (clone Bu15) | Fluidigm | Cat#3147008; RRID:AB_2687850 |
| Mouse monoclonal anti-human CD127 (clone A019D5) | Fluidigm | Cat#3149011; RRID:AB_2661792 |
| Mouse monoclonal anti-human CD1c (clone L161) | BioLegend | Cat# 331501; RRID:AB_1088996 |
| Mouse monoclonal anti-human CD14 (clone M5E2) | Fluidigm | Cat#3175015B; RRID:AB_2811083 |
| Mouse monoclonal anti-human CD7 (clone CD7–6B7) | DVS Sciences | Cat#3153014B; RRID:AB_2811084 |
| Mouse monoclonal anti-human CD3 (clone UCHT1) | DVS Sciences | Cat#3154003B; RRID:AB_2811086 |
| Mouse monoclonal anti-human CD279/PD1 (clone EH12.2H7) | Fluidigm | Cat#3155009B; RRID:AB_2811087 |
| Mouse monoclonal anti-human CD45RA (clone HI100) | BioLegend | Cat#304102; RRID:AB_314406 |
| Mouse monoclonal anti-human FOXP3 (clone 259D/C7) | Fluidigm | Cat#3159028A; RRID:AB_2811088 |
| Rabbit monoclonal anti-human IFNAR1 (clone EP899Y) | Abcam | Cat#ab45172; RRID:AB_775764 |
| Mouse monoclonal anti-human CD16 (clone B73.1) | BioLegend | Cat#360702; RRID:AB_2562693 |
| Mouse monoclonal anti-human CD8a (clone RPA-T8) | Fluidigm | Cat#3162015; RRID:AB_2661802 |
| Mouse monoclonal anti-human CD34 (clone 581) | Fluidigm | Cat#3163014B; RRID:AB_2811091 |
| Mouse monoclonal anti-human CD45RO (clone UCHL1) | Fluidigm | Cat#3164007B; RRID:AB_2811092 |
| Mouse monoclonal anti-human CD27 (clone L128) | Fluidigm | Cat#3167006B; RRID:AB_2811093 |
| Mouse monoclonal anti-human CD25 (clone 2A3) | Fluidigm | Cat#3169003; RRID:AB_2661806 |
| Mouse monoclonal anti-human CD141 (clone M80) | BioLegend | Cat#344102; RRID:AB_2201808 |
| Mouse monoclonal anti-human CD38 (clone HIT2) | Fluidigm | Cat#3172007B; RRID:AB_2756288 |
| Mouse monoclonal anti-human GZMB (clone GB11) | Fluidigm | Cat#3173006B; RRID:AB_2811095 |
| Mouse monoclonal anti-human HLA-DR (clone L243) | Fluidigm | Cat#3174001; RRID:AB_2665397 |
| Mouse monoclonal anti-human CD56 (clone N901) | Fluidigm | Cat#3176009B; RRID:AB_2811096 |
| Mouse monoclonal anti-human COL1 (clone 5D8-G9/Col1) | ThermoFisher Scientific | Cat#CSI 008–01-02; RRID:AB_1073749 |
| Mouse monoclonal anti-human phospho-STAT4 (Tyr693) (clone 38/p-Stat4) | Fluidigm | Cat#3148006A; RRID:AB_2811098 |
| Rabbit monoclonal anti-human phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (clone D13.14.4E) | Cell Signaling Technology | Cat#4370; RRID:AB_2315112 |
| Mouse monoclonal anti-human phospho-STAT3 (Tyr705) (clone 4/P-STAT3) | Fluidigm | Cat#3158005A; RRID:AB_2811100 |
| Rabbit monoclonal anti-human phospho-STAT5 (Tyr694) (clone C11C5) | Cell Signaling Technology | Cat#9359; RRID:AB_823649 |
| Rabbit monoclonal anti-human phospho-4E-BP1 (Thr37 / Thr46) (clone 236B4) | Cell Signaling Technology | Cat#2855; RRID:AB_560835 |
| Rabbit monoclonal anti-human phospho-STAT6 (Tyr641) (clone 18) | Fluidigm | Cat#3168012A; RRID:AB_2811103 |
| Rabbit monoclonal anti-human phospho-STAT2 (Tyr690) (clone D3P2P) | Fluidigm | Cat#88410; RRID:AB_2800123 |
| Rabbit monoclonal anti-human phospho-STAT1 (Tyr701) (clone 58D6) | Cell Signaling Technology | Cat#9167; RRID:AB_561284 |
| Mouse monoclonal anti-human CD45 (clone HI30) | Fluidigm | Cat#3089003; RRID: AB_2661851 |
|
Biological Samples | ||
| Blood from donors | The Crnic Institute Human Trisome Project Biobank | Clinicaltrials.gov Identifier: ; www.trisome.org |
|
Chemicals, Peptides, and Recombinant Proteins | ||
| Human IFNα−2a | PBL Assay Science | Cat#11100–1 |
|
Critical Commercial Assays | ||
| Cell-ID 20-Plex Pd Barcoding Kit | Fluidigm | Cat#PRD023 |
| Transcription Factor Phospho Buffer Set | BD PharMingen | Cat#563239 |
| Cell-ID Intercalator-Ir | Fluidigm | Cat#201192A |
|
Deposited Data | ||
| RNA-seq data | Espinosa lab | GEO: GSE128622 (super series), GEO: GSE128614 (unstranded data), and GEO: GSE128621 (strand-specific). |
| All flow and mass cytometry data | Espinosa lab | https://trisome.shinyapps.io/Waugh_et_al_Cell_Reports_Online_Portal/ |
| Raw CyTOF data for unstimulated samples | Espinosa lab | https://doi.org/10.17605/OSF.IO/JTE35, https://osf.io/jte35/ |
| Raw CyTOF data for stimulated samples | Espinosa lab | https://doi.org/10.17605/OSF.IO/JTE35, https://osf.io/jte35/ |
|
Software and Algorithms | ||
| FlowJo | Tree Star | v10.5.3 |
| Bead normalization of mass cytometry data | Finck et al., 2013 | https://www.cytobank.org/nolanlab |
| Combat | ‘sva’ Bioconductor package | R package v3.26.0 |
| Rtsne | Amir et al., 2013 | R package v0.13 |
| Jenson-shannon divergence | Amir et al., 2013 | |
| PhenoGraph ‘cytof_cluster’ function of cytofkit bioconductor package | Chen et al., 2016 | R package v1.12.0 |
| ‘kde2d’ function | MASS library | R package v7.3.50 |
Highlights.
Individuals with trisomy 21, or Down syndrome, have a unique disease spectrum
Mass cytometry reveals global immune dysregulation affecting key cell types
Changes in myeloid and lymphoid subsets are associated with inflammatory states
Trisomy 21 causes overexpression of IFN receptors and hypersensitivity to IFN-α
ACKNOWLEDGMENTS
We thank members of the Sie Center for Down Syndrome, the Global Down Syndrome Foundation, the Clinical and Translational Research Center, the University of Colorado Cancer Center Flow Cytometry Shared Resource, and the Human Immune Monitoring Shared Resource for their assistance in various aspects of the project. This work was supported by NIH grants R01AI150305, R01AI145988, R01AI141662, T32CA190216, UL1TR002535, and P30CA046934. Additional funding was provided by NSF grant MCB1817582, the Linda Crnic Institute for Down Syndrome, the Global Down Syndrome Foundation, the Anna and John J. Sie Foundation, the Human Immunology and Immunotherapy Initiative, the GI & Liver Innate Immune Program, and a Blumenthal Fellowship to K.A.W.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.038.
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND CODE AVAILABILITY
RNA-seq data were deposited at the Gene Expression Omnibus (National Center for Biotechnology Information) with series accession numbers GEO: GSE128622 (super series), GEO: GSE128614 (unstranded data), and GSE128621 (strand-specific). All mass cytometry and flow cytometry data can be accessed through the online portal: https://trisome.shinyapps.io/Waugh_et_al_Cell_Reports_Online_Portal/. The code supporting the current study have not been deposited in a public repository but are available from the lead contact upon request. Raw mass cytometry data can be obtained at the following Open Science Framework websites with https://doi.org/10.17605/OSF.IO/JTE35 and https://doi.org/10.17605/OSF.IO/P5KFM: https://osf.io/jte35/ and https://osf.io/p5kfm/.
ADDITIONAL RESOURCES
All immune and clinical variables collected are accessible to readers through the following online interactive portal: https://trisome.shinyapps.io/Waugh_et_al_Cell_Reports_Online_Portal/.
All adult participants were enrolled in The Crnic Institute Human Trisome Project Biobank (www.trisome.org, ClinicalTrials.gov Identifier ).
REFERENCES
- Addison EG, North J, Bakhsh I, Marden C, Haq S, Al-Sarraj S, Malayeri R, Wickremasinghe RG, Davies JK, and Lowdell MW (2005). Ligation of CD8alpha on human natural killer cells prevents activation-induced apoptosis and enhances cytolytic activity. Immunology 116, 354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akunuru S, and Geiger H (2016). Aging, clonality, and rejuvenation of hematopoietic stem cells. Trends Mol. Med 22, 701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, and Pe’er D (2013). viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 31, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar AJ, Walker JD, and Frier BM (1998). Type 1 diabetes mellitus and Down’s syndrome: prevalence, management and diabetic complications. Diabet. Med 15, 160–163. [DOI] [PubMed] [Google Scholar]
- Bakkaloglu A, Ozen S, Besbas N, Saatci U, and Balci S (1994). Down syndrome associated with systemic lupus erythematosus: a mere coincidence or a significant association? Clin. Genet 46, 322–323. [DOI] [PubMed] [Google Scholar]
- Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, Lou X, Pavlov S, Vorobiev S, Dick JE, and Tanner SD (2009). Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem 81, 6813–6822. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Biehl A, Gadina M, Hasni S, and Schwartz DM (2017). JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs 77, 521–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemers BL, Van Bleek GM, Kimpen JL, and Bont L (2010). Distinct abnormalities in the innate immune system of children with Down syndrome. J. Pediatr 156, 804–809. [DOI] [PubMed] [Google Scholar]
- Buckner CM, Moir S, Ho J, Wang W, Posada JG, Kardava L, Funk EK, Nelson AK, Li Y, Chun TW, and Fauci AS (2013). Characterization of plasmablasts in the blood of HIV-infected viremic individuals: evidence for nonspecific immune activation. J. Virol 87, 5800–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne KT, and Turk MJ (2011). New perspectives on the role of vitiligo in immune responses to melanoma. Oncotarget 2, 684–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carsetti R, Valentini D, Marcellini V, Scarsella M, Marasco E, Giustini F, Bartuli A, Villani A, and Ugazio AG (2015). Reduced numbers of switched memory B cells with high terminal differentiation potential in Down syndrome. Eur. J. Immunol 45, 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheadle WG (1993). The human leukocyte antigens and their relationship to infection. Am. J. Surg 165 (2A, suppl), 75S–81S. [DOI] [PubMed] [Google Scholar]
- Chen H, Lau MC, Wong MT, Newell EW, Poidinger M, and Chen J (2016). Cytofkit: a bioconductor package for an integrated mass cytometry data analysis pipeline. PLoS Comput. Biol 12, e1005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesney J, Bacher M, Bender A, and Bucala R (1997). The peripheral blood fibrocyte is a potent antigen-presenting cell capable of priming naive T cells in situ. Proc. Natl. Acad. Sci. USA 94, 6307–6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooles FAH, Anderson AE, Skelton A, Pratt AG, Kurowska-Stolarska MS, McInnes I, Hilkens CMU, and Isaacs JD (2018). Phenotypic and transcriptomic analysis of peripheral blood plasmacytoid and conventional dendritic cells in early drug naı¨ve rheumatoid arthritis. Front. Immunol 9, 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossarizza A, Ortolani C, Forti E, Montagnani G, Paganelli R, Zannotti M, Marini M, Monti D, and Franceschi C (1991). Age-related expansion of functionally inefficient cells with markers of natural killer activity in Down’s syndrome. Blood 77, 1263–1270. [PubMed] [Google Scholar]
- de Graaf G, Buckley F, and Skotko BG (2017). Estimation of the number of people with Down syndrome in the United States. Genet. Med 19, 439–447. [DOI] [PubMed] [Google Scholar]
- de Hingh YC, van der Vossen PW, Gemen EF, Mulder AB, Hop WC, Brus F, and de Vries E (2005). Intrinsic abnormalities of lymphocyte counts in children with down syndrome. J. Pediatr 147, 744–747. [DOI] [PubMed] [Google Scholar]
- de Weerd NA, and Nguyen T (2012). The interferons and their receptors—distribution and regulation. Immunol. Cell Biol 90, 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörner T, and Lipsky PE (2004). Correlation of circulating CD27high plasma cells and disease activity in systemic lupus erythematosus. Lupus 13, 283–289. [DOI] [PubMed] [Google Scholar]
- Duval N, Vacano GN, and Patterson D (2018). Rapamycin treatment ameliorates age-related accumulation of toxic metabolic intermediates in brains of the Ts65Dn mouse model of Down syndrome and aging. Front. Aging Neurosci. 10, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold M, and Schneller S (1995). Down syndrome and systemic lupus erythematosus. Clin. Genet. 48, 277. [DOI] [PubMed] [Google Scholar]
- Finck R, Simonds EF, Jager A, Krishnaswamy S, Sachs K, Fantl W, Pe’er D, Nolan GP, and Bendall SC (2013). Normalization of mass cytometry data with bead standards. Cytometry A 83, 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galligan CL, and Fish EN (2013). The role of circulating fibrocytes in inflammation and autoimmunity. J. Leukoc. Biol. 93, 45–50. [DOI] [PubMed] [Google Scholar]
- Garrison MM, Jeffries H, and Christakis DA (2005). Risk of death for children with down syndrome and sepsis. J. Pediatr 147, 748–752. [DOI] [PubMed] [Google Scholar]
- Gerdes AM, Hørder M, Petersen PH, and Bonnevie-Nielsen V (1993). Effect of increased gene dosage expression on the alpha-interferon receptors in Down’s syndrome. Biochim. Biophys. Acta 1181, 135–140. [DOI] [PubMed] [Google Scholar]
- Gogas H, Ioannovich J, Dafni U, Stavropoulou-Giokas C, Frangia K, Tsoutsos D, Panagiotou P, Polyzos A, Papadopoulos O, Stratigos A, et al. (2006). Prognostic significance of autoimmunity during treatment of melanoma with interferon. N. Engl. J. Med 354, 709–718. [DOI] [PubMed] [Google Scholar]
- Hao Y, O’Neill P, Naradikian MS, Scholz JL, and Cancro MP (2011). A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood 118, 1294–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, McQuitty M, Hunter DS, Levy R, Knoops L, et al. (2012). JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med 366, 787–798. [DOI] [PubMed] [Google Scholar]
- Hartley D, Blumenthal T, Carrillo M, Dipaolo G, Esralew L, Gardiner K, Granholm AC, Iqbal K, Krams M, Lemere C, et al. (2014). Down syndrome and Alzheimer’s disease: common pathways, common goals. Alzheimers Dement 11, 700–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasle H, Friedman JM, Olsen JH, and Rasmussen SA (2016). Low risk of solid tumors in persons with Down syndrome. Genet. Med 18, 1151–1157. [DOI] [PubMed] [Google Scholar]
- Hoggatt J, and Pelus LM (2011). Mobilization of hematopoietic stem cells from the bone marrow niche to the blood compartment. Stem Cell Res. Ther 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idborg H, Eketjäll S, Pettersson S, Gustafsson JT, Zickert A, Kvarnström M, Oke V, Jakobsson PJ, Gunnarsson I, and Svenungsson E (2018). TNF-α and plasma albumin as biomarkers of disease activity in systemic lupus erythematosus. Lupus Sci. Med 5, e000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv LB, and Donlin LT (2014). Regulation of type I interferon responses. Nat. Rev. Immunol 14, 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer AM, van Scheppingen J, Milenkovic I, Anink JJ, Adle-Biassette H, Kovacs GG, and Aronica E (2014). mTOR Hyperactivation in down syndrome hippocampus appears early during development. J. Neuropathol. Exp. Neurol 73, 671–683. [DOI] [PubMed] [Google Scholar]
- Karlmark KR, Tacke F, and Dunay IR (2012). Monocytes in health and disease: minireview. Eur. J. Microbiol. Immunol. (Bp.) 2, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson B, Gustafsson J, Hedov G, Ivarsson SA, and Annerén G (1998). Thyroid dysfunction in Down’s syndrome: relation to age and thyroid autoimmunity. Arch. Dis. Child 79, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnell JL, Kumar V, Wang J, Wang S, Voynova E, and Ettinger R (2017). Role of CD11c+ T-bet+ B cells in human health and disease. Cell. Immunol 321, 40–45. [DOI] [PubMed] [Google Scholar]
- Kaur S, Lal L, Sassano A, Majchrzak-Kita B, Srikanth M, Baker DP, Petroulakis E, Hay N, Sonenberg N, Fish EN, and Platanias LC (2007). Regulatory effects of mammalian target of rapamycin-activated pathways in type I and II interferon signaling. J. Biol. Chem 282, 1757–1768. [DOI] [PubMed] [Google Scholar]
- King P, and Tulloh R (2011). Management of pulmonary hypertension and Down syndrome. Int. J. Clin. Pract. Suppl 174, 8–13. [DOI] [PubMed] [Google Scholar]
- Klipper-Aurbach Y, Wasserman M, Braunspiegel-Weintrob N, Borstein D, Peleg S, Assa S, Karp M, Benjamini Y, Hochberg Y, and Laron Z (1995). Mathematical formulae for the prediction of the residual beta cell function during the first two years of disease in children and adolescents with insulindependent diabetes mellitus. Med. Hypotheses 45, 486–490. [DOI] [PubMed] [Google Scholar]
- Kunkel EJ, and Butcher EC (2003). Plasma-cell homing. Nat. Rev. Immunol 3, 822–829. [DOI] [PubMed] [Google Scholar]
- Leal Rojas IM, Mok WH, Pearson FE, Minoda Y, Kenna TJ, Barnard RT, and Radford KJ (2017). Human blood CD1c+ dendritic cells promote Th1 and Th17 effector function in memory CD4+ T cells. Front. Immunol 8, 971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FE, Halliley JL, Walsh EE, Moscatiello AP, Kmush BL, Falsey AR, Randall TD, Kaminiski DA, Miller RK, and Sanz I (2011). Circulating human antibody-secreting cells during vaccinations and respiratory viral infections are characterized by high specificity and lack of bystander effect. J. Immunol 186, 5514–5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekkou A, Karakantza M, Mouzaki A, Kalfarentzos F, and Gogos CA (2004). Cytokine production and monocyte HLA-DR expression as predictors of outcome for patients with community-acquired severe infections. Clin. Diagn. Lab. Immunol 11, 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Simonds EF, Bendall SC, Davis KL, Amir AD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, et al. (2015). Datadriven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162, 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan V, Williams J, and Lear JT (2006). Dermatological manifestations of Down’s syndrome. Clin. Exp. Dermatol 31, 623–629. [DOI] [PubMed] [Google Scholar]
- Maloney KW, Taub JW, Ravindranath Y, Roberts I, and Vyas P (2015). Down syndrome preleukemia and leukemia. Pediatr. Clin. North Am 62, 121–137. [DOI] [PubMed] [Google Scholar]
- Mandelboim O, Malik P, Davis DM, Jo CH, Boyson JE, and Strominger JL (1999). Human CD16 as a lysis receptor mediating direct natural killer cell cytotoxicity. Proc. Natl. Acad. Sci. USA 96, 5640–5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mårild K, Stephansson O, Grahnquist L, Cnattingius S, Söderman G, and Ludvigsson JF (2013). Down syndrome is associated with elevated risk of celiac disease: a nationwide case-control study. J. Pediatr 163, 237–242. [DOI] [PubMed] [Google Scholar]
- Martínez E, Castañeda D, Jaramillo S, Iregui A, Quiñonez T, Rodríguez JA, Herrera E, Gómez AM, Rondón MA, Prieto JC, et al. (2016). Altered immune parameters correlate with infection-related hospitalizations in children with Down syndrome. Hum. Immunol 77, 594–599. [DOI] [PubMed] [Google Scholar]
- Moeller A, Gilpin SE, Ask K, Cox G, Cook D, Gauldie J, Margetts PJ, Farkas L, Dobranowski J, Boylan C, et al. (2009). Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med 179, 588–594. [DOI] [PubMed] [Google Scholar]
- Newby BN, Brusko TM, Zou B, Atkinson MA, Clare-Salzler M, and Mathews CE (2017). Type 1 interferons potentiate human CD8+ T-cell cytotoxicity through a STAT4- and granzyme B-dependent pathway. Diabetes 66, 3061–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte MA, van Olffen RW, van Gisbergen KP, and van Lier RA (2009). Timing and tuning of CD27-CD70 interactions: the impact of signal strength in setting the balance between adaptive responses and immunopathology. Immunol. Rev 229, 216–231. [DOI] [PubMed] [Google Scholar]
- O’Gorman WE, Hsieh EW, Savig ES, Gherardini PF, Hernandez JD, Hansmann L, Balboni IM, Utz PJ, Bendall SC, Fantl WJ, et al. (2015). Single-cell systems-level analysis of human Toll-like receptor activation defines a chemokine signature in patients with systemic lupus erythematosus. J. Allergy Clin. Immunol 136, 1326–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odendahl M, Mei H, Hoyer BF, Jacobi AM, Hansen A, Muehlinghaus G, Berek C, Hiepe F, Manz R, Radbruch A, and Dörner T (2005). Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood 105, 1614–1621. [DOI] [PubMed] [Google Scholar]
- Parker BS, Rautela J, and Hertzog PJ (2016). Antitumour actions of interferons: implications for cancer therapy. Nat. Rev. Cancer 16, 131–144. [DOI] [PubMed] [Google Scholar]
- Pellegrini FP, Marinoni M, Frangione V, Tedeschi A, Gandini V, Ciglia F, Mortara L, Accolla RS, and Nespoli L (2012). Down syndrome, autoimmunity and T regulatory cells. Clin. Exp. Immunol 169, 238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Lakshminarasimhan R, Techner JM, Fong S, Flach J, Binnewies M, and Passegué E (2014). Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J. Exp. Med 211, 245–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portugal S, Obeng-Adjei N, Moir S, Crompton PD, and Pierce SK (2017). Atypical memory B cells in human chronic infectious diseases: An interim report. Cell. Immunol 321, 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachubinski AL, Enriquez-Estrada B, Norris D, Dunnick CA, Boldrick JC, and Espinosa JM (2019). JAK inhibition in Down syndrome: two cases of therapeutic benefit for alopecia areata. JAAD Case Rep. 5, 365–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram G, and Chinen J (2011). Infections and immunodeficiency in Down syndrome. Clin. Exp. Immunol 164, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenspire AJ, and Chen K (2015). Anergic B cells: precarious on-call warriors at the nexus of autoimmunity and false-flagged pathogens. Front. Immunol 6, 580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roura-Mir C, Catálfamo M, Cheng TY, Marqusee E, Besra GS, Jaraquemada D, and Moody DB (2005). CD1a and CD1c activate intrathyroidal T cells during Graves’ disease and Hashimoto’s thyroiditis. J. Immunol 174, 3773–3780. [DOI] [PubMed] [Google Scholar]
- Roy S, Ly D, Li NS, Altman JD, Piccirilli JA, Moody DB, and Adams EJ (2014). Molecular basis of mycobacterial lipid antigen presentation by CD1c and its recognition by αβ T cells. Proc. Natl. Acad. Sci. USA 111, E4648–E4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, and Marrack P (2011). Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood 118, 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsova K, Rubtsov AV, Cancro MP, and Marrack P (2015). Age-associated B cells: a T-bet-dependent effector with roles in protective and pathogenic immunity. J. Immunol 195, 1933–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundberg Nilsson A, Soneji S, Adolfsson S, Bryder D, and Pronk CJ (2016). Human and murine hematopoietic stem cell aging is associated with functional impairments and intrinsic megakaryocytic/erythroid bias. PLoS ONE 11, e0158369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch J, Rohrer TR, Kaestner M, Abdul-Khaliq H, Gortner L, Sester U, Sester M, and Schmidt T (2017). Quantitative, phenotypical, and functional characterization of cellular immunity in children and adolescents with Down syndrome. J. Infect. Dis 215, 1619–1628. [DOI] [PubMed] [Google Scholar]
- Spitzer MH, and Nolan GP (2016). Mass cytometry: single cells, many features. Cell 165, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KD, Lewis HC, Hill AA, Pandey A, Jackson LP, Cabral JM, Smith KP, Liggett LA, Gomez EB, Galbraith MD, et al. (2016). Trisomy 21 consistently activates the interferon response. eLife 5, e16220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KD, Evans D, Pandey A, Hraha TH, Smith KP, Markham N, Rachubinski AL, Wolter-Warmerdam K, Hickey F, Espinosa JM, and Blumenthal T (2017). Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation. Sci. Rep 7, 14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwa A, Hirakata M, Satoh S, Ezaki T, Mimori T, and Inada S (2000). Systemic lupus erythematosus associated with Down syndrome. Clin. Exp. Rheumatol 18, 650–651. [PubMed] [Google Scholar]
- Urban SL, Berg LJ, and Welsh RM (2016). Type 1 interferon licenses naïve CD8 T cells to mediate anti-viral cytotoxicity. Virology 493, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Acker HH, Capsomidis A, Smits EL, and Van Tendeloo VF (2017). CD56 in the immune system: more than a marker for cytotoxicity? Front. Immunol 8, 892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstegen RH, Kusters MA, Gemen EF, and DE Vries E (2010). Down syndrome B-lymphocyte subpopulations, intrinsic defect or decreased T-lymphocyte help. Pediatr. Res 67, 563–569. [DOI] [PubMed] [Google Scholar]
- Welsch K, Holstein J, Laurence A, and Ghoreschi K (2017). Targeting JAK/STAT signalling in inflammatory skin diseases with small molecule inhibitors. Eur. J. Immunol 47, 1096–1107. [DOI] [PubMed] [Google Scholar]
- Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, de Jong A, Harel S, DeStefano GM, Rothman L, et al. (2014). Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat. Med 20, 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeap WH, Wong KL, Shimasaki N, Teo EC, Quek JK, Yong HX, Diong CP, Bertoletti A, Linn YC, and Wong SC (2016). CD16 is indispensable for antibody-dependent cellular cytotoxicity by human monocytes. Sci. Rep 6, 34310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng NY, Wilson K, Wang X, Boston A, Kolar G, Jackson SM, Liu YJ, Pascual V, Capra JD, and Wilson PC (2004). Human immunoglobulin selection associated with class switch and possible tolerogenic origins for C delta class-switched B cells. J. Clin. Invest 113, 1188–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.