

Abstract
The enrichment of biotinylated proteins using immobilized streptavidin has become a staple methodology for affinity purification-based proteomics. Many of these workflows rely upon tryptic digestion to elute streptavidin-captured moieties from the beads. The concurrent release of high amounts of streptavidin-derived peptides into the digested sample, however, can significantly hamper the effectiveness of downstream proteomic analyses by increasing the complexity and dynamic range of the mixture. Here, we describe a strategy for the chemical derivatization of streptavidin that renders it largely resistant to proteolysis by trypsin and thereby dramatically reduces the amount of streptavidin contamination in the sample. This rapid and robust approach improves the effectiveness of mass spectrometry-based characterization of streptavidin-purified samples making it broadly useful for a wide variety of applications. In addition, we show that this chemical protection strategy can also be applied to other affinity matrices including immobilized antibodies against HA epitopes.
Graphical Abstract
INTRODUCTION
The ability to enrich a specific protein or class of peptides or proteins using affinity-based purification techniques is the foundation for of a wide range of biochemical methods. The subsequent characterization of these affinity-purified mixtures is often done using proteomic mass spectrometry which has the capacity to elucidate the composition, abundance, and post-translational modification state of the sample in a largely unbiased manner. Although these workflows are well-established in the field, these enrichment methods still face technical challenges that can limit their overall effectiveness. For example, the salt content, surfactant, or solvent composition required for elution from specific affinity matrices may be incompatible with mass spectrometry necessitating further clean-up of the sample1. Similarly, the elution from certain affinity supports may be inefficient or compromised by the co-elution of contaminants that interfere with the analysis. This is commonly the case when biotinylated proteins are isolated from biological mixtures using immobilized streptavidin2. The extremely high affinity of the biotin-streptavidin interaction prevents facile elution of the proteins of interest and requires either extremely harsh chemical conditions or more commonly the use of trypsin to digest the proteins directly from the beads3. Although effective for elution, this second option releases high amounts of streptavidin-derived peptides into the sample upon tryptic digestion which can compromise the overall effectiveness of the downstream analysis.
To document these technical limitations, we demonstrate that the high levels of streptavidin-derived peptides present in a typical on-bead digestion of streptavidin-bound samples reduces overall peptide identification rates in the region of the chromatography corresponding to their elution. In addition, the elution of these abundant streptavidin-derived peptides leads to local chromatographic disturbances that result in both ion suppression and retention time shifts for co-eluting peptides of interest. To overcome these challenges, we have developed a strategy for the chemical derivatization of streptavidin which renders it largely resistant to trypsinization without affecting its biotin binding character. We show that the use of these derivatized streptavidin beads in standard proteomics workflows prevents the reduction in peptide identification rates and chromatographic shifts observed in purifications using underivatized streptavidin beads. In addition, we show that this chemical derivatization strategy can the limit digestion of antibody-based supports without interfering with target binding using immobilized α-HA antibody as an example. Together, these data suggest that this strategy is robust, generalizable, and has the capacity to improve the effectiveness of a wide range of proteomic workflows.
EXPERIMENTAL PROCEDURES
Plasmids and Cell Culture:
Expression plasmids were generated using the Gateway Cloning System (Invitrogen). Briefly, open reading frames for genes of interest were amplified from the appropriate cDNAs with primers containing flanking AttB1/2 sites using the Phusion TaqDNA polymerase (New England Biolabs) and recombined into the pDONR221 donor plasmid as described previously4. The open reading frames (ORFs) were subsequently recombined from the donor plasmid into in-house generated destination vectors based on the pcDNA3 backbone and encoding either 3×HA-3×FLAG or BioID-FLAG affinity tags. The plasmid containing the MMS19 ORF for amplification was acquired previously while plasmids for PCNA, CIAPIN1, and BOLA2 were purchased from Dharmacon (previously Open Biosystems)4. MMS19 and PCNA were used to generate BioID and BioID2 fusion products, respectively. CIAPIN1 and BOLA2 were tagged with the 3×HA-3×FLAG tag sequence. HEK293 Flp-In T-Rex cells with stable, doxycycline inducible integrands of the various gene fusions mentioned above were cultured in a mixture of Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% Fetal Bovine Serum (FBS), and 2mM glutamine, into which antibiotic-antimycotic (Gibco™ 15240062) was added. Cells were cultured at 37°C in 5% CO2. Induction for expression was carried out by the addition of 500ng/mL of doxycycline into the cell culture media for 24 hours prior to induction. For BioID experiments, the cells were additionally cultured for the duration of induction in the presence of a final concentration of 50μM biotin. Cells were harvested by scraping, and the pellets washed 3 times in 50mL PBS with spins at 800g to pellet in between washes. The cell pellets were snap frozen and stored at −80°C until further use.
Reductive Methylation of Affinity Purification Matrices:
Pierce™ High capacity streptavidin agarose (20359) or Pierce™ α-hemagglutinnin agarose (α-HA, 26181) was reductively methylated using the Hampton Research Reductive Alkylation Kit (HR2–434). Briefly, 1mL of bead slurry was washed and equilibrated 5 times with 1mL of Phosphate Buffered Saline (PBS, Gibco™ 10010023, pH 7.4) on ice. After the final wash, the beads were resuspended in 1mL cold PBS and 20μL of 1M dimethylamine borane complex and 40μL of 1M formaldehyde were added. The beads were placed on a laboratory rotator for 2 hours at 4°C. The addition of dimethylamine borane complex and formaldehyde was repeated, and the beads left for an additional 2-hour incubation on rotation at 4°C. A final addition of 10μL of 1M dimethylamine borane complex was carried out, and the beads left to rotate overnight. Finally, the reaction was quenched with the addition of 125μL of 1M glycine (pH 8.6) and 125μL of 50mM dithiothreitol along with a final 2-hour incubation and rotation. The derivatized beads were washed 10 times with 1mL of 1x PBS and finally resuspended in PBS to a final combined slurry volume of 1mL and stored at 4°C.
Methylglyoxal Derivatization of Affinity Purification Matrices:
Affinity purification bead slurries were resuspended, and 1mL taken, and washed 5 times with 1mL PBS on ice. The beads were exchanged into 1mL of 100mM methylglyoxal (Sigma Aldrich, M0252) in PBS and placed on rotation at 37°C. After 24 hours, the derivatized beads were washed 10x in ice cold PBS and stored at 4°C. For beads modified by both reductive methylation and methylglyoxal derivatization, the reductive methylation was invariably performed first. Prior to each experiment, a fresh preparation of modified beads was generated.
Streptavidin-Biotin Binding Colorimetric Assay:
Derivatized streptavidins were interrogated to determine their biotin binding capabilities by colorimetric assay to determine biotinylated-HRP retention on the beads. For each of the four relevant bead types, 800μL of bead slurry was washed with 1mL of PBS with the beads placed on a laboratory rotator for 3 minutes between each wash. 50μL bead slurry aliquots were moved into separate Eppendorf tubes for each bead type, in duplicates, for each of 5 steps of a 10-fold dilution series. Biotinylated HRP (Pierce™ 29139) was introduced to each aliquot of beads at 1ng, 10ng, 100ng, 1ug, or 10ug at a fixed volume of 200μL and placed on rotation at room temperature for 30 minutes. The beads were washed 5 times with 1 mL PBS, allowing for 5 minutes on rotation between washes. Peroxidase activity was measured using the colorimetric Slow TMB ELISA (Thermo Scientific, 34024) substrate solution at 450nm according to the manufacturer’s directions on a Thermo Scientific NanoDrop 2000 spectrophotometer.
BioID Sample Lysis and Streptavidin Affinity Purification:
BioID-fusion protein expressing cell pellets were lysed in the pellet’s volume equivalent of 8M Urea, 100mM Tris pH 8.0 and thoroughly mixed at room temperature. After complete resuspension, 1μL of Benzonase nuclease was added to reduce sample viscosity via degradation of nucleic acids. Samples were placed on rotation for 30 minutes at room temperature, and spun at 20,000rcf for 15 minutes to pellet any insoluble debris. The soluble fraction for each sample was taken and normalized for protein quantity by a BCA assay.
After normalization, 125μL of each relevant streptavidin bead slurry was equilibrated in the urea lysis buffer via 3× 1mL washes. For each wash, the previous buffer was removed and replaced, and the beads placed on rotation at room temperature for 5 minutes before the beads were pelleted by a slow centrifugation at 31rcf. Normalized lysates were split equally between each of the modified bead types and left on rotation for 30 minutes at room temperature. Samples were centrifuged and washed 5x with 1mL urea lysis buffer, in a similar method to the slurry equilibration. Finally, all liquid was removed from the beads using narrow bore gel-loading tips (Eppendorf, 022351656) and replaced with 50μL of the urea lysis buffer for digestion.
Cell Surface Labeling and Streptavidin Affinity Purification:
HEK293 cells were grown to 90% confluency in 15cm plates, gently washed 3x in cold PBS and then incubated with 0.45 mM (3.75mg for 2e7 cells) Sulfo-NHS-LC-Biotin (EZ-Link™ Sulfo-NHS-LC-Biotin; 21335; Thermo Scientific) for 30 minutes at room temperature. Excess biotinylation reagent was quenched by washing with 100mM Glycine in PBS at room temperature. Labeled cells were collected by centrifugation (500g, 3min, 4°C) and then resuspended in native lysis buffer for 30 minutes (100 mM Tris pH 8, 150 mM NaCl, 5 mM EDTA, 1 mM DTT, 5% glycerol, 0.1% NP-40) containing AEBSF, pepstatin, and leupeptin. After lysis, lysates were collected by centrifugation (16,100 xg, 15 min, 4°C) and subjected to streptavidin purification as described above.
HA Tagged Sample Lysis and Immunoprecipitation:
HA-tagged fusion protein expressing cell pellets were lysed in native lysis buffer containing 2mL 100mM Tris-HCl pH 8.0, 150 mM NaCl, 5% Glycerol, 0.1% NP-40, 1μM leupeptin, 1μM pepstatin, and 1μM AEBSF. Into each sample, 1μL of Benzonase nuclease was added and each sample placed on rotation for 30 minutes at 4°C, and clarified by a 15-minute spin at 15,000rcf with retention of only the soluble supernatant. Sample content was normalized via measurement of absorption at 280nm on a Thermo Scientific NanoDrop 2000. For each sample, 100μL of bead slurry was equilibrated with three buffer exchanges of native lysis buffer, in the same manner as the streptavidin affinity purification beads. Normalized protein extracts were split between each derivatized bead type and bound during a 2-hour rotation at 4°C. Beads were washed thrice with buffer exchanges of 1mL native lysis buffer, and a final wash of native lysis buffer lacking protease inhibitors. After the final wash, all liquid was removed from the beads with Eppendorf gel-loading tips, and the beads resuspended in 50μL of 8M Urea, 100mM Tris pH 8.0.
Sample Digestion and Desalting:
Each sample was reduced and cysteines alkylated via addition of 1.25μL of 200mM TCEP and 1.2μL of 500mM iodoacetamide prior to a 20-minute dark incubation while shaking at 1300rpm at room temperature (Eppendorf ThermoMixer, 022670000). 2.5μL of 0.1μg/μL endoproteinase Lys-C (Wako Chemicals, 125–05061) was added to each sample and allowed to continue to shake in the dark for 4 hours at 37°C. Urea content of each sample was reduced from 8M to 2M via the addition of 150μL of 100mM Tris-HCl pH 8.5, and the addition of 2μL of 100mM CaCl2. Trypsinization was carried out with the addition of 4μL of 0.4μg/μL trypsin per immunoprecipitation, and incubated, shaking, in the dark, at 37°C overnight. Digestion was quenched via the addition of formic acid to bring the final concentration to 5% by volume. Each digestion was desalted via binding to C18 desalting tips, washing twice with 200uL of 5% formic acid, and elution in 50μL of 60% acetonitrile with 5% formic acid. Eluates were dried via SpeedVac and resuspended in 15μL of 5% formic acid prior to chromatographic separation and mass spectrometric acquisition.
Streptavidin LC-MS Acquisition:
Samples generated for the streptavidin affinity purification bead comparison were interrogated by Data Dependent Acquisition (DDA) on a Thermo Q-Exactive classic instrument. Mass spectrometric acquisition was coupled to a nanoflow liquid chromatographic separation delivered by a Thermo easy nLC-1000 over a 30-minute gradient on a 100uM ID, 12cm column home-packed with 1.9μM C18 particles (Dr. Maisch GmbH). For buffer A, water with 0.1% formic acid while buffer B contained acetonitrile with 0.1% formic acid. To both buffer A and B, 3% DMSO was added. Gradient delivery started at a flow rate of 450nl/min and 3% B. Over the first 2 minutes, gradient flow rate was reduced to 300nl/min while the gradient organic content increased to 9% B. Over the 23 subsequent minutes, the gradient increased linearly to 38% B, at which point the gradient rapidly increased to 80% B over 2 minutes time. The column was held at 80% B for the remaining 3 minutes of the gradient delivery, completing in 30 minutes.
During this gradient delivery, peptides were ionized by an electrospray ionization voltage of 2.2kV in the positive mode. The data dependent acquisition included MS1 scans of 70,000 resolution and MS2 scans of 17,500 resolution. Maximum injection time for MS1 and MS2 scans was set to 120ms, with an MS1 and MS2 AGC target of 1e6 and 5e4, respectively. MS1 scan range was set from 400 to 1800 m/z and data acquired in profile mode, while the MS2 scan range set from 200 to 2000 m/z. Precursors were selected for fragmentation provided that they were charge +2 to +6, allowing for fragmentation of multiple charge states, but excluding isotopes. Selected precursors were fragmented in a top-12 cycle. Dynamic exclusion for the shorter 30-minute gradients was set at 12 seconds, with a minimum AGC target of 5e2. The quadrupole isolation width was set to 2.1 m/z and HCD fragmentation collision energy set to 25NCE. Samples for the HEK293 control acquisition using RMMG beads, HEK293 control using WT beads, PCNA-BioID2 using WT beads and MMS19-BioID using WT beads were all acquired with two technical replicate acquisitions. The MMS19-BioID using RMMG beads and PCNA-BioID2 using RMMG beads were only acquired in a single technical replicate acquisition.
α-Hemagglutinin LC-MS Acquisition:
Data acquisition for the α-HA bead comparisons was performed on a Thermo Orbitrap Fusion Lumos mass spectrometer through DDA. Chromatographic gradient delivery was performed by a Thermo Dionex Ultimate 3000 nanoLC ProFlow pump system. Peptides were separated on a 70-minute gradient through a 75uM ID, 18cm C18 column packed with 1.9μM C18 particles (Dr. Maisch GmbH). Buffer compositions matched the chromatographic apparatus utilized for the acquisition of the streptavidin samples. Gradient delivery began at a flow rate of 400nl/min and 1%B. In the first quarter minute, organic content increased to 4%B and 8.2%B at 4 minutes when the gradient flow rate was lowered to 200nl/min. Organic buffer composition increased linearly to 29%B at 65 minutes, and 80%B at 67 minutes. At 68 minutes, the organic buffer composition was dropped to 1%B and held there until the end of the 70-minute chromatographic separation. Before sample loading, columns were washed by introduction of 6μL 60% acetonitrile, 20% 2-propanol, 20% H2O and equilibration to aqueous condition.
Peptides were ionized by the application of 2.0kV ionization voltage, in the positive mode. DDA contained MS1 scans generated in the Orbitrap at 500,000 resolution in profile mode, and MS2 scans acquired in the linear ion trap in the rapid scan mode. Maximum injection time for MS1 scans was set to 100ms and to 35ms for the linear ion trap MS2 scans. The MS1 Orbitrap AGC target was set to 2e5, and the MS2 scans with an AGC target of 2e3. MS1 scan range was set to 400–1600 m/z, using quadrupole isolation, and with the easy-IC internal calibrant turned on. Peptide precursors were selected from charges +2 to +6, with an intensity threshold of 4e3 with monoisotopic precursor selection turned on in a 3 second cycle time between MS1 scans. Quadrupole isolation for MS2 scans was set to a width of 1.6 m/z, with HCD fragmentation utilized with 35% collision energy. Dynamic exclusion was set to 25 seconds, with ±10 ppm tolerances and isotope exclusion turned on. All conditions were acquired in technical replicates on two separate chromatographic columns.
Database Search Parameters and Acceptance Criteria for Identifications:
Each experiments’ LC-MS raw data was converted to mzML format by ProteoWizard’s msconvert (v. 3.0.11348) with vendor peak picking enabled5-6. Each run was searched against the EMBL human reference proteome (UP000005640_9606), appended with the mouse IgG heavy chain sequence (P01868), the Streptavidin sequence (P22629), and the common contaminant fasta provided by MaxQuant; a total of 21,253 sequences. Database searching was carried out by MSGF+ (v. 2016.06.29) considering peptides with a precursor mass tolerance of 15ppm and an allowable isotope error in the range (−1,2), requiring candidate peptides to be within 6–40 amino acids in length and obeying tryptic enzymatic digestion rules at both termini and allowing unlimited missed cleavages7-9. The high resolution MS2 scans from the Streptavidin (QE) dataset was searched with the “Q-Exactive” instrument ID, while the low resolution MS2 scans from the linear ion trap of the Lumos was searched using the High-res LTQ instrument ID in MSGF+ with carbamidomethylation added as a fixed modification on cysteine residues for both experiments. Target/decoy searching was carried out by means of database protein sequence reversal, and separate target/decoy searches10. For each of the two experimental sets, the target and decoy searches for the corresponding runs were combined and fed to the crux (v. 3.1) wrapper of percolator (v. 3.01.nightly-18–1e0fbeb)11-12. The resulting PSMs were fed into the standalone version of FIDO (v. 1.0) to produce protein level probabilities, which were subsequently converted to q-values13. Identifications were filtered at both PSM and protein level q-value thresholds of 0.01. Spectral counts were calculated by the crux spectral-counts function allowing for degenerately mapping peptides to be counted for each protein.
MS1-Intensity Based Data Extraction:
For label free, intensity-based comparisons, confident identifications were converted into spectral libraries and MS1 extracted ion chromatograms generated by Skyline (v. 4.1.0.18169)14. Skyline’s peptide database background was set to a digestion setting of “Trypsin/P”, allowing for no missed cleavage sites, and disallowing ragged-ended peptides. Extracted ion chromatogram windows were generated with a 2-minute retention time tolerance for the Lumos runs containing a longer gradient and 1-minute for the shorter QE datasets. For both experiments, an 8ppm mass tolerance window around three isotopic peaks per analyte was extracted. For each of the experiments’ Skyline analyses, an mProphet peak picking model was trained on all available scores and used to assign confidence to the integrated peaks15.
Gene Ontology and Subcellular Compartment Analysis:
To gauge the efficacy of the cell surface labeling procedure when utilized, confident protein identifications were grouped by the RMMG and WT bead purifications. Uniprot accessions were converted to Gene IDs via the Uniprot mapping web interface. These gene identifiers were queried for gene ontology mappings via g:Profiler16. Proteins which mapped to the membrane GO term (GO:0016020) were considered protein targets of cell surface labeling. The gene identifiers from both RMMG and WT runs were also taken separately and examined for enrichment of subcellular compartment terms through the Enrichr implementation of the COMPARTMENTS database with default analysis parameters utilized17-18.
Experimental Design and Statistical Rationale:
Data generated in the experiments testing protection of streptavidin affinity purification beads by chemical derivatization were made using three cell lines, each in a single biological replicate. The fully modified RMMG beads contained two technical replicate injections from the HEK293 control pulldown, one technical replicate of the MMS19-BioID pulldown, and one technical replicate of the PCNA-BioID pulldown. These four RMMG files were compared against a total of six WT control streptavidin acquisitions across the same three pulldowns, each acquired in technical duplicates. Testing of the α-hemagglutinnin derivatization was carried out from three cell lines. Pulldowns for each of these three cell lines were performed on four bead types in biological singlicates. DDA acquisition was carried out in technical replicate for each of the pulldowns and bead conditions, leading to a total of 24 acquisitions. Each bead type dataset contained six acquisitions to compare the level of protection provided by the different stages of chemical derivatization. Thus, the six control acquisitions of WT α-hemagglutinnin on-bead digestions were compared with the six acquisitions of each of the three other groups.
Peptide intensity values exported by Skyline were filtered by mProphet q-value at a threshold of 0.01, and protein intensities modeled and compared by the MSstats package (v. 3.9.2) after filtering to require that all peptides used for quantitation mapped uniquely within the background proteome and requiring proteins to have two quantifiable peptides19. Protein intensities were summarized by means of the Tukey Median Polish implementation within MSstats, with model-based imputation turned on and the “maxQuantileforCensored” set to NULL. Normalization for the α-HA experiment was set to include all peptides belonging to 5 human proteins selected for their universal identification amongst every acquisition in the dataset: IRS4, THRAP3, GTF2I, BCLAF1, and LSM14A. For the streptavidin comparisons, intensities were normalized by means of median equalization. Statistical differential protein abundance testing was provided by means of the linear mixed model implementation within the MSstats package, and p-values adjusted for multiple hypothesis testing by the Benjamini-Hochberg correction20.
To determine the signal intensity impact of streptavidin-peptide derived chromatographic shifting, mProphet filtered peptide intensity values were grouped by those without chromatographic shifts and those peptides for which the median Skyline determined retention time of the peptide differed between RMMG and WT runs by at least 30 seconds. Only peptides which were confidently detected in both conditions were included in this analysis. The median normalized intensity values were made into log2 transformed ratios comparing the WT and RMMG peptide intensities. The two populations were compared by the Mann-Whitney U test implementation within Python’s (v. 2.7) scipy stats module21. Histograms were rendered within R (v. 3.5.0) using the ggplot2 package, and basepeak chromatograms through the MSnbase interface to mzML files22.
To visualize the impact of reductive methylation on each BioID-fused protein’s detectable peptide ion intensities, a representative file was selected from both the WT and RMMG pulldowns. For these acquisitions, Skyline exported peptide intensities mapping to the BioID fusion proteins were summed across all charge states and isotopes. Peptide signals were filtered to retain only those with mProphet q-values less than or equal to 0.01, and only those confidently detected in both WT and RMMG compared against each other. Comparison of the peptide intensity distributions between these bead types was carried out via ggpubr (v. 0.2) and R’s Wilcoxon signed rank test for the paired peptide intensities.
All raw data acquired and analyzed here are available through the MassIVE repository via the ProteomeXchange identifier “PXD011858”23-24.
RESULTS
The tryptic digestion of protein samples bound to immobilized streptavidin beads is routinely performed during the course of a wide range of proteomic experiments. Despite the fact that the compact structure of streptavidin makes it naturally resistant to trypsin-mediated proteolysis, we have frequently observed high amounts of streptavidin-derived peptides in these samples. A representative basepeak chromatogram from the LC-MS/MS run of one such sample prepared by digesting streptavidin beads with trypsin is shown in Fig. 1A. Multiple high intensity peaks are observed in the chromatogram that we hypothesized were generated by the digestion of streptavidin by trypsin. We confirmed this by LC-MS/MS analysis which identified a large number of peptides mapping to the streptavidin sequence and the subsequent plotting of extracted ion chromatograms (XICs) of those peptides which clearly demonstrated that the dominant peaks observed in the basepeak chromatogram traces correspond to streptavidin-derived peptides (Fig. 1B). Given the prevalence of these peptides in the sample, it seemed likely that they were lowering the quality of these LC-MS/MS datasets by increasing the overall complexity and dynamic range of the sample.
Figure 1:

On-bead tryptic digestion of streptavidin affinity purification samples leads to substantial streptavidin peptide contamination. (A) Basepeak chromatogram from a representative streptavidin affinity purification on-bead digestion demonstrates the high dynamic range in peptide intensities, with a small number of peaks dominating the chromatographic separation. (B) Skyline generated extracted ion chromatograms (XICs) of confidently identified streptavidin-derived peptides were manually integrated and plotted against retention time showing the level of high-intensity contamination which originates from the matrix.
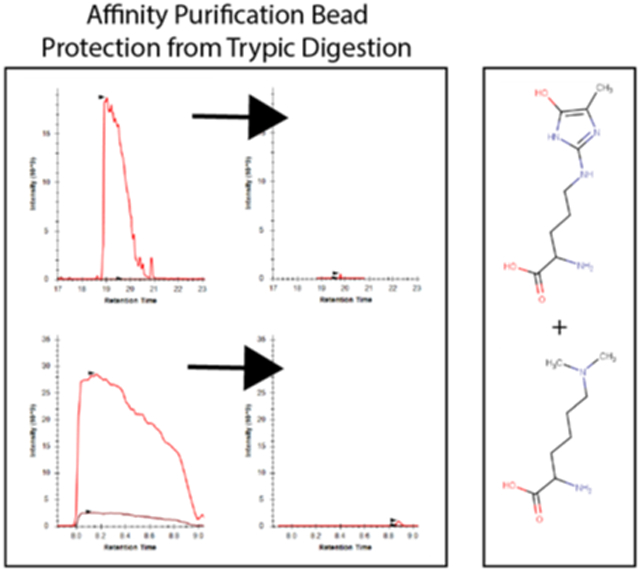
To address this issue of streptavidin peptide contamination, we hypothesized that generating chemically derivatized streptavidin that was resistant to proteolysis by trypsin would improve the coverage of proteomic analyses. To test this idea, we used two chemical derivatization strategies. First, we methylated the lysine residues in streptavidin using standard reductive methylation strategies that utilize dimethylamine borane and formaldehyde (Fig. 2A). We then further modified the reductively methylated streptavidin by treatment with methylglyoxal (MGO) to form dihydroxyimidazolidine or hydroimidazolone adducts on arginine residues (Fig. 2A)25. We tested whether the covalent modification of the lysine and arginine residues in streptavidin in this manner (1) impairs its binding to biotin and (2) renders it more resistant to digestion by trypsin. To test whether biotin binding by the doubly derivatized streptavidin (RMMG) was impaired relative to wildtype, we used biotin-horseradish peroxidase (HRP) to assay its binding activity. Wildtype or modified streptavidin was incubated was biotin-HRP and washed before measuring the amount of biotin-HRP retained on the beads using the Pierce 1-Step Slow TMB ELISA colorimetric assay. Fig. 2B shows that biotin binding was unaffected by modification of the lysines and arginines in streptavidin across a large range of biotin-HRP concentrations. We further validated this observation by performing a streptavidin pulldown using wildtype or modified beads from cell extracts prepared from stable cell lines expressing BioID fused to either MMS19 or PCNA. The BioID-MMS19 and BioID2-PCNA fusion proteins non-specifically biotinylate proteins within their vicinity which can then be affinity purified using streptavidin beads, digested with trypsin, and the analyzed by LC-MS/MS. As shown in Fig. 2C, label-free quantitation of the amount of the BioID-MMS19 or BioID2-PCNA fusion protein was unaffected by modification of the streptavidin. In order to examine whether the derivatization of the lysines and arginines in streptavidin made it more resistant to trypsin digestion, we examined the wildtype and modified beads by LC-MS/MS after proteolytic digestion using our standard trypsin-based workflows. Extracted ion chromatograms of streptavidin-derived peptides generated from wildtype or modified streptavidin samples after tryptic digestion are shown in Fig. 2D. These chromatograms clearly demonstrate a major reduction in streptavidin-derived peptides upon chemical derivatization and support the hypothesis that these modifications impair tryptic digestion. Additionally, label-free quantitation of streptavidin abundance shows a substantial decrease in streptavidin abundance for the modified streptavidin (RMMG) relative to the underivatized (WT) beads (Fig. 2E). Together these data argue that the derivatized streptavidin beads are not measurably impaired in their ability to bind biotinylated proteins but are highly resistant to proteolysis by trypsin.
Figure 2:

Streptavidin derivatization provides protection from enzymatic digestion without disrupting biotin binding. (A) Schematic showing the derivatization of arginine and lysine residues of polypeptides to protect against enzymatic digestion. (B) Biotinylated HRP activity was measured after binding to various forms of derivatized streptavidin. HRP enzymatic activity was not impacted by the lysine and arginine derivatization, suggesting the binding of biotinylated proteins would not be disrupted. Values are given in arbitrary units of absorbance, with error bars showing the standard deviation of replicates for each bead type and HRP mass. (C) Auto-biotinylated BioID fusion proteins’ peptides are detected with similar unnormalized label-free intensities when purified by either derivatized (RMMG) or underivatized (WT) streptavidin beads within representative acquisitions. Differences between the peptide intensity populations for the BioID-MMS19 fusion protein and PCNA-BioID fusion protein are not significant between their respective RMMG and WT pulldowns by the Wilcoxon signed rank test. Box and whiskers illustrate the first and third quartiles, and the maximum and minimum observed values up to 1.5 times the interquartile range beyond the edges of the box. (D) Manually integrated extracted ion chromatograms were generated for uniquely mapping tryptic peptides derived from Streptavidin within the Skyline software. A representative pair of WT/RMMG pulldowns display the impact of derivatization on the detectable streptavidin peptide signals, dramatically reducing the overall streptavidin intensity. (E) Label-free protein intensities comparing the overall streptavidin intensity of the aggregate of all BioID data acquired here with WT beads and RMMG beads. Values displayed are modeled protein intensities, with error bars representing the reported 95% confidence intervals.
Having established the suitability of the modified streptavidin beads for affinity purification experiments, we next compared proteomic analyses of streptavidin pulldowns performed using wildtype and derivatized beads in order to assess their relative contribution to overall proteomic data quality. First, we plotted the normalized PSM identification rate across chromatographic runs for LC-MS/MS analyses of streptavidin pulldowns done using wildtype (WT) or derivatized (RMMG) beads (Fig. 3A, top). Wildtype streptavidin purifications displayed regions of the chromatography in which PSM identifications were reduced. These regions correspond to the elution of major streptavidin-derived tryptic peptides (Fig. 3B, bottom) suggesting that the increased dynamic range generated by the elution of these high abundance contaminating peptides hampers peptide identification in those stretches. Strikingly, this reduction in peptide identifications was restored in purifications done using protected streptavidin beads highlighting the benefit of these derivatized streptavidin beads in limiting the elution of streptavidin-derived peptide contaminants and preventing the masking of signals belonging to non-streptavidin peptide analytes of interest.
Figure 3:

Streptavidin derivatization ameliorates the local chromatographic perturbation due to streptavidin peptide elution. (A) Top: Peptide-Spectrum Matches (PSMs) for the aggregate of all samples prepared using RMMG streptavidin beads and the aggregate of all samples prepared using WT streptavidin beads were plotted as normalized PSM density in histograms with a bin width of 0.1 min. Bottom: An example affinity purification performed using on-bead digestion of WT streptavidin beads shows high intensity streptavidin contamination which correlates in time with the drops in PSM rates for data acquired using the WT streptavidin beads. (B) A Skyline generated plot of a paired set of peptide retention times from affinity purifications performed using WT (y-axis) and RMMG (x-axis) streptavidin beads from 17 to 23 minutes of retention time. The WT streptavidin runs exhibit local perturbation of the chromatography during elution of the high intensity streptavidin peptides. (C) Comparison of the log2 fold-change of peptides compared between the WT and RMMG conditions. Peptides which did not exhibit a chromatographic shift between the two bead types retained an approximately zero fold-change (n=5,872), while the peptides with retention time shifts were suppressed in intensity in the WT runs compared to the RMMG runs (n=1,059). Differences in log transformed fold-change ratios distributions between the two groups were assessed by the Mann-Whitney U test, showing a significant difference (p < 1E-54). Box and whiskers illustrate the first and third quartiles, and the maximum and minimum observed values up to 1.5 times the interquartile range beyond the edges of the box.
We also examined the streptavidin-contaminated samples to determine whether the streptavidin-derived peptides might negatively impact the chromatography. Fig. 3B shows the peptide retention time correlation between analyses of pulldowns using wildtype or protected streptavidin. Strikingly, the elution of major streptavidin species leads to a marked disruption in peptide elution times with streptavidin peptides effectively pushing other peptides out of their typical elution window. Importantly, the population of peptides displaying shifted retention times also display reduced intensity relative to the unshifted peptides suggesting that the streptavidin-derived peptides suppress the ionization of these retention time shifted peptides. Based on these data, we conclude that streptavidin-derived peptides contribute to reduced peptide identification rates, shifted retention times, and ion suppression of co-eluting peptides and that these negative effects are alleviated when digestion-resistant streptavidin beads are used for the affinity purification.
To more comprehensively examine the utility of this method in complex biological samples, we also tested its effectiveness in cell surface labeling experiments. Briefly, HEK293 cells were incubated with NHS-biotin to label cell surface proteins, lysed, and then affinity purified using either standard or protected immobilized streptavidin beads in triplicates. Purified proteins were then subjected to tryptic digestion and LC-MS/MS. As before, the elution of high abundance streptavidin-derived peptides shifted the retention times of co-eluting peptides (Fig. 4A), an effect that was not observed in the chemically protected streptavidin beads (RMMG vs. WT). We also observed an increase in peptide identifications in samples purified using protected streptavidin (RMMG) relative to wildtype streptavidin (WT) (Fig. 4B) and found that these identified proteins were strongly enriched for membrane proteins based on overenrichment of relevant gene ontology (GO) terms in the datasets (Fig. 4C). The identification of more proteins overall and specifically more membrane proteins in affinity purifications conducted using the derivatized streptavidin beads highlights the increase in proteomic depth made possible by this approach. It should be noted, however, that this increase in proteomic depth is strongly dependent on both the degree of streptavidin contamination as well as the complexity of the mixture which can be highly variable depending on the application.
Figure 4:

Analysis of cell surface proteome using protected streptavidin beads improves peptide identification rates. (A) Extracted ion chromatogram overlay around the retention time of the streptavidin peptide NAHSATTWSGQYVGGAEAR for the cell surface biotinylation samples. As before, we observe that elution of excessive streptavidin peptides in WT runs perturbs the retention times of otherwise coeluting peptides in the RMMG runs. (B) Confident peptide spectrum matches (percolator q-value<=0.01) by sample. PSMs which mapped to protein sequences belonging to genes annotated with the membrane gene ontology cellular compartment term (GO:0016020) are shown in light blue. (C) Compartment localization enrichments were calculated from the COMPARTMENTS database through the Enrichr platform. Both RMMG and WT beads capture significant enrichment of extracellular and membrane related localizations.
Given the effectiveness of this protection strategy in reducing contamination in on-bead digested streptavidin pulldowns, we explored the possibility that this approach could be generalizable and potentially extended to other affinity purification matrices. We first tested this using anti-HA antibodies coupled to an agarose support. The anti-HA resin was chemically derivatized using either lysine reductive methylation (RM), methylglyoxal modification of arginines (MG), or both (RMMG). Digestion of these chemically protected beads with trypsin dramatically reduced the signal intensity of IgG-derived peptides in the digest. This is evident in Fig. 5A which shows basepeak chromatograms from WT α-HA and RMMG α-HA digested samples and the prominent loss of high abundance IgG-derived peptides specifically in the derivatized sample. These chromatograms are consistent with the LFQ analysis of these samples shown in Fig. 5B in which both the RM and RMMG protected beads show dramatically reduced Igg abundance in the sample after on-bead tryptic digestion. Interestingly, reductive methylation of lysines appears to be sufficient for this effect with little to no contribution from methylgloxal treatment (MG) being observed. Importantly, we also confirmed that the chemical protection of anti-HA did not significantly impair its ability to immunoprecipitate HA-tagged proteins. Control, RM, MG, or RMMG treated anti-HA beads were used to immunoprecipitate either 3HA-3FLAG-tagged CIAPIN1 or 3HA-3FLAG-tagged BOLA2 from protein lysates generated from HEK293 cell lines stably expressing those fusion proteins. The spectral counts obtained for each bait after LC-MS/MS analysis of the immunoprecipitated sample was used to assess the effectiveness of the immunoprecipitation. Fig. 5C clearly shows that HA-tagged CIAPIN1 and BOLA2 were both similarly enriched in these samples irrespective of whether untreated or protected beads were used.
Figure 5:

The chemical derivatization method is extensible to antibody based affinity purification matrices. (A) Example basepeak chromatograms of a pair of pulldowns generated on WT (top) and derivatized (RMMG, bottom) α-HA beads. (B) IgG heavy chain protein intensity is dramatically reduced upon chemical derivatization of the beads prior to enzymatic digestion. The majority of signal reduction is granted by the reductive methylation and not the methylglyoxal treatment. Values are MSstats modeled protein intensities with error bars representing the 95% confidence interval. (C) α-HA beads retain binding of epitope tagged protein targets despite modification status. Confident spectral counts (SpC) shown for both of the bait proteins from their respective pulldowns. Each pulldown was acquired with two technical replicates on separate chromatographic columns. Values shown as the mean with error bars displaying the standard deviation.
DISCUSSION
The on-bead digestion of streptavidin to elute proteins during affinity purification workflows results in the release of streptavidin-derived peptides into the sample. The high abundance of these peptides limits the subsequent mass spectrometric analysis by suppressing identification of co-eluting peptides and reducing peptide identification rates during their elution. We report a strategy for reducing the production of these peptides by chemically derivatizing lysine and arginine residues in streptavidin to render it largely resistant to trypsinization. Proteomic analysis of affinity purifications performed using these modified beads restores the loss of peptide IDs and aberrant chromatography observed in purifications done using wildtype streptavidin beads. Finally, we also demonstrate that this approach for generating digestion resistant beads is potentially generalizable using the immunoprecipitation of HA-tagged proteins with immobilized anti-HA antibodies as an example.
A wide range of elution strategies are currently employed to facilitate streptavidin-based affinity purifications. These range from engineered streptavidin resins in which their reduced biotin affinity enables elution by excess free biotin, to techniques that separate the biotinylated peptides of interest away from contaminating peptides26-27. Based on the effectiveness and facile implementation of our approach, we anticipate that it will become a robust alternative to these options that that can be incorporated into different workflows as needed based on their analytical requirements. The availability of a suite of sample preparation approaches will offer flexibility and adaptability as new applications are developed.
A key advantage to the presented method is its potential to be generalized to other affinity purification matrices. The ability to elute proteins or other biological analytes directly from affinity supports using trypsin simplifies the sample preparation workflow and minimizes the opportunity for sample loss. Like streptavidin, however, these strategies are difficult for antibody-based affinity resins which will release IgG-derived peptides into the sample after proteolysis. Our results indicate that this chemical derivatization strategy can be adapted for α-HA resin opens up new sample preparation options for immunoaffinity chromatography and highlights the broad utility of this method.
Although we have focused on the utility of these beads in the context of bottom-up proteomics, we anticipate potential uses for the derivatized affinity beads in other experimental workflows. For example, immobilized streptavidin is often used to purify biotinylated nucleic acids from biological mixtures. Using derivatized streptavidin in these workflows would provide an option for deproteinizing these samples without eluting the nucleic acid from the bead. Similarly, methods exist for the purification of specific cell types from mixtures using biotinylated antibodies. Protection of the streptavidin beads in these experiments would enable elution of specific cells without the concurrent proteolysis and release of substantial streptavidin contaminants.
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (GM089778 and GM112763 to JAW). WDB and XF were supported by the Ruth L. Kirschstein National Research Service Award GM007185 from the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS:
- HA
hemagglutinnin
- ORF
Open Reading Frame
- PCNA
Proliferating Cell Nuclear Antigen
- DMEM
Dulbecco’s Modified Eagle’s Media
- FBS
Fetal Bovine Serum
- PBS
Phosphate Buffered Saline
- HRP
Horse Radish Peroxidase
- TMB
3,3',5,5'-Tetramethylbenzidine
- ELISA
Enzyme Linked Immunosorbent Assay
- BCA
Bicinchoninic acid assay
- AEBSF
4-benzenesulfonyl fluoride hydrochloride
- NP-40
Nonidet P-40 (4-Nonylphenyl-polyethylene glycol)
- DDA
Data Dependent Acquisition
- NCE
Normalized Collision Energy
- MG
Methylglyoxal
- RM
Reductive Methylation
- RMMG
Reductive Methylation and Methylglyoxal
- AGC
Automatic Gain Control
- HCD
Higer-energy Collisional Dissociation
- QE
Q-Exactive
- LTQ
Linear Trap Quadrupole
- PSM
Peptide-Spectrum Match
REFERENCES
- 1.Cheah JS; Yamada S, A simple elution strategy for biotinylated proteins bound to streptavidin conjugated beads using excess biotin and heat. Biochem Biophys Res Commun 2017, 493 (4), 1522–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rybak JN; Scheurer SB; Neri D; Elia G, Purification of biotinylated proteins on streptavidin resin: a protocol for quantitative elution. Proteomics 2004, 4 (8), 2296–9. [DOI] [PubMed] [Google Scholar]
- 3.Fukuyama H; Ndiaye S; Hoffmann J; Rossier J; Liuu S; Vinh J; Verdier Y, On-bead tryptic proteolysis: an attractive procedure for LC-MS/MS analysis of the Drosophila caspase 8 protein complex during immune response against bacteria. Journal of proteomics 2012, 75 (15), 4610–9. [DOI] [PubMed] [Google Scholar]
- 4.Vashisht AA; Yu CC; Sharma T; Ro K; Wohlschlegel JA, The Association of the Xeroderma Pigmentosum Group D DNA Helicase (XPD) with Transcription Factor IIH Is Regulated by the Cytosolic Iron-Sulfur Cluster Assembly Pathway. J Biol Chem 2015, 290 (22), 14218–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kessner D; Chambers M; Burke R; Agus D; Mallick P, ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 2008, 24 (21), 2534–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holman JD; Tabb DL; Mallick P, Employing ProteoWizard to Convert Raw Mass Spectrometry Data. Current protocols in bioinformatics 2014, 46, 13 24 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim S; Gupta N; Pevzner PA, Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. Journal of proteome research 2008, 7 (8), 3354–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim S; Mischerikow N; Bandeira N; Navarro JD; Wich L; Mohammed S; Heck AJ; Pevzner PA, The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Molecular & cellular proteomics : MCP 2010, 9 (12), 2840–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim S; Pevzner PA, MS-GF+ makes progress towards a universal database search tool for proteomics. Nature communications 2014, 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elias JE; Gygi SP, Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature methods 2007, 4 (3), 207–14. [DOI] [PubMed] [Google Scholar]
- 11.McIlwain S; Tamura K; Kertesz-Farkas A; Grant CE; Diament B; Frewen B; Howbert JJ; Hoopmann MR; Kall L; Eng JK; MacCoss MJ; Noble WS, Crux: rapid open source protein tandem mass spectrometry analysis. Journal of proteome research 2014, 13 (10), 4488–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.The M; MacCoss MJ; Noble WS; Kall L, Fast and Accurate Protein False Discovery Rates on Large-Scale Proteomics Data Sets with Percolator 3.0. Journal of the American Society for Mass Spectrometry 2016, 27 (11), 1719–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serang O; MacCoss MJ; Noble WS, Efficient marginalization to compute protein posterior probabilities from shotgun mass spectrometry data. Journal of proteome research 2010, 9 (10), 5346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ, Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26 (7), 966–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reiter L; Rinner O; Picotti P; Huttenhain R; Beck M; Brusniak MY; Hengartner MO; Aebersold R, mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nature methods 2011, 8 (5), 430–5. [DOI] [PubMed] [Google Scholar]
- 16.Reimand J; Kull M; Peterson H; Hansen J; Vilo J, g:Profiler--a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic acids research 2007, 35 (Web Server issue), W193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen EY; Tan CM; Kou Y; Duan Q; Wang Z; Meirelles GV; Clark NR; Ma’ayan A, Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC bioinformatics 2013, 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Binder JX; Pletscher-Frankild S; Tsafou K; Stolte C; O’Donoghue SI; Schneider R; Jensen LJ, COMPARTMENTS: unification and visualization of protein subcellular localization evidence. Database : the journal of biological databases and curation 2014, 2014, bau012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi M; Chang CY; Clough T; Broudy D; Killeen T; MacLean B; Vitek O, MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 2014, 30 (17), 2524–6. [DOI] [PubMed] [Google Scholar]
- 20.Benjamini Y; Hochberg Y, Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 1995, 57 (1), 289–300. [Google Scholar]
- 21.Oliphant TE, Python for Scientific Computing. Computing in Science & Engineering 2007, 9 (3), 10–20. [Google Scholar]
- 22.Gatto L; Lilley KS, MSnbase-an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 2012, 28 (2), 288–9. [DOI] [PubMed] [Google Scholar]
- 23.Deutsch EW; Csordas A; Sun Z; Jarnuczak A; Perez-Riverol Y; Ternent T; Campbell DS; Bernal-Llinares M; Okuda S; Kawano S; Moritz RL; Carver JJ; Wang M; Ishihama Y; Bandeira N; Hermjakob H; Vizcaino JA, The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic acids research 2017, 45 (D1), D1100–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jarnuczak AF; Vizcaino JA, Using the PRIDE Database and ProteomeXchange for Submitting and Accessing Public Proteomics Datasets. Current protocols in bioinformatics 2017, 59, 13 31 1–13 31 12. [DOI] [PubMed] [Google Scholar]
- 25.Chumsae C; Gifford K; Lian W; Liu H; Radziejewski CH; Zhou ZS, Arginine modifications by methylglyoxal: discovery in a recombinant monoclonal antibody and contribution to acidic species. Anal Chem 2013, 85 (23), 11401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schiapparelli LM; McClatchy DB; Liu HH; Sharma P; Yates JR 3rd; Cline HT, Direct detection of biotinylated proteins by mass spectrometry. Journal of proteome research 2014, 13 (9), 3966–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Sullivan VJ; Barrette-Ng I; Hommema E; Hermanson GT; Schofield M; Wu SC; Honetschlaeger C; Ng KK; Wong SL, Development of a tetrameric streptavidin mutein with reversible biotin binding capability: engineering a mobile loop as an exit door for biotin. PloS one 2012, 7 (4), e35203. [DOI] [PMC free article] [PubMed] [Google Scholar]