

Abstract
DNA methylation is a class of epigenetic modification essential for coordinating gene expression timing and magnitude throughout normal brain development and for proper brain function following development. Aberrant methylation changes are associated with changes in chromatin architecture, transcriptional alterations and a host of neurological disorders and diseases. This review highlights recent advances in our understanding of the methylome's functionality and covers potential new roles for DNA methylation, their readers, writers, and erasers. Additionally, we examine novel insights into the relationship between the methylome, DNA–protein interactions, and their contribution to neurodegenerative diseases. Lastly, we outline the gaps in our knowledge that will likely be filled through the widespread use of newer technologies that provide greater resolution into how individual cell types are affected by disease and the contribution of each individual modification site to disease pathogenicity.
Introduction
Epigenetic modifications are defined as heritable genomic changes not due to alterations in DNA sequence. Although there are many different classes of epigenetic modifications (e.g. DNA modifications, histone modifications, RNA modifications, RNAi, etc.), each class shares the similar functions of transcriptional or post-transcriptional regulation to alter the prevalence of protein products (1–5). The first methylated cytosine was discovered shortly after 1948 (6), although the functions of these modifications remained elusive until the mid-1970s and 80s (7,8). The discovery that 5-methylcytosine (5mC) functions as a gene expression regulator and is important for cellular differentiation and cell fate led to the first boom in DNA modification studies. However, following this initial boom, research in this field grew more gradually under the premise that the high stability of DNA methylation meant that genomic methylation profiles are relatively stable and the modification irreversible. It was not until roughly 30 years later when the field was reinvigorated with the discovery that 5mC is converted into 5-hydroxymethylcytosine (5hmC), indicating that DNA methylation profiles are dynamic, vary in response to external influences and thus serve as a bridge between environment, genotype, and phenotype (4,9,10). The location and general function of 5mC and 5hmC have been well characterized; however, new advances in the field have identified a novel DNA modification, N6-methyladenine (6mA), in eukaryotes (11). This modification appears to be enriched only in early developmental stages and is responsive to various environmental stressors (12–16). Together, the abundance and location of 5mC, 5hmC, and 6mA comprise the DNA methylome and play a critical role in governing cell-type specific gene expression. In this review, we will focus on recent advances made on characterizing the role of DNA modifications and the proteins that recognize (read), place (write), and remove (erase) them. We will discuss how these interactions work to regulate gene expression, local chromatin environment, and methylome variations in the molecular pathogenesis of neurodegenerative disorders.
Cytosine Modifications (5mC, 5hmC, 5fC and 5caC)
Establishment, maintenance and function of 5mC
5mC is generated through the covalent addition of a methyl group to the fifth carbon of a DNA cytosine by DNA methyltransferases (DNMTs) (17,18). 5mC is the most prevalent DNA modification in humans and functions as a critical player in the regulation of tissue and cell specific gene expression. DNMTs catalyze the covalent addition of a methyl group to the fifth carbon of cytosine (Fig. 1A). There are two classes of DMNTs: de novo (DNMT3A, DNMT3B, DNMT3L) and maintenance (DNMT1). Both work in concert to generate symmetrical cytosine methylation profiles genome wide (19). DNMT3A, DNMT3B and cofactor DNMT3L are de novo DNMTs that methylate DNA during embryogenesis and in differentiated cells and are highly expressed in embryonic stem cells (20). Additionally, DNMT expression is high in neural progenitor cells, post-mitotic and adult neurons. DNMT1 and the accessory protein UHRF1 primarily contribute to the maintenance of DNA methylation during cellular replication and following DNA repair through the recognition and binding of hemi-methylated DNA (21). To alter gene expression, 5mC modifications employ several different functions ranging from the recruitment of methyl-binding proteins (MBPs) to the exclusion of transcription factors (TFs) or chromatin remodeling complexes by altering the morphology of canonical binding sites. The function of 5mC and the proteins it interacts with is context dependent with the local chromatin and TF environment, leveraging a substantial influence on the role of 5mC (22–24). Neri et al. (25) showed that, for certain genes, local repressive chromatin changes precede and facilitate 5mC addition, and at other loci 5mC placement serves as the initial step in creating a repressive chromatin environment. Research from Castillo-Aguilera et al. has shown that, in the presence of the H3K36me3 histone methylation mark in gene bodies, DNMT3B is recruited to place gene body 5mC (26). In addition to chromatin environment, genomic location plays a critical role in determining 5mC function (Fig. 2). When located at enhancer or promoter regions, 5mC is canonically repressive in nature and functions through both the prohibition and recruitment of TFs and MBPs (22). Gene body 5mC is reported to associate with active transcription and functions to reduce spurious transcription of Pol II (27), whereas reduction in gene body 5mC produced via DNMT3B knockdown increases aberrant transcription of intragenic regions (25). Spurious transcription products can contribute to disease state because they can be properly post-transcriptionally modified and translated into dysfunctional protein products by ribosomes (25,28). Mutations lying primarily in the catalytic domain of DNMT3A have been identified in a host of neurodevelopmental disorders including epileptic encephalopathies, autism spectrum disorder (ASD) and intellectual disability (29,30). Reduced DNMT functionality in turn causes alterations in 5mC and the oxidized derivatives, leading to differences in the genomic location of methylation readers. The importance of DNMT3A is not exclusive to development. The relationship between stress and anxiety-like behavior was explored in mice. Following stressful events, there is a reduction in the expression of DNMT3A, resulting in a drop in global DNA methylation in the prefrontal cortex of adult mice (31). These experiments reinforce that the methylome dynamics plays important roles in neuronal function and the capacity for the methylome to function as a bridge between genotype, environmental influences and phenotype.
Figure 1.
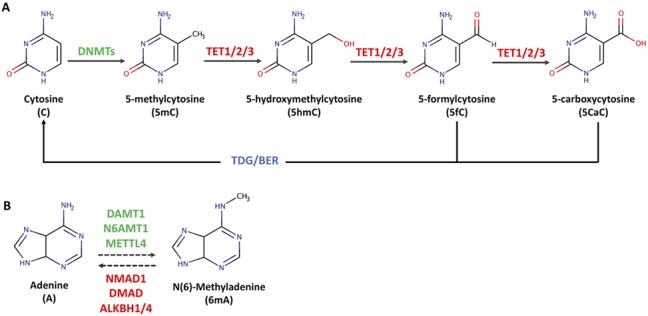
Generation of DNA modifications. (A) Mechanisms of cytosine methylation and demethylation. Cytosine (C) is converted to 5mC by DNMT enzymes. TET enzymes further catalyze the oxidization of 5mC to 5hmC, 5fC and 5CaC. (B) Potential mechanisms of DNA 6mA methylation and demethylation. 6mA is potentially catalyzed by DNA N6-methyl methyltransferase (DAMT1) in worm. N(6) adenine-specific DNA methyltransferase 1 (N6AMT1) and methyltransferase-like protein 4 (METTL4) have been found to catalyze 6mA deposition in mammals. DNA N6-methyl adenine demethylase (NMAD1) has been shown to have 6mA demethylation activity in worm, and 6mA demethylase (DMAD) was identified as a 6mA demethylase in fly. Alpha-ketogluterate-dependent dioxygenase AlkB homolog 1 (ALKBH1) and 4 (ALKBH4) were found to mediate 6mA removal in mammals.
Figure 2.
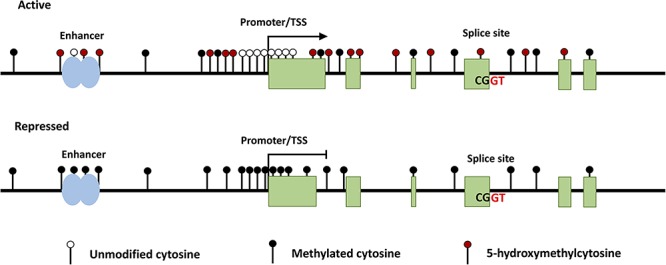
DNA modifications associated with active and repressed transcriptional states. Generally, the enhancer regions of actively transcribed genes contain unmethylated cytosine and hydroxymethylated cytosine residues spanning the enhancer region. The regions flanking the TSS contain both methylated and hydroxymethylated cytosines. In the immediate vicinity of the TSS, the CpG sites are unmethylated. 5hmC and 5mC are dispersed throughout the gene body, with an enrichment of 5hmC at the splice sites of exons to be incorporated in the mRNA transcript. Repressed genes typically contain 5mC at the enhancer, TSS, and throughout the gene body and a depletion of 5hmC.
5mC is read by a variety of MBPs that orchestrate gene expression through TF regulation and modification of chromatin architecture. MBP proteins contain a methyl-CpG-binding domain that enables them to bind unmethylated and methylated DNA (32). One of the most widely studied MBPs is the chromatin modifying protein, MeCP2. MeCP2 is ubiquitously expressed with elevated levels in the brain, especially in the cortex and cerebellum (33,34). The predominant function of MeCP2 in the brain is to maintain synaptic connections between neurons, enabling proper communication. De novo mutations in MeCP2 are commonly found in individuals with Rett syndrome, a severe neurological disorder with symptoms consisting of mental retardation, repetitive behaviors, and learning disabilities (35). Additionally, increased methylation at the MeCP2 locus is associated with reduced MeCP2 expression (36). Through 5mC recognition, MeCP2 is involved in the regulation of transcription, chromatin structure, and RNA splicing, and variation in methylation states alters MeCP2 binding (33,37,38). MeCP2 functions as both a transcriptional activator and repressor depending on the gene and additional protein factors it is associated with. When functioning as transcriptional repressor, MeCP2 can complex with histone deacetylases such as SIN3a to alter chromatin structure (34). To function as a transcriptional activator, MeCP2 can associate with activating TFs or can reduce the expression of genes whose protein products repress transcription, leading to increased gene expression of downstream target genes (37,39). Methyl-CpG-binding domain 1 (MBD1), another MBP, is critical for maintaining the stemness properties of neuronal stem cells through the direct binding of MBD1 to genes responsible for cell differentiation (40). In mice, loss of MBD1 in the hippocampus led to increased expression of genes associated with cellular differentiation and was associated with altered neurogenesis, impaired learning, and autism-like behaviors (40,41). In previous studies, mutations in MBD1 were observed in individuals with ASD (42). Another MBP, SETDB1 is recruited to 5mC by MBD1 and can methylate histone H3 lysine 9 (H3K9) (43,44). The location of SETDB1 in neural development genes overlapped with that of polycomb repressive complex 2 (PRC2) and two proteins, JARID2 and MTF2, that complex with PRC2 (43). The interaction of these proteins indicates that 5mC readers play a significant role in determining gene expression and in regulating cell fate throughout neuronal development.
Establishment, maintenance, and function of 5hmC derivatives (5hmC, 5fC, and 5caC)
The DNA methylation pathway is cyclical: the methyl group in 5mC can be oxidized to 5hmC by ten-eleven translocation (TET) enzymes through the recognition of methylated residues by Tet CXXC DNA binding domains (Fig. 1A). TET enzymatic function requires oxygen, iron (Fe (II)), and α-ketogluterate to oxidize 5mC into 5hmC. The presence of 5hmC is indicative of active demethylation as TET enzymes provide the only mechanism known to produce this modification. Succeeding 5mC conversion, TET enzymes can catalyze the subsequent oxidation of 5hmC into 5-flurocytosine (5fC) and 5-carboxylcytosine (5CaC) which are rare, thought to be intermediates in the cytosine methylation pathway, and confer minimal function (9,10) (Fig. 1A). Following 5hmC oxidation, 5fC and 5CaC can be excised by thymine DNA glycosylase (TDG), and the abasic site is repaired via the canonical base-excision repair pathway.
Of the three Tet enzymes in mammals (TET1/2/3), there is variation in the expression, prevailing function, and location of each variant. However, there is overlap between functional regions of the TET enzymes. TET1 is predominantly located at promoter regions. TET1 interacts with both repressive and active histone modifications and can be repressive or active in nature depending on the location and factors to which it binds (19) Throughout neurodevelopment, the expression of TET enzymes remains relatively constant, suggesting that TET activity could be modified post-transcriptionally or through additional cofactors (45). Further studies support the model of TET working with additional cofactors to modulate TET function. One way that TET1 functions in an active manner is through complexing with histone acetyltransferases (HATs) (e.g. MOF, identified via cooccupancy and co-IP (46)) and modulating H4K16ac, an acetylation mark known to facilitate both gene expression and DNA repair. TET2 is more versatile and located at diverse genomic regions such as enhancers and with the predominant function of oxidation of 5hmC residues into intermediates (5fC and 5caC). Gene body 5hmC plays a role in genomic stability, whereas loss of TET2 and a resulting increase in gene body 5hmC are associated with reduced genomic stability and an increase in mutagenesis (47). This reduction in TET2/5hmC stabilization and increased mutagenesis could explain a portion of genetic mosaicism seen throughout psychiatric disease (48). The mechanism responsible for these observed changes has yet to be elucidated, and it is unknown whether the oxidation and removal of 5hmC mark by TET2 enzymes increases genomic stability or if the presence TET2 and the proteins it associates with are responsible. TET3 is highly expressed in brain, throughout embryonic development, and in epigenetic reprogramming in oocytes (49). TET3 is a critical player driving the activation of neuronal genes and the proper development of neurons and synapses (50). In adult mice, TET3 expression is high in dorsal root ganglion and is associated with increased axon regeneration (51). TET3 is largely located at transcriptional start sites where it catalyzes the conversion of 5hmC to 5fC and 5caC before facilitating the removal of 5caC via TDG-mediated base excision repair (BER) (52).
In concordance with the cell-specific expression and regional functional specificity of TET enzymes, the function of the 5mC derivatives is also closely tied to genomic context. Although 5hmC is present in most tissues and cell types, it is significantly enriched in the brain and in human embryos (53–56). Actively transcribed tissue-specific genes are enriched in 5hmC modifications relative to non-tissue specific genes that have lower expression or housekeeping genes that are highly expressed (57,58). 5hmC has been found in diverse regions, such enhancers, promoters, transcription start sites (TSSs), gene bodies, 3′ UTRs and intragenic regions. However, 5hmC appears to be predominantly enriched near TSSs and in gene bodies associated with increased transcription (55,59–62). Additionally, 5hmC modifications located in enhancer and intergenic regions can be associated with actively transcribed genes (63); however, this is not always true as shown by the identification of genomic regions enriched in 5hmC that are associated with reduced transcriptional activity (56). Lin et al. (55) and Gross et al. (63) found that genes with greater levels of 5hmC within their gene body are expressed at a greater level. Relative to the other cytosine modifications, 5hmC appears to have the greatest contribution to neurological disorders and is linked with many disorders spanning neurodevelopment through neurodegeneration (64–68). Throughout neurodevelopment, 5hmC is dynamic with the majority of methylation changes occurring in proximity to genes involved in neurodevelopment. In the actively expressed genes, 5hmC is generally enriched in gene bodies, in proximity to the TSS, and in regions flaking CpG islands (69). The signal encoded for by 5hmC, as highlighted above, is read by TET enzymes but can also be read by enzymes distinct from TET binding and function. UHRF2 is a 5hmC binding protein expressed in brain and is involved in proper neuronal function but is not required for development. UHRF2 knockout in mice is associated with impaired spatial memory learning and recall. Chen et al. (24) provided evidence that binding of UHRF2 to 5hmC does not alter the function of TET1 enzymatic activity. The exact function of UHRF2 has yet to be elucidated but is hypothesized to be a failsafe for UHRF1 function. UHRF1 is a 5mC and 5hmC reader and is critical for development: knockout of UHRF1 results in embryonic lethality (70). UHRF1 aids in the recruitment of DNMT3A to its binding sites. As the knockout of UHRF1 is lethal, but DNMT3A knockout mice are viable, it is likely that UHRF1 has additional regulatory functions that have yet to be discovered. An additional example of the contribution of 5hmC writers to neuronal development is the function of MeCP2, which binds both 5mC and 5hmC with similar affinity. When bound to 5hmC in active gene bodies, MeCP2 facilitates the recruitment of chromatin remodelers to create a more accessible environment. Loss of MeCP2 is associated with lower levels of 5hmC at DhMRs linked with postnatal neurodevelopment and aging (54). Our current understanding on what defines the function of the different TET enzymes and MBPs is still limited. Deciphering the mechanisms that provide specificity for the function of methylation readers, writers, and erasers is critical for understanding how the methylome is regulated and how deviations may contribute to disease progression.
N6-methyladenine
6mA refers to the methylation of adenosine at its nitrogen-6 position. DNA 6mA was previously considered to be absent in eukaryotes but is the most predominant DNA modifications in prokaryotes. With the technological advances in quantifying base modification using immunoblot, ultra-high performance liquid chromatography tandem mass spectrometry (UHPLC-MS/MS) and single molecule real-time sequencing, 6mA has been found in several metazoans including worm (11), fruit flies (12,71), frog (72), zebrafish and pig (73), as well as mouse (72,74,75) and in humans (16,72,76). Genomic distribution of 6mA differs among species and is dynamically regulated during different developmental stages and under varying environmental conditions. For instance, during early embryogenesis in zebrafish, 6mA peaks are widely distributed across the genome with slight enrichment following TSS and found to be enriched at exons (73). However, 6mA shows exon depletion in the genome of frog (72). 6mA peaks in mouse embryonic stem cells (ESCs) are accumulated in intergenic regions (74) and excluded from gene-coding regions in gDNA from mouse tissues (72). Further analysis showed that 6mA is abundant on X chromosomes and strongly deposited on the unique 5' UTR and ORF1 regions of young LINE-1 elements (74). Substantial depletion of 6mA at TSS region has been found in mouse prefrontal cortices (PFCs) upon environmental stress (75). In contrast to mice, Xiao et al. (76) observed low 6mA density on the X chromosome but high in mitochondria using human blood samples. Various genomic distributions of 6mA indicate its potentially diverse biological functions and mechanisms across species. However, detection of 6mA modification using more sensitive and accurate methods is required to precisely characterize its genomic distribution and the deposition at specific genomic regions.
Currently, methyltransferases and demethylases for 6mA addition/removal in genomic DNA have only been discovered in limited species. DAMT-1, a member of the MT-A70 family of methyltransferases, can potentially mediate DNA 6mA methylation in worm (11). In vivo knock down of DAMT-1 leads to decreased 6mA level in WT and spr-5(by101) mutant worms, and the potential methylation activity is restricted to DAMT-1 (11). The human methyltransferase is N6AMT-1 (76), which has regions of sequence similarity with M.TaqI-like 6mA DNMT in bacteria restriction–modification systems (77). However, replication of N-6 methylation activity of this gene in human glioblastoma cells was unsuccessful (16). One just published research shows that mouse METTL4, belonging to subclade of MT-A70 adenine methyltransferases, has methyltransferases activity for 6mA deposition (78). For identification of demethylases, in vitro biochemical and genetic studies indicate that ALKBH1 and ALKBH4, as homolog of prokaryotic AlkB α-ketogluterate-dependent dioxygenase, are responsible for DNA 6mA erasure in mammals (16,74,76,78). Fly DMAD, the only αKG-dependent dioxygenase homolog in the TET family, was identified as a 6 mA demethylase in germline cells, early development and subsequently confirmed in neurogenesis (12,71). The precise molecular functions of DNA 6mA methyltransferases or demethylases need to be further defined.
Unlike the rapid progress made in understanding 5mC and 5hmC, epigenetic roles of 6mA remain poorly understood and characterized in eukaryotes. 6mA has been shown to dynamically respond to different forms of environmental stress, such as pre-frontal cortex stress (75), hypoxia (16), extinction learning (13) and mitochondrial stress (14). The tightly regulated 6mA could serve as a new “emergency responder” in adult brain in dealing with stress. Several findings suggest that 6mA plays a role in shaping transcription. Fly DMAD can remove 6mA primarily from transposon regions, which is associated with transposon repression in ovaries (71). In mouse ESCs, 6mA is enriched on the X chromosome and acts mainly as a repressive mark to silence LINE transposon expression (74). One biochemical and structural experiment found that the deposition of 6mA in the TSS can cause specific pol II pausing/stalling (79). Importantly, the molecular studies in fly established the potential role of 6mA in regulating development and neuronal function. 6mA level is tightly regulated by DMAD during embryogenesis (71). 6mA-formed epigenetic marks can be read by jumu to mediate maternal-to-zygotic transition through regulating expression of the zelda gene (80). Dynamic changes of 6mA could be also important for fly neuronal functions. DMAD depletion in fly neurons leads to impaired neurodevelopment as well as behavioral abnormalities (12,81–84). 6mA accumulation caused by DMAD depletion can recruit Polycomb proteins to regulate transcriptional repression in fly neurons (12). It is interesting that Kweon et al. (78) identified an adversarial 6mA-sensor network through 6mA-deposition-coupled proteolysis mechanism to regulate Polycomb gene silencing in HEK293T cells, providing insight into that the cooperation between 6mA and Polycomb proteins could also play roles in mammalian neurons. Importantly, epigenetic roles of 6mA in brain function have also been indicated in mice. Genes containing stress-induced 6mA alternations overlap with neuropsychiatric disorders-linked loci, which indicates the participation of 6mA in the pre-frontal cortex's stress-associated pathway (12). Upon fear extinction, 6mA deposits along promoters and coding sequences in activating cortical neurons of mice highlight the role of 6mA in adult brain function (13). Therefore, like 5mC and 5hmC in mammals, 6mA could serve as a DNA anchor for regulatory protein binding. Such coordination could then implement various downstream pathways, alter chromatin landscapes, and regulate gene expression (12,13,72). Further investigations are required to identify epigenetic modifiers of 6mA and potential 6mA-interacting proteins in different cell types, tissues and species.
It is worth noting that very recently O'Brown et al. (85) showed that the use of high performance liquid chromatography (HPLC) analysis to detect 6mA in genomic DNA could overestimate 6mA abundance, which could be due to the potential bacterial DNA contamination of enzyme preparations used to digest DNA in HPLC analysis. After minimizing and subtracting artifacts, the study indicated that 6mA is present at fold lower levels in metazoan DNA than previously published 6mA studies (85). Thus, improved sample preparation and sequencing sensitivity are necessary to accurately characterize genomic features of 6mA and reveal its biological functions. However, before concluding that 6mA is not present in eukaryotic genomes, several factors need to be considered. First, many published studies have conducted 6mA-IP-seq to identify differentially expressed 6mA regions between experimental conditions. Directly comparing control and genetic depletion or stressed samples could define 6mA in eukaryotic DNA with elimination of any bacterial contamination or IgG nonspecific binding. Analyses of differential 6mA signals between DMAD knockout and control fly neuronal cells revealed the alterations of 6mA at distinct genomic loci, and the cells were free of bacterial contamination as determined by bacterial 16S quantitative PCR (12). Therefore, it would be difficult to imagine that all these differential signals were due to bacterial contamination. In addition, 6mA reads from prokaryotic origin can be controlled for by sequencing alignment (86). Second, the HPLC method used by O'Brown et al. (85) as described in the article might not be fully optimized. Offline enrichment, specifically of 6mA, before liquid chromatography tandem mass spectrometry (LC-MS/MS) acquisition will reduce the likelihood of 6mA experiencing signal suppression due to isobaric interference (15). Third, 6mA abundance should differ among various cell types, tissues, developmental stages and environmental circumstances. O'Brown et al. (85) looked at static tissues and cell types, such as human HEK293 and HeLa cells, which have very low levels of 6mA as reported previously. Detectable 6mA signal could be diluted using HPLC analysis of whole brain (15,84). Last but not the least, unlike what has been portrayed in public discussions, O'Brown et al. (85) found rare (0.9–3.7 ppm) 6mA signals above background in DNA samples from gnotobiotic mouse tissues (85), which are non-treated and non-stressed samples. Like 5fC, steady low levels of DNA modification could still play critical roles in specific circumstances. Low levels of 6mA in the adult brain of fly (12) and mice (13,75) are important for neuronal function. Targeting 6mA modification can significantly decrease cancer cell growth and extend survival of tumor-bearing mice (16). Again, these rare DNA modifications could have critical biological functions, such as marking genes or regulation elements and recruiting specific “reader” protein to render their biological functions in a more dynamic manner that may be under-appreciated.
DNA Modifications in Neurodegenerative Disorders
Genetic studies in the past 30 years have made a great contribution to our understanding of numerous neurodegenerative disorders and have identified many genetic loci associated with disease. As the identified genetic variants have not been able to explain the entirety of disease pathology, epigenetics has been gaining favor as one of the key regulators for pathogenesis and progression of several neurodegenerative disorders: aberrant cytosine modifications located at the promoter, surrounding regions of causal genes, flanking regions of causal repeats, and genome-wide level are often linked with disease state. Emerging epigenome association studies have identified many dynamically regulated DNA modification sites, providing novel insights into the diseases. Further functional studies using epigenomic editing are required to reveal the exact epigenetic roles for candidate DNA modification sites. The association of 6mA with brain disorders has yet to be explored and is a strong candidate for identifying novel molecular mechanisms that contribute to neurological disorders. In this section, we present current evidence of DNA modification changes in representative neurodegenerative disorders for highlighting the diseases-associated epigenetic modifications.
Alzheimer's disease
Alzheimer's disease (AD) is caused by the gradual decay of neuronal connections and eventual death of neurons. There are two hallmark pathologies in AD: neurofibrillary tangles and amyloid plaques, both of which contribute to neuronal dysfunction and loss. Sequencing-based studies have identified mutations in many genes that contribute to AD susceptibility, the strongest of which directly contribute to the tangle or plaque pathology (APP, PSEN1, PSEN2, and MAPT) and are often seen in familial AD cases (87,88). In addition to identifying genetic variants, previous work has implicated the methylome as a strong contributor to AD pathology with methylation changes occurring in proximity or in genes with altered expression in AD (e.g. ABCA7, ANK1, BIN1, CELF1, BZRAP1, RHBDF2, and MINK1) (89–91). Strikingly, De Jager et al. (89) found that genetic variants associated with AD explained 13.9% of neuritic plaque burden, whereas the 71 CpGs identified explained 28.7% of amyloid plaque pathology.
In AD, 5mC profiles between brain cell types are relatively similar; however, differences in the location of methylation are present. More strikingly, a difference in both the location and global presence in 5hmC is seen in AD. As AD progresses, a global increase in 5hmC is observed, with differential methylation and gene expression cooccurring in pathways involved in neuronal projection development and neurogenesis (neurogenesis, enzyme-linked receptor protein signaling, synapse organization, regulation of vesicle-mediated transport, and small GTPase-mediated signal transduction) (91). Differentially hydroxymethylated loci associated with gene expression changes were primarily located in intragenic regions. In AD mouse models, the same trend is seen, with an increase in gene body 5hmC associated with increased gene expression of signaling and metabolic pathways implicated in AD (67).
In support of the involvement of the methylome in AD progression, recent studies utilizing deep sequencing reinforced the role of abnormal methylation changes in AD pathology with the identification of DNMT3A and TET2 loss of function variants in individuals with AD (92). Reduced expression of DNMT3A has previously been characterized in aging brains and is associated with memory impairment. Memory impairment can be rescued through the overexpression of DNMT3A2 (93,94). Additionally, the effect of neuronal activity on methylation patterns is disrupted with the injection of DNMT inhibitors in mice which prevent the conversion of short-term memory into long term memories (95). TET2 has been functionally studied in the context of AD. The knockout and knockdown of TET2 resulted in reduced genome-wide levels of 5hmC and increased AD severity (67). In WT aging mice, overexpression of TET2 rescued age-related cognitive decline (96). In addition to TET's role as a hydroxymethylation writer, TET proteins exhibit additional functions. TET1 was shown to complex with lysine acetyltransferase 8 (KAT8 or MOF), a HAT responsible for placing H4K16ac. Nativio et al. (97) showed that, in TET1 knockout MEF cells, there is a significant loss of H4K16ac proximal to genes linked to AD. Loss of H4K16ac is associated with increased genomic instability and DNA damage, highlighting the importance of TET1 in maintaining genomic stability (46). The importance of DNA hydroxymethylation in maintaining genomic stability is beginning to be understood; however, the mechanistic underpinnings of how 5hmC protects against instability have yet to be determined.
Recent advancements have enabled the isolation and profiling of individual cell types in AD. Gasparoni et al. (98) isolated specific neuronal and glial cell types from mouse brain. Their studies found variations in methylation profiles between cell types in the brain and corresponding differences in gene expression changes. This study also identified differential methylation in ADAM17 and APP, two well-known AD-associated disease genes. APP encodes for the amyloid precursor protein responsible for the hallmark amyloid plaque pathology. Further studies could help explain how plaque pathology contributes to the neurodegenerative cascade seen in AD and provide insight into the dynamics of the methylome in each cell type affected in AD which is critical for understanding how AD progresses and for the discovery of AD therapeutics.
Parkinson's disease
The second most common neurodegenerative disorder in the elderly is Parkinson's disease (PD), affecting 1%–2% of the population over 65 years and rising to nearly 5% in the population over the age of 85 (99). Several studies have shed light on the epigenetic machinery in modulating progression of PD, but the role of DNA methylation and its link to PD pathogenesis are still unclear. SNCA, a gene encoding the aplha-synuclein protein, is one of the most important risk genes for PD and is regulated by DNA methylation. Hypomethylation of CpG islands at intron 1 of SNCA can lead to overexpression of α-synuclein in PD and related disorders. Methylation level of SNCA shows a negative correlation with its expression (100,101). The altered methylation level in PD could be mediated by the association of DNMT1 with α-syn, which may sequester DNMT1 in the cytoplasm leading to the global DNA methylation seen in postmortem brains of PD (102). Using DNA from peripheral blood leukocytes, methylation in CpG-2 sites in the SNCA promoter was shown to be significantly decreased in PD patients compared to controls, implicating DNA methylation levels in peripheral blood leukocytes as a potential noninvasive biomarker for PD (103). Moreover, epigenome-association studies identified many other differentially methylated genes or sites in PD that need further validation, such as differentially methylated CpG in the FANCC and TNKS2 genes (104) and 82 altered CpG sites (105). Differential expression levels of total 5mC and 5hmC across different brain regions have been seen for PD. Cortical sections from PD patients contains extensive 5mC while 5hmC is significantly accumulated in cerebellar white matter (106), highlighting the importance of cytosine modification in PD. More extensive studies are still needed in order to better understand the role of 5mC and 5hmC in PD.
Ataxia-related disorders
Friedreich ataxia
Friedreich ataxia (FRDA), the most common heritable ataxia, is an autosomal recessive neurodegenerative disorder predominantly caused by a homozygous GAA repeat expansion mutation within intron 1 of the FXN gene (107). Patients with FRDA usually have at least one allele of FXN gene containing >90 GAA repeats (the majority have 600 to 900 repeats), while normal alleles have repeats between 8 and 33 (108). Impaired DNA replication, recombination and repair have all been considered as possible mechanisms to trigger this disorder. More recently, FRDA repeats leading to aberrant epigenetic modifications have been appreciated. Histone modifications are known to affect flanking regions of GAA repeats, while the influence of DNA modifications are largely unknown. Bisulfite mapping shows that the upstream region adjacent to the repeat is methylated more extensively in patients than in normal individuals (109–111), while downstream of the expansion showed decreased methylation levels (110). A direct correlation between CpG methylation levels and triplet expansion size has been found, suggesting that evaluation of the methylation status of specific CpG sites in FRDA patients could be a convenient biomarker (111). One methylated residue in FRDA patients is located within an E-box site which is important in promoter activity in reporter assays (110). In addition, methylation level of GAA expansion upstream can predict the age of disease onset (111,112). Therefore, it is possible that DNA methylation can affect the progression of this disorder and the epigenetic profiles could be dynamically regulated. It is of note that Al-Mahdawi et al. (110) found that the increased DNA methylation level at the GAA region of FXN is caused by 5hmC in FRDA cerebellum, demonstrating the importance of 5hmC in FRDA and the need for further investigation.
Spinocerebellar ataxias
Spinocerebellar ataxias (SCAs) are a large complex group of autosomal dominant degenerative disorders characterized by cerebellar degeneration with variable involvement of the brain stem and spinal cord (113). Until now, more than 40 different types of SCAs have been discovered. The clinical hallmark for all SCAs is a progressive loss of balance accompanied by slurred speech and reduced life span (114). Genetic causal variants have been identified for over 30 different types of SCAs (115). Many SCAs, including SCA1, 2, 3, 6, 7, 12 and 17 are caused by dynamic repeat mutations, which contains trinucleotide CAG expansions in the coding region of genes leading to poly-Q expanded proteins (114). DNA methylation within the promoter region of repeats or close to the repeats has been identified for many SCAs, but the role of epigenetic regulation remains unknown. Close to the CAG repeats in ATXN7, DNA methylation at the CTCF binding elements could promote repeat instability (116). Emmel et al. (117) proposed that epigenetic control, such as DNA methylation of the ATXN3 promoter region, could contribute to the age of onset in SCA3. Wang et al. (118) found higher methylation in the ATXN3 promoter region in SCA3 patients, which further identified that the first CpG island of the promoter could serve as main regulation region of DNA methylation. CpG methylation of the ATXN2 gene promoter in human shows correlation with pathogenic CAG expansions in spinocerebellar ataxia type 2 (SCA2) cases (119). One recent study found global 5mC level significantly elevated in the patients of SCA1 and SCA2, when compared with age- and sex-matched healthy controls, pointing to a role of cytosine modification in SCAs (120). If epigenetic events indeed cause the SCAs phenotypes, specific regulation factors might incorporate with DNA modifications to trigger downstream pathways, which is worth further study. These methylation sites could be clinical biomarkers for SCAs prediction, diagnostics and therapies.
Fragile X-associated tremor/ataxia syndrome
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late onset (>50 years) neurodegenerative disorder caused by a premutation (50–200 CGG-repeat expansion) in the fragile X mental retardation 1 (FMR1) gene located in Xq27.3 (121,122). The disorder is characterized by intention tremor, cerebella ataxia, parkinsonism, and cognitive decline, as well as peripheral neuropathy (122). However, no abnormal methylation has been found surrounding CpG island and promoter of the repeats in FXTAS (123). In comparison, FMR1 full mutations, with over ~ 200 CGG repeats, are associated with aberrant CpG methylation of the repeat, neighboring CpG island, and gene promoter, leading to silencing of FMR1 and fragile X syndrome (FXS) (124). Such differences indicate distinct molecular mechanisms of pathogenesis between FXTAS and FXS, even though they are both developed from the expanded CGG repeat at 5' UTR of FMR1. In mouse models, CGG repeats can be specifically expressed in Purkinje neurons, and this is sufficient to induce a FXTAS phenotype (125). Considering the high levels of 5hmC seen in Purkinje cells, we determined genome-wide 5hmC profiles using cerebellum from FXTAS (rCGG) mice and wide-type littermates (66). Overall, a global reduction of 5hmC was seen in the rCGG mice. DhMRs are highly associated with genes and TFs that are important for neurogenesis and neurological activities, suggesting that 5hmC-mediated epigenetic modulation plays a role in the onset of FXTAS. In addition, differentially regulated 5hmC has been found mainly on the gene body including FMR1 gene, further highlighting the involvement of 5hmC in early FXTAS pathogenesis. Alternatively, the globally decreased 5hmC in rCGG mice could indicate increased levels of 5mC, which could lead to gene silencing. Genome-wide or locus-specific fine mapping of 5mC in a cell-type specific manner should be applied to reveal dysregulation of 5mC in FXTAS and identify the 5mC cofactors for the pathogenesis and progression.
Possible New Roles of DNA Modifications
Modulators of dynamic TF and DNA interaction
Recent advances in the field of DNA modifications have shed light on the role of DNA modifications and their readers, writers, and erasers in gene expression regulation and in shaping the local chromatin environment (23,126,127). We are just now scratching the surface on understanding how the two most studied DNA modifications (5mC and 5hmC) and their association with three different TET enzymes govern lineage-specific gene expression for roughly 400 different cell types. As previously mentioned, the genomic location and prevalence of DNA methylation varies widely with respect to tissue and cell type (128,129). DNA hydroxymethylation is especially enriched in brain (129,130). 5hmC heterogeneity is observed both in distinct brain regions as well as the individual cell types that inhabit them (129,131,132). The methylation profiles for three major cell types within the prefrontal cortex (GABAergic neurons (inhibitory), glutamatergic neurons (excitatory), and oligodendrocytes were examined and correlated with gene expression profiles. The methylome and gene expression varied between the inhibitory neurons and the excitatory neurons and oligodendrocytes. Most importantly, risk loci associated with neuropsychiatric disease were differentially modified between excitatory and inhibitory neurons. In excitatory neurons, risk loci were enriched in 5hmC-depleted regions, and in inhibitory neurons, risk loci were enriched in 5mC depleted regions (132). These findings indicate that previous studies using whole brain or isolated brain regions to characterize the role of the methylome in disease development did not have the resolution required to identify all differentially methylated regions and differentially hydroxymethylated regions involved in disease pathogenicity. Even further, single-cell studies show that cell types within the same region display significant differences in methylation profiles that are associated with different gene expression patterns (133). The methylation signatures for many cell types are unique enough to justify using new techniques that involve cell-type deconvolution to determine the proportion of cell types in a population-based DNA modification signature at specific loci (134).
Base-pair resolution of DNA methylation is critical for deciphering the nuances in the methylome. EGR1 is a three-finger-binding DNA protein whose expression is upregulated in response to environmental signals encompassing hormones, growth factors, and neurotransmitters (135). WT1 is a TF that contains four zinc-finger DNA binding domains and is responsible for regulating cell differentiation and cell survival (136). The binding domains of EGR1 and WT1 both recognize the 5'-GCG(T/G)GGGCG-3' sequence motif that contains two CpG sites with the capacity to be methylated and oxidized. Surprisingly, the methylation status (unmodified cytosine or 5mC) does not change the binding of EGR1 or WT1; however, the oxidation state does (5hmC, 5fC or 5caC). The presence of 5caC, created by TET enzymes, alters the binding kinetics and prevents the binding of EGR1 but not of WT1 (137). The observed differential recognition of cytosine intermediates suggests a potential biological significance of these modifications that could expand beyond serving as a marker for TDG-mediated excision and BER.
TET enzymes are known to interact with TFs, splicing factors, DNA repair enzymes, histone modifiers, and chromatin remodeling complexes (23,62,91). At intron–exon boundaries, 5mC and 5hmC are recognized by protein complexes containing CTCF, MeCP2 and HP1 that regulate alternative splicing (62). In neuro stem cells (NSCs), TET2 directly binds to Foxo3a to regulate the expression of genes involved in NSC proliferation (127). These studies highlight the importance of TET proteins and indicates that additional efforts should be placed on identifying protein–protein and protein–DNA interactions facilitated by DNA modifications and the genomic context of these interactions. The importance of TET enzyme location lies in the alternate functions associated with methylation variant location. Additionally, as previous studies have largely focused on 5mC and 5hmC modifications, novel insight on the function of an additional DNA modification, 6mA, may further explain how chromatin environments are regulated and increase our understanding of the specificity of DNA modifications in determining cell-specific gene expression.
Brain genomic mosaicism
Impaired DNA repair might play a fundamental role in development of age-related neurodegeneration, but the exact molecular mechanism is unclear (138–141). Defects in cellular DNA repair processes have been linked to genome hypermutation and instability, as well as premature aging syndromes (138). Upon ultra-high depth sequencing (>5000×) with a mathematical model of neurodevelopment, Keogh et al. (92) revealed that human brains are likely to harbor zones of cells containing somatic mutations, including mutations in genes critical for neurodegenerative diseases. The advent of single-cell deep sequencing revealed rapid accumulation of somatic mutation in PFC neurons from patients with progeroid neurodegeneration, while the accumulation of somatic mutations with age in normal human neurons is slow but inexorable (142). The high somatic mutation burden in PFC neurons elucidates defective DNA damage repair and chromosomal instability, and the molecular mechanism is important to identify.
Studies have shown that loss of TET enzymes and 5hmC depletion can contribute to genome instability and inaccurate chromosome segregation. 5hmC could be as an epigenetic marker of DNA damage (143). For example, TET1-deficient cells contained more DNA strand breaks, as well as in cells without exogenous DNA damaging agents (144). In Purkinje cells of Atm−/− mice, TET1-mediated 5hmC production is linked to the DNA damage process (145). Pan et al. (47) found that TET2 loss can lead to hypermutagenicity in hematopoietic stem/progenitor cells. The increased mutation burden was found to be particularly high at genomic sites that gained 5hmC, where TET2 normally binds (47). In HeLa cells, TET2 was found to be necessary for the production of 5hmC foci at endogenous DNA damage sites (143). In addition, TET3-mediated conversion of 5mC to 5hmC can elevate ATR-dependent DNA damage response and regulate DNA repair, suggesting that TET enzymes and active DNA demethylation could play direct role in DNA repair (146). Therefore, a far-reaching epigenetic role for covalent 5hmC by TET enzymes could be promoting DNA repair and genome stability in neurodegeneration (Fig. 3). Integrated analysis using deep sequencing for single neuronal cells and in situ study for somatic mutations could identify critical loci, cells types and brains regions for neurodegenerative disorders in the future (147).
Figure 3.
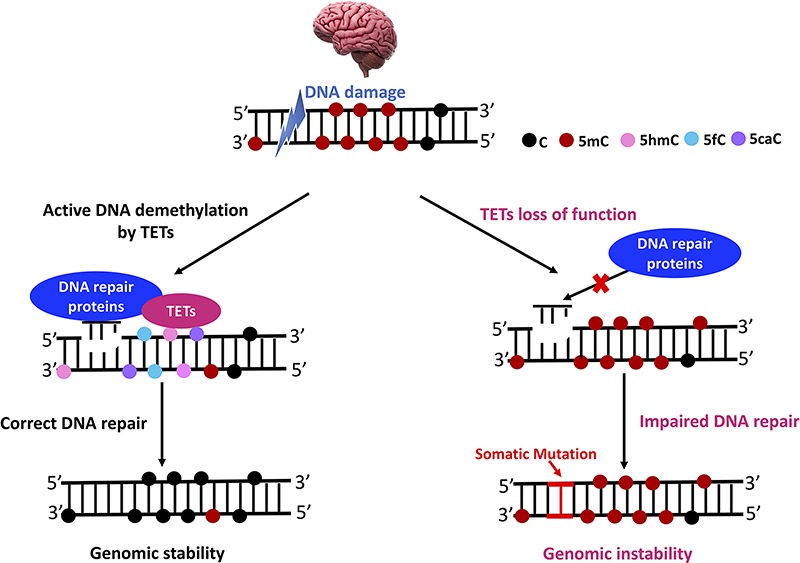
A potential epigenetic role for TET proteins and DNA repair machinery in regulating neurodegeneration. TET proteins play important roles in DNA repair processes, which could be required to active DNA demethylation at neighboring DNA damage sites. Normally, TET proteins could recruit DNA repair proteins at damage sites to promote genome integrity and stability. However, TET loss of function could lead to impaired DNA repair and increased somatic mutation in brain, resulting in neurodegeneration.
Summary
In this review, we discussed how DNA methylation lies at the core of lineage-specific gene expression by establishing an epigenetic code that is dynamically read and regulated by DNA-binding proteins. There is a diverse set of protein–DNA interactions that temporally modulates gene expression by altering the chromatin environment through recruitment of chromatin remodelers, negative and positive TF binding, and splicing complexes. The extent of protein–DNA interactions and a full list of 5mC and 5hmC readers has yet to be discovered. Even less is known about the more recently discovered 6mA modification and the enzymes that read, write, and erase it. The majority of our 6mA understanding comes from association-based studies that highlight the dynamic nature of 6mA in disease and stress response.
Previous technological and cost-preventative limitations are now being addressed with newer studies utilizing single-cell and single base-pair resolution methods rather than capture-based techniques on tissues that contain heterogenous cell populations. We anticipate that the greater resolution provided by single cell studies in combination with assays that provide single-base pair resolution will greatly enhance our understanding on how neurological disorders progress and the individual cell types and methylation changes that drive their progression. As these studies will be largely association-based, functional studies leveraging novel technique such as dCas9 tethered to DNMTs or TET enzymes to target DNA modifications could be very powerful tools for defining the biological significance of methylation and hydroxymethylation changes at individual methylation sites.
Acknowledgement
The authors would like to thank Dr Bing Yao for the discussion and critical comments on the article.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health [NS111602, NS051630, NS097206, HG008935, MH116441, AG052476 and AG062256 to P. J and NS091859 to E.G.A and P.J.].
References
- 1. Pal S. and Tyler J.K. (2016) Epigenetics and aging. Sci. Adv., 2, e1600584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jonkhout N., Tran J., Smith M.A., Schonrock N., Mattick J.S. and Novoa E.M. (2017) The RNA modification landscape in human disease. RNA, 23, 1754–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grassi D., Franz H., Vezzali R., Bovio P., Heidrich S., Dehghanian F., Lagunas N., Belzung C., Krieglstein K. and Vogel T. (2017) Neuronal activity, TGFβ-signaling and unpredictable chronic stress modulate transcription of Gadd45 family members and DNA methylation in the hippocampus. Cereb. Cortex, 27, 4166–4181. [DOI] [PubMed] [Google Scholar]
- 4. Bartel D.P. (2018) Metazoan MicroRNAs. Cell, 173, 20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuehner J.N., Bruggeman E.C., Wen Z. and Yao B. (2019) Epigenetic regulations in neuropsychiatric disorders. Front. Genet., 10, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hotchkiss R.D. (1948) The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J. Biol. Chem., 175, 315–332. [PubMed] [Google Scholar]
- 7. Riggs A.D. (1975) X inactivation, differentiation, and DNA methylation. Cytogenet. Genome Res., 14, 9–25. [DOI] [PubMed] [Google Scholar]
- 8. Keshet I., Yisraeli J. and Cedar H. (1985) Effect of regional DNA methylation on gene expression. Proc. Natl. Acad. Sci. U. S. A., 82, 2560–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ito S., Shen L., Dai Q., Wu S.C., Collins L.B., Swenberg J.A., He C. and Zhang Y. (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science, 333, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nabel C.S., Jia H., Ye Y., Shen L., Goldschmidt H.L., Stivers J.T., Zhang Y. and Kohli R.M. (2012) AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat. Chem. Biol., 8, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Greer E.L., Blanco M.A., Gu L., Sendinc E., Liu J., Aristizábal-Corrales D., Hsu C.-H., Aravind L., He C. and Shi Y. (2015) DNA methylation on N6-adenine in C. elegans. Cell, 161, 868–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yao B., Li Y., Wang Z., Chen L., Poidevin M., Zhang C., Lin L., Wang F., Bao H. and Jiao B. (2018) Active N6-methyladenine demethylation by DMAD regulates gene expression by coordinating with polycomb protein in neurons. Mol. Cell, 71, 848–857. e846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li X., Zhao Q., Wei W., Lin Q., Magnan C., Emami M.R., Wearick-Silva L.E., Viola T.W., Marshall P.R. and Yin J. (2019) The DNA modification N6-methyl-2′-deoxyadenosine (m6dA) drives activity-induced gene expression and is required for fear extinction. Nat. Neurosci., 22, 534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ma C., Niu R., Huang T., Shao L.-W., Peng Y., Ding W., Wang Y., Jia G., He C. and Li C.-Y. (2019) N6-methyldeoxyadenine is a transgenerational epigenetic signal for mitochondrial stress adaptation. Nat. Cell Biol., 21, 319–327. [DOI] [PubMed] [Google Scholar]
- 15. Kigar S.L., Chang L., Guerrero C.R., Sehring J.R., Cuarenta A., Parker L.L., Bakshi V.P. and Auger A.P. (2017) N 6-methyladenine is an epigenetic marker of mammalian early life stress. Sci. Rep., 7, 18078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xie Q., Wu T.P., Gimple R.C., Li Z., Prager B.C., Wu Q., Yu Y., Wang P., Wang Y. and Gorkin D.U. (2018) N6-methyladenine DNA modification in glioblastoma. Cell, 175, 1228–1243. e1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bestor T., Laudano A., Mattaliano R. and Ingram V. (1988) Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells: the carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J. Mol. Biol., 203, 971–983. [DOI] [PubMed] [Google Scholar]
- 18. Yoder J.A., Soman N.S., Verdine G.L. and Bestor T.H. (1997) DNA (cytosine-5)-methyltransferases in mouse cells and tissues. Studies with a mechanism-based probe. J. Mol. Biol., 270, 385–395. [DOI] [PubMed] [Google Scholar]
- 19. Edwards J.R., Yarychkivska O., Boulard M. and Bestor T.H. (2017) DNA methylation and DNA methyltransferases. Epigenetics Chromatin, 10, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bayraktar G. and Kreutz M.R. (2018) The role of activity-dependent DNA demethylation in the adult brain and in neurological disorders. Front Mol. Neurosci., 11, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zeng Y. and Chen T. (2019) DNA methylation reprogramming during mammalian development. Genes (Basel), 10, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moore L.D., Le T. and Fan G. (2013) DNA methylation and its basic function. Neuropsychopharmacology, 38, 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maor G.L., Yearim A. and Ast G. (2015) The alternative role of DNA methylation in splicing regulation. Trends Genet., 31, 274–280. [DOI] [PubMed] [Google Scholar]
- 24. Chen R., Zhang Q., Duan X., York P., Chen G.-D., Yin P., Zhu H., Xu M., Chen P. and Wu Q. (2017) The 5-hydroxymethylcytosine (5hmC) reader UHRF2 is required for normal levels of 5hmC in mouse adult brain and spatial learning and memory. J. Biol. Chem., 292, 4533–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Neri F., Rapelli S., Krepelova A., Incarnato D., Parlato C., Basile G., Maldotti M., Anselmi F. and Oliviero S. (2017) Intragenic DNA methylation prevents spurious transcription initiation. Nature, 543, 72–77. [DOI] [PubMed] [Google Scholar]
- 26. Castillo-Aguilera O., Depreux P., Halby L., Arimondo P. and Goossens L. (2017) DNA methylation targeting: the DNMT/HMT crosstalk challenge. Biomolecules, 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tang Y., Jiang S., Gu Y., Li W., Mo Z., Huang Y., Li T. and Hu Y. (2017) Promoter DNA methylation analysis reveals a combined diagnosis of CpG-based biomarker for prostate cancer. Oncotarget, 8, 58199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wade J.T. and Grainger D.C. (2018) Spurious transcription and its impact on cell function. Transcription, 9, 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tatton-Brown K., Seal S., Ruark E., Harmer J., Ramsay E., Vecchio Duarte S., Zachariou A., Hanks S., O'Brien E. and Aksglaede L. (2014) Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat. Genet., 46, 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu Z., Li Z., Zhi X., Du Y., Lin Z. and Wu J. (2018) Identification of de novo DNMT3A mutations that cause west syndrome by using whole-exome sequencing. Mol. Neurobiol., 55, 2483–2493. [DOI] [PubMed] [Google Scholar]
- 31. Elliott E., Manashirov S., Zwang R., Gil S., Tsoory M., Shemesh Y. and Chen A. (2016) Dnmt3a in the medial prefrontal cortex regulates anxiety-like behavior in adult mice. J. Neurosci., 36, 730–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bogdanović O. and Veenstra G.J.C. (2009) DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma, 118, 549–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liyanage V.R. and Rastegar M. (2014) Rett syndrome and MeCP2. Neuromolecular Med., 16, 231–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fasolino M. and Zhou Z. (2017) The crucial role of DNA methylation and MeCP2 in neuronal function. Genes (Basel), 8, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Song C., Feodorova Y., Guy J., Peichl L., Jost K.L., Kimura H., Cardoso M.C., Bird A., Leonhardt H., Joffe B. et al. (2014) DNA methylation reader MECP2: cell type- and differentiation stage-specific protein distribution. Epigenetics Chromatin, 7, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liyanage V.R.B., Olson C.O., Zachariah R.M., Davie J.R. and Rastegar M. (2019) DNA methylation contributes to the differential expression levels of Mecp2 in male mice neurons and astrocytes. Int. J. Mol. Sci., 20, 1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao Y.T., Goffin D., Johnson B.S. and Zhou Z. (2013) Loss of MeCP2 function is associated with distinct gene expression changes in the striatum. Neurobiol. Dis., 59, 257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sanfeliu A., Hokamp K., Gill M. and Tropea D. (2019) Transcriptomic analysis of Mecp2 mutant mice reveals differentially expressed genes and altered mechanisms in both blood and brain. Front. Psych., 10, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chahrour M., Jung S.Y., Shaw C., Zhou X., Wong S.T., Qin J. and Zoghbi H.Y. (2008) MeCP2, a key contributor to neurological disease, activates and represses transcription. Science, 320, 1224–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jobe E.M., Gao Y., Eisinger B.E., Mladucky J.K., Giuliani C.C., Kelnhofer L.E. and Zhao X. (2017) Methyl-CpG-binding protein MBD1 regulates neuronal lineage commitment through maintaining adult neural stem cell identity. J. Neurosci., 37, 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Allan A.M., Liang X., Luo Y., Pak C., Li X., Szulwach K.E., Chen D., Jin P. and Zhao X. (2008) The loss of methyl-CpG binding protein 1 leads to autism-like behavioral deficits. Hum. Mol. Genet., 17, 2047–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li H., Yamagata T., Mori M., Yasuhara A. and Momoi M.Y. (2005) Mutation analysis of methyl-CpG binding protein family genes in autistic patients. Brain Dev., 27, 321–325. [DOI] [PubMed] [Google Scholar]
- 43. Fei Q., Yang X., Jiang H., Wang Q., Yu Y., Yu Y., Yi W., Zhou S., Chen T. and Lu C. (2015) SETDB1 modulates PRC2 activity at developmental genes independently of H3K9 trimethylation in mouse ES cells. Genome Res., 25, 1325–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Du J., Johnson L.M., Jacobsen S.E. and Patel D.J. (2015) DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol., 16, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Szulwach K.E., Li X., Li Y., Song C.-X., Wu H., Dai Q., Irier H., Upadhyay A.K., Gearing M. and Levey A.I. (2011) 5-hmC–mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci., 14, 1607–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhong J., Li X., Cai W., Wang Y., Dong S., Yang J., Zhang J.a., Wu N., Li Y. and Mao F. (2016) TET1 modulates H4K16 acetylation by controlling auto-acetylation of hMOF to affect gene regulation and DNA repair function. Nucleic Acids Res., 45, 672–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pan F., Wingo T.S., Zhao Z., Gao R., Makishima H., Qu G., Lin L., Yu M., Ortega J.R. and Wang J. (2017) Tet2 loss leads to hypermutagenicity in haematopoietic stem/progenitor cells. Nat. Commun., 8, 15102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nishioka M., Bundo M., Iwamoto K. and Kato T. (2018) Somatic mutations in the human brain: implications for psychiatric research. Mol. Psychiatry, 24, 839–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gu T.-P., Guo F., Yang H., Wu H.-P., Xu G.-F., Liu W., Xie Z.-G., Shi L., He X. and Jin S.-g. (2011) The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature, 477, 606. [DOI] [PubMed] [Google Scholar]
- 50. Zhang J., Chen S., Zhang D., Shi Z., Li H., Zhao T., Hu B., Zhou Q. and Jiao J. (2016) Tet3-mediated DNA demethylation contributes to the direct conversion of fibroblast to functional neuron. Cell Rep., 17, 2326–2339. [DOI] [PubMed] [Google Scholar]
- 51. Weng Y.-L., An R., Cassin J., Joseph J., Mi R., Wang C., Zhong C., Jin S.-G., Pfeifer G.P. and Bellacosa A. (2017) An intrinsic epigenetic barrier for functional axon regeneration. Neuron, 94, 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jin S.-G., Zhang Z.-M., Dunwell T.L., Harter M.R., Wu X., Johnson J., Li Z., Liu J., Szabó P.E. and Lu Q. (2016) Tet3 reads 5-carboxylcytosine through its CXXC domain and is a potential guardian against neurodegeneration. Cell Rep., 14, 493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Globisch D., Münzel M., Müller M., Michalakis S., Wagner M., Koch S., Brückl T., Biel M. and Carell T. (2010) Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One, 5, e15367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mellén M., Ayata P., Dewell S., Kriaucionis S. and Heintz N. (2012) MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell, 151, 1417–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lin I.-H., Chen Y.-F. and Hsu M.-T. (2017) Correlated 5-hydroxymethylcytosine (5hmC) and gene expression profiles underpin gene and organ-specific epigenetic regulation in adult mouse brain and liver. PLoS One, 12, e0170779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wu H., D'Alessio A.C., Ito S., Wang Z., Cui K., Zhao K., Sun Y.E. and Zhang Y. (2011) Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev., 25, 679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shen L. and Zhang Y. (2013) 5-Hydroxymethylcytosine: generation, fate, and genomic distribution. Curr. Opin. Cell Biol., 25, 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Laird A., Thomson J.P., Harrison D.J. and Meehan R.R. (2013) 5-Hydroxymethylcytosine profiling as an indicator of cellular state. Epigenomics, 5, 655–669. [DOI] [PubMed] [Google Scholar]
- 59. Yu M., Hon G.C., Szulwach K.E., Song C.-X., Zhang L., Kim A., Li X., Dai Q., Shen Y. and Park B. (2012) Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell, 149, 1368–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Song C.-X., Yi C. and He C. (2012) Mapping recently identified nucleotide variants in the genome and transcriptome. Nat. Biotechnol., 30, 1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Han D., Lu X., Shih A.H., Nie J., You Q., Xu M.M., Melnick A.M., Levine R.L. and He C. (2016) A highly sensitive and robust method for genome-wide 5hmC profiling of rare cell populations. Mol. Cell, 63, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shi D.-Q., Ali I., Tang J. and Yang W.-C. (2017) New insights into 5hmC DNA modification: generation, distribution and function. Front. Genet., 8, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gross J., Pacis A., Chen G., Drupals M., Lutz P., Barreiro L. and Turecki G. (2017) Gene-body 5-hydroxymethylation is associated with gene expression changes in the prefrontal cortex of depressed individuals. Transl. Psychiatry, 7, e1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang T., Pan Q., Lin L., Szulwach K.E., Song C.-X., He C., Wu H., Warren S.T., Jin P. and Duan R. (2012) Genome-wide DNA hydroxymethylation changes are associated with neurodevelopmental genes in the developing human cerebellum. Hum. Mol. Genet., 21, 5500–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cheng Y., Bernstein A., Chen D. and Jin P. (2015) 5-Hydroxymethylcytosine: a new player in brain disorders? Exp. Neurol., 268, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yao B., Lin L., Street R.C., Zalewski Z.A., Galloway J.N., Wu H., Nelson D.L. and Jin P. (2013) Genome-wide alteration of 5-hydroxymethylcytosine in a mouse model of fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet., 23, 1095–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shu L., Sun W., Li L., Xu Z., Lin L., Xie P., Shen H., Huang L., Xu Q. and Jin P. (2016) Genome-wide alteration of 5-hydroxymenthylcytosine in a mouse model of Alzheimer's disease. BMC Genomics, 17, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stöger R., Scaife P.J., Shephard F. and Chakrabarti L. (2017) Elevated 5hmC levels characterize DNA of the cerebellum in Parkinson's disease. NPJ Parkinsons Dis., 3, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Spiers H., Hannon E., Schalkwyk L.C., Bray N.J. and Mill J. (2017) 5-Hydroxymethylcytosine is highly dynamic across human fetal brain development. BMC Genomics, 18, 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sharif J., Muto M., Takebayashi S.-i., Suetake I., Iwamatsu A., Endo T.A., Shinga J., Mizutani-Koseki Y., Toyoda T. and Okamura K. (2007) The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature, 450, 908–912. [DOI] [PubMed] [Google Scholar]
- 71. Zhang G., Huang H., Liu D., Cheng Y., Liu X., Zhang W., Yin R., Zhang D., Zhang P. and Liu J. (2015) N6-methyladenine DNA modification in Drosophila. Cell, 161, 893–906. [DOI] [PubMed] [Google Scholar]
- 72. Koziol M.J., Bradshaw C.R., Allen G.E., Costa A.S., Frezza C. and Gurdon J.B. (2016) Identification of methylated deoxyadenosines in vertebrates reveals diversity in DNA modifications. Nat. Struct. Mol. Biol., 23, 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu J., Zhu Y., Luo G.-Z., Wang X., Yue Y., Wang X., Zong X., Chen K., Yin H. and Fu Y. (2016) Abundant DNA 6mA methylation during early embryogenesis of zebrafish and pig. Nat. Commun., 7, 13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wu T.P., Wang T., Seetin M.G., Lai Y., Zhu S., Lin K., Liu Y., Byrum S.D., Mackintosh S.G. and Zhong M. (2016) DNA methylation on N 6-adenine in mammalian embryonic stem cells. Nature, 532, 329–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yao B., Cheng Y., Wang Z., Li Y., Chen L., Huang L., Zhang W., Chen D., Wu H. and Tang B. (2017) DNA N6-methyladenine is dynamically regulated in the mouse brain following environmental stress. Nat. Commun., 8, 1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xiao C.-L., Zhu S., He M., Chen D., Zhang Q., Chen Y., Yu G., Liu J., Xie S.-Q. and Luo F. (2018) N6-methyladenine DNA modification in the human genome. Mol. Cell, 71, 306–318. [DOI] [PubMed] [Google Scholar]
- 77. Timinskas A., Butkus V. and Janulaitis A. (1995) Sequence motifs characteristic for DNA [cytosine-N4] and DNA [adenine-N6] methyltransferases. Classification of all DNA methyltransferases. Gene, 157, 3–11. [DOI] [PubMed] [Google Scholar]
- 78. Kweon S.-M., Chen Y., Moon E., Kvederaviciutė K., Klimasauskas S. and Feldman D.E. (2019) An adversarial DNA N6-methyladenine-sensor network preserves polycomb silencing. Mol. Cell, 74, 1138–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang W., Xu L., Hu L., Chong J., He C. and Wang D. (2017) Epigenetic DNA modification N 6-methyladenine causes site-specific RNA polymerase II transcriptional pausing. J. Am. Chem. Soc., 139, 14436–14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. He S., Zhang G., Wang J., Gao Y., Sun R., Cao Z., Chen Z., Zheng X., Yuan J. and Luo Y. (2019) 6mA-DNA-binding factor Jumu controls maternal-to-zygotic transition upstream of Zelda. Nat. Commun., 10, 2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ismail J., Badini S., Frey F., Abou-Kheir W. and Shirinian M. (2019) Drosophila Tet is expressed in midline glia and is required for proper axonal development. Front. Cell. Neurosci., 13, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wang F., Minakhina S., Tran H., Changela N., Kramer J. and Steward R. (2018) Tet protein function during Drosophila development. PLoS One, 13, e0190367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Delatte B., Wang F., Ngoc L.V., Collignon E., Bonvin E., Deplus R., Calonne E., Hassabi B., Putmans P. and Awe S. (2016) Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science, 351, 282–285. [DOI] [PubMed] [Google Scholar]
- 84. Shah K., Cao W. and Ellison C.E. (2019) Adenine methylation in Drosophila is associated with the tissue-specific expression of developmental and regulatory genes. G3, 9, 1893–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. O'Brown Z.K., Boulias K., Wang J., Wang S.Y., O'Brown N.M., Hao Z., Shibuya H., Fady P.-E., Shi Y. and He C. (2019) Sources of artifact in measurements of 6mA and 4mC abundance in eukaryotic genomic DNA. BMC Genomics, 20, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Alderman M.H. and Xiao A.Z. (2019) N (6)-Methyladenine in eukaryotes. Cell. Mol. Life Sci., 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Tanzi R.E. (2012) The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med., 2, a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Frigerio C.S., Wolfs L., Fattorelli N., Thrupp N., Voytyuk I., Schmidt I., Mancuso R., Chen W.-T., Woodbury M.E. and Srivastava G. (2019) The major risk factors for Alzheimer's disease: age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep., 27, 1293–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. De Jager P.L., Srivastava G., Lunnon K., Burgess J., Schalkwyk L.C., Yu L., Eaton M.L., Keenan B.T., Ernst J. and McCabe C. (2014) Alzheimer's disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci., 17, 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yokoyama A.S., Rutledge J.C. and Medici V. (2017) DNA methylation alterations in Alzheimer's disease. Environ. Epigenet., 3, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bernstein A.I., Lin Y., Street R.C., Lin L., Dai Q., Yu L., Bao H., Gearing M., Lah J.J. and Nelson P.T. (2016) 5-Hydroxymethylation-associated epigenetic modifiers of Alzheimer's disease modulate Tau-induced neurotoxicity. Hum. Mol. Genet., 25, 2437–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Keogh M.J., Wei W., Aryaman J., Walker L., van den Ameele J., Coxhead J., Wilson I., Bashton M., Beck J. and West J. (2018) High prevalence of focal and multi-focal somatic genetic variants in the human brain. Nat. Commun., 9, 4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Oliveira A.M., Hemstedt T.J. and Bading H. (2012) Rescue of aging-associated decline in Dnmt3a2 expression restores cognitive abilities. Nat. Neurosci., 15, 1111–1113. [DOI] [PubMed] [Google Scholar]
- 94. Oliveira A.M. (2016) DNA methylation: a permissive mark in memory formation and maintenance. Learn. Mem., 23, 587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Miller C.A., Gavin C.F., White J.A., Parrish R.R., Honasoge A., Yancey C.R., Rivera I.M., Rubio M.D., Rumbaugh G. and Sweatt J.D. (2010) Cortical DNA methylation maintains remote memory. Nat. Neurosci., 13, 664–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Gontier G., Iyer M., Shea J.M., Bieri G., Wheatley E.G., Ramalho-Santos M. and Villeda S.A. (2018) Tet2 rescues age-related regenerative decline and enhances cognitive function in the adult mouse brain. Cell Rep., 22, 1974–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Nativio R., Donahue G., Berson A., Lan Y., Amlie-Wolf A., Tuzer F., Toledo J.B., Gosai S.J., Gregory B.D. and Torres C. (2018) Dysregulation of the epigenetic landscape of normal aging in Alzheimer's disease. Nat. Neurosci., 21, 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gasparoni G., Bultmann S., Lutsik P., Kraus T.F., Sordon S., Vlcek J., Dietinger V., Steinmaurer M., Haider M. and Mulholland C.B. (2018) DNA methylation analysis on purified neurons and glia dissects age and Alzheimer's disease-specific changes in the human cortex. Epigenetics Chromatin, 11, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Trinh J. and Farrer M. (2013) Advances in the genetics of Parkinson disease. Nat. Rev. Neurol., 9, 445–454. [DOI] [PubMed] [Google Scholar]
- 100. Jowaed A., Schmitt I., Kaut O. and Wüllner U. (2010) Methylation regulates alpha-synuclein expression and is decreased in Parkinson's disease patients' brains. J. Neurosci. Res., 30, 6355–6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Matsumoto L., Takuma H., Tamaoka A., Kurisaki H., Date H., Tsuji S. and Iwata A. (2010) CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson's disease. PLoS One, 5, e15522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Desplats P., Spencer B., Coffee E., Patel P., Michael S., Patrick C., Adame A., Rockenstein E. and Masliah E. (2011) α-Synuclein sequesters Dnmt1 from the nucleus a novel mechanism for epigenetic alterations in lewy body diseases. J. Biol. Chem., 286, 9031–9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Tan Y.-y., Wu L., Zhao Z.-b., Wang Y., Xiao Q., Liu J., Wang G. and Ma J.-f. (2014) Methylation of α-synuclein and leucine-rich repeat kinase 2 in leukocyte DNA of Parkinson's disease patients. Parkinsonism Relat. Disord., 20, 308–313. [DOI] [PubMed] [Google Scholar]
- 104. Moore K., McKnight A.J., Craig D. and O'Neill F. (2014) Epigenome-wide association study for Parkinson's disease. Neuromolecular Med., 16, 845–855. [DOI] [PubMed] [Google Scholar]
- 105. Chuang Y.-H., Paul K.C., Bronstein J.M., Bordelon Y., Horvath S. and Ritz B. (2017) Parkinson's disease is associated with DNA methylation levels in human blood and saliva. Genome Med., 9, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kaut O., Kuchelmeister K., Moehl C. and Wüllner U. (2019) 5-Methylcytosine and 5-hydroxymethylcytosine in brains of patients with multiple system atrophy and patients with Parkinson's disease. J. Chem. Neuroanat., 96, 41–48. [DOI] [PubMed] [Google Scholar]
- 107. Cossée M., Schmitt M., Campuzano V., Reutenauer L., Moutou C., Mandel J.-L. and Koenig M. (1997) Evolution of the Friedreich's ataxia trinucleotide repeat expansion: founder effect and premutations. Proc. Natl. Acad. Sci. U. S. A., 94, 7452–7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Campuzano V., Montermini L., Moltò M.D., Pianese L., Cossée M., Cavalcanti F., Monros E., Rodius F., Duclos F. and Monticelli A. (1996) Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science, 271, 1423–1427. [DOI] [PubMed] [Google Scholar]
- 109. Greene E., Mahishi L., Entezam A., Kumari D. and Usdin K. (2007) Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res., 35, 3383–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Al-Mahdawi S., Sandi C., Pinto R.M. and Pook M.A. (2013) Friedreich ataxia patient tissues exhibit increased 5-hydroxymethylcytosine modification and decreased CTCF binding at the FXN locus. PLoS One, 8, e74956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Castaldo I., Pinelli M., Monticelli A., Acquaviva F., Giacchetti M., Filla A., Sacchetti S., Keller S., Avvedimento V.E. and Chiariotti L. (2008) DNA methylation in intron 1 of the frataxin gene is related to GAA repeat length and age of onset in Friedreich ataxia patients. J. Med. Genet., 45, 808–812. [DOI] [PubMed] [Google Scholar]
- 112. Evans-Galea M.V., Carrodus N., Rowley S.M., Corben L.A., Tai G., Saffery R., Galati J.C., Wong N.C., Craig J.M. and Lynch D.R. (2012) FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann. Neurol., 71, 487–497. [DOI] [PubMed] [Google Scholar]
- 113. Koeppen A.H. (2005) The pathogenesis of spinocerebellar ataxia. Cerebellum, 4, 62–73. [DOI] [PubMed] [Google Scholar]
- 114. Klockgether T., Mariotti C. and Paulson H.L. (2019) Spinocerebellar ataxia. Nat. Rev. Dis. Primers., 5, 24. [DOI] [PubMed] [Google Scholar]
- 115. Evans-Galea M.V., Hannan A.J., Carrodus N., Delatycki M.B. and Saffery R. (2013) Epigenetic modifications in trinucleotide repeat diseases. Trends Mol. Med., 19, 655–663. [DOI] [PubMed] [Google Scholar]
- 116. Libby R.T., Hagerman K.A., Pineda V.V., Lau R., Cho D.H., Baccam S.L., Axford M.M., Cleary J.D., Moore J.M. and Sopher B.L. (2008) CTCF cis-regulates trinucleotide repeat instability in an epigenetic manner: a novel basis for mutational hot spot determination. PLoS Genet., 4, e1000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Emmel V., Alonso I., Jardim L., Saraiva-Pereira M. and Sequeiros J. (2011) Does DNA methylation in the promoter region of the ATXN3 gene modify age at onset in MJD (SCA3) patients? Clin. Genet., 79, 100–102. [DOI] [PubMed] [Google Scholar]
- 118. Wang C., Peng H., Li J., Ding D., Chen Z., Long Z., Peng Y., Zhou X., Ye W. and Li K. (2017) Alteration of methylation status in the ATXN3 gene promoter region is linked to the SCA3/MJD. Neurobiol. Aging, 53, 192. e195–192. e192. [DOI] [PubMed] [Google Scholar]
- 119. Laffita-Mesa J.M., Velázquez-Pérez L.C., Falcón N.S., Cruz-Marino T., Zaldívar Y.G., Mojena Y.V., Almaguer-Gotay D., Mederos L.E.A. and Labrada R.R. (2012) Unexpanded and intermediate CAG polymorphisms at the SCA2 locus (ATXN2) in the Cuban population: evidence about the origin of expanded SCA2 alleles. Eur. J. Hum. Genet., 20, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hamzeiy H., Savaş D., Tunca C., Şen N.E., Eken A.G., Şahbaz I., Calini D., Tiloca C., Ticozzi N. and Ratti A. (2018) Elevated global DNA methylation is not exclusive to amyotrophic lateral sclerosis and is also observed in spinocerebellar ataxia types 1 and 2. Neurodegener Dis, 18, 38–48. [DOI] [PubMed] [Google Scholar]
- 121. Jacquemont S., Hagerman R.J., Leehey M., Grigsby J., Zhang L., Brunberg J.A., Greco C., Des Portes V., Jardini T. and Levine R. (2003) Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am. J. Hum. Genet., 72, 869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Berry-Kravis E., Abrams L., Coffey S.M., Hall D.A., Greco C., Gane L.W., Grigsby J., Bourgeois J.A., Finucane B. and Jacquemont S. (2007) Fragile X-associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines. Mov. Disord., 22, 2018–2030. [DOI] [PubMed] [Google Scholar]
- 123. Biancalana V., Glaeser D., McQuaid S. and Steinbach P. (2015) EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders. Eur. J. Hum. Genet., 23, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Pieretti M., Zhang F., Fu Y.-H., Warren S.T., Oostra B.A., Caskey C.T. and Nelson D.L. (1991) Absence of expression of the FMR-1 gene in fragile X syndrome. Cell, 66, 817–822. [DOI] [PubMed] [Google Scholar]
- 125. Hashem V., Galloway J.N., Mori M., Willemsen R., Oostra B.A., Paylor R. and Nelson D.L. (2009) Ectopic expression of CGG containing mRNA is neurotoxic in mammals. Hum. Mol. Genet., 18, 2443–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Pfeifer G. (2018) Defining driver DNA methylation changes in human cancer. Int. J. Mol. Sci., 19, 1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Li X., Yao B., Chen L., Kang Y., Li Y., Cheng Y., Li L., Lin L., Wang Z. and Wang M. (2017) Ten-eleven translocation 2 interacts with forkhead box O3 and regulates adult neurogenesis. Nat. Commun., 8, 15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ehrlich M., Gama-Sosa M.A., Huang L.-H., Midgett R.M., Kuo K.C., McCune R.A. and Gehrke C. (1982) Amount and distribution of 5-methylcytosine in human DNA from different types of tissues or cells. Nucleic Acids Res., 10, 2709–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Nestor C.E., Ottaviano R., Reddington J., Sproul D., Reinhardt D., Dunican D., Katz E., Dixon J.M., Harrison D.J. and Meehan R.R. (2012) Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res., 22, 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Li W. and Liu M. (2011) Distribution of 5-hydroxymethylcytosine in different human tissues. J. Nucleic Acids., 2011, 870726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Meadows J.P., Guzman-Karlsson M.C., Phillips S., Holleman C., Posey J.L., Day J.J., Hablitz J.J. and Sweatt J.D. (2015) DNA methylation regulates neuronal glutamatergic synaptic scaling. Sci. Signal., 8, ra61–ra61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kozlenkov A., Li J., Apontes P., Hurd Y.L., Byne W.M., Koonin E.V., Wegner M., Mukamel E.A. and Dracheva S. (2018) A unique role for DNA (hydroxy) methylation in epigenetic regulation of human inhibitory neurons. Sci. Adv., 4, eaau6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Luo C., Keown C.L., Kurihara L., Zhou J., He Y., Li J., Castanon R., Lucero J., Nery J.R. and Sandoval J.P. (2017) Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science, 357, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Titus A.J., Gallimore R.M., Salas L.A. and Christensen B.C. (2017) Cell-type deconvolution from DNA methylation: a review of recent applications. Hum. Mol. Genet., 26, R216–R224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Duclot F. and Kabbaj M. (2017) The role of early growth response 1 (EGR1) in brain plasticity and neuropsychiatric disorders. Front Behav. Neurosci., 11, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ambu R., Vinci L., Gerosa C., Fanni D., Obinu E., Faa A. and Fanos V. (2015) WT1 expression in the human fetus during development. Eur. J. Histochem., 59, 2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Hashimoto H., Olanrewaju Y.O., Zheng Y., Wilson G.G., Zhang X. and Cheng X. (2014) Wilms tumor protein recognizes 5-carboxylcytosine within a specific DNA sequence. Genes Dev., 28, 2304–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Rass U., Ahel I. and West S.C. (2007) Defective DNA repair and neurodegenerative disease. Cell, 130, 991–1004. [DOI] [PubMed] [Google Scholar]
- 139. Coppedè F. and Migliore L. (2010) DNA repair in premature aging disorders and neurodegeneration. Curr. Aging Sci., 3, 3–19. [DOI] [PubMed] [Google Scholar]
- 140. Madabhushi R., Pan L. and Tsai L.-H. (2014) DNA damage and its links to neurodegeneration. Neuron, 83, 266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Bernstein C. and Bernstein H. (2019), The Role of DNA Repair and the Epigenetic Markers Left after Repair in Neurologic Functions, Including Memory and Learning. In Advances in DNA Repair. IntechOpen.
- 142. Lodato M.A., Rodin R.E., Bohrson C.L., Coulter M.E., Barton A.R., Kwon M., Sherman M.A., Vitzthum C.M., Luquette L.J. and Yandava C.N. (2018) Aging and neurodegeneration are associated with increased mutations in single human neurons. Science, 359, 555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Kafer G.R., Li X., Horii T., Suetake I., Tajima S., Hatada I. and Carlton P.M. (2016) 5-Hydroxymethylcytosine marks sites of DNA damage and promotes genome stability. Cell Rep., 14, 1283–1292. [DOI] [PubMed] [Google Scholar]
- 144. Coulter J.B., Lopez-Bertoni H., Kuhns K.J., Lee R.S., Laterra J. and Bressler J.P. (2017) TET1 deficiency attenuates the DNA damage response and promotes resistance to DNA damaging agents. Epigenetics, 12, 854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Jiang D., Zhang Y., Hart R.P., Chen J., Herrup K. and Li J. (2015) Alteration in 5-hydroxymethylcytosine-mediated epigenetic regulation leads to Purkinje cell vulnerability in ATM deficiency. Brain, 138, 3520–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Jiang D., Wei S., Chen F., Zhang Y. and Li J. (2017) TET3-mediated DNA oxidation promotes ATR-dependent DNA damage response. EMBO Rep., 18, 781–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. D'Gama A.M. and Walsh C.A. (2018) Somatic mosaicism and neurodevelopmental disease. Nat. Neurosci., 21, 1504–1514. [DOI] [PubMed] [Google Scholar]