

Abstract
Abstract
Two hitherto unknown iboga-type indole alkaloids, namely (3R)-7,19-di-epi-3-methoxytabernoxidine (1) and (3R,19R)-19-hydroxy-3-(2-oxopropyl)voacangine (2), together with eight known alkaloids (3–10), were isolated from the twigs and leaves of Tabernaemontana divaricata. Their structures were established on the basis of spectroscopic data interpretation, single crystal X-ray diffraction analysis and circular dichroism spectrum.
Graphic Abstract

Electronic supplementary material
The online version of this article (10.1007/s13659-019-00226-z) contains supplementary material, which is available to authorized users.
Keywords: Tabernaemontana divaricata, Iboga-type indole alkaloid, Single crystal X-ray diffraction
Introduction
The genus Tabernaemontana (Apocynaceae family), comprising 120 species, is a rich source of monoterpenoid indole alkaloids with diverse skeletons, particularly iboga-type alkaloids [1–3]. Tabernaemontana divaricata, distributed in Yunnan and Guangxi Provinces of China [4], is widely used as folk medicine for the treatment of sore throat and hypertension [5], and a series of monoterpenoid indole and bisindole alkaloids were reported [6, 7]. As part of a BioBioPha (https://www.chemlib.cn) objective to assemble a large-scale natural product library valuable in the discovery of new drug leads from nature, the phytochemical investigation on the twigs and leaves of T. divaricata resulted in the isolation of two new iboga-type indole alkaloids, namely (3R)-7,19-di-epi-3-methoxytabernoxidine (1) and (3R,19R)-19-hydroxy-3-(2-oxopropyl)voacangine (2) (Fig. 1), together with eight known indole alkaloids: 19,20-(E)-vallesamine (3) [8, 9], ibogaine (4) [10], conophylline (5) [11], taberdivarine H (6) [12], conodurine (7) [13], hecubine (8) [14], voafinidine (9) [15], and tabernanthine (10) [10, 16]. In this paper, we report the isolation and structure elucidation of new alkaloids 1 and 2.
Fig. 1.

The structures of compounds 1, 2 and tabernoxidine
Results and Discussion
Compound 1, obtained as colorless block crystals from MeOH, possessed a molecular formula of C23H30N2O6, as evidenced by HR-ESI-MS (pos.) at m/z 453.1997 [M + Na]+ (calcd. for C23H30N2O6Na, 453.1996), requiring 10 degrees of unsaturation. The 1H NMR spectrum in chloroform-d (Table 1) displayed one NH proton at δH 7.90 (1H, s), one 1,2,4-trisubstituted benzene ring at δH 7.63 (1H, d, J = 8.5 Hz), 6.54 (1H, dd, J = 8.5, 2.6 Hz), and 6.43 (1H, d, J = 2.6 Hz), two oxygenated methines at δH 4.18 (1H, d, J = 3.3 Hz) and 3.95 (1H, m), one secondary methyl at δH 1.24 (3H, d, J = 6.4 Hz), and three methoxy groups at δH 3.78, 3.33, and 3.19 (each 3H, s). The 13C NMR spectrum (Table 1) displayed a total of 23 carbon resonances, including two overlapping signals (C-5 and C-16). Combined with the DEPT spectrum, different types of carbons were distinguished to be four methyls, four methylenes, eight methines and seven quaternary carbons (including two carbonyls at δC 179.2 and 174.3). The HSQC and 1H-1H COSY spectra (Fig. 2) revealed the connections of C-3/C-14 and C-17/C-14/C-15/C-20/C-19/C-18, which were confirmed by the HMBC correlations (Fig. 2) from H-3 to C-15 and C-17, H-15 to C-17 and C-19, and Me-18 to C-19 and C-20. The above NMR analysis indicated the planar structure of 1 was remarkably similar to that of tabernoxidine (Fig. 1) [17], an iboga-type spiro oxindole alkaloid whose absolute stereochemistry has been established by single crystal X-ray diffraction. The prominent difference between them was the absence of resonance for a methylene (C-3) and the presence of resonances for a downfield oxygenated methine and a methoxy. The oxygenated methine was assigned to C-3 based on the HMBC correlations from H-21 to C-3, and from H-3 to C-5, C-15, and C-17. And the methoxy was positioned at C-3 by the HMBC correlation between the methoxy protons and C-3. In the ROESY spectrum (Fig. 3), the correlations between H-9 and H-21/H-5α revealed R* configuration of the spiro carbon atom at C-7, and the correlations between H-3 and H-5β/H-6β/H-17β indicated R* configuration of C-3. The above deduction was confirmed by single crystal X-ray diffraction using Mo Kα radiation (Fig. 4). Since the absolute stereochemistry of iboga-type indole alkaloids from Apocynaceae had been determined previously by single crystal X-ray diffraction using Cu Kα radiation or electronic circular dichroism (ECD) analysis [1–3, 17], biogenetically, the absolute configuration of 1 could be deduced as 3R,7R,14R,16R,19R,20S,21S. Therefore, the structure of 1 was established and named as (3R)-7,19-di-epi-3-methoxytabernoxidine, as shown in Fig. 1.
Table 1.
1H and 13C NMR spectroscopic data of compounds 1 and 2 in CDCl3 (δH 7.26, δC 77.0 ppm)
| No. | 1 | 2 | ||
|---|---|---|---|---|
| δH | δC | δH | δC | |
| 1 | 7.90 (s) | 7.70 (s) | ||
| 2 | 179.2 (s) | 136.3 (s) | ||
| 3 | 4.18 (d, 3.3) | 91.5 (d) | 3.38 (dd, 8.6, 4.0) | 54.2 (d) |
| 5 |
3.46 (ddd, 14.6, 14.2, 3.5, Hα) 3.05 (br dd, 14.6, 5.1, Hβ) |
48.7 (t) |
3.34 (m, Hα) 3.15 (m, Hβ) |
50.4 (t) |
| 6 |
2.39 (td, 14.2, 5.1, Hβ) 1.28 (br dd, 14.2, 3.5, Hα) |
26.2 (t) |
3.14 (m, Hβ) 3.04 (m, Hα) |
21.7 (t) |
| 7 | 50.6 (s) | 109.5 (s) | ||
| 8 | 123.6 (s) | 128.9 (s) | ||
| 9 | 7.63 (d, 8.5) | 126.9 (d) | 6.89 (d, 2.4) | 100.7 (d) |
| 10 | 6.54 (dd, 8.5, 2.6) | 106.5 (d) | 154.1 (s) | |
| 11 | 160.0 (s) | 6.83 (dd, 8.7, 2.4) | 112.4 (d) | |
| 12 | 6.43 (d, 2.6) | 97.0 (d) | 7.15 (d, 8.7) | 111.2 (d) |
| 13 | 140.7 (s) | 130.6 (s) | ||
| 14 | 2.16 (m) | 30.1 (d) | 1.86 (br s) | 30.5 (d) |
| 15 |
1.64 (m, Hβ) 1.32 (m, Hα) |
21.4 (t) |
1.90 (m, Hβ) 1.63 (m, Hα) |
24.0 (t) |
| 16 | 48.7 (s) | 53.7 (s) | ||
| 17 |
3.18 (m, Hβ) 1.96 (dd, 15.8, 2.9, Hα) |
27.2 (t) |
2.63 (dd, 13.8, 1.8, Hα) 2.09 (ddd, 13.8, 4.0, 3.0, Hβ) |
37.7 (t) |
| 18 | 1.24 (d, 6.4) | 21.9 (q) | 1.28 (d, 6.5) | 22.2 (q) |
| 19 | 3.95 (m) | 70.2 (d) | 3.89 (qd, 6.5, 2.0) | 71.0 (d) |
| 20 | 1.84 (m) | 39.2 (d) | 1.35 (m) | 39.7 (d) |
| 21 | 3.95 (br s) | 50.4 (d) | 4.10 (s) | 55.2 (d) |
| 22 | 174.3 (s) | 174.7 (s) | ||
| 3-OMe | 3.33 (s) | 53.7 (q) | ||
| 10-OMe | 3.84 (s) | 56.0 (q) | ||
| 11-OMe | 3.78 (s) | 55.4 (q) | ||
| 22-OMe | 3.19 (s) | 51.4 (q) | 3.72 (s) | 52.9 (q) |
| 1′ |
2.77 (dd, 17.3, 8.6) 2.69 (dd, 17.3, 4.0) |
45.1 (t) | ||
| 2′ | 207.6 (s) | |||
| 3′ | 2.13 (s) | 31.0 (q) | ||
Fig. 2.

Key 1H–1H COSY (
 ) and HMBC (
) and HMBC (
 ) correlations of 1 and 2
) correlations of 1 and 2
Fig. 3.

Key ROESY (
 ) correlations of 1 and 2
) correlations of 1 and 2
Fig. 4.
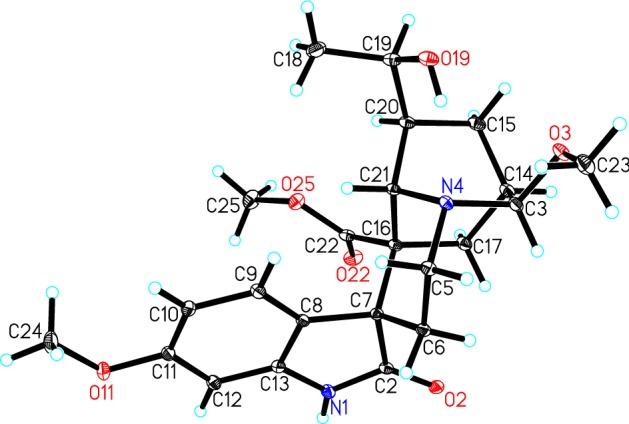
X-ray crystallographic structure of compound 1
Compound 2, white amorphous powder, gave a molecular formula of C25H32N2O5 as determined by positive HRESIMS at m/z 441.2384 [M + H]+ (calcd. for C25H33N2O5, 441.2384) with 11 degrees of unsaturation. The 1H and 13C NMR spectra (Table 1) showed that 2 had a structure similar to that of 3-(2-oxopropyl)voacangine [18], except for the presence of a 1-hydroxyethyl group [δH 3.89 (1H, qd, J = 6.5, 2.0 Hz)/δC 71.0 (d) and δH 1.28 (3H, d, J = 6.5 Hz)/δC 22.2 (q)] in place of an ethyl side chain at C-20, which was confirmed by the HMBC correlations (Fig. 2) from Me-18 to C-19 and C-20. The ROESY correlations (Fig. 3) of H-21 ↔ H-5α/H-6α, H-17α ↔ H-20/H-15α suggested that these protons were α-oriented, and the correlations of H-3 ↔ H-17β, H-1′ ↔ H-15β revealed the stereochemistry of C-3, which indicated that the configurations of C-3, C-14, C-16, C-20 and C-21 were identical to those in 3-(2-oxopropyl)voacangine. Comparison of the 13C NMR chemical shifts of C-15 and C-21 as well as the 1H NMR chemical shift of H-19 with those of 19R/S-hydroxyiboga alkaloids indicated a 19R configuration [19, 20]. Additionally, the Cotton effects in the CD spectrum (See S14, Supplementary Material) of 2 were in good agreement with those for voacangine [18], so the absolute configuration of 2 was assigned as 3R,14R,16S,19R,20S,21S. Hence, the structure of 2 was established and named as (3R,19R)-19-hydroxy-3-(2-oxopropyl)voacangine.
Experimental Section
General Experimental Procedures
X-ray data were collected using a Bruker APEX DUO diffractometer with graphite-monochromated Mo Kα radiation. Optical rotations were measured on a Rudolph Autopol VI automatic polarimeter. UV data were obtained from HPLC online analysis. IR spectra (KBr) were obtained on a Thermo Nicolet iS10 FT-IR spectrometer. NMR spectra were carried out on a Bruker Avance III 600 or DRX-500 spectrometer with deuterated solvent signals as internal standards. ESIMS and HRESIMS were measured using an Agilent G6230 time-of-flight mass spectrometer. Preparative HPLC separation was performed using an Agilent 1260 series HPLC system equipped with a Zorbax SB-C18 column (5 μm, 21.2 × 150 mm). Silica gel (200–300 mesh, Qingdao Marine Chemical Inc., China), Chromatorex C18 (40–75 μm, Fuji Silysia Chemical Ltd., Japan) and Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) were used for column chromatography. Fractions were monitored and analyzed using TLC, in combination with Agilent 1200 series HPLC system equipped by an Extend-C18 column (5 μm, 4.6 × 150 mm).
Plant Material
The twigs and leaves of Tabernaemontana divaricata were collected from Menglun town, Xishuangbanna Dai Autonomous Prefecture in Yunnan Province, China, in January 2014 and identified by Mr. Yu Chen of Kunming Institute of Botany, Chinese Academy of Sciences. A voucher specimen (No. BBP0671) was deposited at BioBioPha Co., Ltd.
Extraction and Isolation
The twigs and leaves of T. divaricata were extracted with 95% aqueous EtOH at room temperature, and the solvent was removed under reduced pressure to give crude extract (ca. 600 g), then dissolved in 2% HCl and filtered. The filtration was basified using 10% ammonia-water to pH 9−10 and then partitioned with EtOAc to give a total alkaloidal extract (ca. 100 g), which was fractionated by silica gel CC successively eluted with CHCl3/MeOH (200:1 → 10:1 gradient) to give six fractions A –E, respectively. Fraction B was separated by silica gel CC (CHCl3/MeOH, 100:1 → 80:1) into fractions B1 –B4. Fraction B1 was purified by silica gel CC using petroleum ether (PE)/acetone (4:1) and Sephadex LH-20 (CHCl3/MeOH, 1:1) to obtain 7 (50 mg). Fraction B2 was separated by silica gel CC (CHCl3/MeOH, 100:1) to give 8 (18 mg) and 9 (4 mg). Fraction C was separated by silica gel (CHCl3/MeOH, 100:1 → 60:1) into fractions C1–C3. Fraction C1 was separated by Sephadex LH-20 (CHCl3/MeOH, 1:1) to obtain compound 5 (17 mg). Fraction C2 was separated by silica gel (CHCl3/MeOH, 60:1), prep-HPLC (MeOH/H2O; 60%) and Sephadex LH-20 (CHCl3/MeOH, 1:1) to obtain compounds 1 (46 mg), 4 (3 mg) and 10 (102 mg). Fraction D was separated by silica gel CC (CHCl3/MeOH, 60:1 → 30:1) into fractions D1–D4. Fraction D2 was further purified by silica gel (CHCl3/MeOH, 80:1) and prep-HPLC (MeOH/H2O; 80%) to give 2 (14 mg). Fraction E was subjected to MPLC (MeOH/H2O, 50%) and then by Sephadex LH-20 (MeOH) to give 3 (33 mg) and 6 (13 mg). The retention times (tR) of 1 and 2 on an analytical HPLC InertSustain C18 column (20% → 100% MeOH in H2O over 8.0 min followed by 100% MeOH to 13.0 min, 1.0 ml/min) were 4.60 and 6.00 min, respectively.
(3R)-7,19-Di-epi-3-methoxytabernoxidine (1)
Colorless block crystals; [α]22D + 25.9 (c 0.10, MeOH); UV (MeOH) λmax: 217, 261, 285 (sh) nm; IR (KBr) νmax: 3427, 2966, 2945, 2936, 2901, 2835, 1720, 1631, 1594, 1507, 1468, 1365, 1314, 1197, 1160, 1101, 829 cm−1; 1H and 13C NMR data: see Table 1; ESIMS (pos.): m/z 453 [M + Na]+; HRESIMS (pos.): m/z 453.1997 [M + Na]+ (calcd. for C23H30N2O6Na, 453.1996).
Crystallographic Data for 1
C23H30N2O6·CH4O, M = 462.53, monoclinic, a = 10.9955(11) Å, b = 6.7753(7) Å, c = 15.2342(15) Å, α = 90.00°, β = 92.975(2)°, γ = 90.00°, V = 1133.4(2) Å3, T = 100(2) K, space group P21, Z = 2, μ(MoKα) = 0.100 mm−1, 12217 reflections measured, 6280 independent reflections (Rint = 0.0225). The final R1 values were 0.0339 (I > 2σ(I)). The final wR(F2) values were 0.0866 (I > 2σ(I)). The final R1 values were 0.0385 (all data). The final wR(F2) values were 0.0893 (all data). The goodness of fit on F2 was 1.043. Flack parameter = 0.3(3).
(3R,19R)-19-Hydroxy-3-(2-oxopropyl)voacangine (2)
White amorphous powder; [α]22D − 39.3 (c 0.10, MeOH); UV (MeOH) λmax: 223, 287 nm; IR (KBr) νmax: 3442, 2934, 2871, 1711, 1627, 1487, 1455, 1225, 1166, 1077, 1033, 828, 798 cm−1; 1H and 13C NMR data: see Table 1; ESIMS (pos.): m/z 441 [M + H]+; HRESIMS (pos.): m/z 441.2384 [M + H]+ (calcd. for C25H33N2O5, 441.2384).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was financially supported by the “Large-scale Compound Library” project of National Development and Reform Commission of China.
Compliance with Ethical Standards
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Andrade MT, Lima JA, Pinto AC, Rezende CM, Carvalho MP, Epifanio RA. Bioorg. Med. Chem. 2005;13:4092–4095. doi: 10.1016/j.bmc.2005.03.045. [DOI] [PubMed] [Google Scholar]
- 2.Achenbach H, Benirschke M, Torrenegra R. Phytochemistry. 1997;45:325–335. doi: 10.1016/S0031-9422(96)00645-0. [DOI] [Google Scholar]
- 3.Van Beek TA, Verpoorte R, Svendsen AB, Fokkens R. J. Nat. Prod. 1985;48:400–423. doi: 10.1021/np50039a008. [DOI] [PubMed] [Google Scholar]
- 4.Editorial Committee of Flora Reipublicae Popularis Sinicae Flora Reipublicae Popularis Sinicae. Academic Press: Beijing. 1977;63:106. [Google Scholar]
- 5.Zhu ZX, Chen HS. J. Pharm. Pract. 2011;29:412–415. [Google Scholar]
- 6.Bao MF, Yan JM, Cheng GG, Li XY, Liu YP, Li Y, Cai XH, Dong XD. J. Nat. Prod. 2013;76:1406–1412. doi: 10.1021/np400130y. [DOI] [PubMed] [Google Scholar]
- 7.Kam TS, Pang HS, Choo YM, Komiyama K. Chem. Biodivers. 2004;1:646–656. doi: 10.1002/cbdv.200490056. [DOI] [PubMed] [Google Scholar]
- 8.Zeches M, Ravao T, Richard B, Massiot G, Le Men-Olivier L, Verpoorte R. J. Nat. Prod. 1987;50:714–720. doi: 10.1021/np50052a023. [DOI] [Google Scholar]
- 9.Atta-ur-Rahman, Alvi KA, Abbas SA, Voelter W. Heterocycles. 1987;26:413–419. doi: 10.3987/R-1987-02-0413. [DOI] [Google Scholar]
- 10.Clivio P, Richard B, Deverre JR, Sevenet T, Zeches M, Le Men-Oliver L. Phytochemistry. 1991;30:3785–3792. doi: 10.1016/0031-9422(91)80111-D. [DOI] [Google Scholar]
- 11.Kam TS, Loh KY, Wei C. J. Nat. Prod. 1993;56:1865–1871. doi: 10.1021/np50101a001. [DOI] [Google Scholar]
- 12.Zhang BJ, Teng XF, Bao MF, Zhong XH, Ni L, Cai XH. Phytochemistry. 2015;120:46–52. doi: 10.1016/j.phytochem.2014.12.025. [DOI] [PubMed] [Google Scholar]
- 13.Medeiros WLB, Vieira IJC, Mathias L, Braz-Filho R, Leal KZ, Rodrigues-Filho E, Schripsema J. Magn. Reson. Chem. 1999;37:676–681. doi: 10.1002/(SICI)1097-458X(199909)37:9<676::AID-MRC513>3.0.CO;2-F. [DOI] [Google Scholar]
- 14.Éles J, Kalaus G, Greiner I, Kajtár-Peredy M, Szabó P, Keserû GM, Szabó L, Szántay C. J. Org. Chem. 2002;67:7255–7260. doi: 10.1021/jo020386r. [DOI] [PubMed] [Google Scholar]
- 15.Kam TS, Anuradha S, Loh KY. Nat. Prod. Lett. 1996;8:49–53. doi: 10.1080/10575639608043239. [DOI] [Google Scholar]
- 16.Chaturvedula VSP, Sprague S, Schilling JK, Kingston DGI. J. Nat. Prod. 2003;66:528–531. doi: 10.1021/np020548e. [DOI] [PubMed] [Google Scholar]
- 17.Joshi BS, Rao PG, Rogers D, Singri BP, Williams DJ. Indian J. Chem. 1984;23B:101–102. [Google Scholar]
- 18.Okuyama E, Gao LH, Yamazaki M. Chem. Pharm. Bull. 1992;40:2075–2079. doi: 10.1248/cpb.40.2075. [DOI] [PubMed] [Google Scholar]
- 19.Perera P, Sandberg F, van Beek TA, Verpoorte R. Phytochemistry. 1985;24:2097–2104. doi: 10.1016/S0031-9422(00)83130-1. [DOI] [Google Scholar]
- 20.Kam TS, Sim KM. J. Nat. Prod. 2002;65:669–672. doi: 10.1021/np0105432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.