

Visual Abstract

Key Words: contraction and relaxation, endothelial cell–derived nitric oxide, empagliflozin, heart failure, oxidative stress
Abbreviations and Acronyms: Ca, calcium; CM, cardiomyocyte; CMEC, cardiac microvascular endothelial cell; DPPH, 1,1-diphenyl-picrylhydrazyl; DM, diabetes mellitus; EC, endothelial cell; eNOS, endothelial nitric oxide synthase; HF, heart failure; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; JNK, Jun N-terminal kinase; L-NAME, N(ω)-nitro-L-arginine methyl ester; LV, left ventricular; NK-κB, nuclear factor-κB; NO, nitric oxide; ROS, reactive oxygen species; SGLT2, sodium glucose transporter 2
Highlights
-
•
CMECs exert a direct positive effect on cardiomyocyte contraction and relaxation, which is mainly mediated by endothelial-derived NO.
-
•
Pro-inflammatory stimulation of CMECs by pre-incubation with TNF-α or interleukin-1β abrogates the positive regulatory function of these cells on cardiomyocyte contractile property.
-
•
Mechanistically, pro-inflammatory activation of CMECs leads to mitochondrial and cytoplasmic ROS accumulation that results in the scavenging of NO.
-
•
Empagliflozin directly restores the beneficial effect of CMECs on cardiomyocyte contraction and relaxation by reducing TNF-α-induced mitochondrial and cytoplasmic ROS accumulation, which leads to reinstatement of CMEC-derived NO delivery.
Summary
The positive findings of the EMPA-REG OUTCOME trial (Randomized, Placebo-Controlled Cardiovascular Outcome Trial of Empagliflozin) on heart failure (HF) outcome in patients with type 2 diabetes mellitus suggest a direct effect of empagliflozin on the heart. These patients frequently have HF with preserved ejection fraction (HFpEF), in which a metabolic risk-related pro-inflammatory state induces cardiac microvascular endothelial cell (CMEC) dysfunction with subsequent cardiomyocyte (CM) contractility impairment. This study showed that CMECs confer a direct positive effect on contraction and relaxation of CMs, an effect that requires nitric oxide, is diminished after CMEC stimulation with tumor necrosis factor-α, and is restored by empagliflozin. Our findings on the effect of empagliflozin on CMEC-mediated preservation of CM function suggests that empagliflozin can be used to treat the cardiac mechanical implications of microvascular dysfunction in HFpEF.
Heart failure with preserved ejection fraction (HFpEF) accounts for 50% of patients with heart failure (HF) (1) and lacks an effective treatment. Therapies that are used in HF patients with reduced EF (HFrEF) have failed to improve primary outcomes in patients with HFpEF 2, 3, 4, 5, which suggests different patho-mechanisms in HFpEF compared with HFrEF. Recently, the paradigm of HFpEF has shifted from a mere cardiomyocyte (CM) disease to a disorder that initially involves cardiac microvascular endothelial cells (CMECs), which subsequently leads to CM dysfunction 6, 7. Comorbidities, such as type 2 diabetes mellitus (DM), obesity, hypertension, and chronic kidney disease, are highly prevalent in patients with HFpEF 8, 9, 10. These metabolic diseases are accompanied by microvascular endothelial dysfunction, including that of CMECs, which are characterized by impaired nitric oxide (NO) generation, increased reactive oxygen species (ROS) production, inflammatory activation 7, 11, 12, 13, and rarefaction of the cardiac microvascular bed (14). Recent findings in patients with breast cancer showed that the relative risk of HFpEF increased with increasing cardiac radiation exposure during breast cancer radiotherapy 15, 16. In adults, CMs do not or rarely proliferate and thus are highly radioresistant 17, 18, 19. Cardiac radiation exposure causes coronary microvascular endothelial cell (EC) damage and inflammation with subsequent coronary microvascular dysfunction and rarefaction that impair myocardial function 19, 20, 21. These studies provide proof of concept as they underpin the essential role of the cardiac microvascular endothelium in determining risk of left ventricular (LV) diastolic dysfunction. Interaction between endocardial ECs and CMs has been shown in earlier studies on cardiac papillary muscles 22, 23. However, a direct causal effect of cardiac microvascular endothelial dysfunction on cardiac contraction and relaxation, which has been proposed based on clinical associations 11, 24, needs to be fundamentally established. If the CMEC-CM axis plays an important role in the pathogenesis of HFpEF, improvement of CMEC function may represent an important target in developing new treatments and prevention strategies for HFpEF.
Empagliflozin, a sodium glucose transporter 2 (SGLT2) inhibitor that is primarily used in patients with type 2 DM to lower blood glucose levels, may represent a novel therapy to treat HFpEF patients. Recent findings of the EMPA-REG OUTCOME trial (Randomized, Placebo-Controlled Cardiovascular Outcome Trial of Empagliflozin) showed an unexpected beneficial effect of empagliflozin on HF outcome in patients with DM and suggested that empagliflozin acts not only on kidney tubular cells but also directly on the heart (25). The significant reduction of cardiovascular mortality and HF hospitalization by empagliflozin treatment 19, 25 indicated that empagliflozin could be useful as treatment of HFpEF because metabolically compromised patients, such as patients with DM, frequently have HFpEF. This suggestion is currently being investigated in a large clinical trial (EMPEROR-Preserved [Empagliflozin Outcome Trial in Patients With Chronic Heart Failure With Preserved Ejection Fraction]) (26). Apart from a recent study that showed that empagliflozin acts directly on sodium and calcium (Ca2+) exchange in isolated CMs (27), it was recently reported that empagliflozin improved diastolic cardiac function by increasing cyclic guanosine monophosphate (cGMP)-dependent titin phosphorylation in human ventricular trabeculae and in a murine model of HFpEF (28). However, the cardiac mechanisms of action for empagliflozin remain largely unexplored, in particular with respect to interactions between CMECs and CMs, which are especially relevant for cardiac dysfunction in HFpEF.
We report on the effect of CMEC on CM function in a novel co-culture system combined with a novel high-throughput analysis of CM function. We demonstrate that CMECs improve CM contraction and relaxation, an effect that is lost after pre-incubation of CMECs with the inflammatory mediator tumor necrosis factor-α (TNF-α). Moreover, we provide evidence that empagliflozin restored this beneficial effect of CMECs by reducing mitochondrial ROS production and cytoplasmic ROS accumulation, which led to restoration of endothelial NO bioavailability and preservation of CM contraction and relaxation. These data provide a new mechanism underlying the effect of empagliflozin on the heart by modulating CMEC-mediated enhancement of CM function.
Methods
CMEC culture
Human CMECs (CC-7030, Lonza Europe, Breda, the Netherlands) were cultured and characterized (CD31, vWF, and VE-cadherin) before being used in the experiments. For co-culture experiments (see the following), CMECs (passage 5 to 7) were cultured on 24-well format, 3 μm filter inserts (ThinCert, 662631, Grenier Bio-one, Monroe, North Carolina) coated with 1% gelatin (104070, Merck, Whitehouse Station, New Jersey) in endothelial growth medium-2MV (CC-3203, Lonza) at 37°C in a 5% carbon dioxide-95% air atmosphere. For the assessment of NO production (see the following), CMECs were grown on 8-well format μ-Slide (80826, Ibidi GmbH, Gräfelfing, Bayern, Germany) with the same medium and culture condition.
Adult rat ventricular CM isolation and culture
The animal experiments were performed in accordance with the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes and were approved by the ethics committees of VU University Medical Center, Amsterdam, the Netherlands.
Adult rat CMs were isolated using liberase digestion of hearts as described previously 29, 30. Briefly, adult wild-type Wistar rats weighing 200 to 250 g were anesthetized with isoflurane (45.112.110, Pharmachemie, Haarlem, the Netherlands) inhalation. The chest was opened, and the heart was injected with cold Ethylene glycol tetraacetic acid (EGTA) solution. Afterward, it was quickly removed, cannulated via the aorta, and perfused in a Langendorff setup with a perfusion buffer for 5 min. Next, it was perfused with enzyme solution until the tissue was digested sufficiently. The atria and right ventricle were removed, and the LV was cut into small pieces and triturated with a plastic Pasteur pipette for 3 min. Subsequently, the cell suspension was filtered through a 300-μm stainless steel autoclaveable filter and resuspended in calcium chloride buffers of increasing Ca2+ concentrations to reach a final concentration of 0.2 mM Ca2+.
Isolated adult CMs were finally resuspended in plating medium containing Medium 199 (BE12-117F, Lonza), 1% penicillin/streptomycin (DE17-602DE, Lonza), and 5% fetal bovine serum (A15-101, PAA Cell Culture Company, Cambridge, United Kingdom), and seeded on 1% laminin (L2020-1MG, Sigma-Aldrich, St. Louis, Missouri) coated plates (24-well format Costar culture plate, 3524, Corning, Corning, New York). One h after plating, cells that were not attached were removed by replacing the plating medium with culture medium endothelial growth medium-2MV. Subsequently, the cells were co-cultured with CMECs pre-seeded on inserts (see the preceding) at 37°C in humidified air with 5% carbon dioxide. Isolation protocol details can be found in the Supplemental Appendix.
Co-culture model of CMECs and CMs
CMECs were seeded overnight on inserts with a seeding density of 2.5 × 104 per insert. Subsequently, the culture medium was renewed, and the cells were either not stimulated or stimulated with 10 ng/ml TNF-α (T6674, Sigma), 10 ng/ml interleukin-1β (200-01B-10 μg, PeproTech, Rocky Hill, New Jersey), 1 μM empagliflozin (HY-15409, MedChem Express, Monmouth Junction, New Jersey), 10 μM butylated hydroxytoluene (PHR1117, Sigma), or a combination thereof, for 6 h before the co-culture procedure. After this pre-stimulation period, the cells were rinsed 3 times, and the medium was renewed to ensure that all treatments were washed away before the co-culture protocol. Co-culture of CMECs was started by placing the inserts with untreated or treated CMECs closely above the CMs cultured separately in another culture plate (Figure 1, Supplemental Figure 1). After 2 h of co-culture, the inserts containing the CMECs were removed, and CM contractile profiles were assessed as described in the following. To inhibit NO production, CMECs were incubated with 100 μM N(ω)-nitro-L-arginine methyl ester (L-NAME) (N5751, Sigma-Aldrich, St. Louis, Missouri) for 1 h before the co-culture procedure, which was continued subsequently for 2 h during the co-culture period with CMs.
Figure 1.

CMEC-CM Co-Culture and High-Throughput MultiCell CM Detection System
(A) Cardiac microvascular endothelial cells (CMECs) were cultured on an insert placed above the cardiomyocytes (CMs) seeded on a well of a culture plate. (B, right panel) After a co-culture procedure, the CMEC compartment was removed, and (C) the CM compartment was placed into a temperature-controlled inverted microscope chamber and visualized. (B, left panel) The objective lens was positioned directly under the well containing CMs (A). (D) CMs were paced, and cell contraction and relaxation profiles were monitored.
Incubation of CMs with endothelial cell-conditioned medium with and without NO scavenging
CMECs were seeded in 1% gelatin-coated 6-well plates (Costar culture plate, 3506, Corning) until it reached confluency. The cell medium was then refreshed, and after 6 h, the CMEC-conditioned medium was pipetted onto the CMs pre-plated in a separate 24-well plate. The contraction profiles were measured after 30 min, 1 h, 1.5 h, and 2 h incubation of CMs with the conditioned medium. In a separate experiment, 10 μm of the NO scavenger carboxy-PTIO (C221, Sigma) was added to the endothelial-conditioned medium before being administered to the CMs. After 30 min of incubation, the contractility of the CMs was measured.
High-throughput multicell CM function evaluation system
To investigate whether CMECs regulated the contractile properties of CMs, we developed an assay in which we assessed CM contraction and relaxation kinetics during a co-culture period with CMECs. After the co-culture procedure, the plate containing CMs was placed in a high-throughput inverted microscope (Olympus 20x 0.75 aperture objective lens, Olympus, Shinjuku, Japan) setup (CytoCypher, Amsterdam, the Netherlands) (Figure 1B), and the cells were visualized (Figure 1C). The camera-based MultiCell microscope system (CytoCypher, Amsterdam, the Netherlands) (Figure 1C) allowed the selection and assay of numerous CMs within a relatively short period of time. Unloaded intact rat CMs were monitored following field stimulation, and sarcomere shortening was measured using the MultiCell CM detection system in combination with the Ionoptix high-speed sarcomere length measuring software (Ionoptix LLC, Westwood, Massachusetts) (Figure 1D). The contractility profiles were analyzed with the automated, batch analysis software Transient Analysis Tools (CytoCypher). Subsequent to co-culture procedure with CMECs or after direct treatment with 1 μM empagliflozin for 2 h, CMs were placed into a temperature-controlled microscope chamber with platinum electrodes to electrically stimulate the cells. Single CMs were selected based on the following criteria: rod-shaped, no spontaneous contractions, and diastolic sarcomere length of at least 1.6 μm. Upon field stimulation (2 Hz, 4 ms, 25 V), cell contraction and relaxation kinetics were monitored.
NO measurement in endothelial cells
NO bioavailability was assessed on confluent CMECs cultured on μ-slides (Ibidi, 80826). A copper-based NO probe (Cu2FL2E; 96-0396, Strem, Newburyport, Massachusetts) was added in a final concentration of 20 μM and incubated for 45 min to allow cellular uptake. Stimulation with 10 μM acetylcholine (A6625-25G, Sigma) was performed for 45 min, at the same time as the incubation with the Cu2FL2E probe. For NO measurement after 6 h of TNF-α, empagliflozin, and butylated hydroxytoluene treatment, the probe was added during the final 45 min of the incubation period. For inhibition of NO synthases, CMECs were pre-incubated with 100 μM L-NAME for 1 h before addition of and during incubation with Cu2FL2E. The cells were then washed 3 times with Hanks’ Balanced Salt Solution (HBSS) (BE10-547F, Lonza); subsequently, live-cell imaging was performed at 37°C and in a 5% carbon dioxide environment on a Zeiss Axiovert 200M Marianas inverted fluorescence microscope (Intelligent Imaging Innovations, Denver, Colorado), equipped with a motorized stage, a turret with diamidino-2-phenylindole, fluorescein isothiocyanate (FITC), Cy3, and Cy5 filter cubes, a cooled CCD camera (Cooke Sensicam SVGA, Cooke Co., Tonawanda, New York), and a 63× oil-immersion objective (Zeiss, Breda, the Netherlands). The camera was linear over its full dynamic range (up to intensities of >4,000), whereas dark and/or background currents (measured by the intensity outside the cells) were typically <100. All fluorescent images were corrected for background (including dark current) and negative controls. Quantification of all fluorescent images was performed using dedicated digital cell masking software (Slidebook 6, Intelligent Imaging Innovations).
Cytoplasmic and mitochondrial ROS measurement in endothelial cells
Cytoplasmic ROS level was assessed on confluent CMECs cultured on Ibidi μ-slides. A fluorescent dye-based ROS probe (CM-H2DCFDA, C6827, ThermoFischer, Waltham, Massachusetts) was added in a final concentration of 5 μM and incubated for 30 min in phosphate-buffered saline (220/12257974/1110, Braun, Kronberg im Taunus, Germany), supplemented with 1 mM calcium chloride, 0.5 mM magnesium chloride, and 5.4 mM D-glucose, to allow cellular uptake. Mitochondrial CMEC ROS level was assessed with 500 nM MitoTracker Red CM-H2Xros (M7513, ThermoFischer). Six h after TNF-α and/or empagliflozin treatment, the cells were washed 1 time with supplemented phosphate buffer saline and subsequently incubated with the probe. The cells were then washed 1 time, followed by live-cell imaging at 37°C and in a 5% carbon dioxide environment on a Zeiss Axiovert 200M Marianas inverted fluorescence microscope (Intelligent Imaging Innovations) with a 63× oil-immersion objective. All fluorescent images were corrected for background and negative controls. Quantification of all fluorescent images was performed using digital cell masking software (Slidebook 6, Intelligent Imaging Innovations).
Determination of antioxidant capacity
To determine whether empagliflozin possesses direct antioxidant capacity, we performed a 1,1-diphenyl-picrylhydrazyl (DPPH) assay. The antioxidant activity was measured in terms of hydrogen donating or radical scavenging ability, using the stable radical DPPH. DPPH (25 μM) (D9132, Sigma) solution in methanol was pipetted into 96-well plate format (Costar, 3599, Corning), and 1 or 10 μM of empagliflozin was added. Butylated hydroxytoluene (0.4 mg/ml) and ascorbic acid (A5960, Sigma) (0.5 mg/ml) served as positive controls, and methanol served as a negative control. The plate was incubated for 30 min in the dark at room temperature. Afterward, the decrease in absorbance was measured at 517 nm with an enzyme-linked immunosorbent assay reader/spectrophotometer (Epoch, Biotek, Winooski, Vermont). The capability of scavenging the DPPH radical, or the direct antioxidant capacity, was calculated by using the following formula: ((A0 − A1)/A0) × 100), where A0 is the absorbance of the negative control reaction, and A1 is the absorbance in the presence of samples or positive control antioxidants.
Cell adhesion molecule measurement
Cell adhesion molecules were assayed by a cell-bound enzyme-linked immunosorbent assay. CMECs were grown in 96-well plates (Costar, 3599, Corning) until confluency was reached. Subsequently, the culture medium endothelial growth medium-2MV was renewed, and the cells were either not stimulated or stimulated with 10 ng/ml TNF-α, or a combination of 10 ng/ml TNF-α and 1 μM empagliflozin for 6 h. After the 6 h stimulation, CMECs were washed with phosphate-buffered saline and fixated with 0.025% glutaraldehyde (G5882-50mL, Sigma). A cell-bound enzyme-linked immunosorbent assay was performed using either mouse anti-VCAM 1:5,000 (Clone P3C4, MAB2144, Millipore B.V., Amsterdam, the Netherlands) or mouse anti−E-selectin 1:5,000 (CD62E, clone 1.2B6, CBL180, Chemicon) antibody, and peroxidase labeled-goat, anti-mouse immunoglobulin-G 1:2000 (P0447, Dako) as the secondary antibody. The reaction was visualized with a 3',3',5,5'-Tetramethylbenzidine (TMB) (980828, Organon Teknika, Durham, North Carolina) solution and stopped by administration of 2 M sulphuric acid (M205 K17208431 1L, Merck). Adhesion molecules were measured in triplicate with an enzyme-linked immunosorbent assay reader (Epoch, Biotek) at 450 nm.
Sodium dodecyl sulfate−polyacrylamide gel electrophoresis and western blotting
Cell lysates were produced in sample buffer containing 187.5-mM TRIS-hydrochloride, pH 6.8, 6% w/v sodium dodecyl sulfate (SDS), 30% glycerol, 150 mM Dithiothreitol (DTT), and 0.03% w/v bromphenol blue. For endothelial nitric oxide synthase (eNOS) dimer and/or monomer assessment, the SDS was adjusted to 2%, and the DTT was omitted from the sample buffer. Subsequently, samples were boiled for 5 min at 95°C. SDS-polyacrylamide gel electrophoresis (PAGE) was performed in 8% gel at room temperature at 120 V followed by transfer of the protein at 200 mA to a nitrocellulose membrane. Western blotting was performed using the Mini-TransBlot Cell (Bio-Rad, Foster City, California). For eNOS dimer and/or monomer assessment, sodium dodecyl sulfate–polyacrylamide gel electrophoresis was performed in 6% gel at 10 mA followed by membrane blotting at 95 mA at 4°C. The blotted membranes were blocked in 5% bovine serum albumin (A9647, Sigma) or 5% milk (170-6404, Bio-Rad Laboratories B.V., Lunteren, the Netherlands) in Tris-buffered saline-Tween. Primary antibody labeling was performed overnight at 4°C, whereas secondary immunoglobulin-G−horseradish peroxidase-conjugated antibodies were applied for 2 h at room temperature. After each antibody incubation, blots were washed for 3 × 5 min in Tris-buffered saline-Tween. Images were generated using ECL Prime (RPN2232, Amersham, GE Healthcare, Buckinghamshire, United Kingdom) and the LAS-3000 documentation system (Fuji Film Life Science, Stamford, Connecticut). Outputs were normalized for loading, and results were expressed as an n-fold increase over the values of the control group in densitometric arbitrary units. Primary antibodies that were used included rabbit monoclonal phospho−Jun N-terminal kinase (JNK) T185/Y185 (4668T, 1:1,000, Cell Signaling, Danvers, Massachusetts), rabbit total JNK (9252T, 1:1,000, Cell Signaling), rabbit monoclonal phospho-eNOS Ser1177 (9570S, 1:1,000, Cell Signaling), mouse total eNOS (Ab95254, Abcam, Cambridge, United Kingdom), rabbit glyceraldehyde-3-phosphate dehydrogenase (1:100.000, Cell Signaling). Secondary antibodies included goat anti-rabbit (P0448, 1:5,000, Dako) and goat anti-mouse immunoglobulin-G-horseradish peroxidase (P0447, 1:5,000, Dako).
Gene expression assessment
RNA was isolated with Direct-zol RNA MiniPrep kit (R2052, Zymo Research, Irvine, California), and cDNA was generated with the iScript cDNA Synthesis Kit (1708890, Bio-Rad). Quantitative real-time polymerase chain reaction was performed using IQ SYBR Green Supermix (170-8886, Bio-Rad) in C1000 Touch Thermal Cycler CFX96 Real Time System (Bio-Rad), and data were analyzed with Bio-Rad CFX manager 3.1 software (Bio-Rad). Transcript quantities were compared using the relative Ct (cycle threshold) method, in which the amount of target was normalized to the amount of reference gene, β2 microglobulin, and calculated relative to the control group based on the 2−ΔCt method. The primers for the genes of interest are listed in Supplemental Table 1.
Statistics
The results are presented as mean ± SD. Statistical analyses were performed using Prism software (GraphPad Software Inc., San Diego, California), and consisted of 1-way analysis of variance followed by Tukey’s multiple comparison test when comparing >2 experimental groups, or unpaired Student’s t-test when comparing 2 experimental groups. Differences were considered significant when p < 0.05.
Results
CMECs improve contraction and relaxation of CMs
To investigate whether CMECs regulate CM function, we co-cultured both cell types for 2 h (Supplemental Figure 1) and subsequently measured the contraction and relaxation kinetics of individual CMs paced at 2 Hz (Figure 1). The presence of CMECs significantly enhanced CM contraction as shown by an increase in the fractional shortening or the proportion of sarcomere length shortening (Figures 2A and 2D). CMECs also improved CM relaxation as indicated by a higher relaxation velocity (Figures 2B and 2E) and a shorter time constant of relaxation (tau) (Figures 2C and 2F), in comparison to the non-co-incubated control CMs. These contractility improving effects were lost when CMECs were pre-exposed to the pro-inflammatory cytokine TNF-α for 6 h before the co-culture period (Figure 2). In these experiments, TNF-α was washed away and omitted during the 2 h co-culture period. The contractility-improving effect of CMECs was also lost when CMECs were pre-incubated with interleukin-1β (Supplemental Figure 2).
Figure 2.

CMECs Enhance CM Function
CM contraction and relaxation were evaluated under basal conditions (control) and after co-culture with untreated (endothelial cells [ECs]) or tumor necrosis factor (TNF)-α pre-treated CMEC (EC+TNF). (A and D) Co-culture of CMs with ECs increased CM contraction (proportion sarcomere shortening), and (B and E) improved CM relaxation as shown by increased speed of relaxation (return velocity) and (C and F) shortened time constant of relaxation (tau). (A and D) Pre-treatment of CMECs with TNF-α (EC+TNF) abolished the beneficial effect of CMECs on CM contraction and (B, C, E, and F) relaxation. (A to C) Graphs representing single CMs isolated from 1 individual rat, distributed into the 3 corresponding experimental conditions; 40 to 45 CMs were measured per condition. (D to F) Data are representative of 8 independent experiments. (D to F) Graphs of combined average values obtained from 8 independent experiments corresponding to 8 individual rats. All data are represented as mean ± SD. Other abbreviations as in Figure 1.
NO mediates CMEC control of cardiomyocyte function
To investigate if EC-derived NO is involved in CMEC-mediated improvement of CM function, we inhibited NO synthases in CMECs with L-NAME, a NO synthase inhibitor, for 1 h before and during 2 h of CMEC-CM co-culture. Inhibition of endothelial NO production completely abolished the beneficial effect of CMECs on CM contraction (Figure 3A, Supplemental Figure 3A) and relaxation parameters (Figures 3B and 3C, Supplemental Figures 3B and 3C). Direct incubation of CMs with L-NAME did not affect CM contraction and relaxation, which suggested no contribution of CM-derived NO.
Figure 3.
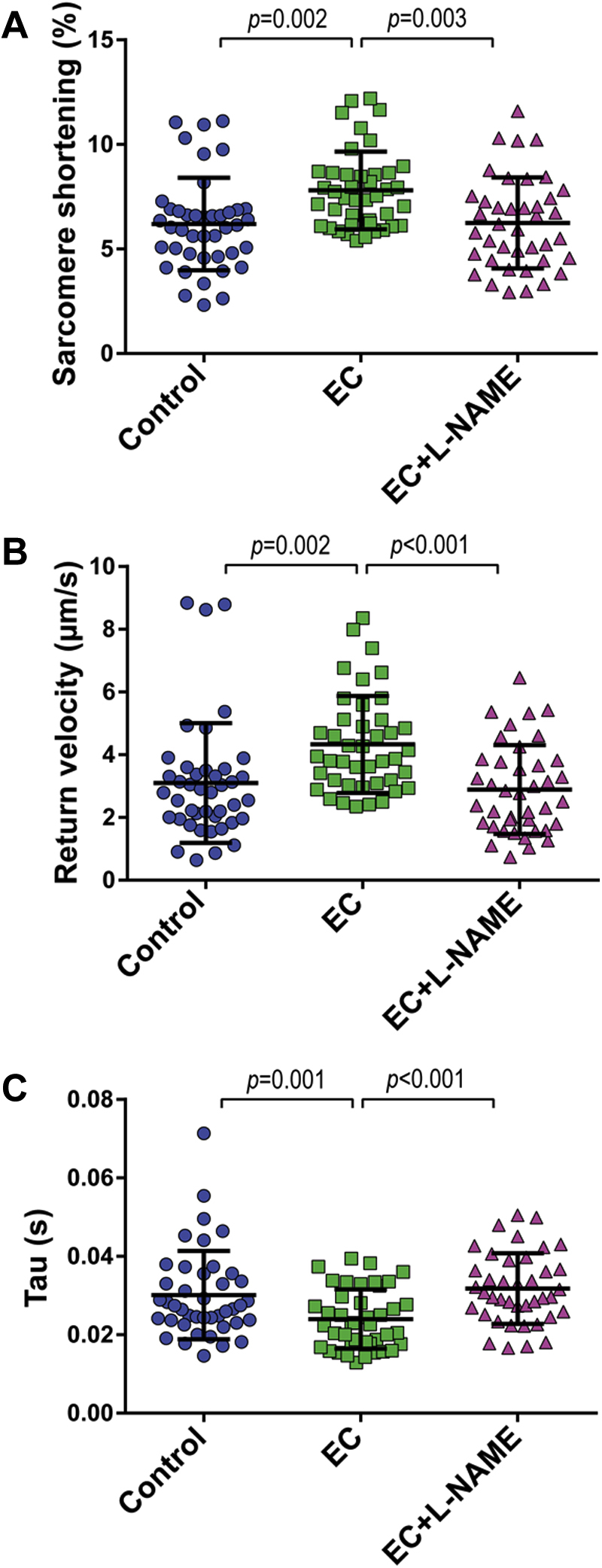
Inhibition of Endothelial NOS Eliminates the Beneficial Effect of CMEC on CM Contraction and Relaxation
(A) Inhibition of CMEC nitric oxide (NO) generation with N(ω)-nitro-L-arginine methyl ester (L-NAME) abrogated the effect of CMECs on CM sarcomere shortening, (B) return velocity, and (C) tau. (A to C) Graphs representing single CMs isolated from 1 individual rat, distributed into the 3 corresponding experimental conditions; 40 to 45 CMs were measured per condition. Data are representative of 3 independent experiments. Data are represented as mean ± SD. NOS = nitric oxide synthase; other abbreviations as in Figures 1 and 2.
Incubation of CMs with CMEC-conditioned medium also enhanced CM contractility performance, an effect that declined during the 2-h evaluation period (Supplemental Figures 3D to 3F). Furthermore, the stimulatory activity of the CMEC-conditioned medium was fully abolished by the NO scavenger carboxy-PTIO (Supplemental Figures 3G to 3I), which indicated that CMEC-derived NO was a major factor mediating the beneficial effect of CMECs on CM contraction and relaxation.
TNF-α reduces CMEC NO bioavailability
The level of NO in CMECs was assayed by live-cell microscopy using a copper-based NO probe. CMECs produced a detectable amount of NO at the basal level (Figures 4A and 4B), which increased by exposure to acetylcholine (Figures 4A and 4B). In contrast, exposure of CMECs to TNF-α largely reduced the NO bioavailability to a level nearly similar to that in the presence of L-NAME (Figures 4A and 4B).
Figure 4.

TNF-α Reduces NO Bioavailability in CMECs
(A and B) NO levels in CMEC. Acetylcholine (Ach)−induced upregulation of NO production in CMECs. Similar to L-NAME treatment, TNF-α reduced CMEC NO levels. (n = 4 to 50, refers to number of cells per group from 3 independent experiments). Data represented as mean ± SD. Other abbreviations as in Figures 1, 2, and 3.
Empagliflozin restores CMEC control of CM function during exposure to TNF-α
We investigated whether empagliflozin could also modulate the effect of CMECs on CM function. CMECs were treated with empagliflozin at a physiological relevant concentration of 1 μM for a 6 h before the co-culture procedure. After several washing steps to ensure elimination of this drug from the CMEC compartment, the cells were co-incubated with CMs. Pre-treatment of CMECs with empagliflozin slightly, but not significantly, enhanced CM sarcomere shortening (Figure 5A, Supplemental Figure 4A) compared with the untreated CMEC group. Moreover, there was a small but significant increase in relaxation velocity in comparison to the effect of CMECs alone (Figure 5B, Supplemental Figure 4B), although tau did not reach a significant difference after empagliflozin pre-treatment (Figure 5C, Supplemental Figure 4C).
Figure 5.

Empa Restores CMEC-Mediated CM Function During TNF-α Exposure
(A) Co-culture of CMEC with CMs increased CM shortening and (B) speed of relaxation, and (C) shortened tau. (A) Pre-treatment of CMECs with empagliflozin (EC+Empa) slightly increased CM contraction compared with CMEC alone (EC). (B) Empa pre-treatment increased CM return velocity but did not affect relaxation time, in comparison to CMEC alone. (D to F) Pre-incubation of CMEC with either TNF-α or a combination of TNF-α and Empa and subsequent co-culture of CMEC with CMs. (D) Co-treatment of TNF-α-stimulated CMEC with Empa prevented reduction of CM contraction in comparison to TNF-α stimulation alone. Co-treatment with Empa also maintained CMEC beneficial effect on (E) CM relaxation velocity and (F) tau. (A to C) Graphs representing single CMs isolated from 1 individual rat, distributed into the 3 corresponding experimental conditions; 30 to 40 CMs were measured per condition. Data are representative of 3 independent experiments. (D to F) Graphs representing single CMs isolated from 1 individual rat, distributed into the 4 corresponding experimental conditions; 30 to 40 CMs were measured per condition. Data are representative of 5 independent experiments; all data are represented as mean ± SD. Abbreviations as in Figures 1, 2, and 3.
Subsequently, we examined whether empagliflozin could modulate the effect of TNF-α-treated CMECs on the contraction and relaxation performance of CMs. We pre-incubated CMECs with either TNF-α or a combination of TNF-α and empagliflozin, and co-incubated the cells with CMs afterward. The presence of empagliflozin during exposure to TNF-α preserved the ability of CMECs to improve CM contraction, which was lost when only TNF-α was added (Figure 5D, Supplemental Figure 4D). Moreover, co-treatment with empagliflozin also maintained the enhancing effects of CMECs on CM diastolic function as shown by a higher relaxation velocity and shorter tau (Figures 5E and 5F, Supplemental Figures 4E and 4F). In addition, empagliflozin restored the beneficial effect of CMECs on CM function, which was inhibited by pre-treatment of CMEC with interleukin-1β (Supplemental Figure 2). These findings indicated that empagliflozin not only acted in concert with endothelium to positively modulate CM function, but also maintained the CMEC-mediated regulation of CM contraction and relaxation that was lost when CMECs were exposed to pro-inflammatory cytokines (e.g., TNF-α and interleukin-1β). The beneficial effects of empagliflozin were abrogated when CMECs were co-treated with L-NAME (Figures 6A to 6C, Supplemental Figures 5A to 5C), which showed that endothelial-derived NO mediates the beneficial effect of empagliflozin on CM function.
Figure 6.

NO Mediates the Beneficial Effect of Empa on CMEC-CM Interaction
(A) Co-treatment with Empa maintained the beneficial effects of CMEC on CM contraction and (B and C) relaxation after TNF-α stimulation. (A to C) The beneficial effect of Empa was abolished when CMECs were co-treated with L-NAME. (A to C) Graphs representing single CMs isolated from 1 individual rat, distributed into the 4 corresponding experimental conditions; 40 to 45 CMs were measured per condition. Data are representative of 3 independent experiments; data are represented as mean ± SD. Abbreviations as in Figures 1, 2, and 3 and 5.
Empagliflozin attenuates TNF-α induced loss of NO availability in CMECs
Empagliflozin has been reported to directly affect CMs (27). We also observed that 1 μM empagliflozin treatment of CMs enhanced CM function (Supplemental Figures 6A and 6D) and relaxation (Supplemental Figures 6B, 6C, 6E, and 6F), which indicated that empagliflozin could directly act on CMs and regulate their contractile profile. However, this direct effect did not explain the results of the co-culture experiments because empagliflozin was only present in the CMEC compartment during the 6 h pre-treatment period and was removed before the onset of co-culture with CMs.
To underscore the effect of empagliflozin on NO production in CMECs, we determined the cellular NO concentration in CMECs. Live-cell microscopy evaluation showed that NO levels in CMECs were reduced by exposure of the cells to TNF-α but was maintained when the cells were incubated with TNF-α and empagliflozin simultaneously (Figures 7A and 7B).
Figure 7.

Empagliflozin Reduced Mitochondrial and Cytoplasmic ROS Level and Restores NO Bioavailability in CMECs After TNF-α Stimulation
(A and B) TNF-α treatment reduced CMEC NO levels. Co-treatment of CMEC with TNF-α and Empa resulted in higher CMEC NO bioavailability in comparison to TNF-α treatment alone (n = 40 to 50, refers to number of cells per group from 3 independent experiments; data represented as mean ± SD). (C and D) TNF-α stimulation led to increased VCAM and E-selectin expression on the cell surface of CMECs, as assayed by a cell-bound enzyme linked immunosorbent assay, and co-treatment with Empa did not change the level of both proteins (n = 4, refers to number of independent experiments; data represented as mean ± SD). (E and F) TNF-α treatment increased cytoplasmic reactive oxygen species (ROS) levels in CMECs, which were attenuated by simultaneous treatment of Empa (n = 40 to 50, refers to number of cells per group from 3 independent experiments; data represented as mean ± SD). (G) Empa did not possess direct antioxidant activity at 1 and 10 μM concentrations, whereas 2 potent antioxidants, butylated hydroxytoluene (BHT) and ascorbic acid, showed a strong radical scavenging effect (n = 3, refers to number of independent experiments; data represented as mean ± SD). (H and I) Empa reduced mitochondrial ROS production after TNF-α stimulation in CMECs (n = 3, refers to number of independent experiments; data represented as mean ± SD). VCAM = vascular cell adhesion molecule; other abbreviations as in Figures 1, 2, and 3 and 5.
Empagliflozin does not act via the nuclear factor−kB pathway and eNOS dimerization or phosphorylation
Several pathways have been shown to be activated by TNF-α, in particular, the nuclear factor-κB (NK-κB) pathway, p38 mitogen-activated protein kinase (MAPK), JNK pathways, and ROS generation 31, 32, which might be attenuated by empagliflozin. A possible role of the NF-κB pathway as a target for the beneficial effect of empagliflozin on CMECs treated with TNF-α was refuted because empagliflozin did not change the transcription (Supplemental Figures 7A and 7B) nor the protein expression (Figures 7C and 7D) of vascular cell adhesion molecule (VCAM)-1 and E-selectin, 2 genes that are commonly induced by TNF-α via NF-κB pathway activation. Similarly, superoxide dismutase 2 (SOD2) mRNA expression, which was strongly enhanced by TNF-α in ECs in an NF-kB dependent manner (33), was not affected by empagliflozin (Supplemental Figure 7C).
Empagliflozin did not change the transcription of eNOS (NOS3), which was attenuated by TNF-α (Supplemental Figure 7D). The eNOS protein in CMECs was mainly encountered as a dimer, which remained stabile after 6 h TNF-α exposure, whereas the small amount of eNOS monomer decreased (Supplemental Figure 8A). Empagliflozin neither affected the amount of eNOS dimer nor altered the eNOS monomer content in control or TNF-α-exposed CMECs (Supplemental Figure 8A). Furthermore, after the 6 h incubation period, empagliflozin did not change the total eNOS protein content or the phosphorylation of eNOS at position Ser1177 in control or TNF-α-treated CMECs (Supplemental Figure 8B).
Empagliflozin restores CMEC NO bioavailability by attenuation of mitochondrial ROS level
Subsequently, we evaluated if empagliflozin could preserve NO levels in CMECs by attenuating TNF-α-induced ROS production. As detected by fluorescent dye-based free radical sensors, TNF-α increased cytoplasmic ROS accumulation in CMECs (Figures 7E and 7F). Because ROS, in particular, as a superoxide, can scavenge NO, we first verified if antioxidants that scavenge ROS could increase the bioavailability of NO in CMECs. Administration of the general antioxidant butylated hydroxytoluene rescued NO bioavailability, which was diminished by exposure to TNF-α (Supplemental Figure 8C). The induction of ROS production by TNF-α was fully blunted by co-treatment with empagliflozin (Figures 7E and 7F). We then examined whether the reduction of ROS with empagliflozin co-treatment was due to a direct antioxidant or radical scavenging capacity of empagliflozin. Using DPPH as a free radical, we observed that empagliflozin at 1 and 10 μM concentration showed no direct antioxidant capability, whereas the antioxidants butylated hydroxytoluene and ascorbic acid displayed a strong direct radical scavenging effect (Figure 7G).
TNF-α−induced ROS generation is complex and involves various pathways, resulting in ROS generation, as well as modulation of ROS scavenging enzymes. TNF-α can induce ROS production via NOX4 31, 34. However, we observed no effects of empagliflozin on NADPH oxidase 4 (NOX4) expression (Supplemental Figure 7E). In addition, empagliflozin did not alter the mRNA expression of the radical scavenging enzymes SOD1 and SOD2 after TNF-α stimulation (Supplemental Figures 7C and 7F). NOX1, NOX2, NOX3, NOX5, and SOD3 mRNA in CMECs remained below detection level in these conditions (data not shown).
Mitochondria are a major source of ROS (31). JNK activation is enhanced by TNF-α and can contribute to mitochondrial ROS production (31). However, the TNF-α-induced JNK phosphorylation was not altered by empagliflozin (Supplemental Figure 8D). However, we observed that TNF-α induced mitochondrial ROS generation in CMECs (Figures 7H and 7I), and remarkably, empagliflozin significantly reduced the mitochondrial ROS production that was enhanced by TNF-α exposure (Figures 7H and 7I). Overall, because empagliflozin showed no direct antioxidant capacity, these data suggested that, in the presence of TNF-α stimulation, empagliflozin could activate intracellular mechanisms that cause a reduction in mitochondrial ROS generation. This resulted in an inhibition of cytoplasmic ROS accumulation, which led to an enhancement of CMEC NO bioavailability.
Discussion
In a co-culture model of CMECs and CMs, we showed that CMECs exerted a direct positive effect on CM contraction and relaxation. These effects were mediated by endothelial-derived NO and lost after pre-exposure of CMECs to TNF-α, predominantly due to NO scavenging by the generation of cytoplasmic and mitochondrial ROS. Moreover, we demonstrated that empagliflozin counteracted the TNF-α-mediated impairment of CMEC-CM interaction. It reduced the mitochondrial ROS level, prevented accumulation of cytoplasmic ROS, and enhanced the bioavailability of NO, both within the CMECs and in its conditioned medium, which resulted in the preservation of both contraction and relaxation of CMs. The latter implied a novel mechanism by which empagliflozin could restore CMEC function in various types of HF with disturbed microvascular function, including HFpEF.
Involvement of endothelial-derived NO in CMEC regulation of CM contraction and relaxation
In the present study, we provided evidence that CMECs directly regulated contraction and relaxation of CMs and that intact cardiac EC function was vital to the regulation of CM function. On the basis of dedicated studies on the role of endocardial endothelium in controlling CM contractility, Brutsaert et al. 23, 35 postulated that intracardiac microvascular endothelial cells would exert a similar effect. In these studies, myocardial contractility was assessed in a papillary muscle from a rabbit right ventricle, with and without damage of endocardial endothelial cells by perfusion with Triton X-100. Subsequent animal intervention studies confirmed the existence of beneficial effects of tissue NO on cardiac contractility, likely derived from ECs (36). Our study added further evidence by using an in vitro co-culture system, which provided a simple and straightforward approach to study direct effects of human CMECs on both the contraction and relaxation performance of CMs. Our observation that L-NAME administration on CMs did not change their basal contractility was in line with other studies 37, 38. It suggests that in isolated CMs, endogenous NO played only a minor role in regulating CM contractile performance and implied that CMEC-derived NO was the more predominant contributor to myocardial contraction and relaxation. How NO exactly modulates CMs warrants further research. Because of the distance between the CMEC and CM compartment in the co-culture setup and the sustained effect of CMEC-derived NO, which spans a 20 min to 30 min interval and is inhibited by carboxy-PTIO, it can be anticipated that NO stabilizes during its transfer from CMECs or that its effect is maintained after reaching the CMs. Further studies are needed to clarify whether stabilization of CMEC-derived NO is due to interaction of NO with sulfhydryl (SH) groups or by protection of NO within vesicles and/or vesicular membranes.
Loss of CMEC-mediated regulation of CM function after TNF-α stimulation is mediated by reduced CMEC NO bioavailability
The beneficial effect of CMECs on CM contraction and relaxation was lost after pre-exposure of CMECs to TNF-α or interleukin-1β. This is of interest for the pathogenesis of HF because Paulus and Tschöpe (7) and others 14, 16 proposed that inflammatory activation of CMECs might be a pivotal initial mechanism in changing the cardiac mechanical properties in HFpEF. Using an NO copper-based probe with high sensitivity for detecting NO at minute levels 39, 40, we confirmed that exposure of CMECs to TNF-α abolished the NO availability in these cells. Most importantly, NO availability in TNF-α-treated CMECs was restored by empagliflozin.
TNF-α causes a reduction in NO bioavailability by 2 major mechanisms: reduction of eNOS mRNA stability and induction of superoxide generation. First, TNF-α reduces eNOS (NOS3) mRNA transcriptionally and post-transcriptionally in ECs 41, 42. Although the transcriptional regulation is dependent on NF-κB, the post-transcriptional regulation proceeds via binding of 52- and 57-kD protein(s) (eEF1A1 and PTB1) to the 3′-untranslated region of NOS3 mRNA, which enhances its degradation 41, 42, 43. However, empagliflozin did not reverse the drop in NOS3 mRNA. Moreover, empagliflozin did not affect the eNOS total protein level and phosphorylation at serine 1177, and it did not alter the dimerization status of eNOS after TNF-α stimulation in CMECs. In addition, empagliflozin did not change the induction of VCAM or E-selectin or the affected SOD2 mRNA that was upregulated by TNF-α, which further excluded a role of NF-kB in the protective effect of empagliflozin.
Second, TNF-α reduces NO by its ability to enhance ROS generation 44, 45, potentially either via an immediate activation of NOX with riboflavin kinase as a connector between the TNF receptor and the p22phox subunit (34), or via impairment of the mitochondrial respiratory chain, which can be achieved via several pathways, including JNK activation 31, 34. Excessive ROS production exerts a deleterious effect on the cells. Superoxide can rapidly react with NO forming peroxynitrite, whereas its conversion to hydrogen peroxide allows generation of highly reactive hydroxyl radicals, which together can cause toxic oxidation of proteins, lipids, and DNA 46, 47. In addition, superoxide can eventually cause eNOS uncoupling, which results in loss of NO production and additional superoxide formation 48, 49. Although empagliflozin completely inhibited the TNF-α−induced production of cytoplasmic and mitochondrial ROS in CMECs, we could not establish a direct ROS scavenging effect of empagliflozin even at a 10-fold higher concentration than used in our experiments. This was in contrast to the observation in a study that used only 1 supra-pharmacological dose of approximately 2 mM of empagliflozin (50). Although several studies, including ours (data not shown), could not demonstrate SGLT2 mRNAs in the whole heart tissue or CMECs 51, 52, recent studies reported on the presence of small amounts of SGLT2 protein in ECs 53, 54. Nevertheless, the mode of action of empagliflozin seems complex and its effect on ECs and their ROS production needs further elucidation. If the effect of empagliflozin on endothelial NO production is also found in arterial and other types of endothelial cells, the impact of these findings may extend to vessels and tissues beyond the heart 54, 55, 56.
Empagliflozin improves CMEC regulation of CM contraction and relaxation
After finding reduced HF hospitalization and mortality in patients with diabetes who were treated with the SGLT2 inhibitor empagliflozin (25), recent studies showed a direct effect of empagliflozin on the heart, despite the absence of detectable SGLT2 in the heart 57, 58. Baartscheer et al. (27) reported that empagliflozin treatment reduced CM cytoplasmic sodium and Ca2+ and increased mitochondrial Ca2+ levels by inhibiting the sodium/hydrogen exchanger. It is interesting to note that the sodium/hydrogen exchanger-1 inhibitors decrease myocardial superoxide production via direct mitochondrial action (59). Whether empagliflozin can affect the sodium/hydrogen exchanger in CMECs needs further investigation and is beyond the scope of our present study. Byrne et al. (60) also reported that empagliflozin prevented further loss of cardiac function in mice with pressure-overload−induced HF. Pabel et al. (28) demonstrated beneficial diastolic effects of empagliflozin in contracting human heart trabeculae, which was accompanied by phosphorylation of titin without changing the Ca2+ influx pattern. Our data provided further proof of a direct effect of empagliflozin on CM contraction and relaxation. Furthermore, our study demonstrated, for the first time, that empagliflozin could act directly on CMECs and thus enhanced the beneficial effect of CMECs on CM function and, most importantly, could preserve CMEC-mediated enhancement of CM contraction and relaxation during pro-inflammatory stimulation.
The beneficial effect of empagliflozin on HF hospitalization was observed in metabolically compromised patients with DM, who are at risk for developing HFpEF (25). HFpEF is characterized by a disturbance in LV diastolic function. However, strain imaging modality also detects impaired systolic function in patients with HFpEF (61). Our data show that CMECs enhanced both CM contraction and relaxation, an effect that was vastly mediated by CMEC-derived NO. NO increases cGMP levels, which can induce a concentration-dependent biphasic contractile response (62). Although at a low concentration cGMP triggers a positive inotropic effect, at a higher concentration it leads to a negative inotropic effect (62). Detachment of CMs from their in vivo environment, which we applied in our co-culture setup, may lead to relatively low cGMP level in the CMs. At a higher, more physiological CM cGMP concentration, CMECs may show a more predominant beneficial effect on the diastolic function of CMs.
Study limitations
Our study shows a beneficial effect of empagliflozin on disturbed endothelial NO delivery to CMs and subsequent improvement of CM contraction and relaxation. Our functional CM measurements were performed in unloaded CMs, which does not reflect the loaded CM as present in the heart. These measurements enable to study diastolic functional parameters such as re-lengthening or relaxation velocity and time constant of relaxation tau. Another limitation regards our co-culture model that combines human CMECs and adult rat CMs. Although this allows the recognition of NO as a major player in mediating the effect of CMEC on CM, it may overlook additional effects of species-specific acting mediators. Future studies using human CMs will clarify this aspect. Finally, most of our studies were performed with TNF-α as an inflammatory mediator and within a limited time period of 6 h. These studies have to be extended with other factors that contribute to heart failure and evaluation of the mutual CMEC-CM interaction over prolonged time periods. Notwithstanding these limitations, the observed mechanism fits with clinical trials on empagliflozin and heart failure.
Conclusions
Our findings showed that CMECs conferred a direct positive effect on CM contraction and relaxation and that impairment of EC function with the pro-inflammatory cytokine TNF-α abolished this beneficial effect. Moreover, we provided the first evidence of a direct effect of empagliflozin on CMECs and on CMEC-enhanced CM function. Our findings on the effect of empagliflozin on EC-mediated preservation of CM contraction and relaxation suggested that empagliflozin would be especially beneficial for treatment of the cardiac mechanical implications of deranged microvascular function in HF.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: HF is an important and growing clinical problem. HFpEF currently accounts for 50% of patients with HF and is expected to become more prevalent than HFrEF in the future. Unlike HFrEF, there is lack of effective treatments for HFpEF. Therapies that are used in HFrEF failed to improve primary outcomes in patients with HFpEF, suggesting different patho-mechanisms of these diseases. The current paradigm of HFpEF proposes that a metabolic risk-related pro-inflammatory state in these patients induces cardiac microvascular dysfunction with subsequent CM contractility impairment. We showed that CMECs positively regulated CM contraction and relaxation. This effect requires NO, is diminished after stimulation of CMECs with pro-inflammatory cytokines, and is restored by empagliflozin by preventing accumulation of ROS, leading to restoration of endothelial NO bioavailability. This suggests that empagliflozin can be beneficial for treatment of the cardiac mechanical implications of microvascular dysfunction in HF, and potentially in HFpEF.
TRANSLATIONAL OUTLOOK: For the clinical translation, our findings should be first evaluated in vivo in an experimental HF model, in particular, an HFpEF model. ZSF1 rats is one of the HFpEF models that have been shown to recapitulate cardiac microvascular and diastolic dysfunction in patients with HFpEF. Further evaluation in larger HFpEF experimental models, such as the swine model of microvascular dysfunction, may be warranted to test the effect of empagliflozin on improving endothelial dysfunction and the subsequent effect on diastolic function. For other types of HF, these findings should be evaluated in their corresponding models. Nevertheless, the notion of empagliflozin as a potential treatment of HF is currently under investigation in a large clinical trial of EMPEROR-Preserved and EMPEROR-Reduced. The outcome of this trial may show whether empagliflozin can be repurposed from being merely a glucose-lowering drug to an effective therapy for HF and possibly HFpEF.
Acknowledgments
The authors acknowledge the support for CVON-RECONNECT from the Netherlands CardioVascular Research Initiative, including the Dutch Heart Foundation, Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research and Development, and the Royal Netherlands Academy of Science. The authors also acknowledge Jeroen Kole, BSc, and Michiel van Wijhe, BSc, for the provided technical support and Etto Eringa, PhD, for his critical reading of the manuscript.
Footnotes
This project is funded by the Netherlands Heart Foundation (CVON-RECONNECT consortium, no. 2014-11). The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
The authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
Appendix
References
- 1.Dunlay S.M., Roger V.L., Redfield M.M. Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2017;65:1–12. doi: 10.1038/nrcardio.2017.65. [DOI] [PubMed] [Google Scholar]
- 2.Carson P.E., Anand I.S., Win S. The hospitalization burden and post-hospitalization mortality risk in heart failure with preserved ejection fraction: results from the I-PRESERVE Trial (Irbesartan in Heart Failure and Preserved Ejection Fraction) J Am Coll Cardiol HF. 2015;3:429–441. doi: 10.1016/j.jchf.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 3.Pitt B., Pfeffer M.A., Assmann S.F. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. 2014;370:1383–1392. doi: 10.1056/NEJMoa1313731. [DOI] [PubMed] [Google Scholar]
- 4.Ahmed A., Rich M.W., Fleg J.L. Effects of digoxin on morbidity and mortality in diastolic heart failure: the Ancillary Digitalis Investigation Group trial. Circulation. 2006;114:397–403. doi: 10.1161/CIRCULATIONAHA.106.628347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Redfield M.M., Chen H.H., Borlaug B.A. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309:1268–1277. doi: 10.1001/jama.2013.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yancy C.W., Jessup M., Bozkurt B. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. J Am Coll Cardiol. 2013;62:e147–e239. doi: 10.1016/j.jacc.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 7.Paulus W.J., Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–271. doi: 10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- 8.Sharma K., Hill T., Grams M. Outcomes and worsening renal function in patients hospitalized with heart failure with preserved ejection fraction. Am J Cardiol. 2015;116:1534–1540. doi: 10.1016/j.amjcard.2015.08.019. [DOI] [PubMed] [Google Scholar]
- 9.Ather S., Chan W., Bozkurt B. Impact of noncardiac comorbidities on morbidity and mortality in a predominantly male population with heart failure and preserved versus reduced ejection fraction. J Am Coll Cardiol. 2012;59:998–1005. doi: 10.1016/j.jacc.2011.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seferovic P.M., Paulus W.J. Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. Eur Heart J. 2015;36:1718–1727. doi: 10.1093/eurheartj/ehv134. 1727a−c. [DOI] [PubMed] [Google Scholar]
- 11.Franssen C., Chen S., Unger A. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. J Am Coll Cardiol HF. 2016;4:312–324. doi: 10.1016/j.jchf.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 12.Gevaert A.B., Shakeri H., Leloup A.J. Endothelial senescence contributes to heart failure with preserved ejection fraction in an aging mouse model. Circ Heart Fail. 2017;10:1–10. doi: 10.1161/CIRCHEARTFAILURE.116.003806. [DOI] [PubMed] [Google Scholar]
- 13.Westermann D., Lindner D., Kasner M. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. 2011;4:44–52. doi: 10.1161/CIRCHEARTFAILURE.109.931451. [DOI] [PubMed] [Google Scholar]
- 14.Mohammed S.F., Hussain S., Mirzoyev S.A., Edwards W.D., Maleszewski J.J., Redfield M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015;131:550–559. doi: 10.1161/CIRCULATIONAHA.114.009625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saiki H., Moulay G., Guenzel A.J. Experimental cardiac radiation exposure induces ventricular diastolic dysfunction with preserved ejection fraction. Am J Physiol Heart Circ Physiol. 2017;313:H392–H407. doi: 10.1152/ajpheart.00124.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saiki H., Petersen I.A., Scott C.G. Risk of heart failure with preserved ejection fraction in older women after contemporary radiotherapy for breast cancer. Circulation. 2017;135:1388–1396. doi: 10.1161/CIRCULATIONAHA.116.025434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y., Boerma M., Zhou D. Ionizing radiation-induced endothelial cell senescence and cardiovascular diseases. Radiat Res. 2016;186:153–161. doi: 10.1667/RR14445.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee C., Moding E., Cuneo K. p53 functions in endothelial cells to prevent radiation-induced myocardial injury in mice. Science Signalling. 2012;5:1–14. doi: 10.1126/scisignal.2002918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lauk S., Kiszel Z., Buschmann J., Diger Trott K. Radiation induced heart disease in rats. Int J Radiation Oncology Biol Phys. 1985;11:801–808. doi: 10.1016/0360-3016(85)90314-1. [DOI] [PubMed] [Google Scholar]
- 20.Fajardo L., Stewart J. Experimental radiation-induced heart disease. Am J Pathol. 1970;59:299–316. [PMC free article] [PubMed] [Google Scholar]
- 21.Fajardo L., Stewart J. Capillary injury preceding radiation-induced myocardial fibrosis. Radiology. 1971;101:429–433. doi: 10.1148/101.2.429. [DOI] [PubMed] [Google Scholar]
- 22.Li K., Rouleau J., Andries L., Brutsaert D. Effect of dysfunctional vascular endothelium on myocardial performance in isolated papillary muscles. Circ Res. 1993:768–777. doi: 10.1161/01.res.72.4.768. [DOI] [PubMed] [Google Scholar]
- 23.Brutsaert D. Cardiac Endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol Rev. 2003:59–115. doi: 10.1152/physrev.00017.2002. [DOI] [PubMed] [Google Scholar]
- 24.Shah S.J., Lam C.S.P., Svedlund S. Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. Eur Heart J. 2018;39:3439–3450. doi: 10.1093/eurheartj/ehy531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fitchett D., Zinman B., Wanner C. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: results of the EMPA-REG OUTCOME(R) trial. Eur Heart J. 2016;37:1526–1534. doi: 10.1093/eurheartj/ehv728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.U.S. National Library of Medicine CG Empagliflozin Outcome Trial in Patients With Chronic Heart Failure With Preserved Ejection Fraction (EMPEROR-Preserved) https://clinicaltrials.gov/ct2/show/NCT03057977 Available at: Accessed February 17, 2017.
- 27.Baartscheer A., Schumacher C.A., Wust R.C. Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia. 2017;60:568–573. doi: 10.1007/s00125-016-4134-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pabel S., Wagner S., Bollenberg H. Empagliflozin directly improves diastolic function in human heart failure. Eur J Heart Fail. 2018;20:1690–1700. doi: 10.1002/ejhf.1328. [DOI] [PubMed] [Google Scholar]
- 29.Kaestner L., Scholz A., Hammer K., Vecerdea A., Ruppenthal S., Lipp P. Isolation and genetic manipulation of adult cardiac myocytes for confocal imaging. J Vis Exp. 2009;31:1–5. doi: 10.3791/1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Deel E.D., Najafi A., Fontoura D. In vitro model to study the effects of matrix stiffening on Ca(2+) handling and myofilament function in isolated adult rat cardiomyocytes. J Physiol. 2017;595:4597–4610. doi: 10.1113/JP274460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blaser H., Dostert C., Mak T.W., Brenner D. TNF and ROS crosstalk in inflammation. Trends Cell Biol. 2016;26:249–261. doi: 10.1016/j.tcb.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 32.Zhou H., Wang S., Zhu P., Hu S., Chen Y., Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018;15:335–346. doi: 10.1016/j.redox.2017.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Visner G.A., Chesrown S.E., Monnier J., Ryan U.S., Nick H.S. Regulation of manganese superoxide dismutase: IL-l and TNF induction in pulmonary artery and microvascular endothelial cells. Biochem Biophys Res Commun. 1992;188:453–462. doi: 10.1016/0006-291x(92)92406-n. [DOI] [PubMed] [Google Scholar]
- 34.Yazdanpanah B., Wiegmann K., Tchikov V. Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature. 2009;460:1159–1163. doi: 10.1038/nature08206. [DOI] [PubMed] [Google Scholar]
- 35.Brutsaert D., Meulemans A., Sipido K., Stanilas U. Effects of damaging the endocardial surface on the mechanical performance of isolated cardiac muscle. Circ Res. 1988:358–366. doi: 10.1161/01.res.62.2.358. [DOI] [PubMed] [Google Scholar]
- 36.Sorop O., Heinonen I., van Kranenburg M. Multiple common comorbidities produce left ventricular diastolic dysfunction associated with coronary microvascular dysfunction, oxidative stress, and myocardial stiffening. Cardiovasc Res. 2018;114:954–964. doi: 10.1093/cvr/cvy038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin C.L., Wu Y.N., Jang J.H. Negligible effect of eNOS palmitoylation on fatty acid regulation of contraction in ventricular myocytes from healthy and hypertensive rats. Pflugers Arch. 2017:1141–1149. doi: 10.1007/s00424-017-1979-x. [DOI] [PubMed] [Google Scholar]
- 38.Smith J.M., Sondgeroth K.B., Wahler G.M. Inhibition of nitric oxide synthase enhances contractile response of ventricular myocytes from streptozotocin-diabetic rats. Mol Cell Biochem. 2007;300:129–137. doi: 10.1007/s11010-006-9376-3. [DOI] [PubMed] [Google Scholar]
- 39.Lim M.H., Xu D., Lippard S.J. Visualization of nitric oxide in living cells by a copper-based fluorescent probe. Nat Chem Biol. 2006;2:375–380. doi: 10.1038/nchembio794. [DOI] [PubMed] [Google Scholar]
- 40.Ghosh M., van den Akker N.M., Wijnands K.A. Specific visualization of nitric oxide in the vasculature with two-photon microscopy using a copper based fluorescent probe. PLoS One. 2013;8:1–14. doi: 10.1371/journal.pone.0075331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson H.D., Rahmutula D., Gardner D.G. Tumor necrosis factor-alpha inhibits endothelial nitric-oxide synthase gene promoter activity in bovine aortic endothelial cells. J Biol Chem. 2004;279:963–969. doi: 10.1074/jbc.M309552200. [DOI] [PubMed] [Google Scholar]
- 42.Yan G., You B., Chen S.P., Liao J.K., Sun J. Tumor necrosis factor-alpha downregulates endothelial nitric oxide synthase mRNA stability via translation elongation factor 1-alpha 1. Circ Res. 2008;103:591–597. doi: 10.1161/CIRCRESAHA.108.173963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yi B., Ozerova M., Zhang G.X., Yan G., Huang S., Sun J. Post-transcriptional regulation of endothelial nitric oxide synthase expression by polypyrimidine tract-binding protein 1. Arterioscler Thromb Vasc Biol. 2015;35:2153–2160. doi: 10.1161/ATVBAHA.115.305750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Buul J., Fernandez-Borja M., Anthony E., Hordijk P. Expression and localization of NOX2 and NOX4 in primary human endothelial cells. Antioxidant Redox Signaling. 2005;7:308–317. doi: 10.1089/ars.2005.7.308. [DOI] [PubMed] [Google Scholar]
- 45.Bedard K., Krause K.H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 46.Zhang H., Park Y., Wu J. Role of TNF-alpha in vascular dysfunction. Clin Sci (Lond) 2009;116:219–230. doi: 10.1042/CS20080196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Y., Vanhoutte P.M., Leung S.W. Vascular nitric oxide: beyond eNOS. J Pharmacol Sci. 2015;129:83–94. doi: 10.1016/j.jphs.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 48.Antoniades C., Shirodaria C., Leeson P. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur Heart J. 2009;30:1142–1150. doi: 10.1093/eurheartj/ehp061. [DOI] [PubMed] [Google Scholar]
- 49.Bendall J.K., Alp N.J., Warrick N. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ Res. 2005;97:864–871. doi: 10.1161/01.RES.0000187447.03525.72. [DOI] [PubMed] [Google Scholar]
- 50.Dhuja D.M.S., Hiah D.N.S.M., Swari D.R.R. Comparative efficacy of canagliflozin versus empagliflozin on oxidative stress – in-vitro method. Int J Pharma Bio Sci. 2017;8:232–236. [Google Scholar]
- 51.Chen J., Williams S., Ho S. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther. 2010;1:57–92. doi: 10.1007/s13300-010-0006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Steenbergen A., Balteau M., Ginion A. Sodium-myoinositol cotransporter-1, SMIT1, mediates the production of reactive oxygen species induced by hyperglycemia in the heart. Sci Rep. 2017;7:41166. doi: 10.1038/srep41166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mancini S.J., Boyd D., Katwan O.J. Canagliflozin inhibits interleukin-1beta-stimulated cytokine and chemokine secretion in vascular endothelial cells by AMP-activated protein kinase-dependent and -independent mechanisms. Sci Rep. 2018;8:5276. doi: 10.1038/s41598-018-23420-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.El-Daly M., Pulakazhi Venu V.K., Saifeddine M. Hyperglycaemic impairment of PAR2-mediated vasodilation: prevention by inhibition of aortic endothelial sodium-glucose-co-transporter-2 and minimizing oxidative stress. Vascul Pharmacol. 2018;109:56–71. doi: 10.1016/j.vph.2018.06.006. [DOI] [PubMed] [Google Scholar]
- 55.Uthman L., Baartscheer A., Bleijlevens B. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation. Diabetologia. 2018;61:722–726. doi: 10.1007/s00125-017-4509-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scognamiglio R., Negut C., De Kreutzenberg S.V., Tiengo A., Avogaro A. Postprandial myocardial perfusion in healthy subjects and in type 2 diabetic patients. Circulation. 2005;112:179–184. doi: 10.1161/CIRCULATIONAHA.104.495127. [DOI] [PubMed] [Google Scholar]
- 57.Vrhovac I., Eror D., Klessen D. Localizations of Na+-D-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflugers Arch Eur J Physiol. 2015:1881–1898. doi: 10.1007/s00424-014-1619-7. [DOI] [PubMed] [Google Scholar]
- 58.Van Steenbergen A., Balteau M., Scholasse J. Identification of a glucose sensor in the heart (abstr.) Eur Heart J. 2015:381. [Google Scholar]
- 59.Garciarena C.D., Caldiz C.I., Correa M.V. Na+/H+ exchanger-1 inhibitors decrease myocardial superoxide production via direct mitochondrial action. J Appl Physiol (1985) 2008;105:1706–1713. doi: 10.1152/japplphysiol.90616.2008. [DOI] [PubMed] [Google Scholar]
- 60.Byrne N.J., Parajuli N., Levasseur J.L. Empagliflozin prevents worsening of cardiac function in an experimental model of pressure overload-induced heart failure. J Am Coll Cardiol Basic Trans Science. 2017;2:347–354. doi: 10.1016/j.jacbts.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kraigher-Krainer E., Shah A.M., Gupta D.K. Impaired systolic function by strain imaging in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2014;63:447–456. doi: 10.1016/j.jacc.2013.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mohan P., Brutsaert D.L., Paulus W.J., Sys S.U. Myocardial contractile response to nitric oxide and cGMP. Circulation. 1996;93:1223–1229. doi: 10.1161/01.cir.93.6.1223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.