

Abstract
Aims
Autonomic dysfunction can promote atrial fibrillation (AF) and results from AF-related remodelling. N-type Ca2+-channels (NTCCs) at sympathetic nerve terminals mediate Ca2+-entry that triggers neurotransmitter release. AF-associated remodelling plays an important role in AF pathophysiology but the effects of NTCC inhibition on such remodelling is unknown. Here, we investigated the ability of a clinically available Ca2+-channel blocker (CCB) with NTCC-blocking activity to suppress the arrhythmogenic effects of AF-promoting remodelling in dogs.
Methods and results
Mongrel dogs were kept in AF by right atrial tachypacing at 600 bpm. Four groups were studied under short-term AF (7 days): (i) Shams, instrumented but without tachypacing (n = 5); (ii) a placebo group, tachypaced while receiving placebo (n = 6); (iii) a control tachypacing group receiving nifedipine (10 mg orally twice-daily; n = 5), an L-type CCB; and (iv) a cilnidipine group, subjected to tachypacing and treatment with cilnidipine (10 mg orally twice-daily; n = 7), an N-/L-type CCB. With cilnidipine therapy, dogs with 1-week AF showed significantly reduced autonomic changes reflected by heart rate variability (decreases in RMSSD and pNN50) and plasma norepinephrine concentrations. In addition, cilnidipine-treated dogs had decreased extracellular matrix gene expression vs. nifedipine-dogs. As in previous work, atrial fibrosis had not yet developed after 1-week AF, so three additional groups were studied under longer-term AF (21 days): (i) Shams, instrumented without tachypacing or drug therapy (n = 8); (ii) a placebo group, tachypaced while receiving placebo (n = 8); (iii) a cilnidipine group, subjected to tachypacing during treatment with cilnidipine (10 mg twice-daily; n = 8). Cilnidipine attenuated 3-week AF effects on AF duration and atrial conduction, and suppressed AF-induced increases in fibrous-tissue content, decreases in connexin-43 expression and reductions in sodium-channel expression.
Conclusions
Cilnidipine, a commercially available NTCC-blocking drug, prevents AF-induced autonomic, electrical and structural remodelling, along with associated AF promotion.
Keywords: Atrial fibrillation, N-type Ca2+ channel, Ca2+ channel blocker, Autonomic nervous system, Cilnidipine, Fibrosis
Graphical Abstract

1. Introduction
Atrial fibrillation (AF) is the most common arrhythmia in clinical practice, and is associated with increased morbidity and mortality.1,2 Classical anti-arrhythmic agents have potential adverse effects and are relatively ineffective. Safer and/or more efficacious drugs are needed for AF treatment.
A wide range of voltage-dependent Ca2+ channels have been described, including L-type Ca2+-channels (LTCCs) responsible for cardiac excitation-contraction coupling, T-type Ca2+-channels (TTCCs) involved in cardiac automaticity, and N-type Ca2+-channels (NTCCs), located predominantly on neurons.3 NTCCs regulate the release of several neurotransmitters, including glutamate, γ-aminobutyric acid, acetylcholine, dopamine, and norepinephrine (reviewed in Ref.4). In the cardiovascular system, NTCCs are particularly involved in the control of sympathetic function.5 Norepinephrine is synthesized in neuronal cell bodies. It is then transported and concentrated in vesicles located in nerve-varicosities adjacent to adrenergic receptors, pending release upon neural depolarization via Ca2+-entry through NTCCs.6–8
Autonomic nervous system (ANS) activity regulates atrial electrophysiology and risk/persistence of AF. ANS dysfunction can promote AF and is a consequence of AF-related remodelling.9 Sympathetic nerve activity is associated with the pathogenesis of AF.9,10 Norepinephrine released from sympathetic nerve terminals activates cardiomyocyte β-adrenergic receptors, causing abnormal Ca2+-handling and arrhythmogenesis.9 Strategies to reduce sympathetic nerve activity may thus have protective value against AF.
Several studies have demonstrated that pharmacological blockade or genetic deletion of NTCCs alters sympathetic nerve activity. Cilnidipine blocks both LTCCs and NTCCs, and is clinically used as an antihypertensive drug. Based on the role of the ANS in AF and atrial remodelling,9 we hypothesized that NTCC blockade might protect against AF-associated remodelling. Accordingly, this study evaluated the effects of NTCC blockade on the development of atrial remodelling and AF vulnerability in dogs with electrically maintained AF.
2. Methods
2.1 Animal model
Animal handling procedures were approved by the local Animal Research Ethics Committee and complied with National Institutes of Health Guidelines for the care and use of laboratory animals. Dogs were initially anesthetized with ketamine (5.3 mg/kg, intravenous), diazepam (0.25 mg/kg, intravenous), and isoflurane (1.5%), intubated, and ventilated. A pacing lead was inserted into the right atrial appendage (RAA) via a jugular vein under fluoroscopic guidance and attached to a subcutaneous pacemaker implanted in the neck. After 72 h for post-operative recovery, AF was maintained continuously by atrial tachypacing (AT-P) at 600 bpm for 7 days (short-term AF groups) or 21 days (longer-term AF groups).
2.2 Experimental groups
Forty-seven mongrel dogs (weight, 23.4–39.4 kg) were studied in the following seven groups (Supplementary material online, Figure S1). Short-term AF dogs: (i) a Sham group, instrumented but without AT-P or drug therapy (n = 5); (ii) a placebo AF group, maintained in AF by AT-P with placebo twice a day (n = 6); (iii) a control AF group receiving nifedipine (10 mg orally twice a day; n = 5); and (iv) a cilnidipine AF group, treated with cilnidipine (10 mg orally twice a day; n = 7). Longer-term AF dogs: (v) a Sham group, instrumented but without AT-P or drug therapy (n = 8); (vi) a placebo group, maintained in AF by AT-P with placebo twice a day (n = 8); and (vii) a cilnidipine AF group, treated with cilnidipine (10 mg orally twice a day; n = 8). Drug therapy was initiated 3 days before tachypacing onset. Nifedipine and cilnidipine were purchased from pharmaceutical companies (Bayer Yakuhin, Ltd, Japan and Mochida Pharmaceutical Co. Ltd, Japan, respectively). The experimenter was blinded to dog therapy-assignment until the experiments were completed and data analysed.
2.3 Open-chest electrophysiological study
Dogs were anesthetized with morphine (2 mg/kg, s.c.) and α-chloralose (120 mg/kg, i.v. load, 29.25 mg/kg/h maintenance), and mechanically ventilated. Body temperature was maintained at 37°C. The pacemaker was deactivated and a median sternotomy performed. In dogs with sustained AF despite pacemaker-cessation on the open-chest study day, AF was direct-current (DC) cardioverted for the electrophysiological study (EPS). Bipolar electrodes were hooked into the RAA and left atrial (LA) appendage for recording and stimulation. In short-term AF dogs, a mapping-electrode array was sutured to the right atrial epicardial surface to measure conduction velocity. LA effective refractory period (ERP) was measured at a basic cycle length of 300 ms with eight basic (S1) stimuli, followed by an S2 with 5-ms decrements (all pulses twice-threshold, 2 ms). The vulnerability to AF induction at each site was defined by the ability of a single S2 to induce, in a reproducible fashion, AF that lasted >1 s. Overall vulnerability in each dog was defined as the percentage of six pacing sites at which AF was inducible. To measure AF duration as an index of AF promotion by remodelling, AF was induced with 2-s burst pacing (25-Hz, 4× threshold current). Mean AF duration in each dog was based on 10 AF inductions. Prolonged AF (>20 min) was terminated by DC cardioversion. A 20-min rest period was allowed before continuing measurements. If prolonged AF was induced twice, no further AF induction was performed. At the end of each experiment, while under continued anaesthesia, dogs were euthanized via exsanguination by cutting open the aorta to remove the heart for further ex-vivo dissection and/or study.
2.4 Heart rate variability
In placebo, nifedipine and cilnidipine short-term AF groups, the ECG was continuously recorded for 24 h with Holter monitoring. We examined variations in heart rate on day 2 (2 days after the start of drug administration, but before AT-P) and during AF on day 9 (after 1 week of AT-P). Heart rate variability (HRV) measurements included: AVNN (average of all NN intervals), RMSSD (root mean square of successive differences), and pNN50 (the proportion of pairs of NNs that differ by more than 50 ms divided by total number of NNs). To obtain these results, we analysed 5-min data segments during each hour (e.g. 8:00–8:05, 9:00–9:05, 10:00–10:05, etc.) and then calculated the average of the results for each three consecutive-hour block (e.g. 5–8 am, 8–11 am, etc.). The first analysable five consecutive minutes of each hour (i.e. free of excessive noise and/or artefacts) were used for analysis.
2.5 Measurement of plasma norepinephrine concentration
In placebo-, nifedipine-, and cilnidipine-treated 1-week AF groups, citrated blood samples from a femoral vein were obtained before drug administration and after 7-day AT-P. Plasma was collected and stored at −80°C. The norepinephrine concentration was measured with an enzyme-linked immunosorbent assay kit (Labor Diagnostika Nord GmbH & Co. KG, Germany).
2.6 Real-time quantitative reverse transcription polymerase chain reaction
Isolated dog LA-samples were homogenized in a lysis buffer, and RNA was isolated with Nucleospin RNA II (Macherey Nagel, Germany), including DNase treatment to prevent genomic contamination. Messenger RNAs (mRNAs) were reverse-transcribed with the High-Capacity Reverse Transcription Kit (Applied Biosystems). Quantitative polymerase chain reaction (PCR) was performed with TaqMan probes and primers from Applied Biosystems for: housekeeping genes HPRT and β2-microglobulin, KCNJ2, collagen-1 (COL1A1 and COL1A2), collagen-3 (COL3A1), fibronectin-1(FBN1), and fibrillin-1 (FN1). SyBr green primers were used to quantify: LTCC (CACNA1C), Kv1.4 (KCNA4), Kv4.3 (KCND3), ryanodine receptors (RYR2), SERCA2A, phospholamban (PLN), the Na+-Ca2+ exchanger (NCX1), NTCC (CACNA1B), connexin-43 (GJA1), and connexin-40 (GJA5). The geometric-mean expression of HPRT and β2-microglobulin was used for normalization. Quantitative PCR reactions were performed with Taqman Gene Expression Master Mix (Applied Biosystems) on a Stratagene MX3000. Gene expression values were calculated by the 2−ΔCt method and normalized based on the Sham group.
2.7 Western blot
Protein samples were separated by electrophoresis on 4–20% sodium dodecyl sulphate polyacrylamide gels and transferred electrophoretically onto polyvinylidene difluoride membranes. Membranes were blocked in a Tris-buffered saline-solution (TBS) containing 0.2% (volume/volume) Tween-20 and 5% (weight/volume) BSA. They were then incubated overnight at 4°C with primary antibodies diluted in TBS containing 0.2% Tween-20 and 1% BSA. After washing with TBS-Tween/1% BSA, membranes were hybridized with horseradish peroxidase-conjugated secondary antibody. Immunoreactive bands were detected by electrochemiluminescence with BioMax MS/MR films. Protein quantification was obtained with Quantity One software (Bio-Rad). All expression data are relative to glyceraldehyde-3-phosphate dehydrogenase staining for the same samples on the same gels. Antibodies used were: GJA1 (AB1727, Merck Millipore), P-Cx43(Ser368) (48-3000, Thermofischer Scientific), and GJA5 (36-4900, Thermofischer Scientific).
2.8 Histology
Sections (6 μm) were cut at room temperature and stained with Masson’s Trichrome. Stained images were digitized and the fibrotic area was analysed with Image Pro 9.3 (Media Cybernetics, Rockville, MD, USA). LA-fibrosis was quantified by an observer blinded to group and expressed as per cent cross-sectional area.
2.9 Echocardiography
Transthoracic echocardiography was performed at baseline and on the last study day prior to euthanasia in longer-term AF dogs. An M3S probe (2.0–4.3 MegaHerz) and a Vivid 7 Dimension system (GE Healthcare Ultrasound, Horten, Norway) were used, under sedation with acepromazine (0.07 mg/kg i.m.). The biplane Simpson method was used to determine LV volumes and LV ejection fraction (LVEF). The average of three to six cardiac cycles was used for each measurement, with the operator blinded to treatment assignment.
2.10 Optical mapping
The LA was dissected free and perfused through its coronary artery with Krebs solution (mM: 120 NaCl, 4 KCl, 1.2 MgSO4 0.7, 1.2 KH2PO4, 25 NaHCO3, 5.5 glucose, 1.25 CaCl2, 95% O2/5% CO2) at 30 mL/min and 37°C. Any leak from arterial branches was ligated with silk thread to maintain adequate perfusion. After 30 min for stabilization and electrical/mechanical decoupling with blebbistatin (15 μM), the heart was loaded with di-4-ANEPPS (Biotium, Inc., Hayward, CA, USA). A charge-coupled device (CardioCCD, RedShirtImaging, LLC, Decatur, GA, USA) was used to record LA free wall fluorescence at 2 kHz focused on a 15 × 15-mm square region. Bipolar electrodes were used to pace the LA appendage. Optical maps were obtained during 1.5× threshold current 2-ms stimulation at a cycle length of 300 ms. Data were analysed with custom-written software algorithms.
2.11 Data analysis
Continuous variables are expressed as mean ± SEM. Multiple group comparisons were obtained with one-way ANOVA and Tukey’s tests (for normally distributed data) or Steel-Dwass test (for non-normally distributed data) for non-repeated measures. Repeated-measures analyses were performed for normally distributed data with two-way ANOVA and Tukey’s tests. Distribution normality was tested with a Shapiro–Wilk test. Two-tailed P < 0.05 indicated statistical significance. Wherever possible, group data are shown in figures as dot-plots of individual-animal results, along with group means and standard errors as horizontal lines. The authors had full access to and take responsibility for the integrity of the data. All authors have read and agreed to the manuscript as written.
3. Results
3.1 Effects of NTCC blockade on remodelling caused by 7 days of AF
3.1.1 Hemodynamic and electrophysiologic changes
The characteristics of experimental animals at end-study are summarized in Table 1. Body weight averaged about 30 kg and did not differ among groups. There were no significant differences in systolic or diastolic blood pressures, whereas LV end-diastolic, LA, and RA pressures were increased in the dogs with 7-day AF.
Table 1.
Characteristics of dogs in various acute-AF groups at end–study
| Sham | Placebo | Nifedipine | Cilnidipine | |
|---|---|---|---|---|
| (n = 5) | (n = 6) | (n = 5) | (n = 7) | |
| Body weight (kg) | 30.4 ± 1.5 | 31.5 ± 1.7 | 32.6 ± 3.3 | 29.1 ± 1.0 |
| Systolic BP (mmHg) | 144 ± 9 | 120 ± 7 | 120 ± 6 | 119 ± 10 |
| Diastolic BP (mmHg) | 79 ± 3 | 80 ± 5 | 71 ± 4 | 75 ± 6 |
| RAP (mmHg) | 3.0 ± 0.7 | 8.0 ± 0.7* | 8.1 ± 1.8* | 7.8 ± 0.8* |
| LAP (mmHg) | 2.8 ± 0.3 | 9.8 ± 0.4* | 10.4 ± 1.7* | 11.0 ± 1.2* |
| LVEDP (mmHg) | 6.5 ± 0.3 | 12.2 ± 0.7* | 11.7 ± 2.0* | 11.2 ± 1.6* |
P < 0.05 vs. Sham.
Placebo-, nifedipine-, and cilnidipine-treated dogs subjected to 7-day AF showed significantly increased vulnerability to AF induction (Figure 1A). There were no statistically significant differences in AF duration among the groups (Figure 1B). Placebo-treated dogs subjected to 7-day AF had significantly reduced ERP (Figure 1C). This effect was unaltered by nifedipine, but was significantly attenuated by cilnidipine (Figure 1C). Conduction velocities in the RA were significantly greater in the nifedipine group vs. the cilnidipine dogs (Figure 1D).
Figure 1.

Effects of cilnidipine on AF features after 1-week AF. Intergroup differences in AF vulnerability (A), AF duration (B), RAA ERP (C), left atrial appendage (LAA) ERP (D), and conduction velocity (E). Cilnidipine significantly attenuated RAA ERP changes caused by AF and quantitatively reduced AF vulnerability. For all graphs, n = 5, 6, 5, and 6 dogs for Sham, AF + Placebo, AF + Nifedipine, and AF + Cilnidipine, respectively; except for AF duration, for which data for 7 dogs were available for AF + Cilnidipine. *P < 0.05, **P < 0.01, ***P < 0.001 vs. Shams, by one-way ANOVA with Tukey’s test. #P < 0.05 vs. AF + cilnidipine, by one-way ANOVA with Tukey’s test.
3.1.2 ANS function
Differences between the effects of nifedipine and cilnidipine were expected to be related to altered autonomic tone associated with NTCC-block. We used HRV as an index of cardiac autonomic function in placebo-, nifedipine-, and cilnidipine-treated AF dogs (Figure 2; Supplementary material online, Figure S2). AVNN was decreased after 1-week AF in all groups. HRV, as assessed by pNN50, and rMSSD was also significantly reduced by AF-induced remodelling. This effect was unaltered by nifedipine, but was attenuated by cilnidipine, which eliminated the differences between baseline and AF conditions. We also found that plasma norepinephrine concentration, an indicator of sympathetic nerve activity, was significantly increased by 1-week AF with or without nifedipine, but significantly reduced by cilnidipine treatment (Figure 3).
Figure 2.

The effect of cilnidipine on HRV. Cilnidipine attenuated AF-induced changes in these indices of autonomic tone (see also Supplementary material online, Figure S2). Left, average of all NN intervals (AVNN). Middle, the proportion of the number of pairs of NNs that differ by more than 50 ms divided by total number of NNs (pNN50). Right, root mean square of the successive differences (rMSSD). The X-axis labels represent 3-h blocks at the times of the day indicated. *P < 0.05, **P < 0.01 by two-way ANOVA (n = 5, 6, 5, and 7 dogs for Sham, AF + Placebo, AF + Nifedipine, and AF + Cilnidipine, respectively).
Figure 3.
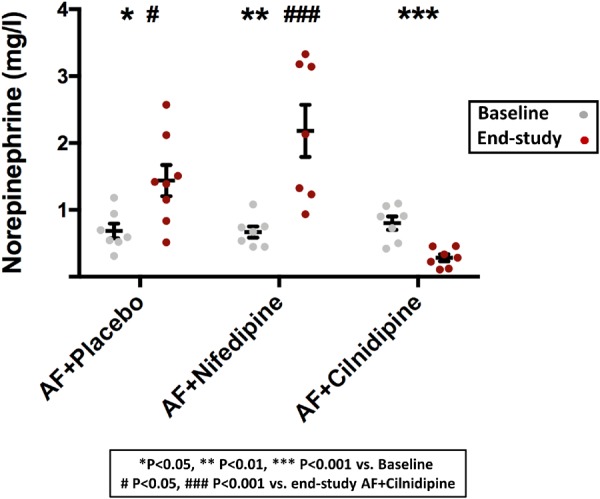
The effect of cilnidipine on plasma norepinephrine concentration. In contrast to AF alone or AF + nifedipine, both of which significantly increased norepinephrine plasma concentration at end-study relative to baseline, cilnidipine significantly reduced it in AF dogs (n = 7 dogs/group). Blood samples were taken before and 7 days after AT-P onset. *P < 0.05, **P < 0.01, ***P < 0.001 for End-study vs. Baseline, by one-way ANOVA with Tukey’s test. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. End-study AF + cilnidipine, by one-way ANOVA with Tukey’s test.
3.1.3 Expression of ion channel- and Ca2+ handling-related genes
Supplementary material online, Figures 3A, B show the expression profiles of a range of ion channel and transporter genes, including K+-channel subunit, NTCC and LTCC subunit, and Ca2+-handling genes. KCNA4, CACNA1C, and RYR2 were down-regulated after 1 week of AF, whether in the absence or presence of cilnidipine or nifedipine. There were no statistically significant differences in the expression levels of KCND3, KCNJ2, CACNA1B, and SERCA2A among groups. NCX1 values were similar for all the AF groups, with a statistically significant decrease vs. Sham for AF + nifedipine.
3.1.4 Extracellular matrix gene expression in the atrium
Supplementary material online, Figure 3C shows the RA expression profiles of the extracellular-matrix (ECM) genes encoding collagens, fibronectin and fibrillin (COL1A1, COL1A2, COL3A1, FN1, and FBN1, respectively). The expression of all ECM genes was significantly increased vs. Sham for nifedipine-treated AF dogs, as was FN1 in the placebo group. Cilnidipine treatment significantly attenuated up-regulation of collagen genes compared with the nifedipine-treated AF group.
3.2 Effects of NTCC blockade on remodelling caused by 21 days of AF
Our 7-day study revealed attenuation of short-term AF-related remodelling by NTCC blockade. In a previous study,11 we showed that 1-week AF increases ECM-gene mRNA expression without increasing collagen content or causing fibrosis. A possible explanation for the lack of fibrosis despite collagen up-regulation is that insufficient time had passed for collagen-protein accumulation. It was therefore of interest to assess whether cilnidipine can prevent the eventual development of AF-related fibrosis, an important determinant of clinical outcome,12 with longer-duration AF. We addressed this issue in a follow-up study comparing 21-day Sham dogs with AF dogs treated with placebo or cilnidipine.
3.2.1 Electrophysiologic and haemodynamic changes
Placebo- and cilnidipine-treated dogs subjected to 21-day AF showed significantly increased vulnerability to AF induction (Figure 4A). Although vulnerability was significantly less in the cilnidpine-group compared with the placebo group, it remained significantly increased vs. Sham. Placebo-treated AF dogs showed significantly increased AF duration (Figure 4B). In the cilnidipine-treated AF group, mean AF duration was almost 60% shorter than in the placebo group, and not significantly different from Sham. Placebo-treated dogs had significantly reduced ERP (Figure 4C, D) and this effect was significantly attenuated by cilnidipine. Optically mapped LA conduction velocities were decreased in the placebo-treated AF group (by 42%); this effect was significantly attenuated (to a 17% decrease) in the cilnidipine group (Figure 4E).
Figure 4.

Effects of cilnidipine on AF substrate and conduction properties after 3-week AF. Intergroup differences in AF-vulnerability (A), AF-duration (B), RAA ERP (C), LAA ERP (D), and conduction velocity (E). Cilnidipine significantly attenuated AF-induced changes in AF vulnerability, atrial ERP, and conduction velocity (n = 8 dogs/group for A–D; 6 for E). *P < 0.05, **P < 0.01, ***P < 0.001 vs. Sham, by one-way ANOVA with Tukey’s test. #P < 0.05, ##P < 0.01 vs. AF + cilnidipine, by one-way ANOVA with Tukey’s test.
LVEF was reduced at end-study by an absolute value of about 15% in AF dogs (Supplementary material online, Figure S4), to an equivalent extent in both placebo- and cilnidipine-treated groups.
3.2.2 Expression of genes potentially involved in conduction changes
We then examined the expression of genes that could be involved in the conduction-slowing caused by AF. Placebo-treated AF dogs showed significantly decreased SCN5A (Na+-channel gene) mRNA expression, an effect fully prevented by cilnidipine (Figure 5A). Connexin-43 (GJA1) mRNA was down-regulated in the placebo AF group, an effect also fully prevented by cilnidipine. Connexin-40 (GJA5) expression was not changed (Figure 5B). Collagen-gene expression was significantly enhanced in AF dogs for both subtypes 1 and 3 (Figure 5C), an effect that was significantly attenuated by cilnidipine. Changes in connexin protein expression (Figure 6) paralleled those in mRNA.
Figure 5.

The effect of cilnidipine on mRNA expression of various genes related to AF-induced conduction changes: (A) sodium-channel gene SCN5A, (B) connexin genes GJA1 and GJA5, (C) collagen genes Col1A1 and Col3A1. AF-induced gene dysregulation was significantly attenuated by cilnidipine (n = 8 dogs/group). *P < 0.05, **P < 0.01, ***P < 0.001 vs. Sham, by one-way ANOVA with Tukey’s test. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. AF + cilnidipine, by one-way ANOVA with Tukey’s test.
Figure 6.

The effects of cilnidipine on connexin protein expression. (A) Examples of original western blots. (B) Mean data for total and phosphorylated connexin-43 (Cx43). (C) Connexin-40 (Cx40). Cilnidipine significantly increased total Cx43 expression in AF dogs vs. placebo (n = 5 dogs/group). #P < 0.05 vs. AF + cilnidipine, by one-way ANOVA with Tukey’s test.
3.2.3 Effects on fibrous-tissue content
Examples of Masson-trichrome stained atrial tissue sections are shown in Figure 7A–C. Fibrous-tissue content was increased in the AF samples. Figure 7D shows overall data and quantitatively confirms the fibrosis associated with AF, with a significant, more than six-fold increase in atrial fibrous-tissue content in AF dogs that showed statistically significant attenuation under cilnidipine therapy.
Figure 7.

The effects of cilnidipine on atrial fibrosis. (A–C) Examples of original photomicrographs. (D) Mean data. Cilnidipine significantly attenuated AF-induced fibrosis (n = 8 dogs/group). *P < 0.05, **P < 0.01, ***P < 0.001 vs. Sham, by one-way ANOVA with Tukey’s test. #P < 0.05, ##P < 0.01 vs. AF + cilnidipine, by one-way ANOVA with Tukey’s test.
4. Discussion
4.1 Main findings
We have found that the N-/L-type Ca2+ channel blocker cilnidipine inhibits the electrophysiological, autonomic and structural consequences of AF-related remodelling and the AF-associated increase in AF vulnerability and AF duration; in contrast, the LTCC-selective blocker nifedipine had no protective effects. The protective effects of cilnidipine on the remodelling consequences of short-term AF are principally manifested by reductions in AF-induced ERP-abbreviation. With longer-term AF, cilnidipine also attenuates conduction velocity reductions, protecting against AF-induced fibrosis and down-regulation of sodium-channel and connexin subunits. Cilnidipine’s anti-remodelling properties were associated with suppression of the changes in autonomic tone caused by AF.
4.2 NTCCs and sympathetic nerve activity
The physiological functions of NTCCs have been studied using ω-conotoxin GVIA, a selective NTCC blocker, and by generating mice lacking CACNA1B, which encodes the α1B subunit of NTCCs.8 NTCCs predominantly regulate the release of norepinephrine at sympathetic nerve terminals.13,14 In the present study, pharmacological blockade of NTCCs by cilnidipine repressed sympathetic activity, as indicated by a reduction in plasma norepinephrine concentrations in AF dogs.
Results obtained with genetically engineered NTCC-deficient mice provide direct evidence for sympathetic nervous system regulation by NTCCs, indicating that the role of NTCCs cannot be filled by other voltage-dependent Ca2+-channels.8 In contrast, parasympathetic nervous activity in NTCC-mutant mice was nearly identical to that of wild-type mice,8 suggesting that channel-types other than NTCCs play a role in controlling parasympathetic activity. In the present study, AF dogs showed significantly altered autonomic balance as reflected by changes in HRV; cilnidipine treatment prevented this change.
4.3 Cilnidipine pharmacology
Cilnidipine is classified as a ‘fourth-generation’ Ca2+-channel blocker (CCB).6 Cilnidipine’s dissociation constant (Kd) for NTCCs is at least an order of magnitude smaller that of nine other CCBs.15 Along with a larger Kd for LTCC blockade than the other agents, this difference provides cilnidipine with ≥20-fold selectivity for NTCCs over LTCCs, vs. 2.3- to 800-fold greater potency for LTCCs over NTCCs for the other agents, indicating about 45 to 1600 fold greater NTCC selectivity for cilnidipine.
Studies in patients and well-controlled animal models suggest that the drug has a variety of unique actions that are mediated by NTCC blockade. These include the suppression of sympathetic nervous overactivity,16,17 protection against the consequences of adverse ventricular remodelling,14,18,19 renal protection,20,21 and improvement of insulin resistance.22 In addition, cilnidipine prevents the increase in plasma angiotensin II levels that occur in spontaneously hypertensive rats23 and in a canine model of a chronic atrioventricular block that exhibits ventricular electrical remodelling.24 These actions contrast with those of the LTCC blocker amlodipine, which increases plasma angiotensin II levels.23 Cilnidipine also directly suppresses aldosterone secretion from adrenocortical cells.25 These results suggest that NTCCs are important regulators of renin-angiotensin-aldosterone system (RAAS) activity.
Accumulating evidence points to a significant role of the RAAS in the development of atrial fibrosis, thereby contributing to AF development.26 Interventions that inhibit the RAAS prevent promotion of atrial fibrosis and fibrillation in animal models independently of haemodynamic actions.27,28 RAAS inhibition by cilnidipine’s NTCC-blocking action might have contributed to attenuating AF-associated atrial ECM-gene activation (Supplementary material online, Figure S3C; Figure 5C) and LA fibrosis (Figure 7). In contrast to cilnidipine, treatment with an LTCC blocker without significant NTCC inhibiting activity (nifedipine) during short-term AF significantly increased plasma norepinephrine concentrations and enhanced collagen-expression vs. short-term AF alone.
4.4 Autonomic remodelling in AF
There is extensive evidence that ANS function is a key determinant of AF. Jayachandran et al.29 first reported that electrically maintained AF is associated with heterogeneous changes in atrial innervation. These changes were subsequently related to regional sympathetic hyperinnervation and nerve sprouting.30 Consistent with these observations, cardiac ganglionated plexus ablation attenuates the enhanced atrial vulnerability resulting from 1 week of atrial tachycardia remodelling in dogs,31 although ganglionated plexus ablation affects both sympathetic and parasympathetic nerves while NTCCs appear to regulate only sympathetic nerve activity.8 The present study adds to this literature by showing that a drug that attenuates the autonomic dysregulation associated with AF-related remodelling by reducing sympathetic outflow suppresses the concomitant electrophysiological, structural, and AF-vulnerability changes.
4.5 Potential implications
To our knowledge, our study is the first to evaluate the effects of a drug with NTCC-blocking properties on arrhythmogenic atrial remodelling. Our results show that cilnidipine suppresses a variety of consequences of AF-related remodelling, including changes in autonomic function, abbreviations in atrial refractoriness, increases in AF-vulnerability, and atrial fibrosis. Cilnidipine is presently used clinically in East Asia, India, and some European countries to treat hypertension and has limited adverse effects. It may thus be an interesting adjunct in the treatment of AF, particularly in patients (like hypertensives) with clinical indications for the drug. Since hypertension is the condition most commonly associated with AF, this consideration may apply to a substantial number of individuals.
Our observation of the suppression of both AF-induced atrial fibrosis and changes in connexin expression by cilnidipine raises the interesting possibility that autonomic modulation may influence not only AF-related electrical remodelling, but also structural remodelling. Kusunose et al.32 showed previously that vagal-nerve stimulation suppresses LA-fibrosis resulting from 2-week ventricular-tachypacing induced cardiomyopathy. Further work to explore the translational importance of these observations might be of value. Since cilnidipine is a clinically available and well-tolerated drug, randomized controlled trials of this agent in the prevention of AF-related remodelling and/or AF recurrence might be of interest. Furthermore, cilnidipine may prove to be a useful lead agent for the development of new upstream drug therapy approaches to AF-management.
4.6 Potential limitations
Although we observed attenuating effects of cilnidipine on AF-induced ERP-shortening, the drug did not affect AF-related down-regulation of LTCC subunits (encoded by CACNA1C), believed to be a central contributor to atrial tachycardia-induced electrical remodelling.33 Rate-related intracellular Ca2+-loading, which activates Ca2+/calmodulin/calcineurin signalling, is a primary signal for CACNA1C-down-regulation. Since CACNA1C-subunit down-regulation was not affected by cilnidipine, the ERP changes that we saw with the drug likely reflect an attenuation of AF-related autonomic remodelling rather than altered ion-current remodelling. Ganglionated plexus ablation has been previously reported to similarly prolong atrial ERP in atrial tachycardia remodelled dogs.31 However, we have not investigated the potential effects of NTCC blockade on other remodelling-affected determinants, like other subunits contributing to IK1, IKACh, ICaL, etc., as well as parasympathetic innervation.
No conduction velocity changes were found with 1 week of AF. This finding is consistent with previous work showing that 7-day atrial tachycardia remodelling does not change atrial conduction.11,34 We previously reported that atrial fibrous-tissue content is unchanged after 7 days of electrically maintained AF in dogs, despite ECM-gene up-regulation and evidence of fibroblast activation.11 It is likely that time is required for the ECM remodelling that produces atrial fibrosis; thus, while the genes involved are activated after 7 days of AF, the accumulation of significant collagen deposits requires at least two additional weeks as shown in the present study.
Nifedipine was not evaluated in the longer-term AF study. We observed no signal for efficacy of nifedipine in preventing any component of AF-associated remodelling at 7 days, and therefore considered it inappropriate to include additional nifedipine-treated animals in the longer-term study.
5. Conclusions
Here, we studied for the first time the effects of a commercially available drug with NTCC-blocking action on AF-related remodelling. The results show that NTCC inhibition is associated with a variety of potentially beneficial effects, including suppression of electrophysiological remodelling, autonomic dysregulation, structural remodelling, and arrhythmia promotion. These observations provide new insights into the mechanisms of AF-related atrial remodelling and have potential therapeutic implications.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Supplementary Material
Acknowledgements
The authors thank Jennifer Bacchi for secretarial help with the manuscript, and Nathalie L’Heureux and Chantal St-Cyr for technical assistance. They also thank Dr. Yoshihiro Sobue for helpful advice regarding EPS performance in the dogs.
Conflict of interest: K.T. is affiliated with the endowed department sponsored by Abbott. J.B.G. received an unrestricted grant in aid of fellowship support from Abbott. The other authors have no potential conflicts to disclose.
Funding
The Canadian Institutes of Health Research [Foundation Grant 148401] and the Heart and Stroke Foundation of Canada [G-16-00012708 to S.N].
Time for primary review: 35 days
References
- 1. Nattel S. New ideas about atrial fibrillation 50 years on. Nature 2002;415:219–226. [DOI] [PubMed] [Google Scholar]
- 2. Brand FN, Abbott RD, Kannel WB, Wolf PA.. Characteristics and prognosis of lone atrial fibrillation. 30-year follow-up in the Framingham Study. JAMA 1985;254:3449–3453. [PubMed] [Google Scholar]
- 3. Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J.. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev 2005;57:411–425. [DOI] [PubMed] [Google Scholar]
- 4. Kuwahara K, Kimura T.. The organ-protective effect of N-type Ca(2+) channel blockade. Pharmacol. Ther 2015;151:1–7. [DOI] [PubMed] [Google Scholar]
- 5. Mori Y, Nishida M.. Shimizu S, Ishii M, Yoshinaga T, Ino M, Sawada K, Niidome T. Ca(2+) channel alpha(1B) subunit (Ca(V) 2.2) knockout mouse reveals a predominant role of N-type channels in the sympathetic regulation of the circulatory system. Trends Cardiovasc. Med 2002;12:270–275. [DOI] [PubMed] [Google Scholar]
- 6. Takahara A. Cilnidipine: a new generation Ca channel blocker with inhibitory action on sympathetic neurotransmitter release. Cardiovasc. Ther 2009;27:124–139. [DOI] [PubMed] [Google Scholar]
- 7. Molderings GJ, Likungu J, Göthert M.. N-Type calcium channels control sympathetic neurotransmission in human heart atrium. Circulation 2000;101:403–407. [DOI] [PubMed] [Google Scholar]
- 8. Ino M, Yoshinaga T, Wakamori M, Miyamoto N, Takahashi E, Sonoda J, Kagaya T, Oki T, Nagasu T, Nishizawa Y, Tanaka I, Imoto K, Aizawa S, Koch S, Schwartz A, Niidome T, Sawada K, Mori Y.. Functional disorders of the sympathetic nervous system in mice lacking the alpha 1B subunit (Cav 2.2) of N-type calcium channels. Proc Natl Acad Sci USA 2001;98:5323–5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen P-S, Chen LS, Fishbein MC, Lin S-F, Nattel S.. Role of the autonomic nervous system in atrial fibrillation: pathophysiology and therapy. Circ Res 2014;114:1500–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sharifov OF, Fedorov VV, Beloshapko GG, Glukhov AV, Yushmanova AV, Rosenshtraukh LV.. Roles of adrenergic and cholinergic stimulation in spontaneous atrial fibrillation in dogs. J Am Coll Cardiol 2004;43:483–490. [DOI] [PubMed] [Google Scholar]
- 11. Harada M, Luo X, Qi XY, Tadevosyan A, Maguy A, Ordog B, Ledoux J, Kato T, Naud P, Voigt N, Shi Y, Kamiya K, Murohara T, Kodama I, Tardif J-C, Schotten U, Van Wagoner DR, Dobrev D, Nattel S.. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation 2012;126:2051–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McGann C, Akoum N, Patel A, Kholmovski E, Revelo P, Damal K, Wilson B, Cates J, Harrison A, Ranjan R, Burgon NS, Greene T, Kim D, DiBella EVR, Parker D, MacLeod RS, Marrouche NF.. Atrial fibrillation ablation outcome is predicted by left atrial remodeling on MRI. Circ Arrhythm Electrophysiol 2014;7:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hirning LD, Fox AP, McCleskey EW, Olivera BM, Thayer SA, Miller RJ, Tsien RW.. Dominant role of N-type Ca2+ channels in evoked release of norepinephrine from sympathetic neurons. Science 1988;239:57–61. [DOI] [PubMed] [Google Scholar]
- 14. Yamada Y, Kinoshita H, Kuwahara K, Nakagawa Y, Kuwabara Y, Minami T, Yamada C, Shibata J, Nakao K, Cho K, Arai Y, Yasuno S, Nishikimi T, Ueshima K, Kamakura S, Nishida M, Kiyonaka S, Mori Y, Kimura T, Kangawa K, Nakao K.. Inhibition of N-type Ca2+ channels ameliorates an imbalance in cardiac autonomic nerve activity and prevents lethal arrhythmias in mice with heart failure. Cardiovasc Res 2014;104:183–193. [DOI] [PubMed] [Google Scholar]
- 15. Uneyama H, Uchida H, Konda T, Yoshimoto R, Akaike N.. Selectivity of dihydropyridines for cardiac L-type and sympathetic N-type Ca2+ channels. Eur J Pharmacol 1999;373:93–100. [DOI] [PubMed] [Google Scholar]
- 16. Takahara A, Koganei H, Takeda T, Iwata S.. Antisympathetic and hemodynamic property of a dual L/N-type Ca(2+) channel blocker cilnidipine in rats. Eur J Pharmacol 2002;434:43–47. [DOI] [PubMed] [Google Scholar]
- 17. Nagahama S, Norimatsu T, Maki T, Yasuda M, Tanaka S.. The effect of combination therapy with an L/N-Type Ca(2+) channel blocker, cilnidipine, and an angiotensin II receptor blocker on the blood pressure and heart rate in Japanese hypertensive patients: an observational study conducted in Japan. Hypertens Res 2007;30:815–822. [DOI] [PubMed] [Google Scholar]
- 18. Varagic J, Susic D, Frohlich ED.. Cilnidipine improves spontaneously hypertensive rat coronary hemodynamics without altering cardiovascular mass and collagen. J Hypertens 2002;20:317–322. [DOI] [PubMed] [Google Scholar]
- 19. Takemori K, Ishida H, Dote K, Yamamoto K, Ito H.. Prophylactic effects of an N- and L-type Ca2+ antagonist, cilnidipine, against cardiac hypertrophy and dysfunction in stroke-prone, spontaneously hypertensive rats. Can J Physiol Pharmacol 2005;83:785–790. [DOI] [PubMed] [Google Scholar]
- 20. Fujita T, Ando K, Nishimura H, Ideura T, Yasuda G, Isshiki M, Takahashi K.. Antiproteinuric effect of the calcium channel blocker cilnidipine added to renin-angiotensin inhibition in hypertensive patients with chronic renal disease. Kidney Int 2007;72:1543–1549. [DOI] [PubMed] [Google Scholar]
- 21. Kojima S, Shida M, Yokoyama H.. Comparison between cilnidipine and amlodipine besilate with respect to proteinuria in hypertensive patients with renal diseases. Hypertens Res 2004;27:379–385. [DOI] [PubMed] [Google Scholar]
- 22. Takeda S, Ueshiba H, Hattori Y, Irie M.. Cilnidipine, the N- and L-type calcium channel antagonist, reduced on 24-h urinary catecholamines and C-peptide in hypertensive non-insulin-dependent diabetes mellitus. Diabetes Res Clin Pract 1999;44:197–205. [DOI] [PubMed] [Google Scholar]
- 23. Konda T, Enomoto A, Aritomi S, Niinuma K, Koganei H, Ogawa T, Nitta K.. Different effects of L/N-type and L-type calcium channel blockers on the renin-angiotensin-aldosterone system in SHR/Izm. Am J Nephrol 2009;30:155–161. [DOI] [PubMed] [Google Scholar]
- 24. Takahara A, Nakamura Y, Wagatsuma H, Aritomi S, Nakayama A, Satoh Y, Akie Y, Sugiyama A.. Long-term blockade of L/N-type Ca(2+) channels by cilnidipine ameliorates repolarization abnormality of the canine hypertrophied heart. Br J Pharmacol 2009;158:1366–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aritomi S, Wagatsuma H, Numata T, Uriu Y, Nogi Y, Mitsui A, Konda T, Mori Y, Yoshimura M.. Expression of N-type calcium channels in human adrenocortical cells and their contribution to corticosteroid synthesis. Hypertens Res 2011;34:193–201. [DOI] [PubMed] [Google Scholar]
- 26. Van Wagoner DR, Nattel S.. Insights into mechanisms linking cardiac hypertrophy and atrial fibrosis: evidence for a role of histone deacetylase in atrial fibrillation pathophysiology and therapy. J Mol Cell Cardiol 2008;45:707–708. [DOI] [PubMed] [Google Scholar]
- 27. Shi Y, Li D, Tardif J-C, Nattel S.. Enalapril effects on atrial remodeling and atrial fibrillation in experimental congestive heart failure. Cardiovasc Res 2002;54:456–461. [DOI] [PubMed] [Google Scholar]
- 28. Shroff SC, Ryu K, Martovitz NL, Hoit BD, Stambler BS.. Selective aldosterone blockade suppresses atrial tachyarrhythmias in heart failure. J Cardiovasc Electrophysiol 2006;17:534–541. [DOI] [PubMed] [Google Scholar]
- 29. Jayachandran JV, Sih HJ, Winkle W, Zipes DP, Hutchins GD, Olgin JE.. Atrial fibrillation produced by prolonged rapid atrial pacing is associated with heterogeneous changes in atrial sympathetic innervation. Circulation 2000;101:1185–1191. [DOI] [PubMed] [Google Scholar]
- 30. Chang C-M, Wu T-J, Zhou S, Doshi RN, Lee M-H, Ohara T, Fishbein MC, Karagueuzian HS, Chen P-S, Chen LS.. Nerve sprouting and sympathetic hyperinnervation in a canine model of atrial fibrillation produced by prolonged right atrial pacing. Circulation 2001;103:22–25. [DOI] [PubMed] [Google Scholar]
- 31. Nishida K, Maguy A, Sakabe M, Comtois P, Inoue H, Nattel S.. The role of pulmonary veins vs. autonomic ganglia in different experimental substrates of canine atrial fibrillation. Cardiovasc Res 2011;89:825–833. [DOI] [PubMed] [Google Scholar]
- 32. Kusunose K, Zhang Y, Mazgalev TN, Van Wagoner DR, Thomas JD, Popović ZB.. Impact of vagal nerve stimulation on left atrial structure and function in a canine high-rate pacing model. Circ Heart Fail 2014;7:320–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qi XY, Yeh Y-H, Xiao L, Burstein B, Maguy A, Chartier D, Villeneuve LR, Brundel BJJM, Dobrev D, Nattel S.. Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circ Res 2008;103:845–854. [DOI] [PubMed] [Google Scholar]
- 34. Li D, Fareh S, Leung TK, Nattel S.. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation 1999;100:87–95. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.