

Abstract
The homeostasis of most organelles requires membrane fusion mediated by soluble N-ethylmaleimide–sensitive factor (NSF) attachment protein receptors (SNAREs). SNAREs undergo cycles of activation and deactivation as membranes move through the fusion cycle. At the top of the cycle, inactive cis-SNARE complexes on a single membrane are activated, or primed, by the hexameric ATPase associated with the diverse cellular activities (AAA+) protein, N-ethylmaleimide-sensitive factor (NSF/Sec18), and its co-chaperone α-SNAP/Sec17. Sec18-mediated ATP hydrolysis drives the mechanical disassembly of SNAREs into individual coils, permitting a new cycle of fusion. Previously, we found that Sec18 monomers are sequestered away from SNAREs by binding phosphatidic acid (PA). Sec18 is released from the membrane when PA is hydrolyzed to diacylglycerol by the PA phosphatase Pah1. Although PA can inhibit SNARE priming, it binds other proteins and thus cannot be used as a specific tool to further probe Sec18 activity. Here, we report the discovery of a small-molecule compound, we call IPA (inhibitor of priming activity), that binds Sec18 with high affinity and blocks SNARE activation. We observed that IPA blocks SNARE priming and competes for PA binding to Sec18. Molecular dynamics simulations revealed that IPA induces a more rigid NSF/Sec18 conformation, which potentially disables the flexibility required for Sec18 to bind to PA or to activate SNAREs. We also show that IPA more potently and specifically inhibits NSF/Sec18 activity than does N-ethylmaleimide, requiring the administration of only low micromolar concentrations of IPA, demonstrating that this compound could help to further elucidate SNARE-priming dynamics.
Keywords: phosphatidic acid, SNARE proteins, membrane fusion, membrane lipid, membrane trafficking, α-SNAP, NSF, Sec17, Sec18
Introduction
Eukaryotic life requires membrane fusion, which is integral to numerous processes involved in cellular homeostasis and distribution of biological molecules. The terminal catalysts of membrane fusion are the soluble NSF3 attachment protein receptor (SNARE) proteins, adding specificity between membranes destined to fuse resulting through SNARE compatibility (1). SNAREs interact in trans (between two membranes) to form parallel four-helix bundles capable of generating the energy needed to merge two membranes into a continuous bilayer. After a fusion event, SNAREs remain as inactive cis-SNARE complexes on this newly formed single bilayer. These complexes need to be disassembled so that a new cycle of fusion may occur. SNAREs are activated, or primed, by the AAA+ protein NSF/Sec18 (Fig. 1A). Sec18 is composed of three domains. The N terminus forms the cap domain while the D1 and D2 nucleotide-binding domains form the two rings of the hexameric complex. Both D1 and D2 domains contain ATP-binding sites; however, the hydrolysis of ATP by D1 generates most of the energy necessary to disassemble inactive SNARE bundles, while the D2 ring binds ATP to stabilize the homohexamer (2).
Figure 1.

Small molecule candidates for Sec18 binding and priming inhibition. A, left: schematic of Sec18 with labeled domains (N, D1, and D2) in its monomeric (mSec18) and hexameric (hSec18) forms. Right: schematic model of Sec18-mediated SNARE activation. Pah1 is a PA phosphatase. Sec17/α-SNAP is the adaptor protein linking Sec18 to inactive cis-SNARE complexes. (Adapted from Ref. 12.) B, structures of IPA (7-methyl-3-(4,5,6-trihydroxy-3-oxo-3H-xanthen-9-yl)bicyclo[2.2.1]hept-5-ene-2-carboxylic acid) and other small molecule candidates.
During activation, Sec18/NSF associates with SNARE complexes through binding the adaptor protein Sec17/α-SNAP that aids in disrupting SNARE complexes into active individual proteins (3). Direct regulation of Sec18/NSF activity remains mostly unknown, although protein kinase C (PKC) has been implicated in negative regulation of NSF association with SNARE complexes (4).
Currently, there are two methods of inhibiting priming in vitro. Sec18 can be blocked with an antibody or covalently modified with the alkylating agent N-ethylmaleimide (NEM), both of which inhibit the ATPase activity of Sec18/NSF (5). The exact mechanism by which alkylating with NEM affects priming is unclear and not specific. To illustrate, the inhibition of fusion by NEM requires a higher concentration than what is needed to inhibit NSF alone, suggesting the presence of multiple NEM-sensitive factors involved in fusion (6). This is due to the lack of specificity by NEM, as it only requires free thiol groups to function. Even though the dosage of NEM required to inhibit Sec18/NSF is in the millimolar range (5, 7), its promiscuity may have even been advantageous (8) to its use in adopting early models identifying the interaction of Sec18/NSF with Sec17/α-SNAP as being crucial to the continual cycle of fusion by membranes within the cell (9).
We previously demonstrated the importance of phosphatidic acid (PA) in regulating Sec18/NSF priming activity (10–12). Deletion of the PA phosphatase PAH1, the yeast orthologue of mammalian lipin1, leads to elevated concentrations of PA on the vacuole that we hypothesized sequesters Sec18 away from cis-SNAREs (10). Even when Pah1 is present, released Sec18 can be inhibited by adding soluble dioctanoyl-PA (C8-PA). Conversely, deleting the diacylglycerol (DAG) kinase DGK1 elevates vacuolar DAG levels at the cost of lowering PA concentrations while resulting in augmented fusion (13). Thus, the temporal regulation of balancing PA and DAG concentrations has a direct effect on progression through the fusion pathway. Subsequent studies showed that PA binding by monomeric Sec18/NSF triggers large conformational changes that appear to be incompatible with the assembly of the active homohexamer needed to bind and prime SNAREs (12). The major site of conformational change, as shown by molecular dynamics, is the predominant PA-binding site between the D1 and D2 domains of NSF.
Although PA serves as a natural regulator of Sec18 function, it has multiple limitations as a tool to further probe the mechanics of priming. The principal limitation with relying on PA as an inhibitor of Sec18 activity is due its insolubility, as it is part of the membrane bilayer, as well as its susceptibility to dephosphorylation by Pah1. Additionally, PA binds other proteins, including the vacuolar SNARE Vam7 (14). Finally, PA is likely to serve both as an inhibitor of Sec18 activity while being a positive regulator through its interactions with Vam7. In fact, reconstituted proteoliposome fusion systems show that PA is essential for fusion to occur when the priming stage is eliminated (15).
Taken together, the lack of NEM specificity and the duality of PA in regulating vacuole fusion was the impetus for finding a specific soluble small molecule inhibitor of NSF/Sec18 function. We used structural data of NSF (16) to computationally screen for compounds that bound to the previously mapped PA-binding site. Through this, we discovered an uncharacterized molecule that we call IPA (Inhibitor of Priming Activity). IPA bound to Sec18 with high affinity and potently blocked SNARE priming and downstream vacuole fusion. Biochemical, biophysical, and molecular dynamics examination of IPA–Sec18 complexes led us to conclude that IPA “locks” NSF/Sec18 into a rigid conformation that it incompatible with SNARE priming presumably by its ability to inhibit NSF/Sec18 binding to PA as shown below.
Results
Identification of a small molecule inhibitor of Sec18 binding to PA
Because PA acts a potent inhibitor of Sec18 function, we used computational modeling to search for small molecules that docked at the previously identified PA-binding regions of Sec18 (12). To accomplish this, we used the cryo-EM–guided resolution of the hexameric structure of NSF bound to SNAREs (17). The Schrodinger SiteMap (18) was then performed on both hexameric and monomeric forms of NSF as well as homology models of Sec18 hexameric and monomeric forms generated using Schrodinger Prime (19, 20). The top resulting binding sites for both NSF/Sec18 hexamer and monomer were docked using all compounds available from the Illinois high-throughput facility initially using Glide HTVS, and the top hits were docked using Glide XP (19). Our screen included compounds from the Illinois high-throughput screening facility, NCI Open, NCI Diversity, and the Chembridge microformat libraries, which were prepared for docking using LigPrep (Schrödinger Release 2018-2: LigPrep, Schrödinger, LLC, New York). Of the boxes examined, the 3rd and 4th had the highest average gscore for binding to PA. Compounds with the best gscore, or lowest predicted ΔG for boxes 3 and 4 using Glide HTVS, were selected to be further docked using the more computationally intensive Schrödinger XP (21). Of these compounds, 19 were selected from the NCI Diversity set according to gscore with corresponding SiteMap sites. In Fig. 1B we show the structures of the top 12 candidates for Sec18 binding, including epirubicin and 7-methyl-3-(4,5,6-trihydroxy-3-oxo-3H-xanthen-9-yl)bicyclo[2.2.1]hept-5-ene-2-carboxylic acid, the compound we have now named the inhibitor of priming activity (IPA) (Fig. 1B). In Fig. 2, A–D, we show epirubicin and IPA binding to boxes 3 and 4 with key interactions depicted by lines between Sec18 residues and the small molecules. From these docking experiments, we found that IPA and epirubicin had the highest gscores of the candidate compounds (Fig. 2E). The interactions between the D1 and D2 domains of NSF with both epirubicin and IPA structures were further examined by using a computational ensemble docking methodology to verify the poses chosen for screening (Fig. 2, F and G). Both epirubicin and IPA showed clustering at the hinge region between the D1 and D2 domains and a second distal location in the D2 domain. Epirubicin showed additional binding to a second region at the D1–D2 hinge.
Figure 2.

IPA and epirubicin binding to Sec18. A, ligand interaction diagram of IPA binding to homology model of mSec18 and receptor grid for Box 3 of homology model of Sec18 corresponding to Schrodinger Sitemap predicted site 3. Interactions are indicated with red arrows showing H-bonding, including the salt bridge between Lys-159 and Asp-374 hydrogen bonding with IPA. B, ligand interaction diagram of IPA binding to mSec18 corresponding to Schrodinger Sitemap predicted site 4. A salt bridge between Ser-378 and IPA is indicated with an arrow. C, ligand interaction diagram of epirubicin binding to receptor grid for Box 3. D, ligand interaction diagram of epirubicin binding to receptor grid for Box 4. E, bar graph depicting gscore of best IPA and epirubicin poses corresponding to Fig. 3, B–E, to boxes 3 and 4 indicated with lowest − ΔG using Schrodinger Glide and exported into GraphPad. F, IPA cluster analysis displayed and edited with VMD for IPA to D1–D2 of NSF with D1 indicated with purple and D2 with green, and the top four most populated clusters represented with dark gray for cluster 1, orange for box 2, light blue for box 3, and light gray for box 4. G, epirubicin cluster analysis displayed and edited with VMD for epirubicin to D1–D2 of NSF as in Fig. 1F.
The group of candidate compounds, including epirubicin and IPA, was further screened for their ability to inhibit vacuole fusion at 100 μm. Compounds that inhibited fusion by >50% were next screened for the inhibition of priming. From the starting pool of chemicals, significant priming inhibition was only observed with epirubicin and IPA, which were further rigorously tested (see below in Fig. 6). Epirubicin was later ruled out as a potential candidate for characterizing the mechanism for PA regulation of priming when it showed no competition for Sec18 to PA liposomes (data not shown), and it exhibited a greater than predicted KD value approaching millimolar concentrations for binding monomeric Sec18 (Fig. 6I). We attributed the ability of epirubicin to alter vacuole fusion to its integration into the membrane, as similar molecules interact with the phospholipid bilayer by insertion (22, 23).
Figure 6.

IPA inhibits vacuole fusion at the SNARE priming stage. A, in vitro vacuole homotypic fusion incubated with a concentration curve of IPA and incubated for 90 min at 27 °C. Fusion was tested by luminal mixing, proPho8 maturation, and conversion of p-nitrophenyl phosphate to p-nitrophenolate measured at 400 nm. B, gain-of-resistance kinetics assays were performed in the presence of 140 μg/ml α-Sec17p IgG, 100 μm IPA, 1 mm NEM, or PS buffer. Data were fit using first-order exponential decay with weights and errors. C, vacuoles from BJ3505 yeast were tested for priming activity as a measure of Sec17 release from the membrane fraction. Fusion reactions were incubated in the presence of buffer, NEM, C8-PA, or IPA. Vacuoles were pelleted by centrifugation at the indicated times, and proteins in the supernatant fraction were resolved by SDS-PAGE and imaged by Western blotting. Densitometry values were normalized against input sample for each condition. D, quantitation of three repeats of C. E, dose-response curve of IPA and the inhibition of priming as detailed in C. F, quantitation of three trials presented in E. G, inhibitor effects of IPA on fusion were reversed with the addition of 5.3 μm recombinant hexameric Sec18–His-8. H, vacuole damage caused by IPA was tested by the release of luminal content. Vacuole fusion reactions were incubated with buffer, 0.2% Triton X-100 (TX), 100 μm IPA, or 100 μm epirubicin (Epi) for 30 min at 27 °C. After incubation, the reactions were fractionated by centrifugation to separate membranes from the supernatant. Membrane and supernatant fractions were resolved by SDS-PAGE and probed for Pep4 and Ypt7 by Western blotting. I, affinity of Sec18 binding to epirubicin determined by labeled MST using Ni-Atto 488 dye indicating a KD of 677 ± 179 μm. J, vacuole fusion reactions were incubated with a dosage curve of epirubicin and processed as described above. Error bars represent S.E. (n = 3). **, p < 0.01.
Sec18 monomer, D1, and D2 domains bind IPA with high affinity
To further determine whether IPA could serve as a specific inhibitor of Sec18, we next measured its dissociation constant. For this purpose, we used label-free microscale thermophoresis (LF-MST), labeled MST, and surface plasmon resonance (SPR). All three techniques yielded a KD value in the low micromolar range for IPA binding to monomeric Sec18 (mSec18). Labeled MST showed that mSec18 bound to IPA with a KD of 3.84 ± 1.3 μm (Fig. 3A and Table 1). Similarly, LF-MST showed that mSec18, labeled with Ni-NTA Atto 488, bound to IPA with a KD of 7.4 ± 3.7 μm (Fig. 3B). Discrepancies between the KD values of mSec18 to IPA using MST can be explained by the presence of the Atto 488 dye in the labeled experiment or as a result of IPA having measurable light absorbance (data not shown). We further verified these affinity measurements using SPR where mSec18–His-8 was linked to an Ni-NTA Biacore chip, after which IPA was flowed and response units (RU) were measured. SPR measurements yielded a KD of 4.08 ± 0.83 μm (Fig. 3C), which was consistent with the MST results. Together, these results indicated that IPA bound Sec18 with high affinity.
Figure 3.

Sec18-binding affinities for IPA. A, fluorescence MST of IPA binding to Sec18–His-8–labeled with Atto 488 Ni-NTA dye with fluorescence converted to fraction-bound M.O. affinity software and exported in Graphpad with KD of 3.84 ± 1.3 μm using log-inhibitor versus response four-parameter equation and error using S.E. (n = 3). B, label-free MST of IPA binding to unlabeled Sec18–His-8 converted to fraction bound as in Fig. 1A with KD of 7.4 ± 3.7 μm μm using log-inhibitor versus response four-parameter equation error using S.E. (n = 3). C, SPR of IPA to Sec18–His-8 linked to a Ni-NTA biosensor chip at ∼2000 RU with response measured subtracting blank reference cell and relevant blank injections. Data were exported from BiaEvaluate and into GraphPad and fit using a one-site specific binding model indicating a KD of ∼4 μm. D, SPR sensorgrams of Ni-NTA captured D1 of ∼2500 RU at corresponding PA injection concentrations with light blue 8 μm, magenta 4 μm, blue 2 μm, and green 1 μm exported from BiaEvaluate and into Graphpad for depiction. KD was measured by Kd/Ka 320 nm with ka of about 1700 m s−1. E, SPR sensorgrams of D2 to IPA as in Fig. 2D with KD ∼1.1 μm with ka of about 1900 m s−1. F, SPR sensorgrams of D1–D2 to IPA as in Fig. 2D with KD ∼960 nm and ka of about 8000 m s−1.
Table 1.
Binding affinities for Sec18 and its domains
mSec18 is monomeric Sec18; Lip. is liposomes.
| Protein | Analyte | Method | KD |
|---|---|---|---|
| mSec18 | IPA | SPR | 4 μm |
| mSec18 | IPA | Label-free MST | 7.5 μm |
| mSec18 | IPA | Labeled MST | 3.8 μm |
| mSec18 | Epirubicin | Labeled MST | 677 μm |
| D1 | IPA | SPR | 320 nm |
| D2 | IPA | SPR | 1.1 μm |
| D1D2 | IPA | SPR | 1 μm |
| mSec18 | 5% PA Lip. 1 μm | SPR | 1.6 μm |
| mSec18 | 5% PA Lip. 4 μm | SPR | 1 μm |
| mSec18 | 5% PA Lip. 8 μm | SPR | 505 nm |
| D1 | 5% PA Lip. 8 μm | SPR | 784 nm |
| D2 | 5% PA Lip. 8 μm | SPR | 867 nm |
Next, we determined which Sec18 domain contributed most to binding IPA. Using SPR, we tested the individual D1 and D2 domains, as well as a D1–D2 continuous polypeptide. These were added to separate channels of a Ni-NTA chip with fresh protein loaded between each injection. Capture of proteins per kinetic injection on the chip ranged from 2050 to 2900 RU for D1, 1500–2500 RU for D2, and 1400–1600 RU for D1–D2. The KD of IPA for D1 was determined from the sensorgrams as 320 nm (Fig. 3D). The KD of IPA for D2 was 1.1 μm (Fig. 3E), and the KD for D1–D2 was 958 nm (Fig. 3F). We reasoned that the improved affinity for the domains was either the result of having measured the affinity via kinetic measurements as opposed to the steady-state measurements used above (Fig. 3, A–C). Importantly, the on-rate for the D1–D2 construct was over 4-fold greater than for either D1 or D2 alone indicating that there may be some cooperativity between the D1 and D2 domains upon the initial recognition of PA. Together, these data show that both Sec18 domains participate in binding IPA similar to PA shown previously (12).
IPA inhibits Sec18 binding to PA liposomes
To test whether IPA inhibits Sec18 binding to PA, we titrated IPA in the presence of PA liposomes and mSec18. Liposomes for floatation experiments (Fig. 4, A and B) were prepared as described previously (12). IPA was added at the concentrations indicated in the presence of mSec18 at a final concentration of 500 nm. This showed that IPA blocked mSec18 binding to PA liposomes with an IC50 of 3.2 μm, which was near the KD values of IPA binding to mSec18 in Fig. 3 (Fig. 4A and Table 2). These data were consistent with previous SPR competition experiments using PA containing nanodiscs (24). To examine whether IPA blocked other PA-binding proteins, we used the DEP domain from the murine protein Dvl2 (25). Previously, we used DEP to bind PA liposomes and vacuolar PA to displace Sec18 from membranes (11). Here, we tested PA liposome binding DEP in the presence of 100 μm IPA. Unlike the competition we observed with Sec18, IPA was unable to compete at greater than 50% inhibition with DEP for PA binding at this IPA concentration (Fig. 4B). This suggested that IPA was specific for the Sec18–PA binding interface. We also tested IPA against the soluble SNARE Vam7, which binds PI3P and PA (14). To ensure that the PI3P-binding domain was not contributing to PA binding we used the Y42A mutation in the PI3P-binding site of the Vam7 PX domain (26). This experiment was performed by SPR where 800 nm of extruded PA liposomes (close to the size of the yeast vacuole) were bound to an L1 Biacore chip, after which Vam7Y42A was injected and bound to saturation (Fig. 4C). When IPA was used at 100 nm, we observed no difference in Vam7Y42A binding to the PA liposomes. In comparison, IPA competed with Sec18 binding to the immobilized PA liposomes considerably at 100 nm (Fig. 4D).
Figure 4.

IPA competes for PA binding. A, Sec18 was incubated with PA liposomes in the presence or absence IPA. After incubation, the liposomes were re-isolated to detect bound and unbound Sec18 by immunoblotting with anti-Sec18 antibody. B, GST–DEP was incubated with PA liposomes in the presence of buffer or IPA. After incubation, the liposomes were re-isolated and bound, and unbound DEP was detected by immunoblotting with anti-GST antibody. C, SPR using an L1 chip captured with ∼3000 RU 5% PA (solid lines) or 2.5% PI (dotted lines) liposomes was titrated with GST-Vam7Y42A. Binding was performed by incubating a fixed protein concentration with a titration of IPA. D, SPR of tagged mSec18–His-8 bound to an L1 Chip as in C. E, His- tagged mSec18, D1, and D2 constructs binding to 0.8 μm PA liposomes at about 4000 RU captured to an L1 biosensor chip exported yielding KD values for D1 (781 ± 110 nm), D2 (866 ± 669 nm), and mSec18 (505 ± 267 nm). F, SPR competition of Sec18 constructs binding to PA liposomes titrated with IPA.
Table 2.
IPA competition for Sec18 binding and function
mSec18 is monomeric Sec18; hSec18 is hexameric Sec18; Lip. is liposomes; NA is not applicable.
| Analyte | Ligand | Competitor | Method | IC50 |
|---|---|---|---|---|
| mSec18 | 5% PA Lip. 8 μm | IPA | SPR | 762 nm |
| D1 | 5% PA Lip. 8 μm | IPA | SPR | 220 nm |
| D2 | 5% PA Lip. 8 μm | IPA | SPR | 641 nm |
| mSec18 | Sonicated Lip. | IPA | Lip. floatation | 3 μm |
| NA | Isolated vacuoles | IPA | Fusion | 50 μm |
| NA | hSec18 | IPA | Fusion priming | 50 μm |
To test whether IPA action was specific to anionic phospholipids, we further checked whether it affected binding of mSec18 and Vam7Y42A to phosphatidylinositol (PI)-containing liposomes. Both mSec18 and Vam7Y42A showed low overall binding to PI liposomes (Fig. 4D). Additionally, the inhibitory effect of IPA was absent when either Vam7Y42A or Sec18 was bound to PI liposomes. Together, these data further verify the specificity of IPA for mSec18 to PA.
We then asked whether D1 or D2 domains of Sec18 were more or less important to IPA competition of PA as both domains were previously shown to bind PA (12). First, we measured the binding of each domain to immobilized 100-nm extruded PA liposomes. In Fig. 4E, we show full-length monomeric Sec18 and the D1 and D2 domains bound with similar affinities to the immobilized PA liposomes. Specifically, the KD was 781 ± 110 nm for D1, 866 ± 669 nm for D2, and 505 ± 267 nm for mSec18. Next, IC50 values for IPA and PA binding were determined using concentrations of mSec18, D1, and D2 near or above the KD values derived from liposome binding using a constant concentration of 250 nm mSec18, 1000 nm D1, and 1000 nm D2 (Fig. 4F).
IPA inhibition of Sec18 binding to PA liposomes is not affected by membrane curvature
Next, we tested whether membrane curvature affected mSec18 binding and the effects of IPA. To this goal, we used three different extrusion filters of 100, 400, and 800 nm containing 5% PA. The differently-sized liposomes bound to an L1 chip in HBS-N buffer for SPR analysis. First, saturation was determined for each liposome type, which showed that mSec18 bound 800-nm liposomes with a KD of ∼670 nm compared with KD values of ∼1600 and ∼1000 nm to the 100- and 400-nm diameter liposomes (Fig. 5A). Although this showed that mSec18 bound more strongly to 800-nm liposomes, the binding affinities to smaller vesicles were still high. To determine whether membrane curvature affected how IPA blocked mSec18 from binding liposomes, we tested the competition of 100 nm IPA with 250 nm mSec18 for liposome binding. Reactions were incubated as before, and liposomes were re-isolated by floatation. This showed that IPA reduced Sec18 (0.5 μm) binding by 90% to 800-nm liposomes, whereas IPA inhibited binding by 80 and 70% to 400- and 100-nm liposomes, respectively (Fig. 5B). Although these results show differences in binding in the presence or absence of IPA, the fact remains that IPA potently inhibits Sec18 from interacting with PA liposomes.
Figure 5.
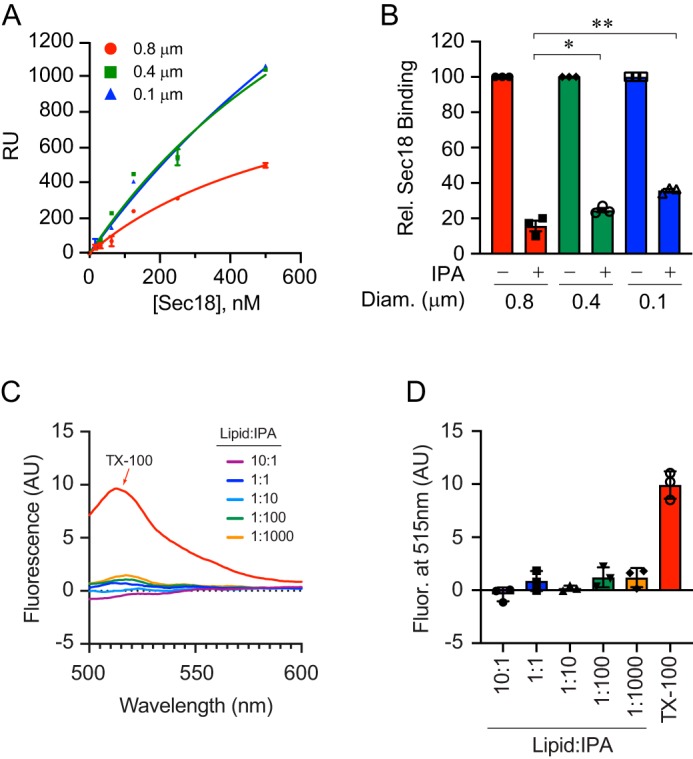
Liposome diameter affects IPA inhibition of Sec18 binding to PA. A, extruded PA liposomes of 0.1, 0.4, and 0.8 μm were bound to L1 biosensor chips. Sec18 was titrated to give a with KD 1600 ± 933, 1020 ± 549, and 673 ± 239 nm for 0.1-, 0.4-, and 0.8-μm diameter liposomes, respectively. Error bars represent S.E. (n = 3). B, as in A, Sec18 was flowed over PA liposomes attached to L1 chips in the presence or absence of 100 μm IPA. The difference in RU of Sec18 binding to liposomes in the presence of IPA was calculated relative to the maximum Sec18 bound in the absence of IPA. The bar graph represents the mean values of inhibition with individual points shown. The error bars represent S.E. (n = 3). C and D, liposome integrity in the presence of IPA was tested by calcein release. PA liposomes were extruded in the presence of 100 mm calcein, a concentration that leads to fluorescence quenching. Calcein containing liposomes were incubated with buffer, Triton X-100, or a dosage curve of IPA. Liposome damage, as measured by content leakage, was detected by the dilution of calcein and gain in fluorescence.
To determine whether IPA competition for Sec18 damages liposomes, we examined membrane integrity by using a calcein dequenching assay. Here, we used extruded 100-nm liposomes in the presence of 100 mm calcein to capture the fluorophore at self-quenching concentrations (27, 28). Calcein-loaded liposomes were treated with buffer, 0.2% Triton X-100, or a dosage curve of IPA. As controls, we found that incubating with buffer had no effect on calcein fluorescence, whereas Triton treatment led to increased fluorescence, indicating that the dye became diluted and de-quenched when the liposome was dissolved (Fig. 5, C and D). When calcein liposomes were incubated with IPA, there was no observable increase in fluorescence, demonstrating that IPA had no effect on the integrity of the liposomes.
Although IPA did not damage liposome integrity, we next asked whether this compound altered liposome diameter or dispersal. To address this, we used dynamic light scattering (DLS) to compare 80/20 PC/PE liposomes to 75/20/5 PC/PE/PA 100-nm extruded liposomes and found that there was no appreciable difference in size as between the two liposome types (Fig. S2A). To test whether IPA affected the recovery of liposomes after floatation, we used DLS and measured the peak intensities (kilocounts/s) of 100-nm extruded liposomes listed above. In the absence of IPA, ∼80% of the starting material was recovered, whereas ∼90% was recovered in the presence of 100 μm IPA (Fig. S2B). Taken together, IPA had no effect on liposome size or floatation.
IPA inhibits fusion and priming
The primary goal of finding a small molecule that specifically binds Sec18 was to use it to block SNARE priming and halt the fusion pathway. To test this, we used a vacuole fusion tester set where half of the vacuoles in a fusion reaction harbored inactive pro-Pho8 (alkaline phosphatase) and lacked the protease Pep4. The second set contained Pep4 but lacked pro-Pho8. Upon membrane fusion and content mixing Pep4 gained access to pro-Pho8 and cleaved the inhibitory pro-peptide to yield active Pho8 that can be tested as proxy for vacuole fusion efficiency. In Fig. 6A, we show that IPA blocked fusion as measured by Pho8 activity with an IC50 of ∼50 μm. The concentration needed to block fusion was notably higher that the dose needed to inhibit binding to PA liposomes. We attribute this to the accessibility of Sec18 interacting with the fusion machinery, and potentially the predominance of hexamer over monomer in this assay. Nevertheless, this is the first demonstration of a specific Sec18 ligand to inhibit membrane fusion. In comparison, inhibiting fusion with NEM requires millimolar concentrations (11).
To verify whether IPA inhibited fusion during the priming stage, we performed multiple tests. First, we tested IPA in a gain-of-resistance assay. Here, individual fusion reactions were treated with buffer as a control, IPA, or antibody against Sec17 to directly block the priming machinery. The reagents were added at different time points to ask whether fusion was still sensitive to the inhibitor. Thus, as a stage of fusion (e.g. priming) is passed, reagents that target the stage lose their efficacy. In Fig. 6B, we found that fusion reactions gained resistance to IPA with similar kinetics to those of anti-Sec17 antibody. In other words, as the bulk of fusion reactions passed the priming stage, both IPA and anti-Sec17 lost their ability to inhibit fusion. The similarity in the rate in which these reagents lost their inhibitory effect indicated that IPA only inhibited fusion at the priming stage. In parallel, untreated reactions were placed on ice to stop maximal fusion at each time point indicated, in order to establish a boundary for the final stage of fusion.
To test the effect of IPA directly on SNARE priming, we used the release of Sec17 as a measure of Sec18 function (3, 11). As Sec18 disassembles cis-SNARE complexes, the co-chaperone Sec17 is released from the membrane and accumulates in the supernatant after membranes are pelleted by centrifugation. We tested IPA compared with NEM and C8-PA and found that IPA blocked Sec17 release as strongly as NEM and C8-PA (Fig. 6, C and D). Both NEM and C8-PA inhibited priming as reported previously (11). However, both NEM and C8-PA lack specificity for Sec18; thus IPA is the first specific inhibitor of SNARE priming. Finally, we determined whether the inhibitory concentration IPA was similar for both overall fusion inhibition and SNARE priming. A range of IPA concentrations was added to vacuoles and incubated for 30 min, after which the membranes were pelleted and the supernatants collected for Western blotting. Sec17 release was blocked by IPA with an IC50 of ∼50 μm (Fig. 6, E and F), which matches the IC50 for content mixing. To determine whether the effect of IPA was reversible, we added exogenous recombinant hexameric Sec18 to fusion reactions first incubated with IPA. In Fig. 6G, we show that IPA inhibited fusion as seen above. When 5.3 μm Sec18 was added, we found that vacuole fusion was restored. Adding Sec18 alone had no effect on vacuole fusion.
We next verified whether the effect of IPA on priming was not due to membrane damage. To this goal, we incubated vacuoles in fusion reaction buffer alone or in the presence of 0.2% Triton X-100, 100 μm IPA, or 100 μm epirubicin. After incubating for 30 min at 27 °C, the reactions were fractionated by centrifugation to separate membranes from solubilized material. To monitor the release of luminal content, we probed for the soluble luminal protease Pep4 by Western blotting. Pep4 remained in the membrane fraction when vacuoles were treated with IPA, epirubicin, or buffer alone (Fig. 6H). As a control, we used Triton X-100 to solubilize the vacuoles and release Pep4 into the supernatant. We also probed for the distribution of the Rab GTPase Ypt7 as a marker for membrane proteins. Similar to what we saw with Pep4, Ypt7 remained in the membrane fractions unless the vacuoles were treated with Triton X-100. Together, this further demonstrates that IPA does not damage vacuoles.
To compare the effects IPA with another small molecule candidate, we tested epirubicin. Although epirubicin was predicted to bind Sec18, we found that it bound poorly with a KD of 677 ± 179 μm (Fig. 6I). We further tested epirubicin by using a dosage curve in fusion reactions. We found that it reduced fusion, albeit with an IC50 >400 μm (Fig. 6J). Because of the poor binding to Sec18, we attributed the inhibition of fusion by epirubicin to its ability to insert into membranes. This was evident by the bright pink coloring of liposomes or vacuoles when incubated with epirubicin (data not shown). Together, these data illustrate that even though epirubicin was initially predicted to bind Sec18 at similar sites as IPA, it was ineffective in altering Sec18 function, thus bolstering the significance of discovering IPA.
IPA does not induce conformational changes in Sec18
To determine how IPA operates, we asked whether it could alter Sec18 conformation in a manner similar to what we previously observed with PA (12). In that work, we showed that PA induced conformational changes in full-length Sec18 allowing for increased proteolytic cleavage, whereas DAG and phosphatidylserine had no effect. Here, we compared the conformational changes induced by PA binding Sec18 with any effects that IPA might have. We incubated Sec18 with a titration curve of thrombin in the absence or presence of 100 μm IPA. As shown in Fig. 7A, IPA did not result in increased thrombin cleavage. Instead, IPA protected Sec18 from cleavage relative to the buffer control. This is opposite to the effect of PA binding to Sec18, which results in enhanced cleavage. As a control, we incubated Sec18 with thrombin and PA, as described previously. As predicted, thrombin cleaved Sec18 more efficiently when C8-PA was present (Fig. 7B). Although IPA reduced Sec18 cleavage, the same major degradation products were found with buffer, IPA, or PA. The cut sites for generating these products were determined by MS. Thrombin cleaved Sec18 at Arg-225 to give a C-terminal 60-kDa fragment (p1) and Lys-378 to give the C-terminal 30-kDa fragment (p2). The 25-kDa fragment was generated by cleaving at Arg-225 and Lys-455 (p3).
Figure 7.

IPA and conformation changes in full-length Sec18. Sec18–His-8 (A and B), N-D1 (C and D), and D1–D2 (E and F) samples were incubated with buffer, IPA, or C8-PA before incubation with increasing concentrations of thrombin for 30 min. Reactions were stopped by adding SDS-PAGE loading buffer. Protein digests were resolved by SDS-PAGE and stained with One-Step Blue® protein gel stain (Biotium). Quantitation was determined for the relative amounts of uncleaved protein. n = 3 for all data shown. Mr indicates relative molecular mass markers. *, p < 0.05; **, p < 0.01; *** p < 0.001; ****, p < 0.0001. Schematics represent Sec18, N-D1, and D1–D2. Cleavage sites are depicted by the amino acid number. Major degradation products are labeled as p1, p2, and p3.
To further test the effects of IPA on Sec18, we next examined proteolytic cleavage of subdomains. In Fig. 7, C and D, we show the effects of IPA and PA on thrombin cleavage of N-D1. In reaction buffer alone, thrombin cleavage produced 31 kDa by cutting at Lys-100 and Arg-360 (p1). The 25-kDa band was a product of cutting at Arg-225 and Lys-455 (p2). As seen with full-length Sec18, we found that IPA reduced the magnitude of cleavage (Fig. 7C). This suggests that IPA either blocks thrombin sites or induces a distinct conformational change that masks cleavage sites. Interestingly, incubating with C8-PA had no effect on the level of proteolysis compared with the buffer control (Fig. 7D). This suggests that the absence of the D2 domain is needed for PA-induced conformational changes that expose thrombin sites. This does not exclude the possibility that conformational changes do not expose cleavage sites.
Next, we tested the D1–D2 continuous domains and found that thrombin cut at Arg-423 produced a C-terminal fragment (p1). A second cleavage occurred at Lys-547 to yield a 23-kDa product. Here, we found that IPA mildly reduced cleavage at the lowest thrombin concentrations (Fig. 7E). This suggests that the N-domain is required for the protective effects of IPA. In comparison, D1–D2 remained sensitive to PA binding as indicated by the increased cleavage shown in Fig. 7F. The inability of PA to induce more cleavage in D1–D2 suggests that its effect requires the N-domain as part of the entire protein to induce the full conformational change.
IPA binding blocks the effect of PA on thrombin cleavage
Because of the overlap in predicted binding sites for IPA and PA, we tested whether binding IPA first would block the effects of PA on Sec18 cleavage by thrombin. We indeed found that 100 μm IPA protected Sec18 from cleavage (Fig. 8). We interpret this as one of two possibilities. One possibility is that IPA directly blocked PA binding. The second possibility is that IPA induces a conformational change distinct from the one induced by PA in which key PA-binding sites were obscured. Because IPA alone had no effect on Sec18 cleavage, it is more likely that the former scenario occurs.
Figure 8.

IPA blocks the effect of PA on Sec18 degradation by thrombin. Full-length Sec18 was incubated with 100 μm C8-PA alone or 100 μm IPA followed by 100 μm C8-PA, after which the samples were incubated with thrombin for 30 min. Samples were mixed with SDS loading buffer, resolved by SDS-PAGE, and visualized with One-Step Blue® Protein Gel Stain. Quantitation was determined for the relative amounts of uncleaved protein. n = 3 for data shown. Mr indicates relative molecular mass markers. **, p < 0.01; ****, p < 0.0001. Major degradation products are labeled as p1, p2, and p3.
To corroborate the limited proteolysis data, we performed ANS fluorescence assays. When ANS binds to exposed hydrophobic areas, it undergoes fluorescence dequenching, and thus changes in ANS fluorescence between different conditions can serve as a reporter for conformational changes (12). Previously, we used ANS fluorescence to show that full-length Sec18 underwent conformational changes when bound to PA, but not DAG or PS. The effects of PA binding on Sec18 were further examined using MD simulations on the D1–D2 domains. The N-domain was excluded due to its promiscuous binding to anionic surfaces. Nevertheless, it remained possible that PA binding would induce changes between the N and D1 domains. This is of particular importance as studies by others have shown that the N-domain undergoes conformational changes with respect to the D1–D2 domains when hexameric Sec18 catalyzes SNARE priming at the expenditure of ATP (16, 29–31).
Here, we used ANS fluorescence assays to test whether the conformational changes were indeed limited to the D1 and D2 domains or whether additional changes occurred between the N and D1 domains. First, we tested N-D1 by incubating it with a dose curve of C8-PA. We observed an increase in fluorescence as C8-PA concentrations increased, suggesting that there was some level of conformational change; however, the difference versus the control without PA was not statistically significant (Fig. 9, A and E). We next tested the effect of C8-PI(4,5)P2 on N-D1 and found very little change compared with the control (Fig. 9B).
Figure 9.

ANS fluorescence of Sec18 domains bound to lipids. N-D1 (0.5 μm) was incubated with dosage curves of C8-PA (A) or C8-PI(4,5)P2 (B) and 5 μm ANS. Fluorescence was measured by exciting at 390 nm and scanning a range of emission wavelengths. C and D, D1–D2 (0.5 μm) was incubated with C8-PA or C8-PI(4,5)P2 and 5 μm ANS. Fluorescence was detected as above. E, quantitation of maximum fluorescence measurements at each concentration of lipid. Error bars represent S.E. (n = 3). *, p < 0.05.
We continued with the D1–D2 construct and PA. Previously, we mapped PA-binding sites to the hinge region between D1 and D2, so we expected to see an increase in ANS fluorescence with the shortened protein when bound to PA (12). This showed a large and statistically significant increase in ANS fluorescence when incubated with C8-PA (Fig. 9, C and E). In contrast, incubating D1–D2 with C8-PI(4,5)P2 had no effect on ANS fluorescence (Fig. 9D).
The lack of a significant change in ANS fluorescence with N-D1 is consistent with the thrombin cleavage patterns seen above. In both cases, the presence of PA had no apparent effect on conformational changes. The lack of ANS fluorescence changes in the presence of PI(4,5)P2 suggests that this lipid interacted poorly with the Sec18 constructs. This is in keeping with our previous report where we measured the KD value for Sec18 to PI(4,5)P2 as >400 μm (12). Together, these results indicated that the conformational changes seen in Sec18 upon PA binding primarily occur between the D1 and D2 domains and that changes were specific to PA binding.
IPA binding and molecular dynamics ensemble docking
To further probe the interactions of Sec18 with PA and IPA, we performed ensemble molecular docking. We next analyzed scores of poses from each cluster, and we selected poses for epirubicin, IPA, and PA from the interquartile region and the highest overall pose corresponding to the lowest ΔG representation for D1–D2 to run MD simulations for 100 ns. These results were further analyzed by determining the water exposure around the predicted thrombin-cut residue for mSec18 of Arg-638 using Arg-628 on NSF (Fig. 10A). We then compared the average RMSD across the entire simulation trajectory for NSF D1–D2 using C8-PA (Fig. 10B), IPA (Fig. 10C), and epirubicin (Fig. 10D) over the course of the 100-ns simulations.
Figure 10.

Molecular dynamics of PA, IPA, and epirubicin. A, exposed waters within 3 Å of arginine 638, the proposed cleavage residue for thrombin as shown in Fig. 5, A and B. B, average RMSD of PA (n = 2) in 100-ns simulations. C, average RMSD of IPA (n = 4) in 100-ns simulations. D, average RMSD of epirubicin (n = 2) in 100-ns simulations.
In these simulations, it appears that IPA hindered initial conformational change in D1–D2 relative to PA, as there is a significant difference in overall RMSD starting at about 20 ns of the MD simulations, where PA has roughly a 15-Å average RMSD versus an ∼6 Å average RMSD for the IPA simulation (Fig. 10, B and C). This conformational difference was explored by determining water accessibility as shown in Fig. 10A, which could limit access for proteases explaining the results in Fig. 10, A and B. We postulate that IPA inhibits mSec18 binding PA by preventing the conformational change necessary for binding the lipid. Additionally, simulations on epirubicin demonstrate an average RMSD of ∼5 Å, over the entire 100-ns simulations (Fig. 10D). The fact that IPA and epirubicin appear to lock the conformation of Sec18, while PA appears to stimulate conformational change, leads to the conclusion that they have different modes of binding, even though IPA appears to competitively inhibit Sec18 binding to PA in biochemical assays.
Discussion
Membrane fusion is required for vesicular trafficking and eukaryotic homeostasis. Although most trafficking pathways have a unique signature of organelle-specific SNARE proteins, every set of SNAREs relies on Sec18/NSF, which is the only protein responsible for catalyzing the disassembly of SNARE complexes at the expense of ATP (32). Thus, it is important to develop tools to enable further study of this mechanism because of its ubiquitous operation on almost all organelle types. In this study, we used structure-based computational drug discovery to find a specific inhibitor of Sec18/NSF function that we call IPA. Although the structure of NSF was used to computationally dock chemical libraries, the candidate compounds were expected to bind Sec18, as these orthologues have been shown to be interchangeable (7, 33, 34).
Previous to the discovery of IPA, there was a complete lack of specific small molecule inhibitors to block SNARE priming. In fact, the only way to specifically inhibit NSF activity was to raise an antibody against it. As its name indicates, NSF (NEM-sensitive factor) was discovered by it sensitivity to the alkylating agent N-ethylmaleimide (NEM), which promiscuously modifies free thiols (35). Although NEM does inhibit SNARE priming in our system, it has downstream effects that are likely due to promiscuous alkylation; for example, NEM could additionally alkylate other proteins involved in SNARE function such as the single Cys on the SNARE Vam7 (11).
In this study, we report that IPA inhibits SNARE priming and subsequent vacuole fusion. Previously, we found that priming could be inhibited by the PA phosphatase inhibitor propranolol (10). Although nonspecific, the results from using propranolol led us to determine that Sec18 bound to PA reservoirs on the vacuole membrane sequestered Sec18 away from cis-SNARE complexes (11). It is only after the PA phosphatase Pah1 converts enough PA to DAG that Sec18 is able to dissociate from the membrane and engage the SNARE complex. Through this, we also found that C8-PA could be added to in vitro reactions and potently block priming by preventing Sec18 from attaching to SNAREs. The ability of C8-PA to inhibit SNARE priming was not entirely specific to priming, as it also inhibited downstream stages of the fusion pathway as indicated by gain-of-resistance assays. We attribute the later effects of C8-PA to interacting with the SNARE Vam7, which binds both PI3P and PA (14). Although both propranolol and C8-PA helped to further understand Sec18 regulation, a more specific molecule such as IPA was used to determine with more specificity the effect of PA on Sec18 priming, and not additional stages of fusion. Unlike propranolol and C8-PA, IPA does not appear to have downstream effects on the fusion machinery. This is due to the specificity of IPA for NSF/Sec18 at the PA-binding interface, whereas the aforementioned propranolol and C8-PA do not specifically bind a single protein. Importantly, the lack of an effect after priming indicates that IPA does not inhibit the newly-discovered priming-independent role of Sec18 in SNARE zippering at the docking stage (36).
Using molecular docking and MD simulations, we found that IPA bound to NSF at the hinge region of D1 linking to the D2 domain. This region of NSF overlaps with regions bound by the lipid PA (12). Consequently, IPA could compete for PA binding by Sec18 on liposomes. This was a specific competition as IPA failed to compete for PA binding against DEP and Vam7Y42A or to significantly interfere with interactions between Sec18 and PI liposomes. With regard to the two binding sites, IPA showed the best competition at the D1 hinge site, which coincides with the key regulatory site of PA binding, and the fact that PA bound D1 better than any other domain of Sec18 alone (10). When PA binds NSF, it induces a conformational change at the hinge region causing the two domains to swing toward and away from each other, creating both closed and open forms of NSF. The “closed” conformation of NSF is incompatible with hexamerization and SNARE-priming activity. We now show that IPA binds in the same hinge region and prevents its change to the closed conformation, presumably inhibiting the conformational change necessary for Sec18 to bind PA and potentially explaining the inhibition of Sec18 binding to soluble SNAREs, as shown previously (10). Instead, IPA appears to make the Sec18 protomers more rigid, which may in turn prevent interactions with Sec17 and cis-SNARE complexes.
In summary, we now report the discovery of a novel small molecule that inhibits SNARE priming through binding Sec18/NSF. Because of the lack of any such inhibitor, IPA will serve as a potent tool to examine the nature of Sec18/NSF function and SNARE priming. Although IPA was potent in the inhibition of Sec18, we expect that it will have a lower KD value and better activity toward NSF, as the molecule was found using the NSF structure. Thus far, only in vitro systems have been thoroughly tested with IPA, but preliminary in vivo tests with mammalian cells indicate that IPA is membrane-permeable and active at nanomolar concentrations.4 This suggests that IPA might not be limited to being an in vitro research tool.
Experimental procedures
Reagents
All reagents were diluted in PS buffer (20 mm PIPES-KOH, pH 6.8, and 200 mm sorbitol) to a working concentration before use in an experiment. Antibody to Sec17 was described previously (37). 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphate, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine, and C8-PA were purchased from Avanti Polar Lipids as chloroform stock solutions and stored at −20 °C. CM7, CM5, Ni-NTA (Standard and S series), and L1 sensor chips and Regeneration buffers (glycine, pH 1–3) were procured from GE Healthcare. Ni-NTA Atto 488 dye, NEM, and calcein were procured from Millipore Sigma. Monolith NT.115 standard-treated capillaries for thermophoresis were purchased from Nanotemper (München Germany). Epirubicin was from Cayman Chemical. 7-Methyl-3-(4,5,6-trihydroxy-3-oxo-3H-xanthen-9-yl)bicyclo[2.2.1]hept-5-ene-2-carboxylic acid was procured via Ambinter under the identification numbers Amb16226271 and Amb4002159.
IPA purity was assessed using NMR and LC/MS. NMR data were recorded on a Bruker spectrophotometer equipped with a CryoProbe (500 MHz, 1H) using deuterated DMSO as a solvent and internal reference (δ = 2.50 ppm). LC/MS data were performed by the University of Illinois Mass Spectrometry Laboratory using a 2.1-mm inner diameter reverse-phase C-18 column and a Waters Synapt G2-Si mass spectrometer (electrospray ionization).
Recombinant proteins
Recombinant expression of C-terminally His-8–tagged full-length Sec18 or its domains (N, D1, D2, D1–D1) were purified from Escherichia coli as described previously (12). Briefly, pSec18–His-8 (or one for a domain derivative) was transformed into Rosetta 2 (DE3) pLysS Competent Cells (Novagen), and Sec18–His-8 expression was carried out using auto-inducing medium (AIM) (38). Cells were grown to stationary phase (37 °C, 18 h, shaking) and harvested by centrifugation. Cells were resuspended in lysis buffer (20 mm HEPES, pH 6.8, 300 mm NaCl, 0.1% Triton X-100, 2 mm 2-mercaptoethanol, 20 mm imidazole, 10% glycerol, 1 mm ATP, 1 mm PMSF, and 1× cOmplete Protease Inhibitor Mixture (Roche Applied Science)) and lysed by French press. Lysates were clarified by centrifugation (50,000 × g, 20 min, 4 °C) and incubated with Ni-NTA resin (Invitrogen) overnight at 4 °C. Next, resin was washed with 100 bed volumes of lysis buffer with 50 mm imidazole, after which protein was eluted in 1-ml fractions (lysis buffer with 250 mm imidazole). Protein was concentrated by centrifugation using a 100-kDa cutoff Centricon before resolving by gel filtration (Superose 6) using size-exclusion buffer (20 mm HEPES, pH 6.8, 300 mm NaCl, 1 mm 2-mercaptoethanol, 10% glycerol). Sec18–His-8 elutes in two peaks corresponding to monomeric and hexameric pools (Fig. S1). Each pool was collected and concentrated before use. The DEP PA-binding domain from murine Dvl2 was purified as a GST fusion as described previously (25). Membrane scaffold protein 1D1 (MSP1D1–His) was prepared as described previously (39). GST-Vam7Y42A was expressed and purified as described previously (40).
Calcein dequenching and membrane integrity
Liposomes were extruded in the presence of 100 mm calcein to encapsulate the dye at self-quenching concentrations (27, 28). Liposomes were dialyzed in 4 liters of TBS, pH 7.4 (10 mm Tris-Cl, pH 7.5, 140 mm NaCl, 10 mm EDTA, pH 8.0, 0.5% SDS), to remove excess unencapsulated dye. Encapsulated calcein liposomes were incubated with buffer, 0.2% Triton X-100, or a dose curve of IPA. Upon lysis, calcein becomes diluted and fluorescence increases (excitation 494 nm and emission 515 nm).
Vacuole isolation and in vitro vacuole fusion assay
Vacuoles were isolated from yeast strains by density gradient floatation as described previously (41). Fusion reactions (30 μl) contained 3 μg each of vacuoles from BJ3505 (pep4Δ PHO8) and DK6281 (PEP4 pho8Δ), fusion assay buffer (125 mm KCl, 5 mm MgCl2, 20 mm PIPES-KOH, pH 6.8, 200 mm sorbitol), ATP-regenerating system (1 mm ATP, 29 mm creatine phosphate, 0.1 mg/ml creatine kinase), 10 μm CoA, and 283 nm Pbi2p. Reactions were incubated at 27 °C for 90 min, and the Pho8 activity was measured in 250 mm Tris-Cl, pH 8.5, 0.4% Triton X-100, 10 mm MgCl2, 1 mm p-nitrophenyl phosphate. Fusion-dependent alkaline phosphatase maturation was measured by the amount of p-nitrophenylate produced. p-Nitrophenylate absorbance was measured at 400 nm.
Liposome preparation and co-floatation assay
Large unilamellar liposomes were prepared using an extrusion method (42). Extruded liposomes were prepared with 1 mm concentration of appropriate lipids 75% PC, 20% PE, and 5% PA, which were heated and passed through an Avanti mini-extruder equipped with filters of sizes 0.1, 0.4, and 0.8 μm filters over 10 times to ensure homogeneous liposome sizes. Small unilamellar liposomes containing various lipid compositions were prepared using the sonication method (15). Briefly, stock lipids in chloroform were mixed to produce a lipid mixture with the desired lipid mole percentages of 2.6 μmol of total phospholipids. The lipid mixture was dried under a gentle stream of nitrogen and dried in a speed vacuum for an additional 60 min. The tubes were placed under vacuum in a desiccator for an additional 14 h. To the dried lipids, 2.6 ml of 1× PBS solution was added. Tubes were covered with parafilm and incubated at room temperature for 1 h. The lipids were resuspended with vortexing and disrupted in a water bath sonicator for 30 min.
To measure protein binding to liposomes, we used a floatation assay as described (43). Briefly, 40 μl of lipid-binding domain/PBS mixture was incubated with 150 μl of the 1 mm liposome suspension for 5 min at 30 °C before 20 μg of recombinant Sec18–His-8 was added to bring up the total volume to 200 μl and give a final concentration of 1.2 μm Sec18. Samples were incubated for an additional 10 min at 30 °C, and 630 μl of 1.65 m sucrose (PBS) was added. Samples were loaded into the bottom of a centrifuge tube and layered with 840 μl of 0.75 m sucrose (PBS), and 1× PBS to the top of the tube. Samples were centrifuged (200,000 × g, 90 min, 4 °C), and 200 μl of floated liposomes were recovered from the top of the 0.75 m sucrose layer. The bottom 100-μl fraction was recovered, and SDS sample buffer was added to sample unbound protein levels. Liposomes were resuspended in 1 ml of 1× PBS and isolated by centrifugation (16,000 × g, 10 min, 4 °C). SDS sample buffer was added to the final liposome pellet, and bound proteins were resolved by SDS-PAGE. The proteins were transferred to nitrocellulose and probed by Western blotting. Images were acquired using a ChemiDoc MP Imaging System (Bio-Rad). Additionally, protein binding and inhibition of protein binding via inclusion of IPA to extruded liposomes were measured using SPR.
Priming assay
Priming activity of Sec18 was assayed as described previously (11). Briefly, vacuoles were harvested from BJ3505. The equivalent of two standard fusion reactions was incubated at 27 °C with buffer, 1 mm NEM, 300 μm C8-PA, or 100 μm IPA. At the indicated times, vacuoles were removed by centrifugation (16,000 × g, 5 min, 4 °C), and SDS sample buffer was added to the supernatants. Samples were heated at 95 °C for 5 min, resolved by SDS-PAGE, transferred to nitrocellulose, and probed by Western blotting.
Surface plasmon resonance
SPR measurements were performed on a Biacore T200 instrument equipped with a Ni-NTA chip (24). Approximately 2000 RU of 5% PA nanodiscs were immobilized noncovalently using 100 mm NiSO4 flowed at 10 μl/s followed by a blank buffer injection of HEPES, pH 7.4, 150 mm NaCl (HBS buffer). Injections were performed in HBS buffer at a flow rate of 30 μl/min with an association time of 90 s and dissociation time of 300 s, and binding was measured in relative response units (RU) as described (44). Regeneration with EDTA was performed at flow rate 30 μl/s for 120 s using 100 μm EDTA buffer. Proteins were injected using a series of 1:1 dilutions from the highest concentration, and steady state was obtained using GE BIAcore T200 evaluation software version 3.0 (BIAevaluate). Proteins were injected using a series of 1:1 dilutions for Sec18 monomer, D1, D2, and Sec18 hexamer with at least one concentration from each titration run in duplicate. Steady-state data were exported using BiaEvaluate software into GraphPad Prism 7.00 for Windows, GraphPad Software (La Jolla, CA), and fit using either a one-site specific binding model or an IC50 model generated using log[IPA] versus response (three parameters) equation.
For SPR using attached liposome an L1 liposome chip was used with liposome attached to a sample flow cell and no liposome to the reference flow cell. Liposomes were attached after conditioning the chip with two injections of CHAPS over both flow cells for 30 s at 30 μl/min. Each liposome capture was regenerated when a different protein was flowed using 30-s injections of 20 mm CHAPS at 5 μl/min to clean the sensor chip. Proteins were attached freshly for each type of protein, where proteins were titrated in the presence and absence of IPA yielding IC50 and KD values. KD values to liposomes were performed at 30 μl/min with association of 70 s and disassociation of 300 s for D1, 60 s association and 300 s dissociation for Sec18 monomer and hexamer, and 60 μl/min for D2 with association 75 s and disassociation of 225 s. Results were exported from BIaEvaluate into Graphpad and fit via one-site–specific binding model for each saturation curve yielding KD values for different constructs.
Microscale thermophoresis
Thermophoresis measurements were performed using a Monolith NT.115-labeled thermophoresis machine (45). Sec18–His-8 was labeled with Ni-NTA Atto 488 according to the manufacturer's protocol mixing 200 nm protein with 100 nm dye and allowing it to sit at room temperature for 30 min followed by centrifugation. M.O. Control software was used for operation of MST. Target protein concentrations were 50 nm for all His-tag–labeled proteins Sec18 monomer, Sec18 hexamer, PA nanodiscs, and PC nanodiscs. LED excitation power was set to 90%, and MST was set to high allowing 3 s prior to MST on to check for initial fluorescence differences, 25 s for thermophoresis, and 3 s for regeneration after MST off. Analysis was performed using M.O. Affinity Analysis Software as the difference between initial fluorescence measure in the first 5 s as compared with thermophoresis at 15 s. All measurements were performed in PBS buffer (137 mm NaCl, 2.7 mm KCl, 8 mm Na2HPO4, and 2 mm KH2PO4, pH 7.4) without Tween, and binding affinity was generated using Graphpad Sigmoidal 4PL fit from points exported from M.O. Affinity Analysis software using a KD model with target concentration fixed at 50 nm generating bound, unbound, and fraction bound for export to Graphpad to estimate the final KD.
1,8-ANS fluorescence spectroscopy
ANS-binding experiments were carried out as described previously (12). Reactions were performed in fluorescence assay buffer with 5 μm ANS (Cayman Chemical). Initial spectra were taken without protein to measure any background fluorescence from buffer or added lipids (excitation 350 nm and emission 390–620 nm). His8-tagged Sec18 truncations containing the N-domain and D1-domains (N-D1) or D1 and D2 domains (D1–D2) were diluted in assay conditions and then added to the assay to the indicated concentration and incubated at 25 °C for 5 min before spectra were obtained. Initial background fluorescence spectra for each lipid concentration were subtracted from final measurements.
Limited proteolysis
Cleavage reactions were carried out as described previously (12). Sec18-His8 (2 μm) was added to proteolysis buffer (20 mm HEPES, pH 7.2, 150 mm NaCl, 2 mm ATP, 2 mm MgCl2) and incubated with the indicated lipid or IPA concentration on ice for 5 min. Thrombin diluted in 1× HBS was added to assay tubes at the indicated concentrations and incubated at 25 °C for 30 min. Cleavage reactions were stopped with the addition of SDS sample buffer containing 1 mm PMSF. Samples were resolved with SDS-PAGE, and gels were stained using Coomassie Blue. Gels were de-stained with methanol/acetic acid solution (50:7%) and imaged using a ChemiDoc MP Imaging System (Bio-Rad).
Mass spectrometry
Cleavage products from limited proteolysis experiments were excised from SDS-polyacrylamide gels and submitted for LC-MS/MS performed and analyzed by Bioinformatics Solutions Inc. (Ontario, Canada).
MD simulations of top poses from ensemble-scoring function
A previously reported model of D1–D2 by Starr et al. (12) was utilized for this study. The model was derived from a cryo-EM structure of ATP-bound NSF complex (Protein Data Bank code 3J94, chain A) containing residues 215–737 (31). Molecular dynamics simulations were done using NAMD 2.12 (46), with the CHARMM36m force field (47). To maintain a constant pressure of 1 atm and temperature of 310.15 K, Langevin dynamics and Langevin piston Nosé-Hoover methods were used, respectively (48, 49). Particle Mesh Ewald (PME) methods were used to calculate long-range electrostatic forces using 1-Å grid spacing (50, 51). van der Waals interactions were evaluated with a cutoff of 12 Å, and after 10 Å we used a force-based switching scheme. Integration time step was set at 2 fs with the SETTLE algorithm (52) applied. VMD 1.9.3 was used for MD trajectory visualization and analysis (53). The D1–D2 monomer was equilibrated for 20 ns using harmonic restraints on the Cα atoms (0.05 kcal/mol/Å2), barring the previously modeled loops. The simulation was continued without restraints to 200 ns.
Probing binding sites of IPA
To identify potential IPA, PA, and epirubicin interactions with D1–D2 monomer, molecular ensemble docking of was done on D1–D2 monomer using AutoDock Vina (54). The previously mentioned equilibrium simulation of D1–D2 was used to fully sample the dynamics of D1–D2 for molecular docking, where snapshots were taken every 1000 ps of the 200-ns trajectory. For each snapshot, an 80 × 94 × 108 Å grid box was used to fully sample the entire structure. Each snapshot was docked with an exhaustiveness of 10, yielding a total of 2000 IPA and PA docked poses, with the affinities of each pose obtained from the resultant log files. These poses where then clustered using a hybrid K-centers and K-medoids clustering algorithm using an RMSD method (55, 56) in which four main clusters where identified. Poses from each cluster provided by the ensemble docking from AutoDock Vina with the highest interquartile scores were selected for simulation. Selected poses were solvated and ionized to a NaCl concentration of 150 mm using the SOLVATE and AUTOIONIZE plugins within VMD, respectively (53). These systems were simulated for 100 ns and were analyzed with VMD as well as MDAnalysis package (57, 58)
Dynamic light scattering
A Malvern Zetasizer Nano ZS was used to measure both 100-nm extruded liposomes and water bath–sonicated liposomes to compare 80:20 PC/PE liposomes to 75:20:5 PC/PE/PA liposomes. Liposomes created for co-floatation and binding experiments were measured exporting a frequency plot for size distribution, taking three separate measurements, and choosing a representative measurement. Liposomes were diluted to 5 μm in TBS (50 mm Tris-Cl, pH 7.5, 150 mm NaCl) using a Zetasizer set with dispersant with a refractive index of 1.331 for TBS, material of DPPC liposomes, and disposable cuvettes. The presence of PA had no effect on the diameter of extruded liposomes (Fig. S2A).
Liposome floatation was performed as described above with 100-nm PA extruded liposomes in the presence or absence of 100 μm IPA. Approximate concentrations of liposome were performed using DLS and measuring kilocounts/s (59, 60). Scattering intensity kilocounts/s values of liposomes before and after floatation were plotted on the y axis for each liposome type (Fig. S2B).
Data analysis and statistics
Results are expressed as the mean ± S.E. Experimental replicates (n) are defined as the number of separate experiments with different batches of protein, liposomes, and nanodiscs. Where appropriate, significant differences were calculated using two-tailed unpaired t tests. p values ≤0.05 were considered significant.
Author contributions
R. P. S., A. S. A., M. L. S., and R. A. F. conceptualization; R. P. S., A. S. A., M. L. S., Z. L. A., L. R. H., D. A. R.-K., C. Z., K. A. H., J. L. J., and R. A. F. data curation; R. P. S., A. S. A., M. L. S., Z. L. A., L. R. H., D. A. R.-K., C. Z., K. A. H., J. L. J., W. C. G., and R. A. F. formal analysis; R. P. S., A. S. A., M. L. S., L. R. H., and R. A. F. investigation; R. P. S., A. S. A., M. L. S., and R. A. F. writing-original draft; R. P. S., A. S. A., M. L. S., and R. A. F. writing-review and editing; A. S. A., M. L. S., J. L. J., W. C. G., E. T., and R. A. F. methodology; E. T. and R. A. F. funding acquisition; W. C. G. software; W. C. G. visualization; E. T. and R. A. F. supervision; E. T. and R. A. F. project administration; R. A. F. resources.
Supplementary Material
Acknowledgments
Computational resources were provided by XSEDE (XSEDE MCA06N060) and Blue Waters (ACI-1440026). SPR was aided by the help of Dr. Jermaine Jenkins at the University of Rochester Structural Biology and Biophysics Facility with support from National Institutes of Health NCRR Grant 1S10 RR027241 and NIAID Grant P30AI078498 and the University of Rochester School of Medicine and Dentistry.
This work was supported by National Institutes of Health Grants R01-GM101132 (to R. A. F.) and P41-GM104601, U01-GM111251, and U54-GM087519 (to E. T.), National Science Foundation Grant MCB 1818310 (to R. A. F.), and Office of Naval Research Grant ONR N00014-16-1-2535 (to E. T.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1 and S2.
R. P. Sparks and R. A. Fratti, unpublished data.
- NSF
- NEM-sensitive factor
- SNARE
- soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- AIM
- autoinducing medium
- α-SNAP
- soluble NSF adaptor protein
- ANS
- 8-anilino-1-naphthalenesulfonic acid
- DAG
- diacylglycerol
- DEP
- Dishevelled, Egl-10, and pleckstrin domain
- C8-PA
- 1,2-dioctanoyl-sn-glycero-3-phosphate
- GST
- glutathione S-transferase
- MD
- molecular dynamics
- MST
- microscale thermophoresis
- Ni-NTA
- nickel-nitrilotriacetic acid
- NEM
- N-ethylmaleimide
- PA
- phosphatidic acid
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- PI
- phosphatidylinositol
- POPE
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine
- PMSF
- phenylmethylsulfonyl fluoride
- RMSD
- root mean square deviation
- SPR
- surface plasmon resonance
- IPA
- inhibitor of priming activity
- RU
- response unit
- LF-MST
- label-free microscale thermophoresis
- PI3P
- phosphatidylinositol 3-phosphate
- DLS
- dynamic light scattering
- PI(4,5)P2
- phosphatidylinositol 4,5-bisphosphate.
References
- 1. Koike S., and Jahn R. (2019) SNAREs define targeting specificity of trafficking vesicles by combinatorial interaction with tethering factors. Nat. Commun. 10, 1608 10.1038/s41467-019-09617-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Whiteheart S. W., Schraw T., and Matveeva E. A. (2001) N-Ethylmaleimide sensitive factor (NSF) structure and function. Int. Rev. Cytol. 207, 71–112 10.1016/S0074-7696(01)07003-6 [DOI] [PubMed] [Google Scholar]
- 3. Mayer A., Wickner W., and Haas A. (1996) Sec18p (NSF)-driven release of Sec17p (α-SNAP) can precede docking and fusion of yeast vacuoles. Cell 85, 83–94 10.1016/S0092-8674(00)81084-3 [DOI] [PubMed] [Google Scholar]
- 4. Matveeva E. A., Whiteheart S. W., Vanaman T. C., and Slevin J. T. (2001) Phosphorylation of the N-ethylmaleimide-sensitive factor is associated with depolarization-dependent neurotransmitter release from synaptosomes. J. Biol. Chem. 276, 12174–12181 10.1074/jbc.M007394200 [DOI] [PubMed] [Google Scholar]
- 5. Tagaya M., Wilson D. W., Brunner M., Arango N., and Rothman J. E. (1993) Domain structure of an N-ethylmaleimide-sensitive fusion protein involved in vesicular transport. J. Biol. Chem. 268, 2662–2666 [PubMed] [Google Scholar]
- 6. Rodriguez L., Stirling C. J., and Woodman P. G. (1994) Multiple N-ethylmaleimide-sensitive components are required for endosomal vesicle fusion. Mol. Biol. Cell 5, 773–783 10.1091/mbc.5.7.773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wilson D. W., Wilcox C. A., Flynn G. C., Chen E., Kuang W. J., Henzel W. J., Block M. R., Ullrich A., and Rothman J. E. (1989) A fusion protein required for vesicle-mediated transport in both mammalian cells and yeast. Nature 339, 355–359 10.1038/339355a0 [DOI] [PubMed] [Google Scholar]
- 8. Mencher S. K., and Wang L. G. (2005) Promiscuous drugs compared to selective drugs (promiscuity can be a virtue). BMC Clin. Pharmacol. 5, 3 10.1186/1472-6904-5-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Söllner T., Bennett M. K., Whiteheart S. W., Scheller R. H., and Rothman J. E. (1993) A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75, 409–418 10.1016/0092-8674(93)90376-2 [DOI] [PubMed] [Google Scholar]
- 10. Sasser T., Qiu Q. S., Karunakaran S., Padolina M., Reyes A., Flood B., Smith S., Gonzales C., and Fratti R. A. (2012) Yeast lipin 1 orthologue pah1p regulates vacuole homeostasis and membrane fusion. J. Biol. Chem. 287, 2221–2236 10.1074/jbc.M111.317420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Starr M. L., Hurst L. R., and Fratti R. A. (2016) Phosphatidic acid sequesters Sec18p from cis-SNARE complexes to inhibit priming. Traffic 17, 1091–1109 10.1111/tra.12423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Starr M. L., Sparks R. P., Arango A. S., Hurst L. R., Zhao Z., Lihan M., Jenkins J. L., Tajkhorshid E., and Fratti R. A. (2019) Phosphatidic acid induces conformational changes in Sec18 protomers that prevent SNARE priming. J. Biol. Chem. 294, 3100–3116 10.1074/jbc.RA118.006552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miner G. E., Starr M. L., Hurst L. R., and Fratti R. A. (2017) Deleting the DAG kinase Dgk1 augments yeast vacuole fusion through increased Ypt7 activity and altered membrane fluidity. Traffic 18, 315–329 10.1111/tra.12479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miner G. E., Starr M. L., Hurst L. R., Sparks R. P., Padolina M., and Fratti R. A. (2016) The central polybasic region of the soluble SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) Vam7 affects binding to phosphatidylinositol 3-phosphate by the PX (Phox homology) domain. J. Biol. Chem. 291, 17651–17663 10.1074/jbc.M116.725366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mima J., and Wickner W. (2009) Complex lipid requirements for SNARE- and SNARE chaperone-dependent membrane fusion. J. Biol. Chem. 284, 27114–27122 10.1074/jbc.M109.010223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. White K. I., Zhao M., Choi U. B., Pfuetzner R. A., and Brunger A. T. (2018) Structural principles of SNARE complex recognition by the AAA+ protein NSF. Elife 7, e38888 10.7554/eLife.38888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao M., and Brunger A. T. (2016) Recent advances in deciphering the structure and molecular mechanism of the AAA+ ATPase N-ethylmaleimide-sensitive factor (NSF). J. Mol. Biol. 428, 1912–1926 10.1016/j.jmb.2015.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Halgren T. A. (2009) Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model 49, 377–389 10.1021/ci800324m [DOI] [PubMed] [Google Scholar]
- 19. Jacobson M. P., Friesner R. A., Xiang Z., and Honig B. (2002) On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 320, 597–608 10.1016/S0022-2836(02)00470-9 [DOI] [PubMed] [Google Scholar]
- 20. Jacobson M. P., Pincus D. L., Rapp C. S., Day T. J., Honig B., Shaw D. E., and Friesner R. A. (2004) A hierarchical approach to all-atom protein loop prediction. Proteins 55, 351–367 10.1002/prot.10613 [DOI] [PubMed] [Google Scholar]
- 21. Friesner R. A., Murphy R. B., Repasky M. P., Frye L. L., Greenwood J. R., Halgren T. A., Sanschagrin P. C., and Mainz D. T. (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49, 6177–6196 10.1021/jm051256o [DOI] [PubMed] [Google Scholar]
- 22. de Wolf F. A., Staffhorst R. W., Smits H. P., Onwezen M. F., and de Kruijff B. (1993) Role of anionic phospholipids in the interaction of doxorubicin and plasma membrane vesicles: drug binding and structural consequences in bacterial systems. Biochemistry 32, 6688–6695 10.1021/bi00077a023 [DOI] [PubMed] [Google Scholar]
- 23. Griffin E. A., Vanderkooi J. M., Maniara G., and Erecińska M. (1986) Anthracycline binding to synthetic and natural membranes. A study using resonance energy transfer. Biochemistry 25, 7875–7880 10.1021/bi00372a013 [DOI] [PubMed] [Google Scholar]
- 24. Sparks R. P., Jenkins J. L., and Fratti R. (2019) Use of surface plasmon resonance (SPR) to determine binding affinities and kinetic parameters between components important in fusion machinery. Methods Mol. Biol. 1860, 199–210 10.1007/978-1-4939-8760-3_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Capelluto D. G., Zhao X., Lucas A., Lemkul J. A., Xiao S., Fu X., Sun F., Bevan D. R., and Finkielstein C. V. (2014) Biophysical and molecular-dynamics studies of phosphatidic acid binding by the Dvl-2 DEP domain. Biophys. J. 106, 1101–1111 10.1016/j.bpj.2014.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheever M. L., Sato T. K., de Beer T., Kutateladze T. G., Emr S. D., and Overduin M. (2001) Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nat. Cell Biol. 3, 613–618 10.1038/35083000 [DOI] [PubMed] [Google Scholar]
- 27. Günther-Ausborn S., Praetor A., and Stegmann T. (1995) Inhibition of influenza-induced membrane fusion by lysophosphatidylcholine. J. Biol. Chem. 270, 29279–29285 10.1074/jbc.270.49.29279 [DOI] [PubMed] [Google Scholar]
- 28. Slepushkin V. A., Simões S., Dazin P., Newman M. S., Guo L. S., Pedroso de Lima M. C., and Düzgüneş N. (1997) Sterically stabilized pH-sensitive liposomes. Intracellular delivery of aqueous contents and prolonged circulation in vivo. J. Biol. Chem. 272, 2382–2388 10.1074/jbc.272.4.2382 [DOI] [PubMed] [Google Scholar]
- 29. Chang L. F., Chen S., Liu C. C., Pan X., Jiang J., Bai X. C., Xie X., Wang H. W., and Sui S. F. (2012) Structural characterization of full-length NSF and 20S particles. Nat. Struct. Mol. Biol. 19, 268–275 10.1038/nsmb.2237 [DOI] [PubMed] [Google Scholar]
- 30. Furst J., Sutton R. B., Chen J., Brunger A. T., and Grigorieff N. (2003) Electron cryomicroscopy structure of N-ethyl maleimide sensitive factor at 11 A resolution. EMBO J. 22, 4365–4374 10.1093/emboj/cdg420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao M., Wu S., Zhou Q., Vivona S., Cipriano D. J., Cheng Y., and Brunger A. T. (2015) Mechanistic insights into the recycling machine of the SNARE complex. Nature 518, 61–67 10.1038/nature14148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao C., Slevin J. T., and Whiteheart S. W. (2007) Cellular functions of NSF: not just SNAPs and SNAREs. FEBS Lett. 581, 2140–2149 10.1016/j.febslet.2007.03.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ko Y. J., Lee M., Kang K., Song W. K., and Jun Y. (2014) In vitro assay using engineered yeast vacuoles for neuronal SNARE-mediated membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 111, 7677–7682 10.1073/pnas.1400036111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Steel G. J., Laude A. J., Boojawan A., Harvey D. J., and Morgan A. (1999) Biochemical analysis of the Saccharomyces cerevisiae SEC18 gene product: implications for the molecular mechanism of membrane fusion. Biochemistry 38, 7764–7772 10.1021/bi990315v [DOI] [PubMed] [Google Scholar]
- 35. Glick B. S., and Rothman J. E. (1987) Possible role for fatty acyl-coenzyme A in intracellular protein transport. Nature 326, 309–312 10.1038/326309a0 [DOI] [PubMed] [Google Scholar]
- 36. Song H., Orr A., Duan M., Merz A. J., and Wickner W. (2017) Sec17/Sec18 act twice, enhancing membrane fusion and then disassembling cis-SNARE complexes. Elife 6, e26646 10.7554/eLife.26646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haas A., and Wickner W. (1996) Homotypic vacuole fusion requires Sec17p (yeast α-SNAP) and Sec18p (yeast NSF). EMBO J. 15, 3296–3305 10.1002/j.1460-2075.1996.tb00694.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Studier F. W. (2014) Stable expression clones and auto-induction for protein production in E. coli. Methods Mol. Biol. 1091, 17–32 10.1007/978-1-62703-691-7_2 [DOI] [PubMed] [Google Scholar]
- 39. Denisov I. G., Grinkova Y. V., Lazarides A. A., and Sligar S. G. (2004) Directed self-assembly of monodisperse phospholipid bilayer nanodiscs with controlled size. J. Am. Chem. Soc. 126, 3477–3487 10.1021/ja0393574 [DOI] [PubMed] [Google Scholar]
- 40. Fratti R. A., and Wickner W. (2007) Distinct targeting and fusion functions of the PX and SNARE domains of yeast vacuolar Vam7p. J. Biol. Chem. 282, 13133–13138 10.1074/jbc.M700584200 [DOI] [PubMed] [Google Scholar]
- 41. Haas A., Conradt B., and Wickner W. (1994) G-protein ligands inhibit in vitro reactions of vacuole inheritance. J. Cell Biol. 126, 87–97 10.1083/jcb.126.1.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mayer L. D., Hope M. J., and Cullis P. R. (1986) Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta 858, 161–168 10.1016/0005-2736(86)90302-0 [DOI] [PubMed] [Google Scholar]
- 43. Matsuoka K., Morimitsu Y., Uchida K., and Schekman R. (1998) Coat assembly directs v-SNARE concentration into synthetic COPII vesicles. Mol. Cell 2, 703–708 10.1016/S1097-2765(00)80168-9 [DOI] [PubMed] [Google Scholar]
- 44. Sparks R. P., Jenkins J. L., Miner G. E., Wang Y., Guida W. C., Sparks C. E., Fratti R. A., and Sparks J. D. (2016) Phosphatidylinositol (3,4,5)-trisphosphate binds to sortilin and competes with neurotensin: implications for very low density lipoprotein binding. Biochem. Biophys. Res. Commun. 479, 551–556 10.1016/j.bbrc.2016.09.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sparks R. P., and Fratti R. (2019) Use of microscale thermophoresis (MST) to measure binding affinities of components of the fusion machinery. Methods Mol. Biol. 1860, 191–198 10.1007/978-1-4939-8760-3_11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot B. L., Grubmüller H., and MacKerell A. D. (2017) CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 10.1038/nmeth.4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Feller S. E., Zhang Y., Pastor R. W., and Brooks B. R. (1995) Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 103, 4613–4621 10.1063/1.470648 [DOI] [Google Scholar]
- 49. Martyna G. J., Tobias D. J., and Klein M. L. (1994) Constant pressure molecular dynamics algorithms. J. Chem. Phys. 101, 4177–4189 10.1063/1.467468 [DOI] [Google Scholar]
- 50. Darden T., York D., and Pedersen L. G. (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 10.1063/1.464397 [DOI] [Google Scholar]
- 51. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 103, 8577–8593 10.1063/1.470117 [DOI] [Google Scholar]
- 52. Miyamoto S., and Kollman P. A. (1992) SETTLE: an analytical version of the SHAKE and RATTLE algorithm for rigid water molecules. J. Comput. Chem. 13, 952–962 10.1002/jcc.540130805 [DOI] [Google Scholar]
- 53. Humphrey W., Dalke A., and Schulten K. (1996) VMD-visual molecular dynamics. J. Mol. Graph. 14, 22–28 [DOI] [PubMed] [Google Scholar]
- 54. Trott O., and Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Beauchamp K. A., Bowman G. R., Lane T. J., Maibaum L., Haque I. S., and Pande V. S. (2011) MSMBuilder2: modeling conformational dynamics at the picosecond to millisecond scale. J. Chem. Theory Comput. 7, 3412–3419 10.1021/ct200463m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pande V. S., Beauchamp K., and Bowman G. R. (2010) Everything you wanted to know about Markov State Models but were afraid to ask. Methods 52, 99–105 10.1016/j.ymeth.2010.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gowers R. J., Linke M., Barnoud J., Reddy T. J. E., Melo M. N., Seyler S. L., Dotson D. L., Domanski J., Buchoux S., Kenney I. M., and Beckstein O. (2016) Proceedings of the 15th Python in Science Conference, July 11–17, 2016, pp. 98–105, Austin, TX 10.25080/majora-629e541a-00e [DOI] [Google Scholar]
- 58. Michaud-Agrawal N., Denning E. J., Woolf T. B., and Beckstein O. (2011) MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 32, 2319–2327 10.1002/jcc.21787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ribeiro L. N. M., Couto V. M., Fraceto L. F., and de Paula E. (2018) Use of nanoparticle concentration as a tool to understand the structural properties of colloids. Sci. Rep. 8, 982 10.1038/s41598-017-18573-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shah V. M., Nguyen D. X., Al Fatease A., Patel P., Cote B., Woo Y., Gheewala R., Pham Y., Huynh M. G., Gannett C., Rao D. A., and Alani A. W. G. (2018) Liposomal formulation of hypoxia activated prodrug for the treatment of ovarian cancer. J. Control Release 291, 169–183 10.1016/j.jconrel.2018.10.021 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.