

Abstract
Recent studies using two cholesterol-binding bacterial toxin proteins, perfringolysin O (PFO) and domain 4 of anthrolysin O (ALOD4), have shown that cholesterol in the plasma membranes (PMs) of animal cells resides in three distinct pools. The first pool comprises mobile cholesterol, accessible to both PFO and ALOD4, that is rapidly transported to the endoplasmic reticulum (ER) to signal cholesterol excess and maintain cholesterol homeostasis. The second is a sphingomyelin (SM)-sequestered pool inaccessible to PFO and ALOD4 but that becomes accessible by treatment with SM-degrading sphingomyelinase (SMase). The third is an essential pool also inaccessible to PFO and ALOD4 that cannot be liberated by SMase treatment. The accessible cholesterol pool can be trapped on PMs of live cells by nonlytic ALOD4, blocking its transport to the ER. However, studies of the two other pools have been hampered by a lack of available tools. Here, we used ostreolysin A (OlyA), which specifically binds SM/cholesterol complexes in membranes, to study the SM-sequestered cholesterol pool. Binding of nonlytic OlyA to SM/cholesterol complexes in PMs of live cells depleted the accessible PM cholesterol pool detectable by ALOD4. Consequently, transport of accessible cholesterol from PM to ER ceased, thereby activating SREBP transcription factors and increasing cholesterol synthesis. Thus, OlyA and ALOD4 both control movement of PM cholesterol, but through different lipid-binding mechanisms. We also found that PM-bound OlyA was rapidly internalized into cells, whereas PM-bound ALOD4 remained on the cell surface. Our findings establish OlyA and ALOD4 as complementary tools to investigate cellular cholesterol transport.
Keywords: cholesterol regulation, sphingolipid, lipid transport, plasma membrane, endoplasmic reticulum (ER), cellular pool, domain 4 of anthrolysin O (ALOD4), membrane organization, ostreolysin A (OlyA), sterol regulatory element-binding protein (SREBP)
Introduction
The cholesterol content of the plasma membranes (PMs)5 of animal cells is tightly regulated by a network of proteins that reside in membranes of the endoplasmic reticulum (ER) (1). When PM cholesterol levels deviate from their optimal range, the ER regulatory network responds rapidly and modulates the activity of SREBP2 transcription factors, which control genes involved in cholesterol synthesis and uptake (2), to restore cholesterol levels back to normal. Proper functioning of this regulatory network requires sensing of cholesterol in PMs and transmission of this information to ER. Many groups have studied these processes (3–8), but they are not yet well-understood.
Recent studies using new tools to directly probe the organization of PM cholesterol have begun to provide some additional insights (9, 10). When PM cholesterol was below a threshold concentration of ∼30 mol % of total PM lipids, it was largely inaccessible to bind to perfringolysin O (PFO), a cholesterol-binding protein (11–13). Above this threshold, PM cholesterol became accessible, and the binding of PFO to PMs increased sharply. In addition to binding PFO, the accessible pool of PM cholesterol was mobile, rapidly moving to the ER to signal cholesterol surplus and shut down activation of SREBPs (9, 10). The mobile pool of accessible cholesterol could be trapped on the PMs of cells at 37 °C by domain 4 of anthrolysin O (ALOD4), a cholesterol-binding protein that is closely related to PFO but that is nonlytic at 37 °C. As a result, transport of accessible PM cholesterol to the ER was halted, triggering activation of SREBP even when PM cholesterol was well-above the threshold set-point (10).
The studies with PFO and ALOD4 showed that cholesterol adopted different forms in PMs and highlighted the signaling function of the accessible form. However, far less is known about the roles played by the other forms of PM cholesterol that are inaccessible to PFO and ALOD4. The earlier studies showed that about half of the inaccessible form of PM cholesterol (∼15 mol % of PM lipids) is sequestered by sphingomyelin (SM), whereas the remainder of inaccessible PM cholesterol is sequestered by other membrane factors and is essential for cellular viability (9). Treatment of cells with sphingomyelinase (SMase), a soluble, membrane-impermeable enzyme that degrades SM, releases the SM-sequestered cholesterol, rendering it capable of binding to PFO and moving to the ER (a finding that is in line with earlier studies (14, 15)). Thus far, further understanding of SM-sequestered cholesterol has been hindered by a lack of experimental tools.
Fortunately, we recently characterized a fungal protein, ostreolysin A (OlyA), that specifically binds SM/cholesterol complexes in membranes (16). Consistent with its dual specificity for SM and cholesterol, OlyA shows no binding to membranes composed of glycerophospholipids and cholesterol or to membranes composed of SM and epicholesterol, a diastereomer of cholesterol (16). The SM/cholesterol complexes that are detected by OlyA likely form the basis of the SM-sequestered pool of PM cholesterol (9, 17–19). An additional attractive feature of OlyA is that it does not lyse cells at 37 °C (20), making it possible to use it to study cholesterol homeostasis in living cells.
Here, we use the newly-described OlyA and the previously-characterized ALOD4 to compare the localization and dynamics of SM-sequestered and accessible pools of cholesterol, respectively, in the PM of live cells. We find that binding of OlyA to SM/cholesterol complexes in the PM depletes the pool of accessible cholesterol in PMs that can transport to the ER, thus activating SREBP2. Our data indicate how controlling the distribution of cholesterol between accessible and inaccessible forms regulates overall cholesterol homeostasis in animal cells.
Results
Recombinant OlyA and ALOD4 bind distinct pools of cholesterol on PMs
We overexpressed and purified recombinant His6-tagged versions of both ALOD4 and OlyA, as described under “Experimental procedures.” The homogeneity of the purified proteins was confirmed by Coomassie staining (Fig. 1A). When CHO-K1 cells were incubated with these proteins at 37 °C, we observed dose-dependent binding of both ALOD4 (Fig. 1B, lanes 1–6) and OlyA (Fig. 1B, lanes 7–12). We next checked whether the binding of these sensor proteins to PMs caused lysis of CHO-K1 cells by monitoring the release of a cytosolic protein, ubiquitin-activating enzyme (E1), into the extracellular medium (Fig. 1C). Addition of ALOD4 and OlyA at concentrations of 30 μm did not result in any cell lysis (Fig. 1C, lanes 2 and 3). In contrast, addition of the full-length version of ALO (ALOFL), which forms large oligomeric pores in cells (21), resulted in complete lysis, as did addition of Nonidet P-40 detergent (Fig. 1C, lanes 4 and 7). Pleurotolysin B (PlyB), a co-factor that is recruited by membrane-bound OlyA to form oligomeric pores in cells (20, 22), did not cause cell lysis when added alone (Fig. 1C, lane 5); however, addition of both PlyB and OlyA resulted in complete lysis (Fig. 1C, lane 6).
Figure 1.

Biochemical characterization and nonlytic properties of OlyA. A, recombinant ALOD4 and OlyA proteins were purified as described under “Experimental procedures.” 2 μg of each protein was subjected to 15% SDS-PAGE and stained with Coomassie. B and C, on day 0, CHO-K1 cells were set up in medium B at a density of 6 × 104 cells per well of 48-well plates. On day 1, media were removed, cells were washed twice with PBS and then subjected to the following treatments. B, dose-curve analysis. Cells were treated with 200 μl of medium B containing the indicated concentrations of either ALOD4 or OlyA. C, release of cytosolic proteins after incubation with sensor proteins. Cells were treated with 200 μl of medium B containing either the indicated proteins (all at a final concentration of 30 μm, except for ALOFL (1 μm) and PlyB (100 nm)) or Nonidet P-40 detergent (1% (v/v)). After incubation for 1 h at 37 °C, media were removed, cells were washed twice with PBS and harvested, and equal fractions of cell lysates (10% of total) (B) or equal aliquots of cell lysates and media (10% of total of each) (C) were subjected to immunoblot analysis as described under “Experimental procedures.”
We then assessed the lipid specificities of nonlytic ALOD4 and OlyA for binding to CHO-K1 cells at 37 °C. For these experiments, cells were set up in lipoprotein-rich FCS. First, we examined their binding dependence on SM by treating cells with SMase (Fig. 2A). We observed that SMase treatment led to a small increase in ALOD4 binding (Fig. 2A, lanes 1 and 2), but completely eliminated OlyA binding (lanes 3 and 4). Mass spectrometry analysis showed that SMase treatment reduced overall cellular SM content by 76% (Fig. 2B). Next, we examined their binding dependence on cholesterol by treating cells with hydroxypropyl-β-cyclodextrin (HPCD), a reagent that extracts cholesterol from membranes (Fig. 2C). When treated with increasing concentrations of HPCD, binding of ALOD4 declined sharply and was completely eliminated after treatment with 1% HPCD (Fig. 2C, lanes 1–6). Binding of OlyA did not decline as sharply and was completely eliminated only at the highest concentration tested of 2% HPCD (Fig. 2C, lanes 7–12). Mass spectrometry analysis showed that treatment with 2% HPCD reduced overall cellular cholesterol content by 42% (Fig. 2D). The observed SM and cholesterol dependences of ALOD4 and OlyA at 37 °C are consistent with previous binding studies that were carried out at 4 °C (16). We also prepared fluorescently labeled versions of ALOD4 and OlyA, as described under “Experimental procedures,” to directly visualize their binding to the cell surface. When CHO-K1 cells were simultaneously incubated with both fluorescent sensors for 5 min, and then observed by Airyscan confocal microscopy after fixation, we detected punctate staining patterns for both probes (Fig. 2E, also see Fig. 6A). An overlay of the individual staining patterns of ALOD4 and OlyA revealed some regions where there was a strong overlap of the two signals and other regions where there was no overlap (Fig. 2E). In this study, we did not further characterize the spatial distribution of ALOD4 and OlyA, but instead we focused on how their binding affected cholesterol distribution and transport.
Figure 2.

Differential lipid specificities of ALOD4 and OlyA for binding to PMs. A–D, on day 0, CHO-K1 cells were set up in medium B at a density of 6 × 104 cells per well of 48-well plates. On day 1, media were removed; cells were washed twice with PBS and then subjected to the following treatments. A and B, sphingomyelin dependence. Cells were treated with 200 μl of medium B without or with SMase (100 milliunits/ml). After incubation for 30 min at 37 °C, media were removed, and cells were washed four times with PBS. Cells were then either treated with 200 μl of medium F containing 3 μm of the indicated protein (A) or scraped off the plate and subjected to quantitative analysis of sphingomyelin and protein content as described under “Experimental procedures” (B). C and D, cholesterol dependence. Cells were treated with 200 μl of medium B containing the indicated amounts of HPCD. After incubation for 1 h at 37 °C, media were removed, and cells were washed four times with PBS. Cells were then either treated with 200 μl of medium F containing 3 μm of the indicated protein (C) or scraped off the plate and subjected to quantitative analysis of cholesterol and protein content as described under “Experimental procedures” (D). A and C, after incubation for 1 h at 37 °C, media were removed, cells were washed twice with PBS and harvested, and equal fractions of cell lysates (10% of total) were subjected to immunoblot analysis as described under “Experimental procedures.” E, on day 0, CHO-K1 cells were set up in medium B at a density of 1.5 × 104 cells per well of an 8-well Lab-Tek II chambered #1.5 cover glass dish. On day 1, cells were washed twice with PBS, and treated with 200 μl of medium B containing 3 μm ALOD4-488 and 3 μm OlyA-555. After incubation for 5 min at 37 °C, media were removed, and cells were washed twice with PBS, fixed, and permeabilized, stained with DAPI, and then imaged immediately with a Zeiss Airyscan 880 as described under “Experimental procedures.” Scale bar, 10 μm.
Figure 6.
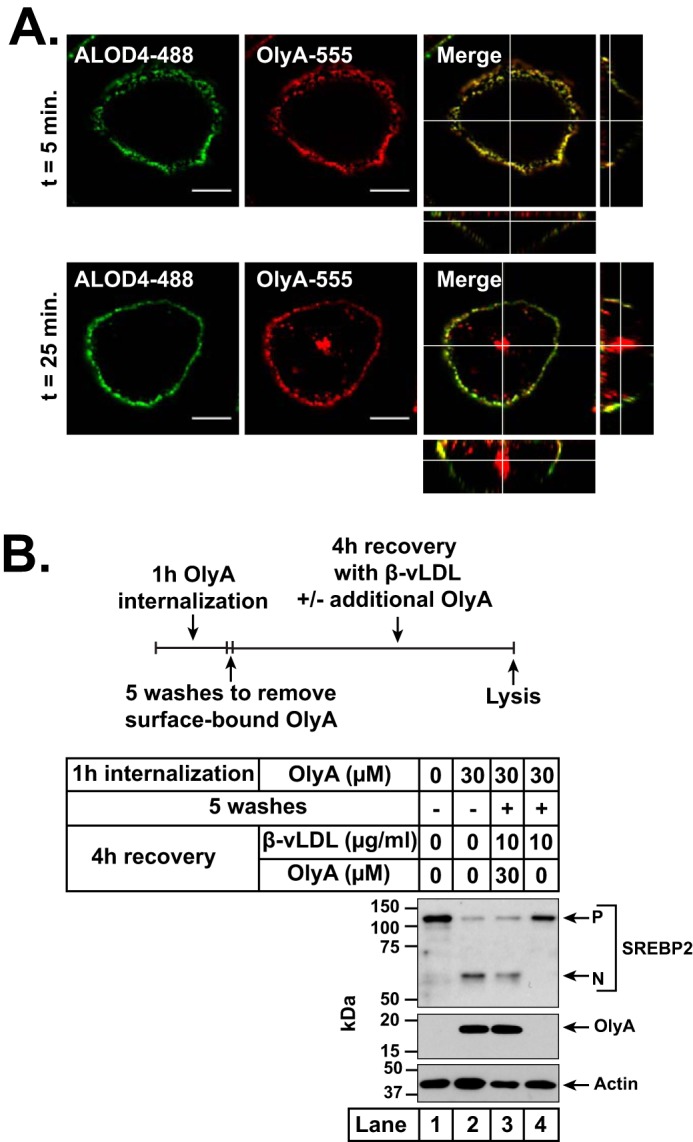
Internalization of OlyA by CHO-K1 cells and effects on intracellular trafficking of lipoprotein-derived cholesterol. On day 0, CHO-K1 cells were set up in medium B at a density of 1.5 × 104 cells per well of an 8-well Lab-Tek II chambered #1.5 coverglass dish (A) or 6 × 104 cells per well of a 48-well plate (B). A, confocal fluorescence microscopy. On day 1, cells were washed twice with PBS and treated with 200 μl of medium B containing 3 μm ALOD4-488 and 3 μm OlyA-555. Fluorescent versions of ALOD4 and OlyA were generated as described under “Experimental procedures.” After incubation for the indicated times at 37 °C, media were removed, and cells were washed twice with PBS, fixed, and imaged within 24 h with a Zeiss Airyscan 880 as described under “Experimental procedures.” Shown are views of the cell center, along with orthogonal views of the merged images. Scale bar, 10 μm. B, effects of internalized OlyA on intracellular cholesterol trafficking. On day 1, media were removed, and cells were washed twice with PBS and then subjected to the treatment protocol illustrated by the schematic in top panel. Cells were first treated with 200 μl of medium B without or with 30 μm OlyA. After incubation for 1 h at 37 °C, cells were either left undisturbed (lanes 1 and 2) or washed five times with PBS to remove all surface-bound OlyA (lanes 3 and 4). The washed cells were then treated with 200 μl of medium F supplemented with 10 μg/ml β-vLDL and the indicated concentrations of OlyA (lanes 3 and 4). After incubation at 37 °C for 4 h, all cells were harvested, and equal fractions of cell lysates (10% of total) were subjected to immunoblot analysis as described under “Experimental procedures.” P, precursor form of SREBP2; N, cleaved nuclear form of SREBP2.
Binding of OlyA depletes accessible cholesterol on PMs
We next sought to determine whether cholesterol can exchange between accessible and SM-sequestered forms by carrying out sequential binding studies with ALOD4 and OlyA. When we first added ALOD4 to cells, we observed the expected dose-dependent increase in ALOD4 binding (Fig. 3A). We then added a constant amount of OlyA to these ALOD4-treated cells and observed that the previously-bound ALOD4 did not affect the subsequent binding of OlyA (Fig. 3A). This result is in line with previous observations that the levels of SM-sequestered cholesterol are relatively constant over a wide range of cholesterol concentrations (9, 16). We then reversed the order of addition and first added OlyA to cells. Again, we observed the expected dose-dependent increase in OlyA binding (Fig. 3B). However, a surprising result was observed when we then added a fixed amount of ALOD4 to these OlyA-treated cells. Robust ALOD4 binding was observed in the absence of OlyA preincubation (Fig. 3B, lane 1), but this binding steadily decreased and was eventually eliminated with increased pre-binding of OlyA (Fig. 3B, lanes 2–6). The elimination of ALOD4 binding occurred even though the cholesterol content of the cells was not altered by pre-treatment with OlyA (Fig. 3C).
Figure 3.

Effects of sequential addition of ALOD4 and OlyA on accessible and SM-sequestered pools of PM cholesterol. A–C, on day 0, CHO-K1 cells were set up in medium B at a density of 6 × 104 cells per well of 48-well plates. A and B, on day 1, media were removed, cells were washed twice with PBS, and treated with 200 μl of medium B containing the indicated concentrations of either ALOD4 (A) or OlyA (B). After incubation for 1 h at 37 °C, each well was supplemented with OlyA (19 μl of protein stock solution, final concentration 10 μm) (A) or ALOD4 (10 μl of protein stock solution, final concentration 1 μm) (B). After further incubation for 30 min at 37 °C, cells were washed twice with PBS and harvested, and equal fractions of cell lysates (10% of total) were subjected to immunoblot analysis as described under “Experimental procedures.” C, quantification of cellular cholesterol after treatment with ALOD4 or OlyA. On day 1, media were removed, cells were washed twice with PBS and treated with 200 μl of medium B without or with 30 μm of the indicated proteins. After incubation for 1 h at 37 °C, cells were washed twice with PBS and harvested as described under “Experimental procedures.” One-half of the cell lysates from each treatment was used for immunoblot analysis (10% of total lysate per lane), and the other half was used for quantification of cholesterol, as described under “Experimental procedures.” Each column represents the mean of cholesterol measurements from 11 independent experiments, and error bars show the standard error (top panel). Immunoblot analysis of lysates from one of the 11 experiments is shown in the bottom panel.
Binding of OlyA lowers ER cholesterol levels
The results of Fig. 3 suggested to us that stabilization of SM/cholesterol complexes by OlyA binding was depleting the pool of accessible cholesterol available for binding to ALOD4. Because accessible PM cholesterol is transported to the ER to shut down lipogenic processes (9, 10), we wondered whether OlyA-induced depletion of accessible PM cholesterol would reduce cholesterol transport to the ER and trigger the activation of lipogenic processes. To monitor the effects of OlyA treatment on ER cholesterol levels, we assayed for the following: (i) proteolytic processing of SREBP2; ii) levels of 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), the rate-limiting enzyme in cholesterol biosynthesis (23); and (iii) activity of acyl-CoA:cholesterol acyltransferase (ACAT), an ER enzyme that esterifies some of the lipoprotein-derived cholesterol that arrives at the ER (24, 25). As a control for these studies, we used ALOD4, which traps accessible PM cholesterol and prevents its transport to the ER, thereby lowering ER cholesterol even though cellular or PM cholesterol does not change (10). As another control, we overexpressed and purified a version of OlyA containing a point mutation (W6A), hereafter designated as OlyA(Mut), that abolishes OlyA's binding to membranes (16).
We first studied the effects of OlyA treatment on cholesterol-replete cells. In one experiment, CHO-K1 cells were set up in lipoprotein-rich FCS, incubated with either ALOD4, OlyA, or OlyA(Mut), and then subjected to immunoblot analysis of SREBP2 (Fig. 4A). ALOD4 bound to the cholesterol-rich PMs of these cells and triggered the processing of SREBP2 from its inactive precursor form to its active nuclear form (Fig. 4A, lanes 1–4), a result that is consistent with previous observations (10). OlyA also bound to PMs and caused proteolytic activation of SREBP2 (Fig. 4A, lanes 5–8), whereas OlyA(Mut), when added at the same levels as OlyA, did not bind to cells or affect SREBP2 processing (lanes 9–12). In another experiment, CHO-K1 cells were again set up in lipoprotein-rich FCS, incubated with either OlyA, OlyA(Mut), or ALOD4, and then subjected to immunoblot analysis of HMGCR (Fig. 4B). In the absence of treatment with any protein, HMGCR protein levels were undetectable (Fig. 4B, lane 1). Incubation with OlyA, but not OlyA (Mut), led to binding to cells and a subsequent increase in HMGCR levels (Fig. 4B, lanes 2–5). ALOD4 also bound to cells and led to an increase in HMGCR levels (Fig. 4B, lane 6). The rise in HMGCR levels after treatment with OlyA or ALOD4 was similar to that observed after extreme cholesterol depletion by HPCD (Fig. 4B, lane 7).
Figure 4.

Effects of binding of OlyA to PMs on ER cholesterol levels. On day 0, CHO-K1 cells were set up in medium B at a density of 6 × 104 cells per well of 48-well plates (A, D, and E) or 2.5 × 105 cells per 60-mm dish (B and C). A, SREBP2 processing in sterol-replete cells. On day 1, media were removed, cells were washed twice with PBS, and treated with 200 μl of medium B with the indicated concentrations of ALOD4, OlyA, or OlyA(Mut). After incubation for 1 h at 37 °C, media were collected and cells were harvested, and equal aliquots of cell lysates and media (10% of total) were subjected to immunoblot analysis as described under “Experimental procedures.” B, HMGCR levels in sterol-replete cells. On day 2, media were removed, and cells were washed once with PBS, and 5 ml of fresh medium B was added. On day 3, media were removed, cells were washed twice with PBS, and treated with either 1 ml of medium B containing the indicated concentrations of OlyA, OlyA(Mut), ALOD4, or 1 ml of cholesterol-depleting medium C. After incubation for 3 h at 37 °C, cells were harvested, and equal fractions of cell lysates (5% of total) were subjected to immunoblot analysis as described under “Experimental procedures.” C, cholesterol esterification after LDL addition to sterol-depleted cells. On day 2, media were removed, and cells were washed twice with PBS and switched to medium F (2 ml). On day 3, cells were washed once with PBS and treated with 1 ml of medium F without or with LDL (100 μg/ml) and the indicated concentrations of OlyA, OlyA(Mut), or ALOD4. After incubation for 1 h at 37 °C, each dish was supplemented with 0.2 mm sodium [14C]oleate and incubated for an additional 2 h at 37 °C, after which the cells were harvested, and levels of [14C]cholesteryl oleate and [14C]triglycerides formed were measured as described under “Experimental procedures.” Each column represents the mean of cholesterol esterification measurements from three independent experiments, and error bars show the standard error. The levels of 14C-labeled triglycerides formed ranged from 9.7 to 14.4 nmol/mg/h. D, treatment of sterol-depleted cells with β-vLDL. On day 1, media were removed and cells were washed twice with PBS, and 200 μl of cholesterol-depleting medium G was added. After incubation for 1 h at 37 °C, media were removed, and cells were washed twice with PBS and treated with 200 μl of medium F without or with β-vLDL (7 μg/ml) and the indicated concentrations of ALOD4 or OlyA. After incubation for an additional 3 h at 37 °C, cells were harvested, and equal fractions of cell lysates (10% of total) were subjected to immunoblot analysis as described under “Experimental procedures.” E, SREBP2 processing in 25HC-treated cells. On day 1, media were removed, and cells were washed twice with PBS and treated with 200 μl of medium B without or with 25HC (5 μm) and the indicated concentrations of ALOD4 or OlyA. After incubation for 1 h at 37 °C, cells were harvested, and equal fractions of cell lysates (10% of total) were subjected to immunoblot analysis as described under “Experimental procedures.” P, precursor form of SREBP2; N, cleaved nuclear form of SREBP2.
We next studied the effects of OlyA treatment on cholesterol-depleted cells. In one experiment, CHO-K1 cells were depleted of cholesterol by incubation in lipoprotein-poor LPDS along with compactin, an inhibitor of cholesterol biosynthesis. We then added back lipoproteins (LDL) along with [14C]oleate, and after 3 h, we processed the cells for measurement of cholesteryl [14C]oleate. As shown in Fig. 4C, we observed an increase in cholesteryl [14C]oleate formation after LDL addition, and this increase was blocked when either OlyA or ALOD4 was included during the incubation with LDL and [14C]oleate. In contrast, incubation with OlyA(Mut) did not significantly affect cholesteryl [14C]oleate formation. In another experiment, CHO-K1 cells were depleted of cholesterol by incubation with cholesterol-extracting HPCD, which triggered the processing of SREBP2 to its active nuclear form (Fig. 4D, lane 1). Addition of β-vLDL, another lipoprotein that is enriched in cholesteryl esters (26), restored cellular cholesterol and suppressed the activation of SREBP2 (Fig. 4D, lane 2), but this suppression was blocked by the addition of ALOD4 (Fig. 4D, lane 3) or OlyA (lanes 4–7), both of which bound to PMs (lanes 3–7) and intercepted the LDL-derived cholesterol directly (ALOD4) or indirectly (OlyA) before it could reach the ER.
While lipoprotein-derived cholesterol suppresses SREBP2 activation by binding to Scap sensors in the ER, oxysterols such as 25-hydroxycholesterol (25HC) also suppress SREBP2 activation by a different mechanism involving binding to Insig proteins in the ER (27, 28). We tested whether ALOD4 or OlyA would affect the actions of 25HC. Consistent with the results of Fig. 4A, in the absence of 25HC, both ALOD4 and OlyA bound to PMs of cholesterol-replete cells and triggered the proteolytic activation of SREBP2 (Fig. 4E, lanes 1–5 and lanes 11–15). In the presence of 25HC, both ALOD4 and OlyA still bound to PMs but 25HC was able to enter cells and suppress SREBP2 activation (Fig. 4E, lanes 6–10 and lanes 16–20). Thus, in contrast to their effects on lipoprotein-derived cholesterol (Fig. 4, C and D), ALOD4 and OlyA did not affect the ability of 25HC to suppress SREBP2 activation.
In summary, triggering of SREBP2 activation, stabilization of HMCGR levels, lowering of ACAT's activity, and inability to block 25HC from suppressing SREBP2 activation, all indicate that OlyA-induced depletion of accessible PM cholesterol reduces cholesterol transport to ER and lowers ER cholesterol levels.
Binding of OlyA does not block endocytosis or processing of lipoproteins
We considered the likelihood that OlyA was lowering ER cholesterol not by depleting accessible cholesterol on PMs, but instead by blocking endocytosis and processing of lipoproteins. To address this possibility, we directly measured the uptake and degradation of LDL in the presence of OlyA. CHO-K1 cells were first depleted of cholesterol by incubation in lipoprotein-poor LPDS along with compactin, after which we added 100 μg/ml 125I-labeled LDL along with increasing concentrations of OlyA. After incubation for 3 h, we measured surface-bound plus internalized 125I-labeled LDL (a measure of LDL uptake) and [125I]monoiodotyrosine released into medium (a measure of LDL degradation). As shown in Fig. 5A, when the incubation was carried out at 37 °C, the uptake of 125I-labeled LDL increased as increasing amounts of OlyA were added (left panel), a result that is likely due to higher expression of the LDL receptor induced by OlyA-mediated activation of SREBP2 transcription factors (Fig. 4A). 125I-Labeled LDL uptake was also unaffected by incubation with OlyA(Mut) or ALOD4 (Fig. 5A, left panel). In addition, the degradation of 125I-labeled LDL was not affected by incubation with OlyA, OlyA(Mut), or ALOD4 (Fig. 5A, right panel). As a negative control, lowering the incubation temperature to 4 °C blocked endocytosis and suppressed both the uptake and degradation of 125I-labeled LDL by more than 85% (Fig. 5A, gray bars).
Figure 5.
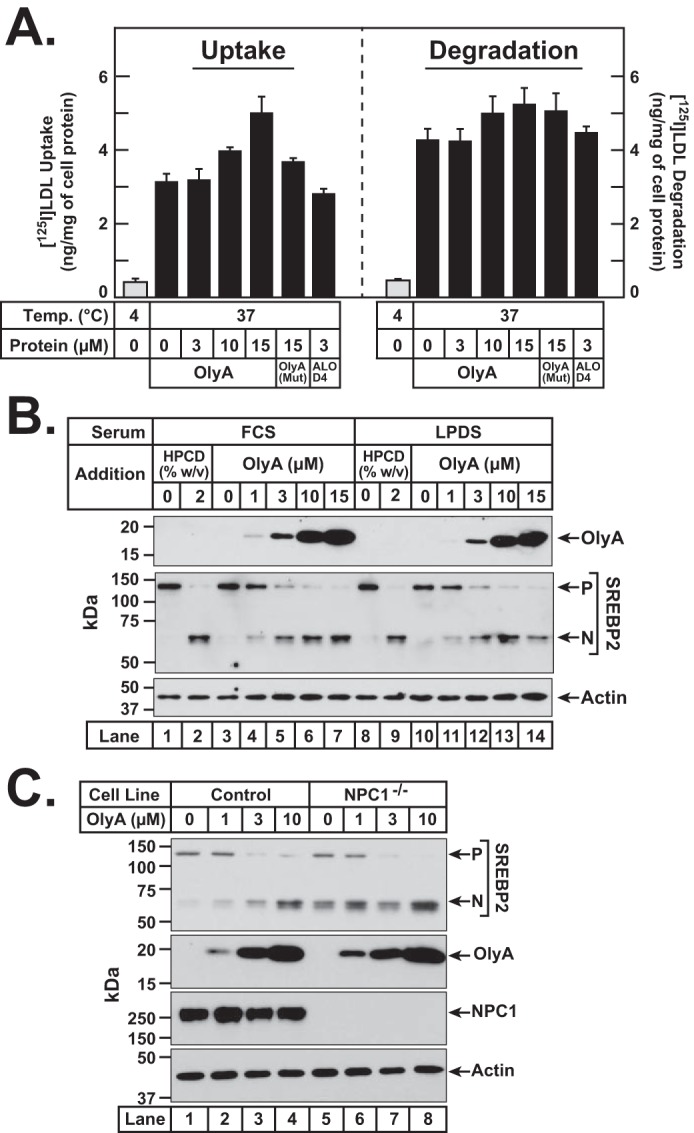
OlyA does not block uptake and processing of lipoproteins. A, LDL uptake and degradation in the presence of OlyA. On day 0, CHO-K1 cells were set up in medium B at a density of 5 × 105 cells per 60-mm dish. On day 1, media were removed, and cells were washed twice with 2 ml of PBS followed by addition of 2 ml of cholesterol-depleting medium F. On day 2, media were removed, and cells were washed with 2 ml of PBS followed by addition of 1 ml of medium F containing 100 μg/ml 125I-labeled LDL (27.4 cpm/ng) in the absence or presence of the indicated proteins. The cells were incubated for 3 h at either 4 or 37 °C, after which LDL uptake and degradation were determined as described under “Experimental procedures.” B, effects of OlyA in lipoprotein-rich and lipoprotein-poor serum. On day 0, CHO-K1 cells were set up in medium B at a density of 6 × 104 cells per well of 48-well plates. On day 1, media were removed, and cells were washed twice with PBS and treated with 200 μl of either lipoprotein-rich medium B (lanes 1–7) or lipoprotein-poor medium D (lanes 8–14) containing the indicated concentrations of either HPCD or OlyA. C, effects of OlyA in control and NPC1−/− cells. On day 0, CHO-K1 cells (lanes 1–4) or NPC1−/− cells (lanes 5–8) were set up in medium B at a density of 6 × 104 cells per well of 48-well plates. On day 1, media were removed, and cells were washed twice with PBS and treated with 200 μl of medium B containing the indicated concentrations of OlyA. B and C, after incubation for 1 h at 37 °C, cells were harvested, and equal fractions of cell lysates (10% of total) were subjected to immunoblot analysis as described under “Experimental procedures.” P, precursor form of SREBP2; N, cleaved nuclear form of SREBP2.
To further assess possible effects of serum lipoproteins on OlyA, we incubated CHO-K1 cells for 1 h with OlyA in lipoprotein-rich FCS or in lipoprotein-poor LPDS and compared the results. As shown in Fig. 5B, we observed similar binding of OlyA to cells (top panel) and similar triggering of SREBP2 activation (middle panel) both in FCS (lanes 3–7) and in LPDS (lanes 10–14). The extent of SREBP2 activation by OlyA treatment was similar to that observed after cholesterol depletion by HPCD (Fig. 5B, lanes 1, 2, 8, and 9). We also tested the effects of OlyA treatment on a mutant CHO-K1 cell line that is deficient in Niemann-Pick C1 (NPC1) (29), a lysosomal membrane protein that is critical for transporting LDL-derived cholesterol out of lysosomes (30–32). As shown in Fig. 5C, we observed similar binding of OlyA and similar triggering of SREBP2 processing in both control cells Fig. 5C, lanes 1–4) and NPC1-deficient cells (lanes 5–8). Combined, these results suggest that the actions of OlyA in reducing ER cholesterol are not simply due to blocking the endocytosis and processing of lipoproteins.
Internalized OlyA does not interfere with intracellular cholesterol trafficking
Because OlyA has been reported to be internalized by cells in a caveolin-mediated manner (20), we addressed the possibility of the internalized OlyA interfering with intracellular cholesterol trafficking to the ER. We first studied the time dependence of OlyA internalization. When CHO-K1 cells were simultaneously incubated with fluorescent versions of OlyA and ALOD4 (3 μm of each), we observed initial binding of both proteins to the cell surface (Fig. 6A, top panel). After incubation for 25 min, some of the surface-bound OlyA, but none of the ALOD4, was internalized into the cells (Fig. 6A, bottom panel). We then tested whether the internalized OlyA would affect the delivery of lipoprotein-derived cholesterol to the ER (Fig. 6B). For this experiment, CHO-K1 cells were set up in FCS and then incubated with 30 μm OlyA for 1 h to allow for maximal internalization of OlyA. As observed previously, this incubation also triggered the activation of SREBP2 to its nuclear form (Fig. 6B, lane 2). We then washed off the PM-bound OlyA and then added back β-vLDL in the presence or absence of OlyA. When OlyA was present in the extracellular medium, it intercepted the β-vLDL–derived cholesterol at the PM and prevented the movement of this cholesterol to ER to block the processing of SREBP2 (Fig. 6B, lane 3). In this case, it is also possible that the internalized OlyA was affecting the intracellular movement of cholesterol. In contrast, when no OlyA was added to the extracellular medium, the β-vLDL–derived cholesterol was able to eventually reach the ER, where it blocked the processing of SREBP2 to its active nuclear form (Fig. 6B, lane 4). This result suggests that the internalized OlyA does not block the movement of lipoprotein-derived cholesterol to the ER.
OlyA and ALOD4 inhibit hemolysis by sequestering accessible cholesterol by different mechanisms
To further establish that OlyA was exerting its effect at the PM, we used red blood cells (RBCs) that have accessible and SM-sequestered pools of cholesterol in their PMs (16, 33) but no internal membranes or internalization processes. Incubation of RBCs with pore-forming ALOFL, which binds accessible cholesterol, led to hemolysis (Fig. 7, left panel). The concentration of ALOFL that resulted in 50% hemolysis (LD50) was ∼0.2 nm. We reasoned that pre-incubation of RBCs with ALOD4 or OlyA, which do not form pores (Fig. 1C), would sequester the accessible RBC cholesterol and make it unavailable to be targeted by ALOFL for pore formation. Indeed, pre-incubation with ALOD4, which binds accessible cholesterol shifted ALOFL's LD50 to ∼2 nm. Pre-incubation with OlyA, which binds to SM-sequestered cholesterol, also shifted ALOFL's LD50 to ∼1 nm, whereas OlyA (Mut), which does not bind membranes, did not affect ALOFL hemolysis. Dose-curve analysis (Fig. 7, right panel) showed that 3 μm ALOD4 could completely inhibit ALOFL hemolysis, whereas inhibition by OlyA was not as complete. This result is in line with the lower potency of OlyA in shifting ALOFL's LD50 (Fig. 7, left panel). In contrast, OlyA(Mut) had no effect on hemolysis by ALOFL, even at the highest tested concentration of 10 μm.
Figure 7.

Effect of pre-incubation with ALOD4 and OlyA on hemolysis by ALOFL. Each reaction mixture, in a final volume of 500 μl, contained 450 μl of rabbit erythrocytes that had been washed and diluted as described under “Experimental procedures” along with the indicated concentrations of ALOD4, OlyA, or OlyA(Mut). After incubation for 15 min at room temperature, each reaction was supplemented with 3 μl of buffer A containing varying amounts of ALOFL to achieve the final indicated concentrations. After further incubation for 15 min at room temperature, the extent of hemolysis was quantified by measuring the release of hemoglobin (absorbance at 540 nm). The amount of hemoglobin released after treatment with 1% (w/v) Triton X-100 detergent was set to 100%, and all values were normalized to this set-point. Each data point represents the mean of hemoglobin measurements from three independent experiments, and error bars show the standard error. When not visible, error bars are smaller than the size of the symbols.
Discussion
This study uses OlyA and ALOD4 as complementary tools to highlight how the interaction between SM and cholesterol in the PM regulates overall cellular cholesterol levels. As illustrated in Fig. 8, cholesterol in the PMs of LDL-treated cells is divided into three distinct pools: (i) an accessible pool that is mobile and transports to the ER to signal cholesterol sufficiency; (ii) a SM-sequestered pool that does not transport to the ER but can be liberated for transport by SMase; and (iii) an essential pool bound to other PM factors that does not transport to the ER and cannot be released by SMase. All of our previous studies used PFO and ALOD4, sensors of the accessible pool of PM cholesterol, to study in detail the role of that pool in maintaining cholesterol homeostasis (9, 10). Here, we focused on the SM-sequestered pool of cholesterol by using OlyA, a newly-identified sensor for this pool of PM cholesterol (16).
Figure 8.
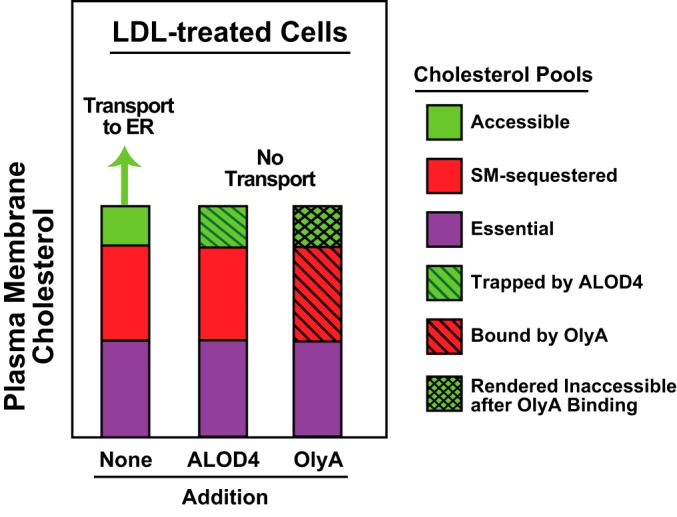
Schematic diagram illustrating the effects of ALOD4 and OlyA on pools of PM cholesterol (adapted from Ref. 9).
When added to the extracellular medium of cells that were replete with cholesterol, we found that OlyA was not just a passive reporter of SM-sequestered cholesterol, but actively reconfigured the distribution of PM cholesterol. Binding of OlyA completely eliminated the pool of accessible PM cholesterol, as judged by a decline in ALOD4 binding, even though there was no change in total cholesterol levels (Fig. 3). Depletion of the mobile pool from the PM triggered the activation of SREBP transcription factors, stabilized HMGCR, and inactivated ACAT, all of which are hallmarks of a fall in ER cholesterol levels (Fig. 4). The effects of OlyA binding to PMs are similar to those observed with binding of ALOD4, but the underlying reasons are different. ALOD4 directly binds to accessible cholesterol and traps it on PMs. In contrast, OlyA directly binds to SM/cholesterol complexes and likely stabilizes them, increasing the equilibrium distribution of cholesterol in complexes with SM and indirectly depleting the uncomplexed accessible cholesterol. After these distinct binding events, the downstream effects converge as both treatments prevent movement of accessible cholesterol to the ER. Thus, movement of accessible PM cholesterol to ER can be blocked either by trapping it on the PM with ALOD4 or by converting it from an accessible form to a sequestered form by OlyA (Fig. 8).
We used OlyA's ability to inhibit PM–to–ER cholesterol transport to investigate the trafficking route of lipoprotein-derived cholesterol after it exits lysosomes. In the presence of OlyA, sterol-depleted cells maintained their ability to internalize LDL and degrade it in lysosomes (Fig. 5A). The LDL-derived cholesterol reached the PM where it was likely rapidly incorporated into SM/cholesterol complexes that were stabilized by OlyA (Figs. 4D and 6B). As a result, the LDL-derived cholesterol failed to reach the ER to stimulate activity of ACAT (Fig. 4C) or suppress activation of SREBP2 (Figs. 4D and 6B). These results are consistent with previous observations made with ALOD4, which directly traps LDL-derived cholesterol on PMs (10). The effects of both OlyA and ALOD4 provide further support for a model where LDL-derived cholesterol moves first from lysosomes to the PM to meet the needs of this cholesterol-rich membrane and subsequently from the PM to regulatory domains of the ER to suppress cholesterol production.
The stabilization of SM/cholesterol complexes on PMs by OlyA was inferred from the complete loss of accessible PM cholesterol after OlyA binding (Fig. 3B). Such stabilization would also predict a loss in free, uncomplexed SM; however, we have so far been unable to test this prediction because none of the SM sensors that we currently have at our disposal (OlyA, lysenin, or equinatoxin II) bind specifically to free SM (16). Similarly, detecting an increase in levels of SM/cholesterol complexes on PMs after OlyA stabilization has been technically challenging, given that we do not have any other sensor (aside from OlyA) for detecting SM/cholesterol complexes, independently of free SM. It is also possible that binding of OlyA leads to sequestration of cholesterol by other PM lipids or proteins. Nonetheless, the OlyA-mediated depletion of accessible cholesterol from the PMs of both cultured CHO-K1 cells and red blood cells (Figs. 3B and 7) indicates that the interconversion of cholesterol between different pools on PMs can dramatically alter intracellular cholesterol trafficking (Fig. 4). It is also noteworthy that binding of ALOD4 to accessible cholesterol on PMs does not lead to a decrease in the subsequent binding of OlyA to SM/cholesterol complexes (Fig. 3A). This result is in line with previous studies showing that SM-sequestered cholesterol levels were constant in the face of significant cholesterol depletion (9, 16) and raises the possibility of an active mechanism mediated by one or more PM proteins that maintains this constancy.
An interesting finding in this study relates to the different fates of PM-bound ALOD4 and OlyA. When we incubated CHO-K1 cells with fluorescently-labeled versions of ALOD4 and OlyA for short times (5 min), we observed punctate staining patterns for both sensors on the cell surface (Figs. 2E and 6A, top panel). However, a dramatic difference was observed when the incubation time was extended to 25 min. At that time point, some of the OlyA had been internalized into cells, whereas ALOD4 remained on the cell surface (Fig. 6A, bottom panel). A previous study showed that OlyA internalization can occur through caveolin-mediated endocytosis (20). Our findings raise the possibility that SM/cholesterol complexes may be enriched in areas of the PM that undergo endocytosis (34) and that accessible cholesterol is excluded from such areas.
The interplay between accessible and SM-sequestered pools of cholesterol that is revealed here through the use of ALOD4 and OlyA points out the need for a sensor that can monitor and modulate the essential pool of cholesterol. This fraction of PM cholesterol is likely complexed to PM proteins or to phospholipids other than SM (18, 35, 36). The discovery and characterization of additional proteins that can detect the essential cholesterol pool will facilitate a more complete understanding of organization of cholesterol in PMs.
Experimental procedures
Materials
We obtained Protein Ark Quick Coomassie stain from Anatrace; Amicon Ultra centrifugal filters (10,000 MWCO) from Millipore; anti-actin antibody, Dulbecco's phosphate-buffered saline (PBS), and sphingomyelinase (SMase) from Sigma; 25HC from Steraloids, Inc.; and Alexa Fluor 488 C5-maleimide, Alexa Fluor 555 C2-maleimide, Amplex red cholesterol oxidase kit, and 8-well Lab-Tek II chambered #1.5 coverglass dishes from Thermo Fisher Scientific. 4′,6-Diamidino-2-phenylindole (DAPI) and 20% (v/v) paraformaldehyde solution were kind gifts from Dr. Marcel Mettlen, University of Texas Southwestern Medical Center (UTSW). mAb IgG–A9 against hamster HMGCR has been described previously (37). Human LDL, newborn calf LPDS, sodium compactin, sodium mevalonate, and all other materials were prepared or obtained from sources as described previously (10).
Buffers and culture media
Buffer A contains 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, and 1 mm tris(2-carboxyethyl)phosphine. Buffer B contains 10 mm Tris-HCl (pH 6.8), 100 mm NaCl, 1% (w/v) SDS, 1 mm EDTA, 1 mm EGTA, 20 μg/ml phenylmethylsulfonyl fluoride, and protease inhibitors (1 tablet/20 ml). Buffer C contains 62.5 mm Tris-HCl (pH 6.8), 100 mm DTT, 8 m urea, 10% (v/v) glycerol, and 15% (w/v) SDS. Buffer D is PBS supplemented with 4% (v/v) paraformaldehyde. Buffer E is PBS supplemented with 2% (v/v) paraformaldehyde and 0.1% (v/v) Triton X-100. Buffer F is PBS supplemented with 2% (v/v) LPDS and 1 mm EDTA. Buffer G is PBS supplemented with 1% Triton X-100. Medium A is a 1:1 mixture of Ham's F-12 and Dulbecco's modified Eagle's medium supplemented with 100 units/ml penicillin and 100 μg/ml of streptomycin sulfate. Medium B is medium A supplemented with 5% (v/v) FCS. Medium C is medium B supplemented with 2% (w/v) HPCD. Medium D is medium A supplemented with 5% (v/v) LPDS. Medium E is medium D supplemented with 2% (w/v) HPCD. Medium F is medium D supplemented with 50 μm compactin and 50 μm sodium mevalonate. Medium G is medium F supplemented with 1% (w/v) HPCD.
Cell culture
Stock cultures of hamster CHO-K1 cells and 10-3 cells (mutant CHO-K1 cells that lack detectable mRNA for NPC1 and hereafter designated as NPC1−/−) (29) were maintained in monolayer culture at 37 °C in 8.8% CO2.
Protein purification and labeling
Recombinant His6–ALOFL (38) and His6–ALOD4 (39), hereafter designated as ALOFL and ALOD4, respectively, were overexpressed in Escherichia coli and purified as described in the indicated references. OlyA–His6 and OlyA–(W6A)–His6, hereafter designated as OlyA and OlyA(Mut), respectively, were overexpressed in E. coli and purified as described previously (16). His8–PlyB, hereafter designated as PlyB, was overexpressed and purified as described previously (22). All purified proteins were subjected to a final gel-filtration step in buffer A, concentrated to 1–5 mg/ml using Amicon Ultracentrifugal filters, stored at 4 °C, and used within 2 weeks. The activity of each batch of ALOD4 and OlyA proteins was tested for their ability to trigger SREBP2 activation and used in experiments only if they showed the dose dependence shown in Fig. 4A. Protein concentrations were measured using a NanoDrop instrument (Thermo Fisher Scientific). For some studies, the lone engineered cysteine on ALOD4 (at amino acid 404 according to the ALOFL sequence) and on OlyA (at amino acid 151) was labeled with Alexa Fluor 488 and Alexa Fluor 555, respectively, as described previously (38). The labeled proteins, designated as ALOD4–488 and OlyA-555, were supplemented with 20% (v/v) glycerol, flash-frozen in liquid nitrogen, stored at −80 °C, and used within 3 months.
Quantification of cellular cholesterol and sphingomyelin
Mass spectrometry analysis of the cellular content of cholesterol was performed as described previously (40), and the measured cholesterol mass was normalized to the protein content. In some experiments, cellular cholesterol content was measured using the Amplex red cholesterol oxidase kit as described previously (10). Mass spectrometry analysis of sphingomyelin was performed as described previously (41) using a Nexera X2 UHPLC system coupled to a Shimadzu LCMS-8050 triple quadrupole mass spectrometer operating the dual ion source in electrospray negative mode. Quantification was performed in selected reaction monitoring mode to detect the common ion fragment of m/z = 168. The relative concentration of each metabolite was determined using the peak/area ratio of analyte versus corresponding internal standard. Data were processed using the LabSolutions (version 5.91) and LabSolutions Insight (version 3.1) program packages (Shimadzu Scientific Instruments). Data are reported as peak area of analyte/peak area of internal standard and normalized to the protein content. Protein content was measured using a bicinchoninic acid (BCA) kit (Thermo Fisher Scientific).
Assays for cholesterol esterification
The rate of incorporation of [14C]oleate into [14C]cholesteryl oleate and 14C-triglycerides in cultured CHO-K1 cells was measured as described previously (24).
Assays for uptake and degradation of 125I-labeled LDL
Human LDL was iodinated as described previously (10). The uptake and proteolytic degradation of labeled LDL was measured as described previously (42).
Immunoblot analysis
After indicated treatments, media were removed, cells were washed twice with PBS and then harvested by the addition of either 200 μl of buffer B to each well of a 48-well plate or 400 μl of a 1:1 mixture of buffers B and C to each 60-mm dish. For the experiment described in Fig. 3C, cells were harvested by the addition of 200 μl of buffer G to each well of a 48-well plate. After incubation for 20 min at room temperature while shaking, the lysed cells were collected, mixed with 5× loading buffer, heated at 95 °C for 10 min, and subjected to either 10 or 15% SDS-PAGE. In the experiment of Fig. 3C, half of the lysed cells were used for lipid extraction followed by quantification of cholesterol as described previously (10). The electrophoresed proteins were transferred to nitrocellulose filters using the Bio-Rad Trans Blot Turbo system and subjected to immunoblot staining with the following primary antibodies: IgG-7D4 (10 μg/ml) to detect SREBP2; IgG-A9 (7 μg/ml) to detect HMGCR; anti-His (1:1,000 dilution) to detect His-tagged ALOD4 and OlyA proteins; anti-actin (1:1,000 dilution); anti-E1 (1:1,000 dilution); and anti-NPC1 (1:1,000 dilution). Bound antibodies were visualized by chemiluminescence (Super Signal Substrate; Pierce) by using a 1:5,000 dilution of donkey anti-mouse IgG (Jackson ImmunoResearch) or a 1:5,000 dilution of anti-rabbit IgG (Jackson ImmunoResearch) conjugated to horseradish peroxidase. Filters were exposed to Phoenix Blue X-Ray Film (F-BX810; Phoenix Research Products) at room temperature for 1–60 s for all cases, except for anti-NPC1 which required an exposure time of 10 min.
Cell treatments for microscopy
After indicated treatments, media were removed, and cells were washed twice with PBS. Some samples were fixed immediately by incubation with 100 μl of buffer D for 15 min and imaged within 24 h. Other samples were first permeabilized by addition of 100 μl of buffer E that had been pre-warmed to 37 °C. After incubation for 2 min, buffer E was removed, and samples were fixed by addition of buffer D for 15 min. Samples were then incubated with PBS containing BSA (2% w/v) and DAPI (1:6,000 dilution) for 1 h at room temperature, after which samples were washed five times with 400 μl of PBS and imaged within 24 h.
Microscopy imaging
Imaging was carried out using a Zeiss 880 Airyscan LSCM (UTSW Live Cell Imaging Facility), which was calibrated before each imaging session. Airyscan images were acquired using the following parameters: zoom ≥ 2.5; pinhole ≥ 1.8 Airy units (AU); line time and speed settings optimized for the zoom factor; and z-direction step size = 0.18 μm for z-stack acquisition. Confocal images were acquired using the following parameters: zoom ≥1 and pinhole size = 1 AU. The z-stacks of cell samples were acquired with step size optimized for the zoom factor (typical value for step size was 1 μm). For both Airyscan and confocal microscopy, the 405, 488, and 561 lasers were used to excite DAPI, Alexa Fluor 488–conjugated proteins, and Alexa Fluor 555–conjugated proteins, respectively. Laser power and gain were adjusted to produce a signal that was 20–30% of the maximum possible signal. Images were analyzed with the Airyscan processing toolbox in the Zen software package.
Hemolysis assay
Fresh rabbit blood was obtained from the Animal Resource Center (UTSW), and RBCs were washed and diluted in buffer F as described previously (38). Reaction mixtures (500 μl) containing 450 μl of RBCs and 50 μl of buffer A with competitor toxins (0–10 μm) were set up in 1.7-ml centrifuge tubes and placed on a rotator. After incubation for 15 min at room temperature, 3 μl of buffer A containing ALOFL (0–10 nm) was added, and reactions were again placed on a rotator. After further incubation for 15 min at room temperature, reactions were subjected to 2,000 × g centrifugation for 15 min at room temperature, after which an aliquot of the supernatant (100 μl) was assayed for released hemoglobin (absorbance at 540 nm).
Reproducibility
All results were confirmed in at least three independent experiments performed on different days with different batches of cells and different batches of recombinant proteins. The only exception was the assay for uptake and degradation of 125I-labeled LDL, which was carried out on 2 different days with different batches of cells.
Author contributions
K. A. J., S. E., R. E. I., and A. R. conceptualization; K. A. J., S. E., R. E. I., and A. R. formal analysis; K. A. J., S. E., D. C. V., and A. R. investigation; K. A. J. and A. R. writing-original draft; S. E. and R. E. I. writing-review and editing; R. E. I. and A. R. project administration and funding acquisition.
Acknowledgments
We thank Daphne Rye and Bilkish Bajaj for excellent technical assistance; Lisa Beatty, Briana Carter, Leticia Esparza, Shomanike Head, and Camille Harry for cell culture assistance; Feiran Lu and Dorothy Williams for assistance with ACAT assays; Linda Donnelly for assistance with 125I-LDL uptake and degradation assays; Ruth Gordillo (Touchstone Diabetes Center, University of Texas Southwestern) for MS analysis of cellular sphingomyelin; Jeff McDonald and Kaitlyn Eckert (Center for Human Nutrition, University of Texas Southwestern) for MS analysis of cellular cholesterol; and Kate Luby-Phelps, Marcel Mettlen, Sandra Schmid, Daniel Kober, and Shimeng Xu for helpful discussions. Confocal imaging was carried out at University of Texas Southwestern Medical Center Live Cell Imaging Facility using an instrument funded by National Institutes of Health 1S10OD021684-01 to Kate Luby-Phelps.
This work was supported by National Institutes of Health Grant HL20948 (to A. R. and R. E. I.), Burroughs Wellcome Fund Grant 1019692 (to R. E. I.), American Gastroenterological Association Grant 2019AGARSA3 (to R. E. I.), and Robert A. Welch Foundation Grant I-1793 (to A. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PM
- plasma membrane
- PFO
- perfringolysin O
- ALOD4
- anthrolysin O domain 4
- SM
- sphingomyelin
- SMase
- sphingomyelinase
- ER
- endoplasmic reticulum
- SREBP
- sterol regulatory element-binding protein
- OlyA
- ostreolysin A
- ALOFL
- anthrolysin O full-length
- PlyB
- pleuorotolysin B
- HMGCR
- 3-hydroxy-3-methylglutaryl-CoA reductase
- ACAT
- acyl-CoA:cholesterol acyltransferase
- FCS
- fetal calf serum
- HPCD
- hydroxypropyl-β-cyclodextrin
- LPDS
- lipoprotein-deficient serum
- NPC1
- Niemann-Pick C1
- DAPI
- 4′,6-diamidino-2-phenylindole
- LDL
- low-density lipoprotein
- vLDL
- very low density lipoprotein
- UTSW
- UT Southwestern Medical Center
- 25HC
- 25-hydroxycholesterol
- AU
- Airy unit
- RBC
- red blood cell.
References
- 1. Brown M. S., Radhakrishnan A., and Goldstein J. L. (2018) Retrospective on cholesterol homeostasis: the central role of scap. Annu. Rev. Biochem. 87, 783–807 10.1146/annurev-biochem-062917-011852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Horton J. D., Goldstein J. L., and Brown M. S. (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 10.1172/JCI0215593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wattenberg B. W., and Silbert D. F. (1983) Sterol partitioning among intracellular membranes. J. Biol. Chem. 258, 2284–2289 [PubMed] [Google Scholar]
- 4. Xu X. X., and Tabas I. (1991) Lipoproteins activate acyl-coenzyme A:cholesterol acyltransferase in macrophages only after cellular cholesterol pools are expanded to a critical threshold level. J. Biol. Chem. 266, 17040–17048 [PubMed] [Google Scholar]
- 5. Neufeld E. B., Cooney A. M., Pitha J., Dawidowicz E. A., Dwyer N. K., Pentchev P. G., and Blanchette-Mackie E. J. (1996) Intracellular trafficking of cholesterol monitored with a cyclodextrin. J. Biol. Chem. 271, 21604–21613 10.1074/jbc.271.35.21604 [DOI] [PubMed] [Google Scholar]
- 6. Underwood K. W., Jacobs N. L., Howley A., and Liscum L. (1998) Evidence for a cholesterol transport pathway from lysosomes to endoplasmic reticulum that is independent of the plasma membrane. J. Biol. Chem. 273, 4266–4274 10.1074/jbc.273.7.4266 [DOI] [PubMed] [Google Scholar]
- 7. Lange Y., Ye J., and Steck T. L. (2004) How cholesterol homeostasis is regulated by plasma membrane cholesterol in excess of phospholipids. Proc. Natl. Acad. Sci. U.S.A. 101, 11664–11667 10.1073/pnas.0404766101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luo J., Jiang L. Y., Yang H., and Song B. L. (2019) Intracellular cholesterol transport by sterol transfer proteins at membrane contact sites. Trends Biochem. Sci. 44, 273–292 10.1016/j.tibs.2018.10.001 [DOI] [PubMed] [Google Scholar]
- 9. Das A., Brown M. S., Anderson D. D., Goldstein J. L., and Radhakrishnan A. (2014) Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis. Elife 3, e02882 10.7554/eLife.02882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Infante R. E., and Radhakrishnan A. (2017) Continuous transport of a small fraction of plasma membrane cholesterol to endoplasmic reticulum regulates total cellular cholesterol. Elife 6, e25466 10.7554/eLife.25466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Das A., Goldstein J. L., Anderson D. D., Brown M. S., and Radhakrishnan A. (2013) Use of mutant 125I-perfringolysin O to probe transport and organization of cholesterol in membranes of animal cells. Proc. Natl. Acad. Sci. U.S.A. 110, 10580–10585 10.1073/pnas.1309273110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sokolov A., and Radhakrishnan A. (2010) Accessibility of cholesterol in endoplasmic reticulum membranes and activation of SREBP-2 switch abruptly at a common cholesterol threshold. J. Biol. Chem. 285, 29480–29490 10.1074/jbc.M110.148254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Flanagan J. J., Tweten R. K., Johnson A. E., and Heuck A. P. (2009) Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry 48, 3977–3987 10.1021/bi9002309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Scheek S., Brown M. S., and Goldstein J. L. (1997) Sphingomyelin depletion in cultured cells blocks proteolysis of sterol regulatory element binding proteins at site 1. Proc. Natl. Acad. Sci. U.S.A. 94, 11179–11183 10.1073/pnas.94.21.11179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Slotte J. P., and Bierman E. L. (1988) Depletion of plasma membrane sphingomyelin rapidly alters the distribution of cholesterol between plasma membranes and intracellular cholesterol pools in cultured fibroblasts. Biochem. J. 250, 653–658 10.1042/bj2500653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Endapally S., Frias D., Grzemska M., Gay A., Tomchick D. R., and Radhakrishnan A. (2019) Molecular discrimination between two conformations of sphingomyelin in plasma membranes. Cell 176, 1040–1053.e17 10.1016/j.cell.2018.12.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Simons K., and Ikonen E. (2000) How cells handle cholesterol. Science 290, 1721–1726 10.1126/science.290.5497.1721 [DOI] [PubMed] [Google Scholar]
- 18. McConnell H. M., and Radhakrishnan A. (2003) Condensed complexes of cholesterol and phospholipids. Biochim. Biophys. Acta 1610, 159–173 10.1016/S0005-2736(03)00015-4 [DOI] [PubMed] [Google Scholar]
- 19. Lönnfors M., Doux J. P., Killian J. A., Nyholm T. K., and Slotte J. P. (2011) Sterols have higher affinity for sphingomyelin than for phosphatidylcholine bilayers even at equal acyl-chain order. Biophys. J. 100, 2633–2641 10.1016/j.bpj.2011.03.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Skočaj M., Resnik N., Grundner M., Ota K., Rojko N., Hodnik V., Anderluh G., Sobota A., Maček P., Veranič P., and Sepčić K. (2014) Tracking cholesterol/sphingomyelin-rich membrane domains with the ostreolysin A-mCherry protein. PLoS ONE 9, e92783 10.1371/journal.pone.0092783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bourdeau R. W., Malito E., Chenal A., Bishop B. L., Musch M. W., Villereal M. L., Chang E. B., Mosser E. M., Rest R. F., and Tang W. J. (2009) Cellular functions and X-ray structure of anthrolysin O, a cholesterol-dependent cytolysin secreted by Bacillus anthracis. J. Biol. Chem. 284, 14645–14656 10.1074/jbc.M807631200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lukoyanova N., Kondos S. C., Farabella I., Law R. H., Reboul C. F., Caradoc-Davies T. T., Spicer B. A., Kleifeld O., Traore D. A., Ekkel S. M., Voskoboinik I., Trapani J. A., Hatfaludi T., Oliver K., Hotze E. M., et al. (2015) Conformational changes during pore formation by the perforin-related protein pleurotolysin. PLoS Biol. 13, e1002049 10.1371/journal.pbio.1002049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brown M. S., and Goldstein J. L. (1990) Regulation of the mevalonate pathway. Nature 43, 425–430 10.1038/343425a0 [DOI] [PubMed] [Google Scholar]
- 24. Goldstein J. L., Basu S. K., and Brown M. S. (1983) Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 98, 241–260 10.1016/0076-6879(83)98152-1 [DOI] [PubMed] [Google Scholar]
- 25. Chang T.-Y., Chang C. Y., Ohgami N., and Yamauchi Y. (2006) Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol. 22, 129–157 10.1146/annurev.cellbio.22.010305.104656 [DOI] [PubMed] [Google Scholar]
- 26. Kovanen P. T., Brown M. S., Basu S. K., Bilheimer D. W., and Goldstein J. L. (1981) Saturation and suppression of hepatic lipoprotein receptors: a mechanism for the hypercholesterolemia of cholesterol-fed rabbits. Proc. Natl. Acad. Sci. U.S.A. 78, 1396–1400 10.1073/pnas.78.3.1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Adams C. M., Reitz J., De Brabander J. K., Feramisco J. D., Li L., Brown M. S., and Goldstein J. L. (2004) Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and insigs. J. Biol. Chem. 279, 52772–52780 10.1074/jbc.M410302200 [DOI] [PubMed] [Google Scholar]
- 28. Radhakrishnan A., Ikeda Y., Kwon H. J., Brown M. S., and Goldstein J. L. (2007) Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc. Natl. Acad. Sci. U.S.A. 104, 6511–6518 10.1073/pnas.0700899104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wojtanik K. M., and Liscum L. (2003) The transport of low density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J. Biol. Chem. 278, 14850–14856 10.1074/jbc.M300488200 [DOI] [PubMed] [Google Scholar]
- 30. Liscum L., and Faust J. R. (1987) Low density lipoprotein (LDL)-mediated suppression of cholesterol synthesis and LDL uptake is defective in Niemann-Pick type C fibroblasts. J. Biol. Chem. 262, 17002–17008 [PubMed] [Google Scholar]
- 31. Pentchev P. G. (2004) Niemann-Pick C research from mouse to gene. Biochim. Biophys. Acta 1685, 3–7 10.1016/j.bbalip.2004.08.005 [DOI] [PubMed] [Google Scholar]
- 32. Kwon H. J., Abi-Mosleh L., Wang M. L., Deisenhofer J., Goldstein J. L., Brown M. S., and Infante R. E. (2009) Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 137, 1213–1224 10.1016/j.cell.2009.03.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chakrabarti R. S., Ingham S. A., Kozlitina J., Gay A., Cohen J. C., Radhakrishnan A., and Hobbs H. H. (2017) Variability of cholesterol accessibility in human red blood cells measured using a bacterial cholesterol-binding toxin. Elife 6, e23355 10.7554/eLife.23355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lakshminarayan R., Wunder C., Becken U., Howes M. T., Benzing C., Arumugam S., Sales S., Ariotti N., Chambon V., Lamaze C., Loew D., Shevchenko A., Gaus K., Parton R. G., and Johannes L. (2014) Galectin-3 drives glycosphingolipid-dependent biogenesis of clathrin-independent carriers. Nat. Cell Biol. 16, 595–606 10.1038/ncb2970 [DOI] [PubMed] [Google Scholar]
- 35. Keller S. L., Radhakrishnan A., and McConnell H. M. (2000) Saturated phospholipids with high melting temperatures form complexes with cholesterol in monolayers. J. Phys. Chem. B 104, 7522–7527 10.1021/jp000958g [DOI] [Google Scholar]
- 36. Lange Y., Tabei S. M., Ye J., and Steck T. L. (2013) Stability and stoichiometry of bilayer phospholipid-cholesterol complexes: relationship to cellular sterol distribution and homeostasis. Biochemistry 52, 6950–6959 10.1021/bi400862q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liscum L., Luskey K. L., Chin D. J., Ho Y. K., Goldstein J. L., and Brown M. S. (1983) Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase and its mRNA in rat liver as studied with a monoclonal antibody and a cDNA probe. J. Biol. Chem. 258, 8450–8455 [PubMed] [Google Scholar]
- 38. Gay A., Rye D., and Radhakrishnan A. (2015) Switch-like responses of two cholesterol sensors do not require protein oligomerization in membranes. Biophys. J. 108, 1459–1469 10.1016/j.bpj.2015.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Endapally S., Infante R. E., and Radhakrishnan A. (2019) Monitoring and modulating intracellular cholesterol trafficking using ALOD4, a cholesterol-binding protein. Methods Mol. Biol. 1949, 153–163 10.1007/978-1-4939-9136-5_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McDonald J. G., Smith D. D., Stiles A. R., and Russell D. W. (2012) A comprehensive method for extraction and quantitative analysis of sterols and secosteroids from human plasma. J. Lipid Res. 53, 1399–1409 10.1194/jlr.D022285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Apostolopoulou M., Gordillo R., Koliaki C., Gancheva S., Jelenik T., De Filippo E., Herder C., Markgraf D., Jankowiak F., Esposito I., Schlensak M., Scherer P. E., and Roden M. (2018) Specific hepatic sphingolipids relate to insulin resistance, oxidative stress, and inflammation in nonalcoholic steatohepatitis. Diabetes Care 41, 1235–1243 10.2337/dc17-1318 [DOI] [PubMed] [Google Scholar]
- 42. Goldstein J. L., and Brown M. S. (1974) Binding and degradation of low density lipoproteins by cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J. Biol. Chem. 249, 5153–5162 [PubMed] [Google Scholar]