

Abstract
We investigated a dilated cardiomyopathy (DCM) mutation (F764L) in human β-cardiac myosin by determining its motor properties in the presence and absence of the heart failure drug omecamtive mecarbil (OM). The mutation is located in the converter domain, a key region of communication between the catalytic motor and lever arm in myosins, and is nearby but not directly in the OM-binding site. We expressed and purified human β-cardiac myosin subfragment 1 (M2β-S1) containing the F764L mutation, and compared it to WT with in vitro motility as well as steady-state and transient kinetics measurements. In the absence of OM we demonstrate that the F764L mutation does not significantly change maximum actin-activated ATPase activity but slows actin sliding velocity (15%) and the actomyosin ADP release rate constant (25%). The transient kinetic analysis without OM demonstrates that F764L has a similar duty ratio as WT in unloaded conditions. OM is known to enhance force generation in cardiac muscle while it inhibits the myosin power stroke and enhances actin-attachment duration. We found that OM has a reduced impact on F764L ATPase and sliding velocity compared with WT. Specifically, the EC50 for OM induced inhibition of in vitro motility was 3-fold weaker in F764L. Also, OM reduces maximum actin-activated ATPase 2-fold in F764L, compared with 4-fold with WT. Overall, our results suggest that F764L attenuates the impact of OM on actin-attachment duration and/or the power stroke. Our work highlights the importance of mutation-specific considerations when pursuing small molecule therapies for cardiomyopathies.
Keywords: myosin, actin, cardiomyopathy, enzyme kinetics, heart failure, omecamtiv mecarbil
Introduction
Genetic mutations in sarcomeric proteins are a significant cause of inherited cardiomyopathies with diverse clinical outcomes (1). Hypertrophic cardiomyopathy (HCM)2 and dilated cardiomyopathy (DCM) are two common phenotypes, affecting more than 1 in 500 and 1 in 2500 of the general population, respectively (2–4). MYH7, encoding human β-cardiac myosin heavy chain (M2β), is one of the most commonly mutated genes associated with inherited cardiomyopathies (3, 5). Identification of more than 300 pathogenic cardiomyopathy point mutations in M2β has raised the expectation for understanding the pathogenic mechanisms of mutations and therefore designing therapeutic interventions (6). Even though clinically HCM and DCM mutations are associated with hyper- and hypo-contractile phenotypes, respectively, the detailed molecular mechanisms are still unclear. HCM mutations in M2β are hypothesized to either enhance myosin motor function and/or increase the number of force-generating myosin heads that interact with actin filaments, whereas DCM mutations are proposed to impair intrinsic myosin motor performance (7, 8). However, variabilities have been observed in how DCM mutations affect motor properties including force/load sensitivity (9).
Myosin is the motor protein that drives cardiomyocyte contraction in the heart by utilizing a conserved actomyosin ATPase cycle that has been well-documented in the literature (Scheme 1) (10, 11). Muscle myosins are hexameric and consist of two heavy chains each containing two associated light chains. The myosin heavy chain contains an N-terminal motor domain, a neck domain or lever arm that binds the essential and regulatory light chains, and a C-terminal tail that facilitates dimerization and filament formation. The myosin ATPase cycle consists of weak actin-binding (M·ATP and M·ADP·Pi) and strong actin-binding (AM·ADP·Pi, AM·ADP, and AM) states, which are dependent on the nucleotide bound to the active site. During the strong actin-binding states a 70° lever arm swing referred to as the myosin power stroke occurs to generate contractile force. The period of time that myosin spends in the strong actin-binding states relative to the entire ATPase cycle, referred to as the duty ratio, is an important determinant of the number of myosin motors capable of generating force in a contractile unit.
Scheme 1.

Scheme of myosin ATPase cycle.
A recent report that examined the impact of several DCM-associated mutations in recombinantly expressed human M2β short subfragment-1 (M2β-sS1, containing the motor domain and essential light chain) concluded that many of the mutations reduced the myosin duty ratio (12). Interestingly, the mutations impaired different aspects of the myosin ATPase cycle, whereas they all reduced duty ratio to different extents under unloaded conditions and they were predicted to have a more significant decrease under loaded conditions. Furthermore, DCM mutations examined with single molecule mechanical approaches displayed impaired load-dependent detachment kinetics (9). These results were consistent with the hypothesis that DCM mutations reduce contractile function (ensemble force and/or shortening velocity). One DCM mutation studied previously is the F764L mutation located in the converter domain, which is a crucial domain that impacts communication between the actin-/nucleotide-binding sites and the lever arm. A mouse model that introduced the F764L mutation into α-cardiac myosin demonstrated a decrease in actin-activated ATPase activity and sliding velocity (13). Although the maximal force was comparable with WT, and a similar step size was determined from optical trapping studies (13, 14). The Ujfalusi et al. (12) study performed kinetic simulations that suggested F764L in human M2β-sS1 reduces the duty ratio 25% in unloaded conditions. In the presence of 5 pN load, the duty ratio of WT was predicted to increase 30%, whereas F764L was relatively unchanged. However, this study did not investigate the impact of F764L on in vitro motility that could shed light on how this mutation impairs unloaded shortening velocity in cardiac muscle. Overall, the ability to study the impact of inherited cardiomyopathy mutations is leading to novel insights into how the mutations specifically impair myosin function and opening the door to specific therapeutic developments.
Omecamtiv mecarbil (OM) is a positive cardiac myosin allosteric modulator that is in phase III clinical trials to treat systolic heart failure (15, 16). In both animal models and humans, OM improves cardiac performance as it increases left ventricular shortening, prolongs ejection fraction, and enhances ensemble force generation at intermediate Ca2+ concentrations (17, 18). Additionally, OM has been reported to be able to partially rescue myofibril contractile function caused by a DCM mutation in tropomyosin in a mouse model (19). Recent work found that OM can reverse the faster detachment rate of a DCM mutant introduced into M2β-sS1 (9). Interestingly, OM was demonstrated to enhance contractility through a unique mechanism in which it inhibits the power stroke but prolongs the time myosin is attached to actin and cooperatively activates the thin filament (20). Because OM is the only known inhibitor of the myosin power stroke, understanding the structural mechanism may lead to important insights into allosteric mechanisms that mediate the power stroke in myosin. In addition, it is unclear how OM will impact the performance of myosin containing DCM mutations because OM may alter allosteric pathways impaired by the mutations.
Here, we examined the impact of the F764L mutation in the converter domain on in vitro motility as well as steady-state and transient kinetics of the actomyosin ATPase cycle using recombinant human M2β-S1 (containing the motor domain and entire lever arm, both the essential and regulatory light chains). Our data suggest that the F764L mutation slightly impairs the motor properties of M2β under unloaded conditions. Additionally, we investigated the effect of OM on F764L motility and transient kinetics parameters. Our results show that OM impacts the function of F764L differently than WT, which provides important insight into the structural basis of OM and allosteric mechanisms of the myosin power stroke.
Results
M2β-S1 F764L and WT were purified from C2C12 cells on the same day, which allowed us to perform most experiments on F764L and WT in parallel. We obtained 0.5–1 mg of M2β-S1 protein from 20 p150 plates of infected C2C12 cells per protein preparation. The purified M2β-S1 was similar to our previous work (21) and the mouse essential and regulatory light chains present in the preparation were previously determined by MS (21). MOPS 20 buffer (10 mm MOPS, 20 mm KCl, 1 mm EGTA, 1 mm MgCl2, 1 mm DTT) was used for all solution experiments, except where noted. Unpaired Student's t tests were performed to determine significant differences between WT and F764L or OM and vehicle control (DMSO) on key experiments.
Steady-state ATPase
The actin-activated ATPase activity of M2β-S1 WT and F764L was examined in 5–6 different protein preparations. The ATPase rates were plotted as a function of actin concentration (V0 = ATPase in the absence of actin), and fit to the Michaelis-Menten equation to determine the maximum rate of ATPase (kcat) and the actin concentration at which ATPase is one-half of maximum (KATPase) (Fig. 1A, Table 1). Our results demonstrate a similar KATPase in both WT and F764L (77.5 ± 16.2 and 69.3 ± 24.1 μm, respectively), whereas the kcat for F764L was slightly reduced (6.4 ± 1.4 s−1) compared with WT (8.3 ± 1.1 s−1). However, the kcat difference was found to be not statistically significant (p value equals 0.31).
Figure 1.
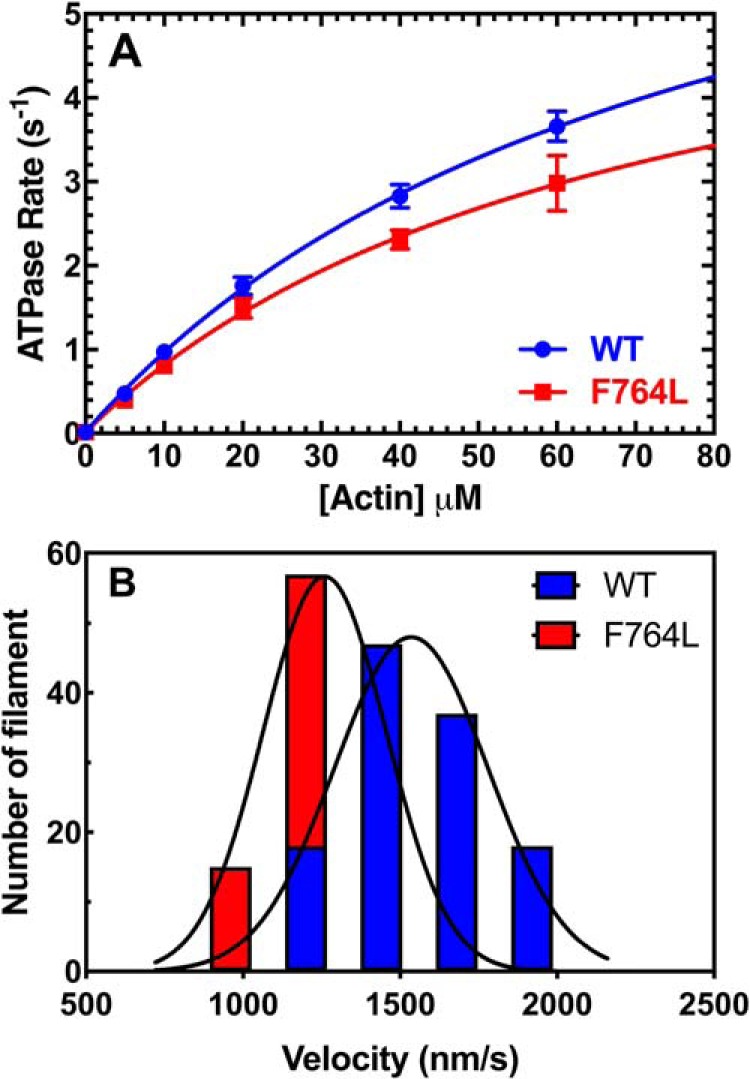
Steady-state ATPase and in vitro motility. A, the actin-activated ATPase activity of purified M2β-S1 WT and F764L in MOPS 20 buffer at 25 °C was determined as a function of actin concentration. The data at each actin concentration represents the average ± S.E. from 5 to 6 separate preparations. B, the sliding velocity of M2β-S1 WT and F764L in MOPS 20 buffer was analyzed manually via ImageJ. 120 filaments from 4 different protein preparations (30 filaments from each, 0.24–0.4 μm M2β-S1 loading concentration) were pooled together and fit to the Gaussian function. The average velocity is 1547 ± 19 nm/s for WT, and 1300 ± 17 nm/s for F764L.
Table 1.
Summary of steady-state ATPase parameters
| Condition | V0 | kcat | KATPase | EC50 |
|---|---|---|---|---|
| s−1 | μm | |||
| WT, buffer (n = 6) | 0.02 ± 0.02 | 8.3 ± 1.1 | 77.5 ± 16.2 | |
| WT, DMSO (n = 3) | 0.02 ± 0.01a | 7.5 ± 0.9a | 56.5 ± 13.9a | |
| WT, OM (n = 3) | 0.04 ± 0.01a | 1.6 ± 0.1a,b | 1.9 ± 0.4a,b | 1.5 ± 2.6 |
| F764L, buffer (n = 5) | 0.01 ± 0.01 | 6.4 ± 1.4 | 69.3 ± 24.1 | |
| F764L, DMSO (n = 3) | 0.02 ± 0.03 | 7.6 ± 1.0 | 59.6 ± 13.8 | |
| F764L, OM (n = 3) | 0.01 ± 0.01 | 3.7 ± 0.2c | 8.3 ± 1.7c | 1.6 ± 1.8 |
a Acquired from our previous work (21).
b 10 μm OM condition compared to 0.1% DMSO, p < 0.05.
c F764L compared to WT condition, p < 0.05.
In vitro motility assay
The actin sliding velocities generated by F764L and WT M2β-S1 were examined with the unloaded in vitro motility assay (Fig. 1B). We examined 120 filaments in total, 30 filaments each from four different protein preparations (0.24–0.4 μm densities), to determine the average sliding velocity (Table 2). We found that F764L (1300 ± 17 nm/s) has about a 15% slower sliding velocity compared with WT (1547 ± 19 nm/s, p < 0.0001). Velocities were also binned and fit to a Gaussian function, which shows that F764L causes a clear shift in the distribution of velocities (Fig. 1B). We confirmed the results by analyzing the velocities with the automated tracking program FAST (22) (Fig. S1, Table S1). We also examined the actin sliding velocities at different myosin surface densities. We found that F764L was slower than WT at all densities examined (Fig. S2). Thus, in unloaded conditions the steady-state motor properties of M2β-S1 F764L were slightly altered compared with WT.
Table 2.
Summary of in vitro motility sliding velocities
| Construct | Velocity (nm/s) |
EC50 (μm) (n = 3) c | ||||
|---|---|---|---|---|---|---|
| Buffer (n = 120)a | DMSO (n = 90)b | OM (n = 90) b | DMSO (n = 90)c | OM (n = 90) c | ||
| WT | 1547 ± 19 | 1771 ± 46 | 8.0 ± 0.2d,e | 1824 ± 26 | 9.9 ± 0.3d,e | 0.074 ± 0.002 |
| F764L | 1300 ± 17d,f | 1539 ± 24d,f | 29.4 ± 0.8d,e,f | 1733 ± 31f,g | 44 ± 1.0d,e,f | 0.246 ± 0.008d,f |
a Four protein preparations at 0.24–0.4 μm density.
b Three protein preparations at 0.24 μm density with 20 mm KCl.
c Three protein preparations at 0.4 μm density with 100 mm KCl.
d p < 0.001.
e 10 μm OM condition compared to 0.1% DMSO.
f F764L compared to WT condition.
g p < 0.05.
Impact of OM on ATPase activity
OM is known as a human cardiac muscle activator, whereas it decreases the ATPase and motility dramatically when it is bound to M2β. To investigate how OM can potentially impact the DCM-causing F764L mutant, we examined the steady-state motor properties of F764L in the presence of saturating OM (10 μm), while DMSO (0.1%) was used as a control. For the steady-state ATPase assay, OM caused a significant inhibition of kcat and decrease in KATPase in F764L, but the relative change in F764L was not as dramatic as was observed with WT (Fig. 2A, Table 1). We also examined the ATPase activity in response to varying OM concentrations at 20 μm actin with 3 different protein preparations (Fig. 2B). Our data demonstrated that at all OM concentrations tested, the ATPase activity of F764L was enhanced at 20 μm actin. This is in contrast to the results with WT where we found OM inhibited actin-activated ATPase in a dose-dependent manner at this actin concentration. Our results are consistent with our other steady-state ATPase activity measurements for F764L (Fig. 2A) and previous work with WT (21) at 20 μm actin. We found no significant difference in the EC50 for OM in WT (1.5 ± 2.6 μm) and F764L (1.6 ± 1.8 μm) based on the dose-dependent ATPase measurements (Table 1).
Figure 2.

Impact of OM on steady-state motor properties. A, the actin-activated ATPase of purified M2β-S1 F764L in the presence of 0.1% DMSO or 10 μm OM. The data at each actin concentration represents the average ± S.D. from three separate preparations. B, the ATPase activity of purified M2β-S1 WT and F764L was determined as a function of OM concentration at 20 μm actin. Traces were fit to a dose-response curve to determine the EC50. The data at each OM concentration represents the average ± S.E. of three separate preparations. C, in vitro motility was performed in the presence of varying concentrations of OM (0–10 μm in 0.1% DMSO) to determine the EC50 with 0.4 μm M2β-S1 WT or F764L. The relative velocities of M2β-S1 WT and F764L are normalized to the velocities at 0 μm OM from three separate preparations (90 filaments). Error bars represent mean ± S.E. D, in vitro motility was performed in the presence of varying concentrations of OM (0–200 μm in 1% DMSO). Relative velocities of M2β-S1 WT or F764 were normalized to the velocities at 0 μm OM from two different protein preparations. Inset highlights the difference in velocity at high OM concentrations (velocity at 200 μm OM: WT, 4.4 ± 0.2 nm/s; F764L, 12.8 ± 0.6 nm/s). Error bars represent mean ± S.E. In the OM titration experiments shown in C and D, additional KCl was added to the activation buffer to bring it to 100 mm KCl.
Impact of OM on in vitro motility
At 0.24 μm M2β-S1 density, the presence of 10 μm OM decreased the sliding velocity more than 200-fold for WT (DMSO, 1771 ± 46 nm/s; OM, 8.0 ± 0.2 nm/s), but only 50-fold for F764L (DMSO, 1539 ± 24 nm/s; OM, 29.4 ± 0.8 nm/s). The motility experiments in the presence of OM were performed in MOPS 20 buffer with additional KCl added (100 mm). The 200-fold decrease in WT M2β-S1 was similar to our previous results (21) but larger than observed by other groups (9, 23), which may be a result of differences in surface attachment methods, constructs examined, and buffer conditions (21). Interestingly, our data fit well to a model developed by Woody et al. (20), which demonstrates that our in vitro motility results are consistent with the optical trapping studies. The sliding velocity as a function of OM concentration (0–10 μm in 0.1% DMSO) was examined and plotted for both WT and F764L with 3 different protein preparations at 0.4 μm M2β-S1 density (Fig. 2C). OM caused a slowing down of the sliding velocity of both WT and F764L, but the inhibitory effect in F764L was not as large as WT at all tested OM concentrations. The difference in velocity between WT (4.4 ± 0.2 nm/s) and F764L (12.8 ± 0.6 nm/s) was still observed even at extremely high OM concentrations (200 μm OM with 1% DMSO, Fig. 2D). Additionally, the velocities of WT and F764L were examined at different myosin loading concentrations (0.12, 0.24, 0.36, and 0.48 μm) in the presence of 0.1% DMSO and 10 μm OM, and we found the difference in velocity was similar at all M2β-S1 densities examined (Fig. S2). The dose dependence of in vitro motility revealed that F764L has a 3-fold higher EC50 for OM compared with WT. The percent of stuck filaments was unaffected by OM and determined to be 2–8% for all conditions. Together, our results revealed that OM may impact F764L differently than WT, which we explored mechanistically with transient kinetic analysis.
Impact of F764L and/or OM on the transient kinetics of M2β-S1
The myosin family shares a highly conserved ATPase cycle to convert chemical energy into mechanical force. Scheme 1 represents the established ATPase cycle and displays the predominant pathway by highlighting the key biochemical states in brackets. In this scheme we used the convention that the forward rate constants occur from left to right. We investigated the impact of the F764L mutation on the transient kinetics of many key steps in the actomyosin ATPase cycle. After determining many of the rate and equilibrium constants with transient kinetic methods, we developed a kinetic model of the overall ATPase cycle and used kinetic simulations to compare the model to our experimental steady-state ATPase measurements. Concentrations listed in the description of experiments are final unless otherwise noted. All errors in the transient kinetic experiments are reported as standard error of the fits.
We examined the impact of the F764L mutant on the individual steps in the myosin ATPase cycle. In addition, we examined the impact of OM on F764L and compared these results to parallel measurements of WT. Therefore, the impact of the mutation and/or the drug were analyzed using 0.1% DMSO as the no drug control. In our previous work, we characterized the impact of OM on 2′/3′-O-(N-methyl-anthraniloyl)-(mant)-ATP single turnover measurements in the presence of varying actin concentrations (21). We found that the fluorescence transients were best fit by a two-exponential function and the fast phase was similar to the steady-state actin-activated ATPase rates, whereas the slow phase was proposed to be an off-pathway intermediate. In the current work, we performed similar single turnover experiments on M2β-S1 WT and F764L in parallel (Fig. 3, A–C). Sub-stoichiometric mant-ATP (0.2 μm) was mixed with 0.25 μm M2β-S1, aged 10 s to allow hydrolysis to occur, and then mixed with varying concentrations of actin (2.5 to 50 μm) to follow actin-activated product release. We observed a two-exponential fluorescence decrease at all actin concentrations. The rate constants associated with the fast phase were plotted as a function of actin concentration and fit to a hyperbolic function to determine the maximal rate constant (Fig. 3, A and B). The impact of the F764L mutation on the maximum rate constant of the single turnover was not determined because the actin-dependence of the rate constants was almost linear and did not saturate at the highest actin concentrations, whereas the value for WT was determined to be 18.9 ± 8.6 s−1 (Table 3). Although the maximum rate of the single turnover should be similar to the steady-state ATPase maximum rate, it is possible that the mant-labeled nucleotide slightly alters the product release steps. The initial slope of the hyperbolic fit was used to compare differences in actin-activated product release, which was 2-fold slower in the F764L compared with WT (0.17 ± 0.01 and 0.35 ± 0.01 μm−1 s−1, respectively). In the presence of OM, the maximum single turnover rate constants were reduced in WT (4.2 ± 0.3 s−1) and F764L (8.4 ± 0.5 s−1), further demonstrating the OM inhibition in F764L is not as large as WT. The K0.5, actin concentration at which the rate constant is one-half maximal, in WT (0.4 ± 0.6 μm) was much lower than F764L (6.1 ± 1.4 μm) in the presence of OM.
Figure 3.

Single turnover measurements. Single turnover sequential mix stopped-flow experiments were performed by mixing 0.25 μm M2β-S1 with 0.2 μm mant-ATP, aged for 10 s, and then mixed with varying concentrations of actin (2.5–50 μm) in the presence of 0.1% DMSO or 10 μm OM. Data were collected from 3 to 4 different protein preparations. The fast phase of the fluorescence transients was plotted as a function of actin concentration and fit to a hyperbolic function for WT (A) and F764L (B). C, representative fluorescence transients in the presence of 40 μm actin (average of 2–3 transients) fit to a two-exponential function (values for the fast phase rate constant and relative amplitude for WT DMSO, k = 6.24 ± 0.03 s−1; A = 0.90; WT OM, k = 4.23 ± 0.03 s−1; A = 0.90; F764L DMSO, k = 5.68 ± 0.02 s−1, A = 0.89; F764L OM, k = 6.91 ± 0.03 s−1, A = 0.91).
Table 3.
Transient kinetics parameters of M2β-S1 F764L, and WT
All parameters were measured in MOPS 20 buffer in the absence (0.1% DMSO) or presence of OM (10 μm).
| Rate constant | WT (DMSO) | WT (OM) | F764L (DMSO) | F764L (OM) |
|---|---|---|---|---|
| Single turnover,a actin-activated product release, n = 3–4 | ||||
| K0.5 (μm) | 38.4 ± 31.5 | 0.4 ± 0.6 | NDb | 6.1 ± 1.4 |
| Actin-dep. (μm−1s−1) | 0.35 ± 0.01 | 1.07 ± 0.01 | 0.17 ± 0.01 | 0.74 ± 0.03 |
| Maximum rate (s−1) | 18.9 ± 8.6 | 4.2 ± 0.3 | ND | 8.4 ± 0.5 |
| AFast at 50 μm actin | 0.88 | 0.86 | 0.82 | 0.84 |
| ATP binding/hydrolysis (myosin),c n = 2 | ||||
| K0.5 (μm) | 48 ± 6 | 49 ± 5 | 79 ± 6 | 93 ± 7 |
| K1Tk+2T (μm−1s−1) | 2.8 ± 0.1 | 2.5 ± 0.2 | 1.9 ± 0.1 | 1.6 ± 0.1 |
| k+H + k−H (s−1) | 182 ± 6 | 169 ± 5 | 166 ± 6 | 148 ± 7 |
| ATP binding to actomyosin,d n = 1 | ||||
| KIT′k+2T′ (μm−1s−1) | 9.6 ± 0.2 | 9.5 ± 0.3 | 9.4 ± 0.2 | 9.7 ± 0.3 |
| 1/KIT′ (μm) | 31.3 ± 8.8 | 62.5 ± 15.6 | 24.4 ± 8.3 | 43.5 ± 13.2 |
| k+2T′ (s−1) | 512 ± 47 | 793 ± 72 | 409 ± 42 | 645 ± 63 |
| Actin-activated Pi release at 30 μm actin,e n = 1 | ||||
| k+Pi(fast)′ (s−1) | 20.7 ± 0.9 | 69.5 ± 1.4 | 16.4 ± 1.7 | 32.1 ± 0.9 |
| k+Pi(slow)′ (s−1) | 1.86 ± 0.02 | 1.52 ± 0.02 | 1.68 ± 0.01 | 1.64 ± 0.02 |
| AFast | 0.24 | 0.66 | 0.08 | 0.38 |
| Kassoc·k+Pi(fast)′ (μm−1s−1) (2.5–30 μm actin), n = 2 | ND | 2.4 ± 0.1 | ND | 1.2 ± 0.1 |
| ADP-release from actomyosin | ||||
| k+D′ (s−1),a n = 1 | 347 ± 7 | 349 ± 7 | 232 ± 3 | 252 ± 3 |
| k+D′ (s−1),f n = 1 | 206 ± 4 | ND | 158 ± 1 | ND |
| k+D′ (s−1),d n = 2 | 175 ± 16 | 191 ± 10 | 136 ± 9 | 131 ± 4 |
a Mant-ADP.
b ND, not determined.
c Tryptophan fluorescence.
d Pyrene actin.
e MDCC-PBP.
f MOPS 20 buffer, 0.1% DMSO with 5 mm MgCl2.
The ATP-binding/hydrolysis steps were examined by monitoring the tryptophan fluorescence enhancement observed upon mixing 1 μm M2β-S1 WT or F764L with varying concentrations of ATP (5 to 1000 μm) (Fig. 4, A–C). The fluorescence transients were fit to a two-exponential function. The rate constants of the fast phase depended on ATP concentration and were fit to a hyperbolic function to determine the maximum rate of ATP hydrolysis, k+H + k−H (s−1), and ATP concentration dependence, K0.5 (μm) (Fig. 4, A and B). F764L contained a slightly slower maximum rate constant (165.5 ± 6.1 s−1) compared with WT (181.5 ± 6.3 s−1), while the K0.5 (μm) was 2-fold higher than WT (WT, 48 ± 6 μm; F764L, 79 ± 6 μm), indicating a weaker ATP-binding affinity. At lower concentrations of ATP, the fast phase was linearly dependent on ATP concentration, which allowed determination of the second-order binding constant for ATP binding to myosin (WT, 2.8 ± 0.1 μm−1 s−1; F764L, 1.9 ± 0.1 μm−1 s−1). The slow phase of the tryptophan fluorescence signal was ∼10% of the total amplitude and independent of ATP concentration and similar for both WT and F764L (∼1.4 s−1). The addition of OM did not have a major impact on the ATP-binding and -hydrolysis rate constants of WT and F764L (Table 3).
Figure 4.

ATP binding and hydrolysis. Tryptophan fluorescence enhancement was used to monitor ATP binding/hydrolysis by mixing 1 μm M2β-S1 WT or F764L with varying concentrations of ATP. The fluorescence transients were fit to a two-exponential function. The observed fast phase was plotted as a function of ATP concentration and fit to a hyperbolic function to determine maximum rate of ATP hydrolysis and apparent affinity for ATP in the presence of 0.1% DMSO (A) or 10 μm OM (B). C, representative fluorescence transients (average of 2 transients) in the presence of 25 μm ATP are shown fit to a two-exponential function (values for the fast phase rate constant and relative amplitude for WT DMSO, k = 61.95 ± 0.86 s−1; A = 0.86; WT OM, k = 47.20 ± 0.78 s−1; A = 0.92; F764L DMSO, k = 39.7 ± 0.69 s−1, A = 0.83; F764L OM, k = 27.98 ± 0.60 s−1, A = 0.82).
We examined ATP-induced dissociation from actomyosin by mixing a complex of M2β-S1:pyrene actin (0.375 μm M2β-S1 and pyrene actin) with a series of ATP concentrations (4 to 250 μm) (Fig. 5, A–C). The fluorescence transients were fit to a two-exponential function and the rate constant of the fast phase was fit to a hyperbolic function (kobs = K1′k+2T′ × [ATP]/(1 + K1′ × [ATP])] to determine the maximum rate k+2T′ (s−1) and equilibrium constant for the initial interaction between ATP and actomyosin (K1′)(24) (Fig. 5, A and B). The maximum rate was similar for WT (512 ± 47 s−1) and F764L (409 ± 42 s−1) in the presence of DMSO (Table 3). The presence of OM increases the maximal rate (∼50%) for both WT and F764L (WT, 793 ± 72 s−1; F764L, 645 ± 63 s−1). The second-order rate constant K1T′k+2T′ (μm−1 s−1) determined from the linear fit of the fast rate constant at lower ATP concentrations (up to 15 μm) was similar in WT and F764L in the presence and absence of OM. The slow phase of the pyrene fluorescence signal was more prominent at higher ATP concentrations (relative amplitude of 30% at 250 μm ATP), hyperbolically dependent on ATP concentration, and similar for both WT and F764L (maximum rate constant 50–100 s−1).
Figure 5.

ATP binding to acto-myosin. ATP-induced dissociation from pyrene actin was performed by mixing a complex of M2β-S1:pyrene actin (0.375 μm M2β-S1 and pyrene actin) with a series of ATP concentration (4 to 250 μm). The fluorescence transients were fit to a two-exponential function at all ATP concentrations. The observed fast rate constant was hyperbolically dependent on ATP concentrations in the presence of 0.1% DMSO (A) or 10 μm OM (B). C, representative fluorescence transients in the presence of 25 μm ATP (average of 2 transients) were fit to a two-exponential function (values for the fast phase rate constant and relative amplitude for WT DMSO, k = 222.6 ± 5.0 s−1; A = 0.92; WT OM, k = 213.2 ± 7.3 s−1; A = 0.94; F764L DMSO, k = 207.1 ± 4.8 s−1, A = 0.91; F764L OM, k = 232 ± 8.8 s−1, A = 0.88).
The actin-activated phosphate release rate constants were monitored by mixing M2β-S1 (0.225–1 μm) with substoichiometric ATP, aged for 10 s to allow for ATP binding and hydrolysis to occur, and then mixed with varying concentrations of actin in the presence of phosphate-binding protein (4 μm MDCC-PBP) (Fig. 6, A and B). The phosphate release fluorescence transients were best fit by a two-exponential function (Fig. 6A). We considered the fast phase to be the actin-activated phosphate release rate constant, whereas the slow phase was assumed to be steady-state ATPase and/or pre-hydrolysis intermediates. The slow phase of phosphate release was similar in WT and F764L in the presence and absence of OM (1–2 s−1). The relative amplitude of the fast phase was 2–3-fold higher for WT compared with F764L in the presence and absence of OM (Table 3). Although the presence of OM enhances the phosphate release rate constant k+Pi′ (s−1) for both WT and F764L (30 μm actin), the rate constant of F764L was slower compared with WT under both DMSO (WT, 20.7 ± 0.9 s−1; F764L, 16.4 ± 1.7 s−1) and OM (WT, 69.5 ± 1.4 s−1; F764L, 32.1 ± 0.9 s−1) conditions (Table 3). The larger amplitude of phosphate burst in the presence of OM allowed us to examine actin-activated phosphate release at a series of actin concentrations (Fig. 6B). We found the phosphate release rate constant was linearly dependent on actin concentration in the presence of OM and was 2-fold faster in WT (2.4 ± 0.1 μm−1 s−1) compared with F764L (1.2 ± 0.1 μm−1 s−1).
Figure 6.

Actin-activated phosphate release. The actin-activated phosphate release rate constants in the presence of OM were examined by mixing M2β-S1 (0.225–1 μm) with substoichiometric ATP, aged for 10 s, and mixed with varying actin concentrations. A, the MDCC-PBP fluorescence transients (average of 2–3 transients) in 10 μm actin were fit to a two-exponential function (values for the fast phase rate constant and amplitude for WT, k = 25.1 ± 0.9 s−1, A = 0.32 ± 0.01; and F764L, k = 18.4 ± 0.8 s−1, A = 0.21 ± 0.01) (inset shows the entire time course). B, the rate constant of the fast phase of the fluorescence transients were plot as a function of actin concentration and fit to a linear relationship to compare actin-activated phosphate release in F764L and WT in the presence of OM. The phosphate release rate constants (WT versus F764L) were also measured in the presence of a single actin concentration (30 μm) in the absence of OM (see Table 3).
We examined the ADP release rate constant by mixing a pyrene actomyosin-ADP complex (0.225 μm of both M2β-S1 and pyrene actin, 50 μm ADP) with varying ATP concentrations (10 to 750 μm) (Fig. 7A). Pyrene fluorescence transients were fit to a single exponential function and the rate constants were fit to a hyperbolic equation (kobs = k+D′/(1 + Kapp/[ATP]) to determine the ADP release rate constant (k+D′) (Fig. 7A). The fluorescence transients were also fit to a kinetic model, which incorporated the rates of ATP binding (determined in Fig. 5) and ADP binding (estimated from Liu et al. (23)), and similar results were obtained (Fig. S3, Table S2). The observed rate constants may be slightly different because some assumptions of the hyperbolic equation are inaccurate (e.g. the maximum rate of the ATP-induced dissociation is much faster than ADP release). We also examined the ADP release rate constant by mixing actomyosin mant-ADP (0.5 μm M2β-S1, 1 μm actin, and 10 μm mant-ADP) with 1 mm ATP at 1 mm (Fig. 7B) and 5 mm MgCl2. With both methods, F764L demonstrated a slower ADP release rate constant (∼25% decrease) compared with WT (Table 3). Consistent with previous work, OM does not affect the ADP release rate constant of either WT or F764L (Table 3) (21).
Figure 7.

ADP release from acto-myosin. A, the ADP release rate constant was examined by mixing pyrene actomyosin·ADP (0.225 μm M2β-S1 and pyrene actin, 50 μm ADP) with varying ATP concentrations (10 to 750 μm) in the presence of 0.1% DMSO or 10 μm OM. Fluorescent transients were fit to a single exponential function. The rate constants were fit to hyperbolic function (kobs = k+D′/(1 + Kapp/[ATP])) to determine the ADP release rate constant (k+D′). B, the ADP release rate constant was also examined with mant-labeled ADP by mixing a complex of 0.5 μm M2β-S1, 1 μm actin, and 10 μm mant-ADP with 1 mm ATP. Representative fluorescence transients of mant-ADP release from actomyosin (average of 5 transients) fit to a single exponential function are shown (rate constant and amplitude at each condition; WT DMSO, k = 347.4 ± 7.6 s−1, A = 0.38; WT OM, k = 348.9 ± 6.1 s−1, A = 0.45; F764L DMSO, k = 232.1 ± 3.2 s−1, A = 0.51; F764L OM, k = 251.6 ± 3.0 s−1, A = 0.53).
Finally, we developed a kinetic model of the overall ATPase cycle and used parameters from transient kinetics to simulate the overall ATPase rate and duty ratio (see Table S3 for values used for simulation). Our simulated ATPase data at different actin concentrations in the presence of DMSO or OM for both WT and F764L matches our steady-state ATPase measurements fairly well (Fig. S4, A and B). Our results also demonstrate that OM can alter the steady-state ATPase parameters (kcat and KATPase) of cardiac myosin by accelerating Pi-release and then slowing another transition after Pi-release (e.g. kD′, or entry into an off-pathway intermediate). In the absence of OM the duty ratio was calculated from the fraction of myosin in the strong binding states relative to the entire ATPase cycle for both WT and F764L (Fig. S4C). Our simulation suggests that F764L has a similar unloaded duty ratio compared with WT (WT, 0.057 ± 0.002; F764L, 0.063 ± 0.002). We did not model the duty ratio with OM because it is still controversial if M2β populates off-pathway intermediates in the presence of the drug.
Discussion
Our work characterized the motor properties of a clinically reported DCM mutation, F764L, in human M2β. We found that F764L only causes relatively minor changes in ATPase kinetics and unloaded in vitro motility sliding velocity. We conclude the mutant has a similar duty ratio as WT under unloaded conditions, whereas the parameters might be dramatically altered in the presence of load. Additionally, we found the myosin allosteric regulator, OM, has a more moderate impact on F764L ATPase and motility compared with WT. A major difference is that OM does not increase the phosphate release rate constant or slow the ATPase cycle time as much in F764L compared with WT. Our findings provide new insight into the understanding of the molecular mechanism of inherited cardiomyopathy mutations, which may improve future therapeutic interventions for treating patients with various mutations.
Comparison of transient kinetic analysis of human cardiac myosin with previous studies
Our transient kinetic analysis of human cardiac myosin S1 has many similarities but also some differences compared with previous work. We have summarized some of the previous studies (Table S4), which highlights that the differences may be due to different buffer conditions, experimental temperatures, and the specific constructs examined. The impact of ionic strength (25) and temperature (26) on the myosin ATPase cycle is well-documented in the literature. The rate constants found to be most different are ATP hydrolysis and ADP-release from actomyosin. One major factor that may account for the differences in ATP hydrolysis is the light chain content. Interactions between the converter domain and essential light chain have been demonstrated (27, 28), and myosins that vary only by the light chain content were found to have different ATP hydrolysis rate constants (29, 30). Interestingly, the light chain content of the C2C12 cell expressed/purified myosin could vary depending on how long the cells are differentiated before harvest (e.g. 7–10 days in the current study and 4 days in Deacon et al. (31)). A major factor that may explain the ADP release rate constant differences could be the free Mg2+ concentration. We and others have previously found that Mg2+ can impact the ADP release rate constant, with higher free Mg2+ concentrations slowing ADP release (32–34). Indeed, our current results demonstrate that higher Mg2+ also slows M2β-S1 ADP-release (Table 3). Overall, our transient kinetic analysis of WT human cardiac S1 is consistent with previous studies given the different constructs used and the different buffer conditions.
Impact of F764L on M2β
We performed a thorough characterization of the impact of the F764L mutation on M2β-S1 motor properties and our results are relatively similar to a study that examined F764L in M2β-sS1 (12). Although there was no significant difference in steady-state ATPase, we observed a 15% decrease in unloaded sliding velocity and a 25% decrease in the ADP release rate constant. The decreased ADP release rate constant is in disagreement with the M2β-sS1 results, where they reported no change in ADP-release in their analysis of F764L (12). Our results which demonstrate a slightly reduced actin-activated phosphate release (kcat and k+Pi′) and reduced ADP release rate constant, suggest that F764L causes a slowing down of the actin-activated product release steps. The direct ATP binding experiments utilizing the tryptophan fluorescence signal demonstrated that F764L has a weaker ATP binding affinity than WT, which was also reported in the M2β-sS1 study (12). The tryptophan fluorescence signal typically correlates with the recovery stroke rate constant (35, 36). Interestingly, disrupting the recovery stroke can alter the number of myosin heads available for entry into the force-generating states. Our study did not demonstrate a change in duty ratio (Fig. S4C), whereas the M2β-sS1 study demonstrated a slight decrease in duty ratio. We hypothesize that the F764L mutation may have a more significant impact in the presence of load under physiological conditions. The converter domain is the key region for load-sensing and it bears most of the elastic distortion during conformational changes in the ATPase cycle (37, 38). Phe-764 is located in the converter domain, in the converter-relay hydrophobic interface. The relay/converter domain interface has been shown in Drosophila indirect flight muscle myosin to be important for fine-tuning myosin kinetics (39, 40). Palmer et al. (41) observed that cardiac muscle from heterozygous F764L knockout mice exhibited significantly reduced cross-bridge stiffness, and therefore proposed that the F764L leads to a more compliant converter domain and myosin cross-bridge. Our recent work investigated the force-generating properties of the corresponding mutation (F750L) in myosin V, and found that this mutant is less able to overcome frictional loads compared with WT in the loaded in vitro motility assay (30). The F750L mutation also alters the recovery stroke rate constant and shifts the equilibrium between conformational states of the lever arm in several nucleotide states. However, extrapolating the myosin V results to cardiac myosin is difficult because myosin V has very different motor properties (e.g. high duty ratio). Nevertheless, the myosin V results suggest it is important to examine structural dynamics of the lever arm and load dependence in the future studies to fully understand the disease-causing mechanisms.
Implications for understanding how F764L leads to a DCM phenotype
Patients with DCM present with systolic heart failure, myocardial wall thinning, and left ventricular dilation (5, 42, 43). Thus, DCM mutations in M2β are proposed to depress contractile function, whereas specific molecular mechanisms may vary based on the mutation. Because the F764L mutation had a relatively minor impact on unloaded myosin ATPase kinetics, duty ratio, and sliding velocity, the implications for understanding how the mutation leads to a DCM phenotype are unclear. One explanation could be that the impact of the F764L mutation is more significant in the presence of load as discussed above. Another possibility is that F764L enhances the formation of the super-relaxed state (SRX), which is crucial for regulating the number of myosin heads available to generate force (44, 45). Interestingly, Phe-764 is not at the interface that is predicted to mediate head-head and head-tail interactions associated with the SRX state (46, 47). However, F764L may induce allosteric changes in the converter domain that alter SRX state interactions. In addition, if the ability to form the SRX state is related to populating the pre-power stroke state and/or controlling lever arm compliance (46, 47), the mutation may indirectly alter the formation of this important regulatory state.
Impact of OM on WT and F764L
Recent studies surprisingly revealed that OM is the first small molecule inhibitor of the myosin power stroke (20, 48), whereas this drug also enhances cardiac muscle force generation at submaximal calcium concentrations (15, 18, 19, 21). OM binds to an allosteric site near the converter domain, and stabilizes the pre-power stroke position of the lever arm, accelerates phosphate release, and is proposed to inhibit the power stroke (15, 20, 47–49). Interestingly, our work demonstrates that OM does not dramatically enhance the affinity for actin in the M·ADP·Pi state, as was also found in a study on porcine cardiac myosin (23), suggesting the drug directly alters the conformational changes that control phosphate release (Fig. 6). In our kinetic simulations we found that acceleration of phosphate release, without dramatically altering actin affinity, and slowing a post-phosphate release step (e.g. transition between actomyosin·ADP states, k+D'′) fits the ATPase data fairly well (Fig. S4, Table S3). Overall, in the presence of OM myosin remains attached to actin for longer periods of time, which enhances calcium sensitivity and cooperative activation of the thin filament. In addition, our results demonstrate that OM has a different impact on the ATPase cycle of F764L compared with WT. To explain the less inhibitory impact of OM on F764L motor properties, we propose three potential mechanisms.
OM binding affinity
It is possible that F764L has a lower affinity for OM compared with WT, which alters the ability of OM to impact the ATPase cycle and motility. A close-up of the OM-binding pocket in the M2β crystal structure shows that Phe-764 is nearby but not directly part of the OM-binding pocket (Fig. 8C). Even though it does not directly participate in OM binding, the Phe to Leu substitution may allosterically alter the OM-binding pocket. We observed a similar EC50 of WT and F764L in the actin-activated ATPase assay, and a 3-fold increase in EC50 of F764L in the in vitro motility assay (see Table 1). The EC50 of WT in the ATPase assay is different from the value determined from our previous work (21). This is likely because we chose to use a different actin concentration (20 μm) for our current dose-dependent ATPase measurements, because this concentration was ideal for observing drug-induced changes in WT and F764L.
Figure 8.
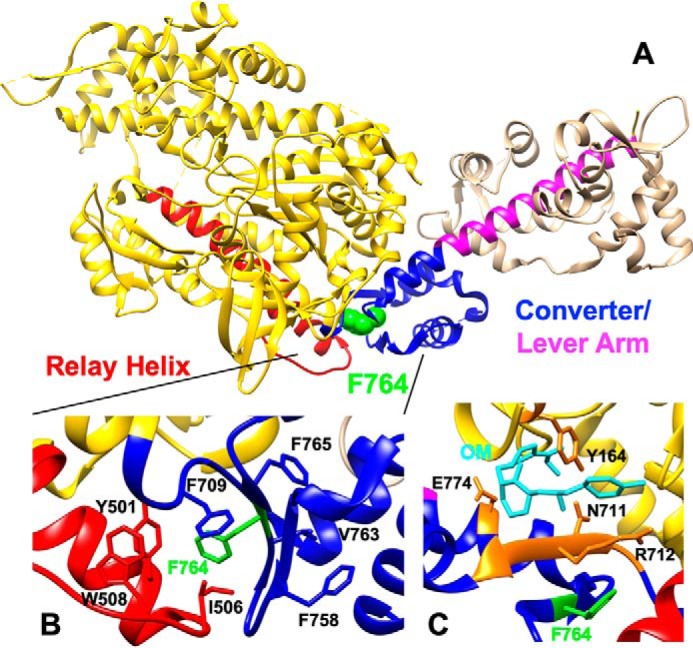
Structure of M2β-sS1 (PDB ID 5N69) highlighting the Phe-764 residue and OM-binding site. A, Phe-764 (F764) (green) is located at the interface of the relay helix (red) and converter domain (blue). B, F764 is involved in forming a hydrophobic interaction at the relay helix/converter domain interface. C, location of F764 in the M2β-sS1 OM-bound state. OM is shown in cyan, residues involved in direct OM binding interaction are shown in orange (27).
The EC50 difference between F764L and WT in the motility assay could be related to the attached time difference of the OM-bound myosin heads (discussed further below) as opposed to a simple binding affinity difference. In an ensemble motility assay the myosin heads unbound to OM have to work against the tethering force of the OM-bound myosin heads. If the OM-bound myosin heads stay attached for a shorter period of time they will be less efficient at inhibiting the motility. Thus, it will take more OM-bound myosin heads to slow the velocity to a similar value (e.g. 50%) in F764L than WT, which could explain the observed differences in EC50. Furthermore, if binding affinity is the only factor causing the different inhibitory effect, we should observe a similar sliding velocity at high saturating OM concentrations. To further investigate the binding affinity hypothesis, we examined the sliding velocity of WT and F764L at extremely high OM concentrations (i.e. up to 200 μm). Surprisingly, we observed nonzero sliding velocities of F764L even at extremely high OM concentrations (velocity saturates at ∼8 nm/s), compared with WT (∼0.4 nm/s) (see Fig. 2D). Overall, our results indicate that binding affinity alone cannot explain the differential effects of OM on M2β-S1 WT and F764L.
Actin-attachment duration
It has been proposed that OM locks the lever arm in a “primed” actin-attached pre-power stroke conformation, which prevents the lever arm swing associated with the power stroke (27, 48). In our previous work, we used single turnover stopped-flow measurements to demonstrate that OM slows a post-hydrolysis transition through the ATPase cycle (21). We concluded that this slower transition was evidence for a prolonged actin-attached state after phosphate release (e.g. AM′·ADP). Woody et al. (20) proposed that OM-bound myosin slowly detaches from an ADP bound or apo off-pathway state. In support of this argument they demonstrated that the detachment step is not dependent on ATP-binding or load. Our current studies demonstrate that OM does not have a major impact on nucleotide binding affinity (ATP binding and ADP release) in WT or F764L, which is consistent with previous work (21, 23). Additionally, OM accelerates the phosphate release step in both WT and F764L, whereas the increase is more pronounced in WT. We found F764L has a 2-fold decrease in the linear dependence of actin-activated phosphate release (Fig. 6) in the presence of OM, resulting in a slower transition into the strong actin-binding states. Our single turnover data suggests the attached lifetime (post-hydrolysis transition) of F764L is two times shorter than WT in the presence of OM. Assuming the step size remains the same, the sliding velocity of F764L should be 2-fold faster than WT as well, whereas the actual velocity is more than 3-fold different in the presence of 10 μm OM (Table 2). Taken together, OM-bound F764L could have a shorter “trapped” on-actin state compared with WT, but this does not completely explain the motility data. Interestingly, if F764L has a shorter actin attachment duration in the presence of OM then the drug will be less effective at cooperatively activating the thin filament and increasing contractile force in cardiac muscle. However, patients are typically heterozygous for the inherited cardiomyopathy mutations and it may only require a small percentage of WT cardiac myosin motors to bind OM and facilitate the increase in force.
Step size
The previous optical trapping studies demonstrated that the step size of WT is completely inhibited by OM (≤0.4 nm) (20), whereas the step size might not be completely inhibited in F764L. As we discussed above, previous work suggested F764L leads to a more compliant converter domain and cross-bridge (41). The shorter side chain of leucine compared with phenylalanine may provide a less rigid hydrophobic interface between the relay helix and converter domain. Thus, the F764L mutation may attenuate the inhibitory effects of OM on the power stroke and hence the step size. Our measurements of the sliding velocity of WT and F764L at saturating OM (Fig. 2D, 0.4 and 8 nm/s, respectively) allow us to estimate the impact of OM on step size. If the attached time (ton) of WT (ton = 0.1s) is two times that of F764L (ton = 0.05 s) (estimated from Woody et al. (20) and the 2-fold difference in kcat) then the estimated step size (duni) would be 0.04 nm for WT and 0.4 nm for F764L (using the equation duni = V × ton). Therefore, the ability of F764L to generate a small step size in the presence of OM can explain our results fairly well.
Overall, we favor that a combination of step size and attached time contributes to the differential impact of OM on F764L compared with WT. Future experiments with single molecule optical trapping, molecular dynamics simulations, FRET-based structural kinetics measurement, as well as crystallography would be helpful to fully characterize the underlying mechanisms that define the impact of OM on F764L and WT cardiac myosin.
Conclusion
Our work highlights the critical nature of the converter domain in providing allosteric communication between the active site/actin-binding regions and the lever arm domain. The relatively small shift in the ATPase kinetics and unloaded sliding velocity in the F764L DCM mutant suggests that examining the impact of load may be crucial for determining genotype-phenotype relationships. Our work further characterizes the impact of OM on human cardiac myosin motor ATPase kinetics by demonstrating that the drug accelerates phosphate release without dramatically altering actin binding affinity. Examining the impact of OM on F764L motor function indicates that small molecule drugs can have differential effects on mutant myosins. Therefore, it is important to examine how small molecule regulators impact specific myosin mutants, which may allow therapies to be precisely designed for cardiomyopathy patients.
Experimental procedures
Reagents
ATP and ADP were freshly prepared from powder (Sigma) and absorbance measurements at 259 nm (ϵ259 = 15,400 m−1 cm−1) were used to determine concentrations. Omecamtiv mecarbil (CK-1827452) was purchased from AdooQ Bioscience, dissolved in DMSO, and stored at 50 mm. The mant-ATP and -ADP were purchased from Jena Biosciences. Pyrene iodoacetamide was purchased from Invitrogen. In most experiments with OM, 0.1% DMSO was present and used as a vehicle control, whereas at high OM concentrations (>20 μm) it was necessary to increase DMSO (1% and also used as a vehicle control) to maintain OM solubility (Fig. 2D).
Protein construction, expression, and purification
The cDNA of M2β-S1 (841 aa) was subcloned into the pshuttle vector containing a C-terminal Avi tag and N-terminal FLAG tag (DYKDDDDK). The F764L point mutation was introduced with QuikChange site-directed mutagenesis (Stratagene). The AdEasy system was used to produce high-titer recombinant adenovirus as previously described (21, 49). The final virus titer was 1010 to 1011 pfu/ml, and was used to infect C2C12 cells at 5 × 108 pfu/ml. Cells were harvested 7–10 days after infection and M2β-S1 was purified as previously described (21). The purified M2β-S1 contains mouse essential and regulatory light chains, and their sequences were determined using LC-MS/MS as described in our previous work (21). M2β-S1 used for the in vitro motility assay was incubated with BirA (10 μg/ml, Avidity), biotin (0.5 mm), and ATP (10 mm) for 1 h at room temperature to allow biotinylation, followed by ammonium sulfate precipitation and dialyzed into MOPS 20 buffer. Actin was purified from rabbit skeletal muscle using an acetone powder method (50) and labeled with pyrene iodoacetamide (51) when appropriate. Proteins were dialyzed in MOPS 20 buffer overnight before experiments.
Steady-state ATPase measurements
Actin-activated ATPase experiments were performed with 0.1 μm M2β-S1 at varying actin concentrations (0, 5, 10, 20, 40, and 60 μm) using a NADH-coupled ATP-regenerating system as previously described (21, 52). Experiments were performed at 25 °C and data were collected for 200 s (0.2-s intervals) using an Applied Photophysics (Surrey, UK) stopped-flow apparatus. The ATPase rate was plotted as a function of actin concentration and fit to the Michaelis-Menten equation to determine the kcat and KATPase. Results were reported as the average of data collected from 3 to 6 protein preparations. Statistical analysis was done with an unpaired Student's t test to determine the impact of mutation or drug.
In vitro motility
In vitro motility assays were performed as previously described (21). Coverslips were coated with 1% nitrocellulose, followed by streptavidin (0.1 mg/ml) and blocked with BSA (2 mg/ml). Biotinylated M2β-S1 WT or F764L was then added to be attached to the surface at different loading concentrations. Unlabeled actin (2 μm) followed by ATP (2 mm) was used to block the dead myosin heads on the surface. The activation buffer contained MOPS 20 buffer, 0.35% methylcellulose, 0.45 mm phosphoenolpyruvate, 45 units/ml of pyruvate kinase, 0.1 mg/ml of glucose oxidase, 5 mg/ml of glucose, 0.018 mg/ml of catalase, and 2 mm ATP. DMSO or OM were added into the activation buffer just before initiating motility. For OM titration experiments, extra KCl was added to the activation buffer to bring it up to 100 mm KCl. Alexa 488 phalloidin-labeled actin was visualized with a Nikon TE2000 fluorescence microscope. Videos were collected for 2 min at 1-s intervals or every 5 to 15 s for 10 min in the absence or presence, respectively, of varying OM concentrations. The velocities were analyzed manually by tracking actin filaments using ImageJ with the MTrackJ plug-in (53). We also used the program FAST to analyze actin filament gliding velocities when examining the motility in the absence of OM (22). An unpaired Student's t test was performed to compare the WT and F764L mutant and the impact of drug.
Transient kinetic measurements
Experiments were performed using an Applied Photophysics stopped-flow equipped with a 1.2-ms dead-time and a 9.3-nm band pass. Tryptophan fluorescence was examined with 290 nm excitation and a 320-nm long pass emission filter. The mant fluorescence of mant-ADP and mant-ATP was examined with 290 nm excitation and a 395-nm long pass filter. Pyrene-actin fluorescence was monitored with 365 nm excitation and a 395-nm long pass filter. MDCC-PBP was excited with 380 nm, and the emission examined with a 425-nm long pass. Fluorescence transients were fit using software provided with the stopped-flow or GraphPad Prism. Kinetic modeling was performed using Kintek Explorer (54, 55). The forward and reverse rate constants used for ATPase and duty ratio modeling are listed in Table S3.
Author contributions
W. T. and C. M. Y. conceptualization; W. T., W. C. U., R. D., and C. M. Y. data curation; W. T. and C. M. Y. formal analysis; W. T. and C. M. Y. methodology; W. T. writing-original draft; W. T. and C. M. Y. writing-review and editing; W. C. U. and R. D. resources; C. M. Y. supervision; C. M. Y. funding acquisition; C. M. Y. project administration.
Supplementary Material
This work was supported by National Institutes of Health Grant HL127699 (to C. M. Y.) and American Heart Association Grant 19PRE34380569 (to W. T.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4 and Tables S1–S4.
- HCM
- hypertrophic cardiomyopathy
- DCM
- dilated cardiomyopathy
- OM
- omecamtive mecarbil
- SRX
- super-relaxed state
- mant
- 2′/3′-O-(N-methyl-anthraniloyl)
- PBP
- phosphate-binding protein
- MDCC
- N-[2-(1-maleimidyl)ethyl]-7-(diethylamino)coumarin-3-carboxamide.
References
- 1. Watkins H., Ashrafian H., and Redwood C. (2011) Inherited cardiomyopathies. N. Engl. J. Med. 364, 1643–1656 10.1056/NEJMra0902923 [DOI] [PubMed] [Google Scholar]
- 2. McNally E. M., and Mestroni L. (2017) Dilated cardiomyopathy: genetic determinants and mechanisms. Circ. Res. 121, 731–748 10.1161/CIRCRESAHA.116.309396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harvey P. A., and Leinwand L. A. (2011) Cellular mechanisms of cardiomyopathy. J. Cell Biol. 194, 355–365 10.1083/jcb.201101100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maron B. J., Ommen S. R., Semsarian C., Spirito P., Olivotto I., and Maron M. S. (2014) Hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 64, 83–99 10.1016/j.jacc.2014.05.003 [DOI] [PubMed] [Google Scholar]
- 5. Hershberger R. E., Hedges D. J., and Morales A. (2013) Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 10, 531–547 10.1038/nrcardio.2013.105 [DOI] [PubMed] [Google Scholar]
- 6. Moore J. R., Leinwand L., and Warshaw D. M. (2012) Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ. Res. 111, 375–385 10.1161/CIRCRESAHA.110.223842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Spudich J. A. (2014) Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys. J. 106, 1236–1249 10.1016/j.bpj.2014.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spudich J. A. (2015) The myosin mesa and a possible unifying hypothesis for the molecular basis of human hypertrophic cardiomyopathy. Biochem. Soc. Trans. 43, 64–72 10.1042/BST20140324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu C., Kawana M., Song D., Ruppel K. M., and Spudich J. A. (2018) Controlling load-dependent kinetics of β-cardiac myosin at the single-molecule level. Nat. Struct. Mol. Biol. 25, 505–514 10.1038/s41594-018-0069-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Geeves M. A. (2016) Review: the ATPase mechanism of myosin and actomyosin: the ATPase mechanism of myosin and actomyosin. Biopolymers 105, 483–491 10.1002/bip.22853 [DOI] [PubMed] [Google Scholar]
- 11. Málnási-Csizmadia A., and Kovács M. (2010) Emerging complex pathways of the actomyosin powerstroke. Trends Biochem. Sci. 35, 684–690 10.1016/j.tibs.2010.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ujfalusi Z., Vera C. D., Mijailovich S. M., Svicevic M., Yu E. C., Kawana M., Ruppel K. M., Spudich J. A., Geeves M. A., and Leinwand L. A. (2018) Dilated cardiomyopathy myosin mutants have reduced force-generating capacity. J. Biol. Chem. 293, 9017–9029 10.1074/jbc.RA118.001938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schmitt J. P., Debold E. P., Ahmad F., Armstrong A., Frederico A., Conner D. A., Mende U., Lohse M. J., Warshaw D., Seidman C. E., and Seidman J. G. (2006) Cardiac myosin missense mutations cause dilated cardiomyopathy in mouse models and depress molecular motor function. Proc. Natl. Acad. Sci. U.S.A. 103, 14525–14530 10.1073/pnas.0606383103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Debold E. P., Schmitt J. P., Patlak J. B., Beck S. E., Moore J. R., Seidman J. G., Seidman C., and Warshaw D. M. (2007) Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse α-cardiac myosin in the laser trap assay. Am. J. Physiol. Heart Circ. Physiol. 293, H284–H291 10.1152/ajpheart.00128.2007 [DOI] [PubMed] [Google Scholar]
- 15. Malik F. I., Hartman J. J., Elias K. A., Morgan B. P., Rodriguez H., Brejc K., Anderson R. L., Sueoka S. H., Lee K. H., Finer J. T., Sakowicz R., Baliga R., Cox D. R., Garard M., Godinez G., Kawas R., et al. (2011) Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science 331, 1439–1443 10.1126/science.1200113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Teerlink J. R., Felker G. M., McMurray J. J. V., Solomon S. D., Adams K. F., Cleland J. G. F., Ezekowitz J. A., Goudev A., Macdonald P., Metra M., Mitrovic V., Ponikowski P., Serpytis P., Spinar J., Tomcsányi J., et al. (2016) Chronic oral study of myosin activation to increase contractility in heart failure (COSMIC-HF): a phase 2, pharmacokinetic, randomised, placebo-controlled trial. Lancet 388, 2895–2903 10.1016/S0140-6736(16)32049-9 [DOI] [PubMed] [Google Scholar]
- 17. Mamidi R., Gresham K. S., Li A., dos Remedios C. G., and Stelzer J. E. (2015) Molecular effects of the myosin activator omecamtiv mecarbil on contractile properties of skinned myocardium lacking cardiac myosin binding protein-C. J. Mol. Cell. Cardiol. 85, 262–272 10.1016/j.yjmcc.2015.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nagy L., Kovács Á., Bódi B., Pásztor E. T., Fülöp G. Á., Tóth A., Édes I., and Papp Z. (2015) The novel cardiac myosin activator omecamtiv mecarbil increases the calcium sensitivity of force production in isolated cardiomyocytes and skeletal muscle fibres of the rat. Br. J. Pharmacol. 172, 4506–4518 10.1111/bph.13235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Utter M. S., Ryba D. M., Li B. H., Wolska B. M., and Solaro R. J. (2015) Omecamtiv mecarbil, a cardiac myosin activator, increases Ca2+ sensitivity in myofilaments with a dilated cardiomyopathy mutant tropomyosin E54K. J. Cardiovas. Pharmacol. 66, 347–353 10.1097/FJC.0000000000000286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Woody M. S., Greenberg M. J., Barua B., Winkelmann D. A., Goldman Y. E., and Ostap E. M. (2018) Positive cardiac inotrope omecamtiv mecarbil activates muscle despite suppressing the myosin working stroke. Nat. Commun. 9, 3838 10.1038/s41467-018-06193-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Swenson A. M., Tang W., Blair C. A., Fetrow C. M., Unrath W. C., Previs M. J., Campbell K. S., and Yengo C. M. (2017) Omecamtiv mecarbil enhances the duty ratio of human β-cardiac myosin resulting in increased calcium sensitivity and slowed force development in cardiac muscle. J. Biol. Chem. 292, 3768–3778 10.1074/jbc.M116.748780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aksel T., Choe Yu E., Sutton S., Ruppel K. M., and Spudich J. A. (2015) Ensemble force changes that result from human cardiac myosin mutations and a small-molecule effector. Cell Rep. 11, 910–920 10.1016/j.celrep.2015.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu Y., White H. D., Belknap B., Winkelmann D. A., and Forgacs E. (2015) Omecamtiv mecarbil modulates the kinetic and motile properties of porcine β-cardiac myosin. Biochemistry 54, 1963–1975 10.1021/bi5015166 [DOI] [PubMed] [Google Scholar]
- 24. De La Cruz E. M., and Michael Ostap E. M. (2009) Kinetic and equilibrium analysis of the myosin ATPase. Methods Enzymol. 455, 157–192 10.1016/S0076-6879(08)04206-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chalovich J. M., Stein L. A., Greene L. E., and Eisenberg E. (1984) Interaction of isozymes of myosin subfragment 1 with actin: effect of ionic strength and nucleotide. Biochemistry 23, 4885–4889 10.1021/bi00316a011 [DOI] [PubMed] [Google Scholar]
- 26. Yengo C. M., Takagi Y., and Sellers J. R. (2012) Temperature dependent measurements reveal similarities between muscle and non-muscle myosin motility. J. Muscle Res. Cell Motil. 33, 385–394 10.1007/s10974-012-9316-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Planelles-Herrero V. J., Hartman J. J., Robert-Paganin J., Malik F. I., and Houdusse A. (2017) Mechanistic and structural basis for activation of cardiac myosin force production by omecamtiv mecarbil. Nat. Commun. 8, 190 10.1038/s41467-017-00176-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shuman H., Greenberg M. J., Zwolak A., Lin T., Sindelar C. V., Dominguez R., and Ostap E. M. (2014) A vertebrate myosin-I structure reveals unique insights into myosin mechanochemical tuning. Proc. Natl. Acad. Sci. U.S.A. 111, 2116–2121 10.1073/pnas.1321022111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De La Cruz E. M., Wells A. L., Sweeney H. L., and Ostap E. M. (2000) Actin and light chain isoform dependence of myosin V kinetics. Biochemistry 39, 14196–14202 10.1021/bi001701b [DOI] [PubMed] [Google Scholar]
- 30. Gunther L. K., Rohde J. A., Tang W., Walton S. D., Unrath W. C., Trivedi D. V., Muretta J. M., Thomas D. D., and Yengo C. M. (2019) Converter domain mutations in myosin alter structural kinetics and motor function. J. Biol. Chem. 294, 1554–1567 10.1074/jbc.RA118.006128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deacon J. C., Bloemink M. J., Rezavandi H., Geeves M. A., and Leinwand L. A. (2012) Identification of functional differences between recombinant human α and β cardiac myosin motors. Cell. Mol. Life Sci. 69, 2261–2277 10.1007/s00018-012-0927-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hannemann D. E., Cao W., Olivares A. O., Robblee J. P., and De La Cruz E. M. (2005) Magnesium, ADP, and actin binding linkage of myosin V: evidence for multiple myosin V-ADP and actomyosin V-ADP states. Biochemistry 44, 8826–8840 10.1021/bi0473509 [DOI] [PubMed] [Google Scholar]
- 33. Swenson A. M., Trivedi D. V., Rauscher A. A., Wang Y., Takagi Y., Palmer B. M., Málnási-Csizmadia A., Debold E. P., and Yengo C. M. (2014) Magnesium modulates actin binding and ADP release in myosin motors. J. Biol. Chem. 289, 23977–23991 10.1074/jbc.M114.562231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rosenfeld S. S., Houdusse A., and Sweeney H. L. (2005) Magnesium regulates ADP dissociation from myosin V. J. Biol. Chem. 280, 6072–6079 10.1074/jbc.M412717200 [DOI] [PubMed] [Google Scholar]
- 35. Trivedi D. V., Muretta J. M., Swenson A. M., Davis J. P., Thomas D. D., and Yengo C. M. (2015) Direct measurements of the coordination of lever arm swing and the catalytic cycle in myosin V. Proc. Natl. Acad. Sci. U.S.A. 112, 14593–14598 10.1073/pnas.1517566112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nesmelov Y. E., Agafonov R. V., Negrashov I. V., Blakely S. E., Titus M. A., and Thomas D. D. (2011) Structural kinetics of myosin by transient time-resolved FRET. Proc. Natl. Acad. Sci. U.S.A. 108, 1891–1896 10.1073/pnas.1012320108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baumketner A. (2012) The mechanism of the converter domain rotation in the recovery stroke of myosin motor protein. Proteins 80, 2701–2710 10.1002/prot.24155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Billington N., Revill D. J., Burgess S. A., Chantler P. D., and Knight P. J. (2014) Flexibility within the heads of muscle myosin-2 molecules. J. Mol. Biol. 426, 894–907 10.1016/j.jmb.2013.11.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bloemink M. J., Melkani G. C., Bernstein S. I., and Geeves M. A. (2016) The relay/converter interface influences hydrolysis of ATP by skeletal muscle myosin II. J. Biol. Chem. 291, 1763–1773 10.1074/jbc.M115.688002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Glasheen B. M., Ramanath S., Patel M., Sheppard D., Puthawala J. T., Riley L. A., and Swank D. M. (2018) Five alternative myosin converter domains influence muscle power, stretch activation, and kinetics. Biophys. J. 114, 1142–1152 10.1016/j.bpj.2017.12.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Palmer B. M., Schmitt J. P., Seidman C. E., Seidman J. G., Wang Y., Bell S. P., Lewinter M. M., and Maughan D. W. (2013) Elevated rates of force development and MgATP binding in F764L and S532P myosin mutations causing dilated cardiomyopathy. J. Mol. Cell. Cardiol. 57, 23–31 10.1016/j.yjmcc.2012.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Weintraub R. G., Semsarian C., and Macdonald P. (2017) Dilated cardiomyopathy. Lancet 390, 400–414 10.1016/S0140-6736(16)31713-5 [DOI] [PubMed] [Google Scholar]
- 43. Japp A. G., Gulati A., Cook S. A., Cowie M. R., and Prasad S. K. (2016) The diagnosis and evaluation of dilated cardiomyopathy. J. Am. Coll. Cardiol. 67, 2996–3010 10.1016/j.jacc.2016.03.590 [DOI] [PubMed] [Google Scholar]
- 44. Hooijman P., Stewart M. A., and Cooke R. (2011) A new state of cardiac myosin with very slow ATP turnover: a potential cardioprotective mechanism in the heart. Biophys. J. 100, 1969–1976 10.1016/j.bpj.2011.02.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McNamara J. W., Li A., Dos Remedios C. G., and Cooke R. (2015) The role of super-relaxed myosin in skeletal and cardiac muscle. Biophys. Rev. 7, 5–14 10.1007/s12551-014-0151-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Alamo L., Ware J. S., Pinto A., Gillilan R. E., Seidman J. G., Seidman C. E., and Padrón R. (2017) Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. Elife 6, e24634 10.7554/eLife.24634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robert-Paganin J., Auguin D., and Houdusse A. (2018) Hypertrophic cardiomyopathy disease results from disparate impairments of cardiac myosin function and auto-inhibition. Nat. Commun. 9, 4019 10.1038/s41467-018-06191-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rohde J. A., Thomas D. D., and Muretta J. M. (2017) Heart failure drug changes the mechanoenzymology of the cardiac myosin powerstroke. Proc. Natl. Acad. Sci. U.S.A. 114, E1796–E1804 10.1073/pnas.1611698114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Winkelmann D. A., Forgacs E., Miller M. T., and Stock A. M. (2015) Structural basis for drug-induced allosteric changes to human β-cardiac myosin motor activity. Nat. Commun. 6, 7974 10.1038/ncomms8974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pardee J. D., and Spudich J. A. (1982) Purification of muscle actin. Methods Enzymol. 85, 164–181 10.1016/0076-6879(82)85020-9 [DOI] [PubMed] [Google Scholar]
- 51. Pollard T. D. (1984) Polymerization of ADP-actin. J. Cell Biol. 99, 769–777 10.1083/jcb.99.3.769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. De La Cruz E. M., Sweeney H. L., and Ostap E. M. (2000) ADP Inhibition of myosin V ATPase activity. Biophys. J. 79, 1524–1529 10.1016/S0006-3495(00)76403-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Meijering E., Dzyubachyk O., and Smal I. (2012) Methods for cell and particle tracking. Methods Enzymol. 504, 183–200 10.1016/B978-0-12-391857-4.00009-4 [DOI] [PubMed] [Google Scholar]
- 54. Johnson K. A., Simpson Z. B., and Blom T. (2009) Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29 10.1016/j.ab.2008.12.024 [DOI] [PubMed] [Google Scholar]
- 55. Johnson K. A., Simpson Z. B., and Blom T. (2009) FitSpace explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem. 387, 30–41 10.1016/j.ab.2008.12.025 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.