

Abstract
The DNA-binding protein PU.1 is a myeloid lineage–determining and pioneering transcription factor due to its ability to bind “closed” genomic sites and maintain “open” chromatin state for myeloid lineage–specific genes. The precise mechanism of PU.1 in cell type–specific programming is yet to be elucidated. The melanoma cell line B16BL6, although it is nonmyeloid lineage, expressed Toll-like receptors and activated the transcription factor NF-κB upon stimulation by the bacterial cell wall component lipopolysaccharide. However, it did not produce cytokines, such as IL-1β mRNA. Ectopic PU.1 expression induced remodeling of a novel distal enhancer (located ∼10 kbp upstream of the IL-1β transcription start site), marked by nucleosome depletion, enhancer-promoter looping, and histone H3 lysine 27 acetylation (H3K27ac). PU.1 induced enhancer-promoter looping and H3K27ac through two distinct PU.1 regions. These PU.1-dependent events were independently required for subsequent signal-dependent and co-dependent events: NF-κB recruitment and further H3K27ac, both of which were required for enhancer RNA (eRNA) transcription. In murine macrophage RAW264.7 cells, these PU.1-dependent events were constitutively established and readily expressed eRNA and subsequently IL-1β mRNA by lipopolysaccharide stimulation. In summary, this study showed a sequence of epigenetic events in programming IL-1β transcription by the distal enhancer priming and eRNA production mediated by PU.1 and the signal-dependent transcription factor NF-κB.
Keywords: macrophage, interleukin 1 (IL-1), transcription factor, chromatin modification, gene expression, melanoma, enhancer, eRNA, PU.1, IL-1beta, enhancer, DNA looping, histone acetylation, melanoma cells
Introduction
The nuclear protein PU.1 (encoded by the gene SPI1 in humans or Spi1 in mice) is an E26 transformation–specific (Ets)3-family transcription factor that plays a crucial role in the development of myeloid and lymphoid lineage cells (1–3). PU.1 is induced at high levels during myeloid development, ranging up to >200 copies of mRNA per cell, even above the “housekeeping gene” glyceraldehyde-3-phosphate dehydrogenase (4, 5). This high amount of PU.1 is required for myeloid cell development (6) and optimal transcription of inflammatory response genes upon stimulation in macrophages (7). PU.1 has been regarded as a lineage-determining and pioneering transcription factor for its capability to bind to “closed” chromatin in a sequence-specific manner and remodel it to an “open” state that allows recruitment of signal-dependent transcription factors/co-factors to enhancers and promoters of cell type–specific genes (8–10). Indeed, forced expression of PU.1, in the presence or co-expression of the leucine zipper–type transcription factor CCAAT/enhancer-binding protein (C/EBP)α/β, reprograms lymphocytes, such as pre-T cells (11) and pre-B cells (12), and nonhematopoietic cells, such as primary fibroblasts and NIH 3T3 cells (13), into macrophage-like cells. C/EBPα and PU.1 cooperate to reprogram cells by activating prospective macrophage enhancers as a pioneer factor or a secondary factor, depending on gene types (14). Similarly, PU.1 can interact with other pioneering factors, such as Runx1 (15) and GATA-1 (16), to program different lineage cell types.
Enhancers are distal regulatory elements that are activated by common and cell type–specific regulators and characterize cell type–specific gene expression (17, 18). Binding of PU.1 to enhancers induces multiple nucleosomal changes, including nucleosome depletion, deposition of active enhancer histone marks such as histone H3 lysine 4 monomethylation (H3K4me1) and lysine 27 acetylation (H3K27ac) (19–22), and enhancer-promoter looping formation, all of which are mediated by interacting with different proteins and protein complexes (23, 24). Active enhancers also recruit the RNA polymerase II–containing transcription initiation complex and RNA capping machinery, yielding transcripts that are referred to as enhancer RNAs (eRNAs) (25–27). eRNAs enhance mRNA transcription; however, their mechanism in regulating gene transcription is unclear. For example, eRNAs were shown to enhance enhancer-promoter looping formation (28), whereas they were also shown to participate in the process of releasing paused RNA polymerase II without changing enhancer-promoter looping (29). eRNAs were also shown to enhance chromatin accessibility of transcription machinery (30); however, it was also shown that active enhancer transcription leads to enhancer remodeling independent of eRNA transcription (31). Therefore, how PU.1 regulates enhancers and how eRNAs participate in gene regulation remain to be determined.
One of the key characteristics of myeloid cells such as macrophages is rapid and robust production of inflammatory cytokines in response to microbial stimulation. Enhancers and promoters of these genes are in poised status, harboring active histone marks and transcriptional machinery that is waiting to be activated immediately by extracellular signals (31, 32). Among those cytokines, interleukin (IL)-1β (derived from the proteolytic processing of pro-IL-1β gene product) is a potent inflammatory cytokine that is rapidly induced by activated Toll-like receptors (TLRs), such as TLR4, activated by the Gram-negative bacterial component lipopolysaccharide (LPS). PU.1 is crucial for priming enhancers and robust pro-IL-1β (herein referred to as IL-1β) transcription in macrophages (33–35). In human macrophages, PU.1 is crucial for establishing poised promoter architecture and, particularly, looping between the promoter and a remote enhancer located about −3,000 bp from the transcription start site (TSS) of IL-1β gene (36, 37). Although both promoter and enhancer elements are expected to be similar in mice, the distal regulatory elements and mechanism of PU.1 in regulating IL-1β transcription in mice are less well-understood.
To examine the detailed mechanisms of PU.1 in programming IL-1β transcription, we used the murine melanocyte cell line B16BL6 (B16). Melanocytes, best known for their role in skin and hair pigmentation, are differentiated from melanoblasts that are originated from pluripotent neural-crest cells (38). These cells express various innate immune receptors, such as TLRs, and exhibit similar phagocytic, antigen-presenting, and inflammatory response capabilities with macrophages (39). Unlike melanocytes, we found that B16 cells lacked PU.1 and failed to produce IL-1β mRNA by TLR stimulation. However, ectopic expression of PU.1 led these cells to express IL-1β. We identified a novel enhancer located ∼10 kbp from the IL-1β TSS. Binding of PU.1 in the enhancer induced nucleosome depletion, enhancer-promoter looping, and H3K27ac. The PU.1-dependent enhancer-promoter looping and H3K27ac were independently required for subsequent signal-dependent and co-dependent events: NF-κB recruitment and further H3K27ac, both of which were required for the eRNA and subsequent IL-1β mRNA transcription.
Results
Ectopic expression of PU.1 reprograms B16 cells to express IL-1β mRNA in response to TLR activation
Human melanocytes and melanoma cells were shown to express TLRs, and activation of these receptors induces expression of inflammatory cytokines, such as IL-1β (40, 41). Unexpectedly, B16 mouse melanoma cells failed to express IL-1β mRNA in response to lipoteichoic acid (LTA), polyinosinic-polycytidylic acid (poly(I:C)), LPS, and CpG dinucleotides, which are TLR2, -3, -4, and -9 ligands, respectively (Fig. 1A). However, these cells activated ERK and p38 mitogen-activated protein kinases (MAPKs) and NF-κB in response to LPS (Fig. 1B), suggesting that these cells are defective in processing signaling downstream to these pathways. Lineage-determining (pioneering) transcription factors, such as C/EBPα, C/EBPβ, PU.1, and Runt-related transcription factor 1 (RUNX1), play key roles in programming myeloid cells for their characteristics in expressing cytokines (42). Therefore, we examined expression of these transcription factors in these cells. Based on RT-qPCR analysis, these cells constitutively expressed C/EBPβ and RUNX1, but not C/EBPα and PU.1 (Fig. S1). Consistent with these results, Western blotting also failed to detect PU.1 and C/EBPα proteins in these cells (Fig. 1C). However, in B16 cells transfected with C/EBPα and PU.1, robust transcription of IL-1β mRNA was detected in response to TLR2, -4, and -9 ligands (Fig. 1A) without affecting the levels of ERK, p38, and NF-κB activation (Fig. 1B). Because C/EBPα, together with PU.1, was shown to be involved in establishing monocyte-specific enhancers (43), we further examined whether both C/EBPα and PU.1 were required for programming IL-1β mRNA transcription. Unexpectedly, PU.1 alone promoted IL-1β mRNA expression to the same extent as detected in cells expressing both PU.1 and C/EBPα (Fig. 1D). Ectopic expression of PU.1 was also shown to induce macrophage colony–stimulating factor receptor (M-CSFR) expression, which is involved in reprogramming nonmyeloid lineage cells into macrophage-like cells (11–13). As expected, ectopic expression of PU.1 together with or without C/EBPα induced expression of M-CSFR mRNA at low levels (Fig. 1E, left). In cells transfected with PU.1 + C/EBPα, M-CSF induced low levels of IL-1β mRNA expression at basal state; however, the extent of IL-1β mRNA induction by LPS was not affected by M-CSF treatment (Fig. 1E, right). These data suggest that PU.1 programs B16 cells to express IL-1β mRNA in response to TLR activation through a mechanism independent of M-CSF.
Figure 1.

Ectopic expression of PU.1 reprograms B16 cells to express IL-1β mRNA in response to TLR activation. B16 cells were transfected with pcDNA3 (Vector) or pcDNA3-HA-PU.1 (PU.1) and pMSCV-C/EBPα (CEBPα) for 24 h as described under “Experimental procedures.” A, after replating and culturing in a fresh dish for an additional 16–18 h, cells were stimulated with TLR4, -2, -9, and -3 ligands LPS (100 ng/ml), LTA (10 μg/ml), mouse CpG (20 μg/ml), and poly(I:C) (10 μg/ml), respectively, for 6 h. IL-1β mRNA transcription was then measured using RT-qPCR. Data are expressed as means ± S.D. (n = 2). B, cells were treated with LPS (100 ng/ml) for the time indicated, and phosphorylation levels of p38, ERK, and inhibitor-κB (IκB) were analyzed by immunoblotting. β-Actin immunoblotting was used as a loading control. One representative blot of three independent experiments is shown. C–E, similarly, cells were transfected with pcDNA3 (Vector) or PU.1 and C/EBPα expression vectors for ∼48 h. C, expression of PU.1 and C/EBPα was analyzed by immunoblotting using anti-PU.1 and anti-C/EBPα antibodies, respectively, in B16 cells transfected with vector, PU.1, and/or C/EBPα (left) and in WT B16 and RAW cells (right). Western blots shown are representative of three independent experiments. D, cells were treated with LPS (100 ng/ml) for 6 h, and IL-1β mRNA transcription was analyzed by RT-qPCR. Data are expressed as means ± S.D. (n = 3). E, after transfection, cells were further cultured for 16–18 h, and expression of M-CSFR mRNA was analyzed and compared with that of a housekeeping gene, GAPDH (left). These cells were also cultured in the presence or absence of murine M-CSF (20 ng/ml) and stimulated with LPS (100 ng/ml) for 6 h. IL-1β mRNA transcription was measured by RT-qPCR. Data are expressed as means ± S.D. (n = 2; right).
Ectopically expressed PU.1 associates with distal IL-1β enhancer and proximal promoter regions and enhances H3K27ac levels and DNase I accessibility
PU.1 was shown to remodel promoters and, particularly, distal regulatory elements of cell-specific and signal-dependent genes (8). To identify potential regulatory elements (enhancers) of the IL-1β gene, we first examined key histone remodeling markers, such as H3K27ac and histone H3 Lys-4 methylation (20, 22, 44), located within 100 kbp of the IL-1β locus using the ENCODE mouse (mm9) genome database (http://genome.ucsc.edu/ENCODE/;4 Fig. 2A). Two regions, located ∼10 and ∼2 kbp upstream of the TSS, showed strong H3K27ac signals in bone marrow–derived macrophages. These regions (hereafter referred to as E1 for the enhancer located ∼2 kbp upstream and E2 for ∼10 kbp upstream of IL-1β TSS) were also very high in H3K4me1, but low in histone H3 Lys-4 trimethylation (H3K4me3), signals (top two panels). Particularly, these H3K27ac signals were unique to macrophages and not found in other nonmyeloid cell types (bottom panel). Within and proximal to these regions, five putative PU.1-binding sites (GGAAGTG (45); −513, −2,285, −9,476, −9,870, and −10,124 bases upstream of the TSS) were found (Fig. 2A). Because only 1% of all potential PU.1-binding motifs found within the human genome are actually occupied by PU.1 (45), we examined PU.1 binding to these regions by ChIP-qPCR analysis and found that PU.1 was indeed recruited to these sites upon ectopic expression of PU.1 in B16 cells (Fig. 2B, top). PU.1 was also shown to recruit the histone acetyltransferases cAMP-response element-binding protein-binding protein (CBP) and the related p300 to specific genomic regions (46, 47). As expected, PU.1 expression increased H3K27ac levels in these enhancer regions (Fig. 2B, bottom). In addition, ectopic expression of PU.1-enhanced chromatin accessibility of DNase I to the regions coincided with the PU.1 recruitment and H3K27ac signal region of the enhancer and also 5′ downstream of the IL-1β promoter (Fig. 2C). Collectively, these results suggest that PU.1 is recruited to these enhancers and promoter and induces H3K27ac and nucleosome-free chromatin conformation in these regions.
Figure 2.

Ectopically expressed PU.1 associates with distal IL-1β enhancer and proximal promoter regions and enhances H3K27ac levels and DNase I accessibility. A, screen snapshots of H3K4me1, and H3K4me3 ChIP-Seq signals in murine bone marrow–derived macrophages (BMDM; top two panels) and H3K27ac ChIP-Seq signals in bone marrow–derived macrophages, ES-Bruce4, and mouse embryonic fibroblasts (MEF) (bottom panel) from the ENCODE-UCSC database. The black vertical arrows indicate locations where primers are targeting for qPCR analysis, and numerical values indicate locations of the first reverse primer nucleotide from the IL-1β TSS. The triangles show locations of putative PU.1-binding sites; the diamonds show locations of putative NF-κB–binding sites; the black bar indicates the target site of the GapmeR ASO. B and C, B16 cells were transfected with pcDNA3 (Vector) or pcDNA3-HA-PU.1 (PU.1) as described in the legend to Fig. 1. B, ChIP, using anti-PU.1 (top) or anti-H3K27ac (bottom), followed by qPCR using the primer sets indicated, was performed as described under “Experimental procedures.” Data are presented as percentage of enrichment with the precipitated target sequence compared with input DNA from two independent experiments. *, p < 0.05 (Student's t test). C, nuclei were prepared and digested using DNase I as described under “Experimental procedures.” Digested DNAs were then purified and analyzed by qPCR using the primer sets indicated. DNase I sensitivity was expressed as qPCR cycle threshold values subtracted from the cycle threshold values of nondigested nuclei.
eRNA transcription induced by NF-κB is required for optimal IL-1β mRNA transcription
Active enhancers are transcribed in a cell type–specific manner and regulate transcription of nearby genes (25). The transcripts, referred to as eRNAs, are involved in histone acetylation and mRNA transcription (31). Therefore, we examined whether distal enhancers produced eRNAs that regulated IL-1β mRNA transcription. Upon LPS treatment with cells transfected with PU.1, eRNAs encoded by a region upstream of the PU.1-binding site in the E2 enhancer were clearly detected (Fig. 3A). The E2 eRNA transcription peaked 4 h after LPS treatment, preceding the peak transcription of IL-1β mRNA at 12 h (Fig. 3B). Knocking down the eRNA using GapmeR antisense oligonucleotides (ASO; targeting negative stranded DNA) inhibited IL-1β mRNA transcription (Fig. 3C). These data indicate that the E2 eRNA is functional in regulating IL-1β transcription. To further reveal signaling cascades involved in eRNA transcription, we examined eRNA and mRNA transcription after treating cells with various chemical inhibitors targeting different MAPK isoforms and NF-κB at maximum nontoxic doses. Among them, NF-κB inhibitor (NFκBi) potently inhibited transcription of both IL-1β eRNA and mRNA (Fig. 3D). Both ERK1/2 (U0126; U0) and p38 (SB203580; SB) inhibitors significantly prevented IL-1β mRNA transcription, but not eRNA. The JNK inhibitor (JNKi) had no effects on IL-1β eRNA and mRNA transcription. NF-κB was also recruited to the 5′ regions of both the enhancer and promoter upon LPS stimulation in PU.1-expressing cells, but not in WT cells (Fig. 3E), supporting a key role of NF-κB in transcription of IL-1β eRNA and mRNA.
Figure 3.

E2 eRNA transcription induced by NF-κB is required for optimal IL-1β mRNA transcription. B16 cells were transfected with pcDNA3 (Vector) or pcDNA3-HA-PU.1 (PU.1) and stimulated with LPS (100 ng/ml) for 6 h as described in the legend to Fig. 1. A, E2 eRNA expression levels were analyzed by RT-qPCR using the primer sets indicated. Data are expressed as mean values from three independent experiments. B, similarly, B16 cells were stimulated with LPS for the time indicated, and E2 eRNA and mRNA transcription was analyzed by qPCR. Data are expressed as means ± S.D. (n = 2). C, B16 cells transfected with PU.1 expression vector were further treated with scrambled or E2 eRNA-specific ASO (250 pmol) for 24 h. Cells were then stimulated with LPS for 6 h, and expression levels of E2 eRNA and mRNA were analyzed by RT-qPCR. D, cells were pretreated with MAPK inhibitors (U0126 (10 μm), SB203580 (10 μm), and JNK inhibitor (10 μm)) and NFκBi (5 μm) for 1 h, and after LPS (100 ng/ml; 6 h) stimulation, IL-1β mRNA and E2 eRNA transcription was analyzed by qPCR. Data are expressed as means ± S.D. (n = 3); *, p < 0.01; **, p < 0.05 (Student's t test). E, ChIP analysis was performed in B16 cells stimulated with or without LPS (4 h) using anti-NF-κB (p65). Purified DNAs were then analyzed by qPCR using the primers indicated. Data are presented as percentage of enrichment with the precipitated target sequence compared with input DNA (n ≥ 2); *, p < 0.05 (Student's t test).
eRNA is required for IL-1β mRNA expression in macrophages
To confirm that E2 eRNA also plays a key role in macrophages that already express high levels of PU.1 (5, 48), transcription of E2 eRNA and its role in IL-1β transcription in response to LPS stimulation were examined in RAW cells. Because qPCR analysis can be misleading when high levels of eRNA were produced at basal levels, we used a droplet digital PCR (ddPCR) technique that analyzes absolute quantities of DNAs (49). As shown in Fig. 4A (top), ddPCR analysis clearly showed an increase of droplet signals in LPS-treated cells when compared with those of control cells. The ddPCR results were also very similar to those of qPCR analysis (bottom), indicating that qPCR analysis accurately represented absolute quantities of the eRNA. Similar to B16 cells, RAW cells strongly produced E2 eRNA by LPS stimulation with faster kinetics, where E2 eRNA and IL-1β mRNA peaked in 90 and 180 min, respectively, after LPS treatment (Fig. 4B). As in B16 cells, knocking down eRNA by ASO in RAW cells also significantly suppressed IL-1β mRNA expression (Fig. 4C). These results suggest that E2 is a functional enhancer for IL-1β transcription in RAW cells.
Figure 4.

E2 eRNA is also required for IL-1β mRNA expression in RAW macrophages. RAW cells were cultured with or without LPS (100 ng/ml) for the time indicated. Expression of E2 eRNA and IL-1β mRNA was analyzed by RT-qPCR as described under “Experimental procedures.” A, the absolute quantity of E2 eRNA was measured using ddPCR, and fluorescence signals were illustrated. The y axis represents fluorescence amplitude of one droplet, and the x axis shows 20,000 droplets per column (sample). The dots that are positioned above the threshold line represent positive droplets. These results were compared with qPCR results (bottom). Concentration of eRNA in copies/μl (solid line) from each sample is derived from the number of positive droplets shown in the dot graphs above the panel. The dotted line represents the -fold changes of eRNA in LPS-stimulated samples quantified via RT-qPCR. B, transcription of E2 eRNA and IL-1β mRNA was analyzed in RAW cells stimulated with LPS for the time indicated. C, cells transfected with scrambled or E2 eRNA-specific ASO for 24 h were stimulated with LPS (100 ng/ml) for 90 min, and transcription of E2 eRNA and IL-1β mRNA was analyzed by RT-qPCR. The data are expressed as means ± S.D. (n = 3); *, p < 0.05; **, p < 0.01 (Student's t test).
PU.1 induces E2-promoter looping formation prerequisite for E2 eRNA and IL-1β mRNA transcription by LPS
Spatial interaction between enhancer and promoter through looping is a mechanism by which enhancers regulate transcription of target genes (50). Therefore, we examined whether PU.1 binding to the E2 enhancer induced looping between the IL-1β enhancer and promoter through chromatin conformation capture (3C)-TaqMan qPCR analysis (51). Using the IL-1β promoter as anchor, interactions between the enhancer and promoter were examined, using the constant (forward orientation) primer and TaqMan probe (starting −912 and −890 bases, respectively, from the TSS) and 16 test (reverse orientation) primers targeting different DpnII-generated fragments within 15 kbp from TSS (Fig. 5A). In B16 cells, we detected high levels of interactions with seven fragments, which did not change in frequency between vector- and PU.1-transfected cells (Fig. 5A, top, dotted lines). In PU.1-transfected cells, six new interactions (reverse primers 9, 10, 11, 12, 13, and 15, starting −7,993, −8,640, −9,014, −9,882, −10822, and −13,584 bases from the TSS, respectively), four of which were located within the E2 enhancer, were detected (Fig. 5A, middle, solid lines). Upon LPS stimulation, these interactions were increased ∼2-fold (gray versus black bars). In RAW cells, which constitutively express PU.1 at high levels, enhancer-promoter interactions (reverse primers 9, 10, 11, and 13) were detected in nontreated cells (Fig. 5A, bottom). As in B16 cells, the enhancer-promoter interactions were increased ∼2-fold by LPS treatment (gray versus black bars). To further examine the role of PU.1 in the enhancer-promoter interactions, regions containing the PU.1-binding site (from −9,824 to −10,833: CRISPR-PU.1) and eRNA-coding region (−10,833 to −11,843: CRISPR-eRNA) were deleted using the CRISPR vector system in B16 cells (Fig. S2). Both CRISPR-PU.1 and CRISPR-eRNA cells produced significantly lower amounts of IL-1β mRNA than control (CRISPR vector–transfected) cells by LPS treatment (Fig. 5B, top). No differences were detected in producing mRNAs of other cytokines, such as CXCL9, in these cells (Fig. 5B, bottom). Similar to IL-1β mRNA transcription, neither CRISPR-PU.1 nor CRISPR-eRNA cells were able to produce the eRNA (Fig. 5C). The enhancer-promoter looping, however, was intact in CRISPR-eRNA cells but defective in CRISPR-PU.1 cells (Fig. 5D). These results suggest that recruitment of PU.1 to the 5′ upstream region of E2 is required for enhancer-promoter looping formation and subsequent E2 and IL-1β mRNA transcription.
Figure 5.

PU.1 induces E2-promoter looping formation prerequisite for E2 eRNA and IL-1β mRNA transcription. A, nuclei were extracted from WT B16 cells (top), B16 cells transfected with PU.1 with or without LPS stimulation (6 h; middle), and RAW macrophages with or without LPS stimulation (3 h; bottom). Genomic DNAs were purified after digestion with DpnII and subsequently ligated and de-cross-linked. Proximal localization of DNA fragments of E2 and IL-1β promoter was analyzed by chromatin conformation capture assay, followed by TaqMan probe qPCR as described under “Experimental procedures.” The numbers below the lines (1–16) indicate the test (reverse orientation) primers used to probe the digested DNA fragments. Each test primer (◀) was utilized in combination with the promoter-recognizing constant (forward orientation) primer (▶) and TaqMan probe (▾). The dotted lines (top) indicate basal ligation between the promoter and DNA fragments detected in all samples regardless of PU.1 expression or LPS stimulation. The solid lines represent newly detected interactions after PU.1 expression in B16 cells (middle) or RAW cells with/without LPS stimulation (bottom). The bars (gray, untreated; black, LPS-treated) in each diagram represent the frequencies of ligation, compared with the ligation frequency of the promoter window and adjacent DNA fragment (primer 1). B–D, E2-deleted (CRISPR-eRNA; Δ11843–10833) and PU.1-binding region–deleted (CRISPR-PU.1; Δ10833–9824) B16 cells were generated using the CRISPR/Cas9 gene-editing system as described under “Experimental procedures” and in Fig. S2. CRISPR vector, CRISPR-eRNA, and CRISPR-PU.1 cells were transfected with pcDNA3-HA-PU.1 plasmid for 48 h as described under “Experimental procedures.” Cells were then stimulated with LPS (100 ng/ml) for 6 h, and transcription of IL-1β and CXCL9 mRNAs (B) and E2 eRNA (C) was examined by RT-qPCR. The proximal localization of E2 and IL-1β promoter was analyzed, and cumulative values of primer sets 9, 10, and 11 were plotted (D). Data are expressed as means ± S.D. (error bars) (B and C, n = 3; D, n = 2).
E2-promoter looping and H3K27ac are mediated by distinct regions of PU.1
PU.1 protein has three distinct functional domains (52): the transactivation domain (amino acids 2–100), the PEST domain (amino acids 118–167), and the DNA-binding Ets domain (amino acids 171–267) (Fig. 6A). The transactivation domain harbors two regions that include acidic (2–74 amino acids) and glutamine-rich (74–100 amino acids) domains (53). Within the acidic domain, two acid regions (I (amino acids 2–30) and II (amino acids 33–74)) were further identified (Fig. 6A). To examine the role of each domain in enhancer-promoter looping and eRNA/mRNA transcription, different deletion and amino acid substitution mutants were constructed and transfected into B16 cells (Fig. S3). B16 cells transfected with the PU.1 mutants, lacking either acidic or glutamine-rich domains or containing R230A/R233A substitutions, failed to produce the E2 eRNA and IL-1β mRNA, whereas PEST domain deletion (Δ118–167) had no effects on this transcription (Fig. 6B, top two panels). Deletion of the acidic domain II (Δ33–74) or the glutamine-rich domain (Δ74–100) had no effects on the formation of enhancer-promoter looping. However, the enhancer-promoter looping was defective in cells transfected with PU.1 mutant lacking the acidic domain I (Δ2–30 and Δ1–100) or containing R230A/R233A substitutions (Fig. 6B, bottom). PU.1 was shown to interact with the histone acetyltransferase CBP through the transactivation domain (47) that targets histone H3 Lys-27 (54). Thus, we examined whether histone acetylation was required for enhancer-promoter looping. As shown in Fig. 6C, the Δ2–30 mutant had no defects in H3K27ac, whereas the Δ33–74 mutant was not able to induce H3K27ac in both the E2 and E1 enhancer regions, suggesting that PU.1-dependent enhancer-promoter looping and H3K27ac are independent events mediated by two different acidic regions of PU.1. To further confirm that histone acetylation is not required for enhancer-promoter looping, the bromodomain and extra-terminal motif (BET) family protein (histone acetyl-lysine reader, BRD) 2/4 inhibitor JQ1 (55) was pretreated in B16 cells. JQ1 had no effects on the enhancer-promoter looping induced by PU.1 (Fig. 6D). These data further suggest that histone acetylation and enhancer-promoter looping are independently induced by PU.1.
Figure 6.

E2-promoter looping and H3K27ac were mediated by distinct domains of PU.1. A, schematic presentation of PU.1 domains and constructs of PU.1 mutants. B, B16 cells were transfected with PU.1 WT or PU.1 mutant (PU.1Δ2–30, PU.1Δ33–74, PU.1Δ75–100, PU.1Δ1–100, PU.1Δ118–167, and PU.1R230A/R233A) plasmids (1 μg) for 48 h as described under “Experimental procedures,” and cells were stimulated with LPS (100 ng/ml) for 6 h. The expression of IL-1β mRNA (top) and E2 eRNA (middle) was analyzed by RT-qPCR. The proximal localization of E2 and the promoter was analyzed as described under “Experimental procedures” (bottom). Data are expressed as means ± S.D. (n = 2). C, B16 cells were transfected with PU.1 WT or PU.1 mutants, and ChIP assays were performed using anti-H3K27ac as described under “Experimental procedures.” Purified DNAs were then analyzed by qPCR using the primers indicated. Data are presented as percentage of enrichment with the precipitated target sequence compared with input DNA (n ≥ 2). D, proximal localization of DNA fragments of E2 and IL-1β promoter was analyzed in B16 cells transfected with vector or HA-PU.1 plasmids with or without JQ1 (1 μm) treatment. Cumulative values of primer sets 9–13, as shown in Fig. 5A, were plotted. Data are expressed as means ± S.D. (n = 2).
H3K27ac and NF-κB recruitment to the IL-1β enhancer and promoter regions by LPS are mutually dependent events that are independent of eRNA transcription
eRNAs play an important role in gene transcription (25) and IL-1β mRNA transcription (Figs. 3C and 4D). Histone acetylation enhances accessibility of transcription factors through recruiting chromatin remodelers, such as BET-binding proteins (56). At the same time, signal-dependent transcription factors, such as NF-κB, induce histone acetylation (57). Thus, we examined the relationship between H3K27ac and NF-κB recruitment and the role of eRNA in the IL-1β transcription in B16 cells. LPS further increased H3K27ac levels above the levels induced by PU.1 in both E2 and E1 regions, which was prevented by NF-κBi (Fig. 7A). Inversely, JQ1 inhibited recruitment of NF-κB to the enhancer and promoter (Fig. 7B), which resulted in inhibition of LPS-induced IL-1β mRNA/eRNA transcription (Fig. 7C). Interestingly, cells specifically deleted in the IL-1β eRNA sequences (CRISPR-eRNA) induced similar levels of H3K27ac in E2 and E1 regions as those of control cells (CRISPR-vector; Fig. 7D), suggesting that eRNA is not required for H3K27ac in E2 and E1 regions. In line with these results and Fig. 6B, B16 cells transfected with Δ2–30 (lacking E2-promoter looping) and Δ33–74 (lacking CBP interaction) mutants failed to further induce H3K27ac in response to LPS treatment (Fig. 7E). Collectively, these results suggest that E2-promoter looping is required for NF-κB recruitment and H3K27ac induced by LPS stimulation.
Figure 7.

H3K27ac and NF-κB recruitment to the IL-1β enhancer and promoter regions by LPS are mutually dependent events that are independent of eRNA transcription. A–C, B16 cells were transfected with pcDNA3 (Vector) or pcDNA3-HA-PU.1 (PU.1) for 48 h and then exposed to drug vehicle, NF-κBi (5 μm), or BRD2/4 inhibitor (JQ1; 1.5 μm). Cells were stimulated with LPS (100 ng/ml) for 4 h, and ChIP-qPCR assays were performed using anti-H3K27ac (A) or anti-NF-κB (B) antibodies. C, similarly, cells were stimulated with LPS (100 ng/ml) for 6 h. Transcription of IL-1β mRNA (top) and E2 eRNA (bottom) was analyzed by RT-qPCR. Data are expressed as means ± S.D. (error bars) (n ≥ 2; *, p < 0.05). D, CRISPR-vector and CRISPR-eRNA B16 cells were transfected with pcDNA3-HA-PU.1 (PU.1) and then stimulated with LPS (100 ng/ml) for 4 h. ChIP-qPCR assays were performed using anti-H3K27ac. ChIP efficiency is represented as percentage of input DNA recovered by immunoprecipitation (N.S., not significant; Student's t test, n ≥ 2). E, B16 cells were transfected with PU.1 WT or PU.1 mutants (PU.1Δ2–30 and PU.1Δ33–74) for 48 h, and cells were stimulated with LPS (100 ng/ml) for 4 h. ChIP assays were performed using anti-H3K27ac as described under “Experimental procedures.” Purified DNAs were then analyzed by qPCR using the primers indicated. Data are presented as percentage of enrichment with the precipitated target sequence compared with input DNA (n = 2; *, p < 0.05).
Discussion
Here, we examined the role of PU.1 in programming IL-1β expression, using murine B16 and RAW cells. Among several lineage-determining transcription factors, C/EBPβ and PU.1 are particularly involved in IL-1β transcription (37), where PU.1 directly binds to genomic DNA and recruits transcription machineries through interacting with C/EBPβ (36, 58). This study mainly used B16 cells, which were selectively deficient in PU.1 expression, to examine the detailed epigenetic mechanism of PU.1 in regulating IL-1β expression.
PU.1 primes the remote regulatory element and eRNA production required for IL-1β expression
Previous studies have identified several cis-acting elements that regulate IL-1β transcription in murine and human macrophages. In murine macrophages, proximal (−100 to −50 bp upstream of the TSS) and distal (−2,586 to −2,106; herein, referred to as E1) regulatory elements that contain C/EBPβ-binding sites have been described (59, 60). In addition to the E1 element, we identified a potential regulatory element located further upstream (∼10 kbp) of the TSS (E2), based on the H3K27ac, H3K4me1, and H3K4me3 profiles in the ENCODE database (Fig. 2A). Here, we demonstrated that the E2 element is a functional and distal enhancer for IL-1β. In B16 cells, ectopically expressed PU.1 was recruited to the E2 region and induced H3K27ac and nucleosome depletion (Fig. 2, B and C). The E2 element also transcribed RNAs in both PU.1-transfected B16 and WT RAW cells in response to LPS treatment, preceding IL-1β mRNA transcription (Figs. 3B and 4B). Importantly, deletion of the PU.1-binding site in the E2 region by CRISPR (Fig. 5B) or knocking down E2 eRNA by ASO targeting negative strand of E2 significantly and specifically prevented IL-1β transcription (Fig. 3C and 4C). It appears that the size of E2 eRNA is ∼5,000 bases long in both B16 and RAW cells, which is longer than usual eRNAs, which range from 500 to 2,000 bases (61). However, >3-kb-wide enhancers have been identified in so-called “stretch enhancers” and “superenhancers” (62). Also, considering the locations of PU.1– and NF-κB–binding sites, which should be located 5′ upstream of transcripts, E2 eRNA is likely a negative-strand unidirectional eRNA in B16 cells (Fig. 3A). In RAW cells, the E2 element appears to render bidirectional eRNAs, but the positive-strand eRNA is shorter than the negative-stranded eRNA (Fig. 4A). Further detailed studies are required to address the exact size, direction, and role of these transcripts in IL-1β expression.
Distinct regions of PU.1 are involved in priming events for IL-1β expression
PU.1 has multiple domains that manifest cell type– and gene–specific programming activities (52). We found that both the transactivation and Ets domains, but not the PEST domain, were essential for E2 eRNA and IL-1β mRNA transcription (Fig. 6B). These results are in line with previous studies highlighting an essential role of the Ets DNA-binding domain, but not the PEST domain, in maximal gene transactivation (53) and IL-6 transcription (63). The 3C analysis we used here was not able to determine whether PU.1 induced looping between E1 and promoter due to high basal interactions (Fig. 5A, top). Because E1 is located close to the promoter, it may communicate with the promoter through a “linking model” (64). This model proposed that enhancers recruit the transcription preinitiation complex and progressively extend along the chromatin fiber from enhancer to promoter without looping formation. Our 3C assay, however, found that the E2 element formed a loop with the IL-1β promoter upon ectopic expression of PU.1 in B16 cells or constitutively in RAW cells (Fig. 5A). Importantly, establishment of the E2-promoter looping by PU.1 was an essential step for the following events induced by LPS stimulation: H3K27ac (Fig. 7E) and transcription of E2 eRNA and IL-1β mRNA (Figs. 5 (B and C) and 8). The E2-promoter looping required binding of PU.1 at the E2 region (between −10,833 and −9,824 sequences from the TSS, which contains three putative PU.1-binding sites; Fig. 5D). We further found that the E2-promoter looping required both the acidic region I in the acidic domain and arginine residues at 230/233 in the Ets domain, but not the PEST domain (Fig. 6B). The residues Arg-230 and Arg-233 have been shown to be crucial for GGAA sequence–specific DNA binding and nuclear localization (65, 66); therefore, it is not surprising that R230A/R233A mutant failed to induce the looping. However, unlike our results, the immunoglobulin κ 3′ enhancer, located 8.5 kbp downstream of the Igκ gene (67), utilizes the PEST domain, but not the acidic domain, to communicate with the promoter in B cells (68). Because enhancer-promoter looping is mediated by various transcription factors (69) and chromatin remodelers, such as Mediator, Cohesin (70), CTCF proteins (71), and YY1 (72), it is possible that the acidic and PEST domains interact with different factors to establish enhancer-promoter interactions in a gene– and cell type–specific manner.
Figure 8.
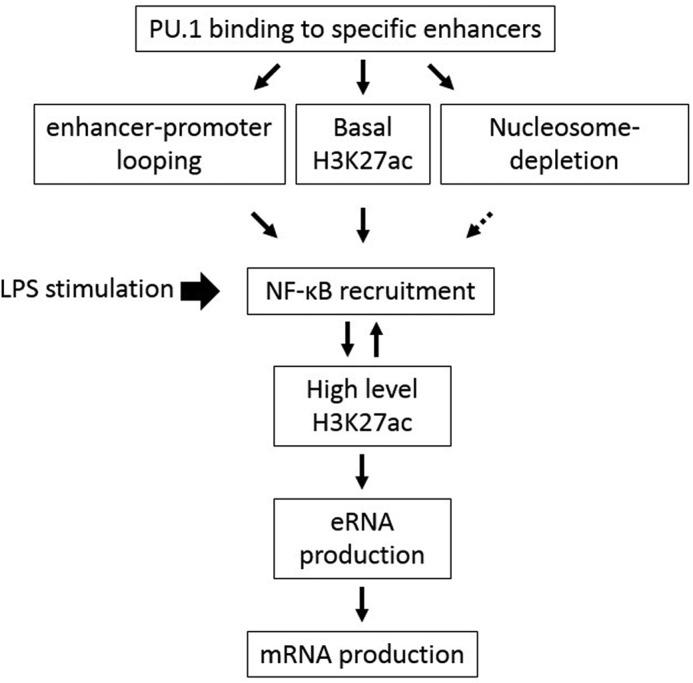
A sequence of events proposed to be involved in programming murine IL-1β transcription through remodeling distal enhancer by the myeloid lineage–determining transcription factor PU.1.
Ectopic expression of PU.1 also induced significant H3K27ac in the E2 region without LPS treatment, which rendered H3K27ac signals similar to those observed in basal bone marrow–derived macrophages (Fig. 2B, bottom). A previous in vitro study showed that PU.1 directly interacts with CBP through residues 74–122, which mainly comprise the glutamine-rich domain (47). However, the study did not examine involvement of the acidic domain in the interaction. We found that the acidic region II was also involved in H3K27ac associated with the E2 and E1 regions (Fig. 6C). These results suggest that E2-promoter looping and H3K27ac are mediated by two different domains of PU.1. In line with these results, E2-promoter looping was intact in B16 cells transfected with Δ33–74 PU.1 (lacking H3K27ac), and H3K27ac was not compromised in cells transfected with Δ2–30 (low in E2-promoter looping). Also, the histone acetylation reader inhibitor JQ1 had no effects on E2-promoter looping (Fig. 6D). Collectively, these results suggest that basal E2-promoter looping and H3K27ac are independent events mediated by two different domains of PU.1.
PU.1 is required for LPS-induced chromatin modifications and eRNA production
Upon stimulation, enhancers and promoters undergo various epigenetic changes for maximal transcription: looping stabilization, eRNA transcription, and histone acetylation that allows recruitment of signal-dependent transcription factors and cofactors. We found that LPS stimulation induced increased E2-promoter looping (Fig. 5A), transcription of E2 eRNA (Figs. 3A and 4A), high H3K27ac levels in the E2 region (Fig. 7A), and NF-κB recruitment to the E2 region (Fig. 7B). Interestingly, LPS stimulation–induced H3K27ac required NF-κB activation (Fig. 7A), and, conversely, NF-κB recruitment required histone acetylation (Fig. 7B), suggesting that both of these events are interdependent. NF-κB directly interacts with the histone acetyltransferases CBP and p300 (73), which are required for inflammatory gene transactivation (74). PU.1 induced H3K27ac without NF-κB activation (Fig. 2B). In LPS-stimulated cells, inhibition of NF-κB was able to decrease H3K27ac only to the levels of PU.1-induced (Fig. 7A). Therefore, we speculate that H3K27ac is mediated by two mechanisms: basal low-level acetylation by PU.1 and stimulation-dependent high-level acetylation by NF-κB (Fig. 8). In addition, we found that defects in PU.1-induced H3K27ac (Δ33–74 PU.1 mutant in Fig. 6C) also led to the absence of LPS-induced acetylation (Fig. 7A), indicating that PU.1-dependent basal acetylation is prerequisite for NF-κB-dependent (stimulation-induced) H3K27ac (Fig. 8). In human monocytic cell line THP-1 cells, LPS-induced NF-κB activation also enhances looping formation between enhancer (located ∼3,000 bp upstream of TSS) and promoter (37). Similarly, we also detected that LPS stimulation further increased E2-promoter looping in B16 cells (Fig. 5A). Therefore, we speculate that, unlike PU.1-induced basal enhancer-promoter looping, stimulation-induced enhancer-promoter looping requires NF-κB activation and histone acetylation in mouse and likely in human cells. However, further detailed experiments are required to address the role of enhanced or stabilized looping in gene transcription.
E2 eRNA is not involved in enhancer-promoter looping and H3K27ac but is involved in IL-1β e/mRNA production
To date, the biological relevance of eRNA has been demonstrated; however, the mechanisms of eRNA in transcription have been controversial (64). Some studies have suggested that eRNAs are involved in enhancer-promoter looping by recruiting Cohesin, Mediator, and other factors to enhancers (28, 75, 76), whereas others have shown that they enhance transcription by increasing chromatin accessibility (77) and H3K27ac levels by directly activating the histone acetyltransferases CBP/p300 (78) or inducing RNA polymerase II elongation through binding to the negative elongation factor complex and releasing paused RNA polymerase II (29). We demonstrated that the E2 eRNA played an essential role in IL-1β transcription (Figs. 3C, 4C, and 5B). Depletion of E2 eRNA by CRISPR had no effects on the establishment of enhancer-promoter looping (Fig. 5D) and H3K27ac (Fig. 7D), ruling out the involvement of E2 eRNA in the establishment of enhancer-promoter looping and enhancement of chromatin accessibility. Therefore, it is possible that the E2 eRNA increases IL-1β transcription through releasing the negative elongation factor complex from the promoter or still unknown mechanisms. We also showed that synthesis of both IL-1β eRNA and mRNA required NF-κB activation (Fig. 3D) and acetylation reader proteins, such as BRD2/4 (Fig. 7C). These results are in line with previous studies shown that NF-κB binds to cis-regulatory elements at enhancers (79) and BRD4 binds to NF-κB (p65; acetylated at Lys-310), which is required for its transactivation activity (80).
Overall, this study demonstrated that PU.1 induced enhancer-promoter looping and H3K27ac, with distinct regions of the activation domain. Both of these events were independently necessary for the subsequent signal-dependent and mutually dependent events: recruitment of NF-κB and induction of further H3K27ac, both of which were required for eRNA production and subsequent IL-1β mRNA transcription (Fig. 8). PU.1 has various developmental and pathogenic functions through different domains and mechanisms (52, 81). Especially, improper regulation of inflammatory cytokines, such as IL-1β transcription, has been shown to be involved in endotoxin shock, sepsis, and infection (7, 48). Identification of other epigenetic events and factors involved in the paradigm proposed herein warrants further studies. Also, unraveling the role and mechanism of PU.1 in specific gene regulation will be crucial for understanding the multifaced developmental and pathophysiological function of PU.1.
Experimental procedures
Materials and reagents
LPS (from E. coli O111:B4) and LTA (from S. aureus) were purchased from List Biological Laboratories and Sigma-Aldrich, respectively; poly(I:C) and CpG oligonucleotides were from InvivoGen. The ERK inhibitor U0126, p38 inhibitor SB203580, JNK inhibitor, NF-κB inhibitor, and BRD2/4 inhibitor (JQ1) were obtained from APExBIO Technology, Selleck Chemicals, EMD Millipore, Calbiochem, and Cayman chemicals, respectively. Antibodies for phospho-p38, phospho-ERK, and phospho-IκB were from Cell Signaling Technology; β-actin antibody was from Rockland Inc.; and pan-histone H3 antibody was from Bio Vision. Antibodies raised against PU.1 C terminus were obtained by Dr. DeKoter, and the PU.1 N terminus and C/EBPα were obtained from Santa Cruz Biotechnology, Inc. H3K27ac antibody was from Active Motif, and anti-NF-κB (p65) was from eBioscience. Mouse PU.1 plasmids used in this study, pcDNA3-HA-PU.1, MIG-PU.1 (WT), MIG-PU.1 (Δ2–30), MIG-PU.1 (Δ33–74), MIG-PU.1 (Δ75–100), MIG-PU.1 (Δ1–100), MIG-PU.1 (Δ118–167), and DNA binding inactive PU.1 (pLVX-human PU.1 (R230A/R233A)) were constructed using standard cloning techniques. Murine transcription factor C/EBPα-encoded plasmid (p67_pMSCV_C/EBPα–IRES-hCD4) was donated by Dr. Thomas Graf (Center for Genomic Regulation, Barcelona, Spain). Scrambled (product 300610; AACACGTCTATACGC) and eRNA-specific locked nucleic acid ASO (product 300600; CAATCCTGGTTGATGA) against pro-IL-1β eRNA were obtained from Exiqon. DpnII, T4 DNA ligase, and T4 DNA ligase buffer, used for 3C analysis, were from New England Biolabs. RNase A was from Qiagen. Proteinase K, phenol:chloroform:isoamyl alcohol, and protein G Dynabeads were purchased from Invitrogen. Halt protease inhibitor mixture, cOmpleteTM EDTA-free protease inhibitor mixture, and phosphatase inhibitor mixture (phosSTOP) tablets were obtained from Thermo Scientific and Roche Applied Science.
Cell culture and transfection
B16 and RAW cells were cultured in RPMI 1640 and Dulbecco's modified Eagle's medium, respectively, supplemented with 10% fetal bovine serum (Sigma-Aldrich), 10 mm minimal essential medium nonessential amino acids, 1 mm sodium pyruvate, 100 IU/ml penicillin, and 100 μg/ml streptomycin. Plasmid transfection was carried out using PolyJet (SignaGen Laboratories) according to the manufacturer's instructions. Briefly, 5.0 × 105 B16 cells were plated on 6-well plates and cultured 1 day prior to transfection. Cells were replaced with fresh culture medium and transfected with plasmids for 5 h. Cells were further cultured for 16–18 h with an additional cell culture medium. Cells were then replated and cultured for an additional 24 h, followed by cell treatments. For ASO transfection, B16 cells transfected with pcDNA3-HA-PU.1 or RAW cells were retransfected with scrambled or eRNA-specific ASO (250 pmol) for 20–24 h using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. Cells were then replated and stimulated with LPS (100 ng/ml) for the time indicated.
Immunoblotting
Preparation of total cell lysates and immunoblotting were performed as reported previously (82).
RT-qPCR
eRNA or mRNA expression was quantified via/by qPCR as reported previously (83). qPCR analysis was performed with the Rotor-Gene RG3000 quantitative multiplex PCR instrument (Montreal Biotech) using PowerUPTM SYBR® Green Master Mix (Applied Biosystems), and gene expression levels were estimated using a ΔΔCt method. Data were expressed as -fold change compared with the lowest detected sample value in each experiment, unless otherwise described. For 3C assay quantification, dual-labeled TaqMan probe (5′-/JOETM/TCGTTCACC/ZENTM/ACCTTTGCACTGTGCAAC/BkFQTM/-3′; synthesized by Integrated DNA Technologies) and Hot Start Taq DNA polymerase (New England Biolabs) were used. Primer sequences used for qPCR are listed in Table S1.
ddPCR
ddPCR was carried out using a Bio-Rad system. First, total RNAs were reverse-transcribed using Moloney murine leukemia virus reverse transcriptase (New England Biolabs) and random hexamers. Oil droplets were then generated using QX200TM ddPCRTM EvaGreen supermix (Bio-Rad) and droplet generation oil for EvaGreen (Bio-Rad) and QX200TM droplet generator (Bio-Rad) according to the manufacturer's instructions. The generated oil droplets were transferred onto a ddPCRTM 96-well plate (Bio-Rad), and subsequently PCR was carried out using a C1000 TouchTM thermal cycler (Bio-Rad). EvaGreen fluorescence was acquired and analyzed using a QX200 droplet reader (Bio-Rad) and QuantaLife software.
ChIP assay
ChIP analysis was conducted as described previously (84). Briefly, cells transfected with plasmids were replated in 10-cm cell culture dishes and cultured for 16–18 h. Cells exposed with or without LPS (100 ng/ml) for 4 h were fixed with 1% formaldehyde, lysed, sonicated, and immunoprecipitated with N terminus PU.1 (Santa Cruz Biotechnology, Inc.), H3K27ac (Active Motif), and NF-κB (p65; eBioscience) antibodies. DNAs were purified after de-cross-linking by overnight heating at 65 ºC and then subjected to qPCR analysis. Primers used for ChIP-qPCR are listed in Table S1.
CRISPR/Cas9 plasmid construction
Genome editing using a CRISPR/Cas9 system and designing guide RNA sequences was carried out as described previously (85). The oligonucleotides used to construct guide RNAs for deleting the eRNA and PU.1-binding sites are listed in Table S1. Annealed oligonucleotides for each CRISPR target were inserted into the BbsI restriction site of the guide RNA expression vector pSpCas9(BB)-2A-puro (Addgene, catalog no. 48138).
CRISPR-eRNA and CRISPR-PU.1 cell preparation and validation
B16 cells were transfected with CRISPR constructs. CRISPR/Cas9 constructs targeting −11,843 and −10,833 bp from the IL-1β TSS were used to delete the eRNA encoding region (CRISPR-eRNA cells); CRISPR/Cas9 constructs targeting −10,833 and −9,824 bp were used to delete the PU.1-binding sites (CRISPR-PU.1 cells). B16 cells transfected with the pSpCas9 (BB) empty plasmid were used as control cells. Cells transfected with these constructs were cultured for 24 h and exposed to puromycin (2 μg/ml) for 72 h. Surviving cells were pooled, and gene editing was validated. For CRISPR gene-editing validation, genomic DNAs were prepared, and PCR was carried out by standard PCR protocols.
DNase I hypersensitivity assay
Nuclei preparation and DNase I digestion were carried out as described previously (86, 87) with minor modifications. Briefly, 3.0 × 106 cells were lysed on ice in 3 ml of ice-cold lysis buffer (10 mm Tris, pH 8.0, 10 mm NaCl, 5 mm MgCl2, 10 mm EDTA, 0.5 mm EGTA, 0.5 mm spermidine, 0.1% Nonidet P-40) supplemented with cOmpleteTM EDTA-free protease inhibitor mixture for 10 min. Cell lysates were centrifuged at 500 × g for 10 min, and pellets were washed with 2 ml of DNase I reaction buffer (10 mm Tris, pH 7.5, 2.5 mm MgCl2, 0.1 mm CaCl2) and divided into two. After recentrifugation, nuclei pellets were resuspended in DNase I reaction buffer (100 μl) containing 20 units of DNase I (Thermo Scientific). After incubating at 37 ºC for 20 min, an equal volume of stop buffer (100 mm EDTA, 0.5% SDS) was added. RNase A (15 μg) was then added to each sample and incubated at 54 ºC for 15 min. These samples were further incubated together with proteinase K (30 μg) for 5 h. DNAs were extracted by standard phenol:chloroform:isoamyl alcohol protocols. DNA pellets were resuspended in Tris-EDTA buffer and quantified using a NanodropTM spectrophotometer. Accessibility of DNase I was analyzed by qPCR using the primer sequences listed in Table S1.
3C assay
3C analysis was conducted as described previously (51, 88) with minor modifications. Briefly, ∼5.0 × 106 B16 cells and ∼1.0 × 107 RAW cells were harvested to cross-link DNA with formaldehyde (final 1.2%) for 10 min at room temperature. Cross-linking was then terminated by glycine (final 0.125 m) on ice. Cells were then lysed by lysis buffer (ice-cold, 10 mm NaCl, 10 mm Tris (pH 8.0), 0.2% Nonidet P-40, and cOmpleteTM EDTA-free protease inhibitor mixture), and nuclei pellets were prepared by centrifugation, followed by snap freezing. Frozen nuclei were thawed in 500 μl of the DpnII restriction enzyme buffer containing 0.3% SDS and incubated at 37 ºC for 1 h with shaking, followed by the addition of Triton X-100 (final concentration 2.0%) and further incubation for 1 h. An aliquot (10 μl) of undigested DNA was saved for determining digestion efficiency. The remainder was exposed to 400 units of DpnII per sample at 37 ºC with shaking. After 24 h, an aliquot (10 μl) of digested DNA was saved for determining digestion efficiency, and the remainder was incubated at 65 ºC for 25 min to inactivate the restriction enzyme. Digested DNAs were then diluted with 6.125 ml of 1.15× ligase buffer containing Triton X-100 (final concentration 1.0%) and exposed to 800 units of T4 DNA ligase at 16 ºC for 72 h. After inactivating the ligase by EDTA, DNAs were de-cross-linked by proteinase K at 65 ºC for 16–18 h and additional fresh proteinase K treatment for 2 h. DNAs were then purified using phenol:chloroform:isoamyl alcohol and subsequently with chloroform. The DNA pellets were resuspended in 10 mm Tris-HCl (pH 8.0) after washing with 70% ethanol and incubated with RNase A for 45 min at 37 ºC. After purifying and washing DNAs, 3C library DNAs were analyzed for interactions between enhancer and promoter by TaqMan qPCR. The constant primer and TaqMan probe targeted the promoter (or constant) fragment, and the test primers targeted the 16 candidate fragments (Fig. 5A). These primer sequences are listed in Table S1.
Statistical analysis
Data were analyzed using GraphPad Prism 4.0 (GraphPad Software). The results are presented as the mean ± S.D. of two or three independent repeats. Statistical significance was defined as p < 0.05 (*).
Author contributions
S.-D. H., W. C., and S. O. K. data curation; S.-D. H. and W. C. formal analysis; S.-D. H. and S. O. K. investigation; S.-D. H. and W. C. methodology; S.-D. H., W. C., R. P. D., and S. O. K. writing-review and editing; R. P. D. resources; S. O. K. conceptualization; S. O. K. supervision; S. O. K. funding acquisition; S. O. K. writing-original draft; S. O. K. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Thomas Graf (Center for Genomic Regulation, Barcelona, Spain) for providing the murine transcription factor C/EBPα-encoded plasmid (p67_pMSCV_C/EBPα-IRES-hCD4).
This work was supported by Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant RGPIN-2018-05514 (to S. O. K.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Table S1 and Figs. S1–S3.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- Ets
- E26 transformation–specific
- qPCR
- quantitative real-time PCR
- ddPCR
- droplet digital PCR
- C/EBP
- CCAAT/enhancer-binding protein
- eRNA
- enhancer RNA
- IL
- interleukin
- TLR
- Toll-like receptor
- LPS
- lipopolysaccharide
- TSS
- transcription start site
- LTA
- lipoteichoic acid
- ERK
- extracellular signal-regulated kinase
- MAPK
- mitogen-activated protein kinase
- M-CSF
- macrophage colony–stimulating factor
- M-CSFR
- M-CSF receptor
- CBP
- cAMP-response element-binding protein-binding protein
- ASO
- antisense oligonucleotide(s)
- NFκBi
- NF-κB inhibitor
- BET
- bromodomain and extra-terminal motif
- 3C
- chromatin conformation capture
- H3K4me1 and H3K4me3
- histone H3 Lys-4 mono- and trimethylation, respectively
- H3K27ac
- histone H3 Lys-27 acetylation.
References
- 1. Scott E. W., Simon M. C., Anastasi J., and Singh H. (1994) Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science 265, 1573–1577 10.1126/science.8079170 [DOI] [PubMed] [Google Scholar]
- 2. Anderson K. L., Smith K. A., Conners K., McKercher S. R., Maki R. A., and Torbett B. E. (1998) Myeloid development is selectively disrupted in PU.1 null mice. Blood 91, 3702–3710 [PubMed] [Google Scholar]
- 3. DeKoter R. P., Walsh J. C., and Singh H. (1998) PU.1 regulates both cytokine-dependent proliferation and differentiation of granulocyte/macrophage progenitors. EMBO J. 17, 4456–4468 10.1093/emboj/17.15.4456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen H. M., Zhang P., Voso M. T., Hohaus S., Gonzalez D. A., Glass C. K., Zhang D. E., and Tenen D. G. (1995) Neutrophils and monocytes express high levels of PU.1 (Spi-1) but not Spi-B. Blood 85, 2918–2928 [PubMed] [Google Scholar]
- 5. Warren L., Bryder D., Weissman I. L., and Quake S. R. (2006) Transcription factor profiling in individual hematopoietic progenitors by digital RT-PCR. Proc. Natl. Acad. Sci. U.S.A. 103, 17807–17812 10.1073/pnas.0608512103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. DeKoter R. P., and Singh H. (2000) Regulation of B lymphocyte and macrophage development by graded expression of PU.1. Science 288, 1439–1441 10.1126/science.288.5470.1439 [DOI] [PubMed] [Google Scholar]
- 7. Karpurapu M., Wang X., Deng J., Park H., Xiao L., Sadikot R. T., Frey R. S., Maus U. A., Park G. Y., Scott E. W., and Christman J. W. (2011) Functional PU.1 in macrophages has a pivotal role in NF-κB activation and neutrophilic lung inflammation during endotoxemia. Blood 118, 5255–5266 10.1182/blood-2011-03-341123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., and Glass C. K. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghisletti S., Barozzi I., Mietton F., Polletti S., De Santa F., Venturini E., Gregory L., Lonie L., Chew A., Wei C. L., Ragoussis J., and Natoli G. (2010) Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity 32, 317–328 10.1016/j.immuni.2010.02.008 [DOI] [PubMed] [Google Scholar]
- 10. Barozzi I., Simonatto M., Bonifacio S., Yang L., Rohs R., Ghisletti S., and Natoli G. (2014) Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol. Cell 54, 844–857 10.1016/j.molcel.2014.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Laiosa C. V., Stadtfeld M., Xie H., de Andres-Aguayo L., and Graf T. (2006) Reprogramming of committed T cell progenitors to macrophages and dendritic cells by C/EBP α and PU.1 transcription factors. Immunity 25, 731–744 10.1016/j.immuni.2006.09.011 [DOI] [PubMed] [Google Scholar]
- 12. Xie H., Ye M., Feng R., and Graf T. (2004) Stepwise reprogramming of B cells into macrophages. Cell 117, 663–676 10.1016/S0092-8674(04)00419-2 [DOI] [PubMed] [Google Scholar]
- 13. Feng R., Desbordes S. C., Xie H., Tillo E. S., Pixley F., Stanley E. R., and Graf T. (2008) PU.1 and C/EBPα/β convert fibroblasts into macrophage-like cells. Proc. Natl. Acad. Sci. U.S.A. 105, 6057–6062 10.1073/pnas.0711961105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van Oevelen C., Collombet S., Vicent G., Hoogenkamp M., Lepoivre C., Badeaux A., Bussmann L., Sardina J. L., Thieffry D., Beato M., Shi Y., Bonifer C., and Graf T. (2015) C/EBPα activates pre-existing and de novo macrophage enhancers during induced pre-B cell transdifferentiation and myelopoiesis. Stem Cell Reports 5, 232–247 10.1016/j.stemcr.2015.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hoogenkamp M., Lichtinger M., Krysinska H., Lancrin C., Clarke D., Williamson A., Mazzarella L., Ingram R., Jorgensen H., Fisher A., Tenen D. G., Kouskoff V., Lacaud G., and Bonifer C. (2009) Early chromatin unfolding by RUNX1: a molecular explanation for differential requirements during specification versus maintenance of the hematopoietic gene expression program. Blood 114, 299–309 10.1182/blood-2008-11-191890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Du J., Stankiewicz M. J., Liu Y., Xi Q., Schmitz J. E., Lekstrom-Himes J. A., and Ackerman S. J. (2002) Novel combinatorial interactions of GATA-1, PU.1, and C/EBPϵ isoforms regulate transcription of the gene encoding eosinophil granule major basic protein. J. Biol. Chem. 277, 43481–43494 10.1074/jbc.M204777200 [DOI] [PubMed] [Google Scholar]
- 17. Ong C. T., and Corces V. G. (2011) Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 12, 283–293 10.1038/nrg2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heintzman N. D., Hon G. C., Hawkins R. D., Kheradpour P., Stark A., Harp L. F., Ye Z., Lee L. K., Stuart R. K., Ching C. W., Ching K. A., Antosiewicz-Bourget J. E., Liu H., Zhang X., Green R. D., et al. (2009) Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112 10.1038/nature07829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mayran A., and Drouin J. (2018) Pioneer transcription factors shape the epigenetic landscape. J. Biol. Chem. 293, 13795–13804 10.1074/jbc.R117.001232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Creyghton M. P., Cheng A. W., Welstead G. G., Kooistra T., Carey B. W., Steine E. J., Hanna J., Lodato M. A., Frampton G. M., Sharp P. A., Boyer L. A., Young R. A., and Jaenisch R. (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 107, 21931–21936 10.1073/pnas.1016071107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rada-Iglesias A., Bajpai R., Swigut T., Brugmann S. A., Flynn R. A., and Wysocka J. (2011) A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283 10.1038/nature09692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zentner G. E., Tesar P. J., and Scacheri P. C. (2011) Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 21, 1273–1283 10.1101/gr.122382.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Riel B., and Rosenbauer F. (2014) Epigenetic control of hematopoiesis: the PU.1 chromatin connection. Biol. Chem. 395, 1265–1274 10.1515/hsz-2014-0195 [DOI] [PubMed] [Google Scholar]
- 24. Ivashkiv L. B., and Park S. H. (2016) Epigenetic regulation of myeloid cells. Microbiol. Spectr. 10.1128/microbiolspec.MCHD-0010-2015 10.1128/microbiolspec.MCHD-0010-2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lam M. T., Li W., Rosenfeld M. G., and Glass C. K. (2014) Enhancer RNAs and regulated transcriptional programs. Trends Biochem. Sci. 39, 170–182 10.1016/j.tibs.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim T. K., Hemberg M., and Gray J. M. (2015) Enhancer RNAs: a class of long noncoding RNAs synthesized at enhancers. Cold Spring Harb. Perspect. Biol. 7, a018622 10.1101/cshperspect.a018622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hon C. C., Ramilowski J. A., Harshbarger J., Bertin N., Rackham O. J., Gough J., Denisenko E., Schmeier S., Poulsen T. M., Severin J., Lizio M., Kawaji H., Kasukawa T., Itoh M., Burroughs A. M., et al. (2017) An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 543, 199–204 10.1038/nature21374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li W., Notani D., Ma Q., Tanasa B., Nunez E., Chen A. Y., Merkurjev D., Zhang J., Ohgi K., Song X., Oh S., Kim H. S., Glass C. K., and Rosenfeld M. G. (2013) Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498, 516–520 10.1038/nature12210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schaukowitch K., Joo J. Y., Liu X., Watts J. K., Martinez C., and Kim T. K. (2014) Enhancer RNA facilitates NELF release from immediate early genes. Mol. Cell 56, 29–42 10.1016/j.molcel.2014.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mousavi K., Zare H., Dell'orso S., Grontved L., Gutierrez-Cruz G., Derfoul A., Hager G. L., and Sartorelli V. (2013) eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol. Cell 51, 606–617 10.1016/j.molcel.2013.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kaikkonen M. U., Spann N. J., Heinz S., Romanoski C. E., Allison K. A., Stender J. D., Chun H. B., Tough D. F., Prinjha R. K., Benner C., and Glass C. K. (2013) Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol. Cell 51, 310–325 10.1016/j.molcel.2013.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Natoli G. (2010) Maintaining cell identity through global control of genomic organization. Immunity 33, 12–24 10.1016/j.immuni.2010.07.006 [DOI] [PubMed] [Google Scholar]
- 33. Marecki S., Riendeau C. J., Liang M. D., and Fenton M. J. (2001) PU.1 and multiple IFN regulatory factor proteins synergize to mediate transcriptional activation of the human IL-1 β gene. J. Immunol. 166, 6829–6838 10.4049/jimmunol.166.11.6829 [DOI] [PubMed] [Google Scholar]
- 34. Kominato Y., Galson D., Waterman W. R., Webb A. C., and Auron P. E. (1995) Monocyte expression of the human prointerleukin 1 beta gene (IL1B) is dependent on promoter sequences which bind the hematopoietic transcription factor Spi-1/PU.1. Mol. Cell. Biol. 15, 58–68 [PMC free article] [PubMed] [Google Scholar]
- 35. Liang M. D., Zhang Y., McDevit D., Marecki S., and Nikolajczyk B. S. (2006) The interleukin-1β gene is transcribed from a poised promoter architecture in monocytes. J. Biol. Chem. 281, 9227–9237 10.1074/jbc.M510700200 [DOI] [PubMed] [Google Scholar]
- 36. Yang Z., Wara-Aswapati N., Chen C., Tsukada J., and Auron P. E. (2000) NF-IL6 (C/EBPβ) vigorously activates il1b gene expression via a Spi-1 (PU.1) protein-protein tether. J. Biol. Chem. 275, 21272–21277 10.1074/jbc.M000145200 [DOI] [PubMed] [Google Scholar]
- 37. Adamik J., Wang K. Z., Unlu S., Su A. J., Tannahill G. M., Galson D. L., O'Neill L. A., and Auron P. E. (2013) Distinct mechanisms for induction and tolerance regulate the immediate early genes encoding interleukin 1β and tumor necrosis factor α. PLoS One 8, e70622 10.1371/journal.pone.0070622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin J. Y., and Fisher D. E. (2007) Melanocyte biology and skin pigmentation. Nature 445, 843–850 10.1038/nature05660 [DOI] [PubMed] [Google Scholar]
- 39. Gasque P., and Jaffar-Bandjee M. C. (2015) The immunology and inflammatory responses of human melanocytes in infectious diseases. J. Infect. 71, 413–421 10.1016/j.jinf.2015.06.006 [DOI] [PubMed] [Google Scholar]
- 40. Mattei S., Colombo M. P., Melani C., Silvani A., Parmiani G., and Herlyn M. (1994) Expression of cytokine/growth factors and their receptors in human melanoma and melanocytes. Int. J. Cancer 56, 853–857 10.1002/ijc.2910560617 [DOI] [PubMed] [Google Scholar]
- 41. Swope V. B., Sauder D. N., McKenzie R. C., Sramkoski R. M., Krug K. A., Babcock G. F., Nordlund J. J., and Abdel-Malek Z. A. (1994) Synthesis of interleukin-1 α and β by normal human melanocytes. J. Invest. Dermatol. 102, 749–753 10.1111/1523-1747.ep12376970 [DOI] [PubMed] [Google Scholar]
- 42. Medzhitov R., and Horng T. (2009) Transcriptional control of the inflammatory response. Nat. Rev. Immunol 9, 692–703 10.1038/nri2634 [DOI] [PubMed] [Google Scholar]
- 43. Jin F., Li Y., Ren B., and Natarajan R. (2011) PU.1 and C/EBPα synergistically program distinct response to NF-κB activation through establishing monocyte specific enhancers. Proc. Natl. Acad. Sci. U.S.A. 108, 5290–5295 10.1073/pnas.1017214108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Heintzman N. D., Stuart R. K., Hon G., Fu Y., Ching C. W., Hawkins R. D., Barrera L. O., Van Calcar S., Qu C., Ching K. A., Wang W., Weng Z., Green R. D., Crawford G. E., and Ren B. (2007) Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39, 311–318 10.1038/ng1966 [DOI] [PubMed] [Google Scholar]
- 45. Pham T. H., Minderjahn J., Schmidl C., Hoffmeister H., Schmidhofer S., Chen W., Längst G., Benner C., and Rehli M. (2013) Mechanisms of in vivo binding site selection of the hematopoietic master transcription factor PU.1. Nucleic Acids Res. 41, 6391–6402 10.1093/nar/gkt355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Janknecht R., and Hunter T. (1996) Transcription: a growing coactivator network. Nature 383, 22–23 10.1038/383022a0 [DOI] [PubMed] [Google Scholar]
- 47. Yamamoto H., Kihara-Negishi F., Yamada T., Hashimoto Y., and Oikawa T. (1999) Physical and functional interactions between the transcription factor PU.1 and the coactivator CBP. Oncogene 18, 1495–1501 10.1038/sj.onc.1202427 [DOI] [PubMed] [Google Scholar]
- 48. Zhang G., Zhou B., Li S., Yue J., Yang H., Wen Y., Zhan S., Wang W., Liao M., Zhang M., Zeng G., Feng C. G., Sassetti C. M., and Chen X. (2014) Allele-specific induction of IL-1β expression by C/EBPβ and PU.1 contributes to increased tuberculosis susceptibility. PLoS Pathog. 10, e1004426 10.1371/journal.ppat.1004426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gutiérrez-Aguirre I., Rački N., Dreo T., and Ravnikar M. (2015) Droplet digital PCR for absolute quantification of pathogens. Methods Mol. Biol. 1302, 331–347 10.1007/978-1-4939-2620-6_24 [DOI] [PubMed] [Google Scholar]
- 50. Bulger M., and Groudine M. (2011) Functional and mechanistic diversity of distal transcription enhancers. Cell 144, 327–339 10.1016/j.cell.2011.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hagège H., Klous P., Braem C., Splinter E., Dekker J., Cathala G., de Laat W., and Forné T. (2007) Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat. Protoc. 2, 1722–1733 10.1038/nprot.2007.243 [DOI] [PubMed] [Google Scholar]
- 52. Gupta P., Gurudutta G. U., Saluja D., and Tripathi R. P. (2009) PU.1 and partners: regulation of haematopoietic stem cell fate in normal and malignant haematopoiesis. J. Cell Mol. Med. 13, 4349–4363 10.1111/j.1582-4934.2009.00757.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Klemsz M. J., and Maki R. A. (1996) Activation of transcription by PU.1 requires both acidic and glutamine domains. Mol. Cell. Biol. 16, 390–397 10.1128/MCB.16.1.390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jin Q., Yu L. R., Wang L., Zhang Z., Kasper L. H., Lee J. E., Wang C., Brindle P. K., Dent S. Y., and Ge K. (2011) Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 30, 249–262 10.1038/emboj.2010.318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W. B., Fedorov O., Morse E. M., Keates T., Hickman T. T., Felletar I., Philpott M., Munro S., McKeown M. R., Wang Y., Christie A. L., et al. (2010) Selective inhibition of BET bromodomains. Nature 468, 1067–1073 10.1038/nature09504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Swygert S. G., and Peterson C. L. (2014) Chromatin dynamics: interplay between remodeling enzymes and histone modifications. Biochim. Biophys. Acta 1839, 728–736 10.1016/j.bbagrm.2014.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Belkina A. C., Nikolajczyk B. S., and Denis G. V. (2013) BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J. Immunol. 190, 3670–3678 10.4049/jimmunol.1202838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pulugulla S. H., Workman R., Rutter N. W., Yang Z., Adamik J., Lupish B., Macar D. A., El Abdouni S., Esposito E. X., Galson D. L., Camacho C. J., Madura J. D., and Auron P. E. (2018) A combined computational and experimental approach reveals the structure of a C/EBPβ-Spi1 interaction required for IL1B gene transcription. J. Biol. Chem. 293, 19942–19956 10.1074/jbc.RA118.005627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Godambe S. A., Chaplin D. D., Takova T., and Bellone C. J. (1994) Upstream NFIL-6-like site located within a DNase I hypersensitivity region mediates LPS-induced transcription of the murine interleukin-1 β gene. J. Immunol. 153, 143–152 [PubMed] [Google Scholar]
- 60. Godambe S. A., Chaplin D. D., Takova T., and Bellone C. J. (1994) An NFIL-6 sequence near the transcriptional initiation site is necessary for the lipopolysaccharide induction of murine interleukin-1 β. DNA Cell Biol. 13, 561–569 10.1089/dna.1994.13.561 [DOI] [PubMed] [Google Scholar]
- 61. Ørom U. A., and Shiekhattar R. (2013) Long noncoding RNAs usher in a new era in the biology of enhancers. Cell 154, 1190–1193 10.1016/j.cell.2013.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Smith E., and Shilatifard A. (2014) Enhancer biology and enhanceropathies. Nat. Struct. Mol. Biol. 21, 210–219 10.1038/nsmb.2784 [DOI] [PubMed] [Google Scholar]
- 63. Nishiyama C., Nishiyama M., Ito T., Masaki S., Masuoka N., Yamane H., Kitamura T., Ogawa H., and Okumura K. (2004) Functional analysis of PU.1 domains in monocyte-specific gene regulation. FEBS Lett. 561, 63–68 10.1016/S0014-5793(04)00116-4 [DOI] [PubMed] [Google Scholar]
- 64. Meng H., and Bartholomew B. (2018) Emerging roles of transcriptional enhancers in chromatin looping and promoter-proximal pausing of RNA polymerase II. J. Biol. Chem. 293, 13786–13794 10.1074/jbc.R117.813485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kodandapani R., Pio F., Ni C. Z., Piccialli G., Klemsz M., McKercher S., Maki R. A., and Ely K. R. (1996) A new pattern for helix-turn-helix recognition revealed by the PU.1 ETS-domain-DNA complex. Nature 380, 456–460 10.1038/380456a0 [DOI] [PubMed] [Google Scholar]
- 66. Listman J. A., Wara-aswapati N., Race J. E., Blystone L. W., Walker-Kopp N., Yang Z., and Auron P. E. (2005) Conserved ETS domain arginines mediate DNA binding, nuclear localization, and a novel mode of bZIP interaction. J. Biol. Chem. 280, 41421–41428 10.1074/jbc.M509143200 [DOI] [PubMed] [Google Scholar]
- 67. Pongubala J. M., and Atchison M. L. (1991) Functional characterization of the developmentally controlled immunoglobulin kappa 3′ enhancer: regulation by Id, a repressor of helix-loop-helix transcription factors. Mol. Cell. Biol. 11, 1040–1047 10.1128/MCB.11.2.1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pongubala J. M., and Atchison M. L. (1997) PU.1 can participate in an active enhancer complex without its transcriptional activation domain. Proc. Natl. Acad. Sci. U.S.A. 94, 127–132 10.1073/pnas.94.1.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ogata K., Sato K., and Tahirov T. (2003) Eukaryotic transcriptional regulatory complexes: cooperativity from near and afar. Curr. Opin Struct. Biol. 13, 40–48 10.1016/S0959-440X(03)00012-5 [DOI] [PubMed] [Google Scholar]
- 70. Kagey M. H., Newman J. J., Bilodeau S., Zhan Y., Orlando D. A., van Berkum N. L., Ebmeier C. C., Goossens J., Rahl P. B., Levine S. S., Taatjes D. J., Dekker J., and Young R. A. (2010) Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435 10.1038/nature09380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Splinter E., Heath H., Kooren J., Palstra R. J., Klous P., Grosveld F., Galjart N., and de Laat W. (2006) CTCF mediates long-range chromatin looping and local histone modification in the β-globin locus. Genes Dev. 20, 2349–2354 10.1101/gad.399506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Weintraub A. S., Li C. H., Zamudio A. V., Sigova A. A., Hannett N. M., Day D. S., Abraham B. J., Cohen M. A., Nabet B., Buckley D. L., Guo Y. E., Hnisz D., Jaenisch R., Bradner J. E., Gray N. S., and Young R. A. (2017) YY1 is a structural regulator of enhancer-promoter loops. Cell 171, 1573–1588.e28 10.1016/j.cell.2017.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gerritsen M. E., Williams A. J., Neish A. S., Moore S., Shi Y., and Collins T. (1997) CREB-binding protein/p300 are transcriptional coactivators of p65. Proc. Natl. Acad. Sci. U.S.A. 94, 2927–2932 10.1073/pnas.94.7.2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ghizzoni M., Haisma H. J., Maarsingh H., and Dekker F. J. (2011) Histone acetyltransferases are crucial regulators in NF-κB mediated inflammation. Drug Discov. Today 16, 504–511 10.1016/j.drudis.2011.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lai F., Orom U. A., Cesaroni M., Beringer M., Taatjes D. J., Blobel G. A., and Shiekhattar R. (2013) Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 494, 497–501 10.1038/nature11884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hsieh C. L., Fei T., Chen Y., Li T., Gao Y., Wang X., Sun T., Sweeney C. J., Lee G. S., Chen S., Balk S. P., Liu X. S., Brown M., and Kantoff P. W. (2014) Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc. Natl. Acad. Sci. U.S.A. 111, 7319–7324 10.1073/pnas.1324151111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hah N., Murakami S., Nagari A., Danko C. G., and Kraus W. L. (2013) Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 23, 1210–1223 10.1101/gr.152306.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bose D. A., Donahue G., Reinberg D., Shiekhattar R., Bonasio R., and Berger S. L. (2017) RNA binding to CBP stimulates histone acetylation and transcription. Cell 168, 135–149.e22 10.1016/j.cell.2016.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pierce J. W., Lenardo M., and Baltimore D. (1988) Oligonucleotide that binds nuclear factor NF-κB acts as a lymphoid-specific and inducible enhancer element. Proc. Natl. Acad. Sci. U.S.A. 85, 1482–1486 10.1073/pnas.85.5.1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Huang B., Yang X. D., Zhou M. M., Ozato K., and Chen L. F. (2009) Brd4 coactivates transcriptional activation of NF-κB via specific binding to acetylated RelA. Mol. Cell. Biol. 29, 1375–1387 10.1128/MCB.01365-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kastner P., and Chan S. (2008) PU.1: a crucial and versatile player in hematopoiesis and leukemia. Int. J. Biochem. Cell Biol. 40, 22–27 10.1016/j.biocel.2007.01.026 [DOI] [PubMed] [Google Scholar]
- 82. Ha S. D., Solomon O., Akbari M., Sener A., and Kim S. O. (2018) Histone deacetylase 8 protects human proximal tubular epithelial cells from hypoxia-mimetic cobalt- and hypoxia/reoxygenation-induced mitochondrial fission and cytotoxicity. Sci. Rep. 8, 11332 10.1038/s41598-018-29463-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ha S. D., Reid C., Meshkibaf S., and Kim S. O. (2016) Inhibition of IL-1β expression by anthrax lethal toxin is reversed by HDAC8 inhibition in murine macrophages. J. Biol. Chem. 291, 8745–8755 10.1074/jbc.M115.695809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ha S. D., Han C. Y., Reid C., and Kim S. O. (2014) HDAC8-mediated epigenetic reprogramming plays a key role in resistance to anthrax lethal toxin-induced pyroptosis in macrophages. J. Immunol. 193, 1333–1343 10.4049/jimmunol.1400420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Dorschner M. O., Hawrylycz M., Humbert R., Wallace J. C., Shafer A., Kawamoto J., Mack J., Hall R., Goldy J., Sabo P. J., Kohli A., Li Q., McArthur M., and Stamatoyannopoulos J. A. (2004) High-throughput localization of functional elements by quantitative chromatin profiling. Nat. Methods 1, 219–225 10.1038/nmeth721 [DOI] [PubMed] [Google Scholar]
- 87. Follows G. A., Janes M. E., Vallier L., Green A. R., and Gottgens B. (2007) Real-time PCR mapping of DNaseI-hypersensitive sites using a novel ligation-mediated amplification technique. Nucleic Acids Res. 35, e56 10.1093/nar/gkm108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Stadhouders R., Kolovos P., Brouwer R., Zuin J., van den Heuvel A., Kockx C., Palstra R. J., Wendt K. S., Grosveld F., van Ijcken W., and Soler E. (2013) Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nat. Protoc. 8, 509–524 10.1038/nprot.2013.018 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.