

Abstract
Methylation of the small ribosome subunit rRNA in the ribosomal decoding center results in exceptionally high-level aminoglycoside resistance in bacteria. Enzymes that methylate 16S rRNA on N7 of nucleotide G1405 (m7G1405) have been identified in both aminoglycoside-producing and clinically drug-resistant pathogenic bacteria. Using a fluorescence polarization 30S-binding assay and a new crystal structure of the methyltransferase RmtC at 3.14 Å resolution, here we report a structure-guided functional study of 30S substrate recognition by the aminoglycoside resistance-associated 16S rRNA (m7G1405) methyltransferases. We found that the binding site for these enzymes in the 30S subunit directly overlaps with that of a second family of aminoglycoside resistance-associated 16S rRNA (m1A1408) methyltransferases, suggesting that both groups of enzymes may exploit the same conserved rRNA tertiary surface for docking to the 30S. Within RmtC, we defined an N-terminal domain surface, comprising basic residues from both the N1 and N2 subdomains, that directly contributes to 30S-binding affinity. In contrast, additional residues lining a contiguous adjacent surface on the C-terminal domain were critical for 16S rRNA modification but did not directly contribute to the binding affinity. The results from our experiments define the critical features of m7G1405 methyltransferase–substrate recognition and distinguish at least two distinct, functionally critical contributions of the tested enzyme residues: 30S-binding affinity and stabilizing a binding-induced 16S rRNA conformation necessary for G1405 modification. Our study sets the scene for future high-resolution structural studies of the 30S-methyltransferase complex and for potential exploitation of unique aspects of substrate recognition in future therapeutic strategies.
Keywords: antibiotic resistance, ribosomal ribonucleic acid (rRNA) (ribosomal RNA), ribosome, RNA methylation, RNA methyltransferase, aminoglycoside antibiotics, m1A1408, m7G1405, RmtC
Introduction
Methylation of 16S rRNA has been identified as a prominent mechanism of self-protection in aminoglycoside-producing bacteria and is emerging as a new threat to the clinical efficacy of aminoglycoside antibiotics (1, 2). Both the intrinsic methyltransferases of drug producers and acquired enzymes of human and animal pathogens chemically modify the aminoglycoside-binding site in the decoding center of the bacterial 30S subunit to block drug binding and confer exceptionally high-level resistance. Regarding the acquired enzymes specifically, of most concern is that these resistance determinants have been identified on various mobile genetic elements, often in conjunction with other resistance enzymes (2–4). As such, the aminoglycoside-resistance methyltransferases can make the bacteria expressing them pan-resistant to entire subclasses of aminoglycosides (2, 5), including even the most recent generation drugs like plazomicin (6, 7). More broadly, given the extensive modification of bacterial rRNAs, especially in functionally critical regions like the decoding center, understanding rRNA methyltransferase–ribosome subunit interactions has relevance to both fundamental bacterial physiology and mechanisms of antimicrobial resistance.
The aminoglycoside-resistance 16S rRNA methyltransferases are functionally divided into two subfamilies that modify the ribosome at either the N7 position of 16S rRNA nucleotide G1405 (m7G1405) or the N1 position of A1408 (m1A1408). Although enzymes from both subfamilies are found in aminoglycoside-producing bacteria, the m7G1405 methyltransferases (Fig. 1A) are far more clinically prevalent than their m1A1408 methyltransferase counterparts (2, 8). In contrast to the single m1A1408 methyltransferase NpmA that was clinically isolated from Escherichia coli strain ARS3 in Japan (9), the m7G1405 methyltransferases are globally disseminated and have been found in many different human pathogens (2).
Figure 1.

The m7G1405 methyltransferase family and binding site on 30S subunit. A, phylogenetic tree of m7G1405 methyltransferase enzymes including acquired genes in gammaproteobacterial and Gram-negative pathogenic bacteria, aminoglycoside producing bacteria, and uncharacterized homologs belonging to the chloroflexi. Pathogen-associated genes (color-coded regions) are further divided into three subclades represented in this work by RmtA/RmtB, RmtD/RmtD2, and RmtC. B, structure of the bacterial 30S subunit bound to NpmA (purple) (PDB code 4OX9) showing the proximity of nucleotides G1405 (red) and A1408 (orange) at the top of h44 (yellow) in the ribosome decoding center (dc). Other 30S features indicated are the head (h), body (b), platform (p), and spur (s). C, schematic of the competition FP assay for 30S-methyltransferase binding using the NpmA* probe (purple) and application to RmtC binding (red). At low competitor concentration (left of plot), high FP signal arises because of NpmA* interaction with 30S; displacement of the probe by RmtC results in lower FP signal from the free probe (right of plot). D and E, competition FP binding experiments with NpmA* and five different unlabeled WT Rmt enzymes. The RmtC curve in D (red dotted line) is the same as in C and is shown for comparison. Binding affinities (Ki) and associated 95% confidence interval were obtained from fits to the data shown in C and D. Error bars represent S.D. of the measurements.
Both free and 30S-bound m1A1408 methyltransferases, including NpmA, have been extensively characterized, revealing the molecular basis of their specific substrate recognition and modification mechanisms (10–15). These enzymes exploit a conserved 16S rRNA tertiary surface adjacent to helix 44 (h44)2 to dock on the 30S, explaining the requirement for intact 30S as their substrate. Two extended regions that connect the fifth/sixth and sixth/seventh β-strands of the methyltransferase core fold (β5/6 and β6/7 linkers, respectively) position key residues for recognition and stabilization of A1408 in a flipped conformation for methylation (10, 13).
Structures of the m7G1405 methyltransferases RmtB (16), which has been identified in multiple Gram-negative pathogens, and Sgm (17) from the producer of sisomicin derivative G52, Micromonospora zionensis, have revealed a distinct methyltransferase architecture. Specifically, these enzymes possess a significantly larger N-terminal extension but no extended sequences within the methyltransferase core fold comparable with those in the m1A1408 methyltransferases. A likely role for the unique N-terminal domain in 30S interaction by the m7G1405 methyltransferases has been suggested, and some functionally critical residues within this domain have been previously identified (16–19). However, to date, no direct binding analysis to allow dissection of important residues in binding or stabilization of a catalytically competent state of the enzyme–substrate complex has been performed. As such, there is a critical gap in our understanding of m7G1405 methyltransferases 30S substrate recognition, despite the potential threat these enzymes pose for clinical aminoglycoside resistance.
Here, we have extended the use of a 30S-binding assay previously developed in our lab for studies of NpmA (13) to the m7G1405 methyltransferases. From these direct 30S binding measurements and a structure-guided mutagenesis strategy based on a new structure of a m7G1405 methyltransferase family member (RmtC), we develop a new model for 30S substrate recognition by the m7G1405 methyltransferases. We identify a molecular surface in the N-terminal domain that is critical for 30S docking, whereas numerous residues on an adjacent surface of the C-terminal domain (CTD) do not contribute to binding affinity but likely control critical conformational changes necessary for catalysis of rRNA modification.
Results
m7G1405 methyltransferases bind 30S with similar affinity and at a site overlapping that of the m1A1408 methyltransferases
We previously developed a competition fluorescence polarization (FP) assay to measure the binding affinity of WT and variant NpmA proteins to define this methyltransferase's mechanism of 30S substrate recognition and m1A1408 modification (13). We speculated that the close proximity of nucleotides A1408 and G1405 in h44 (Fig. 1B) might also make this assay applicable to direct quantification of m7G1405 methyltransferase–30S interactions. In this assay, a fluorescein-labeled, single-Cys variant (E184C) of NpmA (NpmA*) is prebound to 30S (high FP state) and a range of concentrations of unlabeled competitor protein is added to displace the NpmA* probe (shown schematically in Fig. 1C), allowing determination of the methyltransferase 30S-binding affinity (Ki). We first applied the assay to analysis of 30S–RmtC interaction and observed a RmtC concentration–dependent decrease in FP. The resulting data were fit to obtain a Ki of 89.5 nm (Fig. 1, C and E). This value is comparable with the 60 nm affinity previously measured for the m1A1408 methyltransferase NpmA (13). Binding measurements were also performed with RmtA, RmtB, RmtD, and RmtD2, which together with RmtC represent each of the three subclades in the m7G1405 methyltransferase phylogenetic tree (Fig. 1A). All binding affinities for these methyltransferases were comparable within a ∼2.5-fold range from 48 to 118 nm (Fig. 1, D and E).
These results confirm our established assay using the NpmA* probe as suitable for direct binding measurements of m7G1405 methyltransferases to 30S and thus as a tool to provide a deeper analysis of their substrate recognition mechanism. These data also reveal that the binding site of the m7G1405 methyltransferases on the 30S subunit does indeed overlap with that of the m1A1408 methyltransferases, suggesting they may also exploit the same conserved rRNA tertiary surface for specific substrate recognition. We chose to use RmtC for further structural and functional studies of 30S–m7G1405 methyltransferase interaction for several reasons. Most importantly, there has been no such analysis of RmtC to date, and this enzyme is both in the same subclade as ArmA and most distant from RmtB (Fig. 1A), two commonly observed pathogen-acquired m7G1405 methyltransferases. The selection of RmtC thus offers the opportunity to identify conserved features of the 30S recognition mechanism across all m7G1405 methyltransferases.
Structure of the RmtC–SAH complex
The X-ray crystal structure of RmtC bound to S-adenosylhomocysteine (SAH), the methylation reaction by-product, was determined and refined at 3.14 Å resolution (Table 1). RmtC adopts a fold consistent with those of other m7G1405 methyltransferases RmtB and Sgm (16, 17), as expected. Specifically, RmtC possesses a large N-terminal domain (NTD) appended to its CTD methyltransferase fold (Fig. 2, A and B). The NTD is structurally divided into two subdomains, N1 and N2, each comprised of three α-helices. N1 forms a globular three-helical bundle, whereas the three helices of N2 are extended across the N-terminal half of the CTD (Fig. 2A). The CTD adopts a canonical class I methyltransferase fold with a seven-stranded β-sheet core containing a central topological switch point that forms the SAM-binding pocket (Fig. 2B).
Table 1.
X-ray data collection and structure refinement statistics
a.s.u., asymmetric unit.
| RmtC + SAH | |
|---|---|
| PDB code | 6PQB |
| Space group | P61 |
| Resolution (Å) | 40.9–3.14 (3.25–3.14)a |
| Cell dimensions | |
| a, b, c (Å) | 163.5, 163.5, 122.5 |
| α, β, γ (°) | 90, 90, 120 |
| Molecules a.s.u. | 4 |
| Wavelength, Å | 0.987 |
| Rpim | 0.047 (0.867) |
| CC½ | 0.998 (0.419) |
| I/σI | 10.81 (1.34) |
| Completeness (%) | 100 (100) |
| Redundancy | 8.7 (8.2) |
| Total reflections | 283,111 |
| Unique reflections | 32,566 |
| Rwork/Rfreeb | 20.0/23.3 |
| Atoms | |
| Protein | 8538 |
| SAH | 104 |
| Water | 9 |
| Average B-factor | 134 |
| Ramachandran (%) | |
| Favored/allowed | 99.8 |
| Disallowed | 0.2 |
| RMSD | |
| Bond lengths (Å) | 0.002 |
| Bond angles (°) | 0.474 |
a The values in parentheses are for the highest resolution shell.
b Rwork = Σhkl|Fo(hkl) − Fc(hkl)|/Σhkl|Fo(hkl), where Fo and Fc are the observed and calculated structure factors, respectively. Rfree applies to the 5% of reflections chosen at random to constitute the test set.
Figure 2.

Structure of the RmtC–SAH complex. A, crystal structure of the RmtC–SAH complex highlighting (red) the extended N-terminal domain characteristic of the aminoglycoside-resistance 16S rRNA (m7G1405) methyltransferases. The N-terminal domain is divided into two subdomains, N1 and N2. The locations of the bound SAH (yellow sticks) and a partially disordered loop (Loop237–246) adjacent to the opening to the SAM-binding pocket are also indicated. B, the same view of the RmtC structure as A (right) but highlighting the seven β-strand core (red) of the C-terminal methyltransferase fold (with N1 and N2 shown as semitransparent cartoon). C, two orthogonal detailed views of the interactions made with SAH in the SAM-binding pocket. D, alignment of RmtC (red) with the structures of RmtB (PDB code 3FRH; orange) and Sgm (PDB code 3LCV; blue), shown in two orthogonal views (top), reveals potential flexibility in the position of the N1 domain relative to the N2/CTD domains via a hinge region between N1 and N2 (zoomed view).
In the RmtC–SAH complex, the SAH is bound in a pocket lined by numerous conserved residues, with many hydrogen bonding and hydrophobic interactions. Two highly conserved residues, Arg111 and Asp160, anchor the SAH carboxylate group and ribose hydroxyl groups, respectively (Fig. 2C), whereas Asp188 and Gln212 position the base via hydrogen bonds to the adenine amino group and ring N7. The SAH ribose and adenine moieties are also surrounded by a collection of hydrophobic side chains on each side that define the shape of the binding pocket. Overall, the interactions made by RmtC with SAH in the SAM-binding pocket are consistent with previous structures of RmtB and Sgm bound to cosubstrate (16, 17). During this work, a structure of apo RmtC (PDB code 6CN0) was also deposited by the Center for Structural Genomics of Infectious Diseases. Comparison of the RmtC–SAH complex with this structure reveals the residue positions within the SAM-binding pocket to be mostly maintained. However, some potential conformational flexibility is apparent in residues Tyr60 and Ser107. These residues line the opening to the SAM-binding pocket and may assist in positioning G1405 close to the SAM methyl group for modification (17).
Structural alignment of our RmtC structure with those of RmtB and Sgm confirms that these proteins are structurally similar overall (average RMSDs of 2.62 and 3.05 Å, respectively). However, a substantial difference in the orientation of the N1 subdomain relative to the remainder of the protein is apparent in alignments made using only the CTD of each structure (Fig. 2D), reducing the average RMSDs to 1.59 and 1.50 Å, respectively. Additionally, at least two residues in all four copies of RmtC in the crystal are disordered in the sequence that links N1 and N2 (between positions 62 and 64). Together, these observations suggest that the potential for flexibility in N1 subdomain position relative to the remainder of the protein and the sequence between N1 and N2 may act as a hinge that allows movement of this subdomain (Fig. 2D). Given the essential role of the N1 domain in substrate binding (see below), such mobility between the N1 and N2 domains may be an important aspect of specific 30S substrate recognition.
Identification of potential 16S rRNA-binding residues in RmtC
Previous studies of Sgm, ArmA, and RmtB identified the importance of the m7G1405 methyltransferase NTD in substrate recognition and have also suggested a specific role in 30S binding for some residues within both protein domains (16–19). The likely importance of conserved positive surface charges in the NTD are further supported by our structure of RmtC in which residues of the N1 and N2 subdomains form an extended, contiguous positively charged surface that could interact with 16S rRNA (Fig. 3A). Previous structure-guided mutagenesis of RmtB coupled with tobramycin minimum inhibitory concentration (MIC) assays identified several residues potentially important for 30S binding (16), including highly conserved residues within a structurally disordered loop (corresponding to RmtC residues 237–246; Loop237–246). In the SAH-bound structure of RmtC, like the previously determined structures of RmtB and Sgm (16, 17), there is weak or no density visible for most Loop237–246 residues, including the highly conserved Lys236 and Arg241. The functional importance of these and other conserved residues in the absence of an obvious role in Rmt protein structure or SAM binding is suggestive of an important contribution to 30S substrate recognition. However, to date, no measurements of 30S binding have been made for any m7G1405 methyltransferase to directly test the roles of these important residues.
Figure 3.
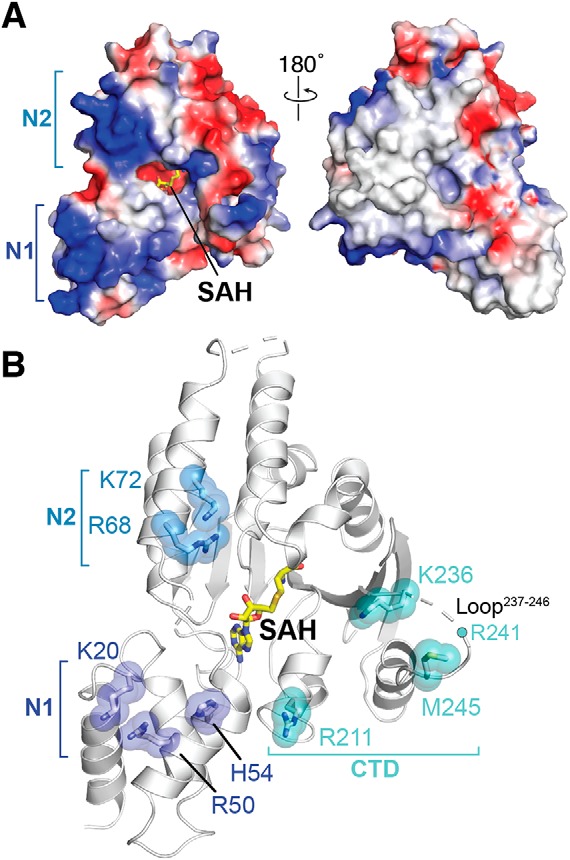
Selection of mutants defining the 30S interaction surface. A, the electrostatic surface potential of the RmtC structure reveals the N1 and N2 domains to be rich in positively charged residues (blue). B, locations of positively charged residues in the N1 and N2 domain and other conserved or putative functionally critical residues for 30S interaction. All residues shown as sticks with semitransparent spheres, as well as Arg241 (located in the partially disordered Loop237–246), were substituted to test their role in 30S recognition (see main text and Tables 2 and 3 for details).
To gain deeper insight into 30S recognition by RmtC and other m7G1405 methyltransferases, we therefore selected nine individual residues for site-directed mutagenesis, based on insights from both our RmtC structure and the previous studies of other enzymes (Table 2 and Fig. 3B). Four basic residues in the N1 and N2 domains (Lys20, Arg50, Arg68, and Lys72) were substituted with Glu to assess the contribution of the positive surface they collectively form (Fig. 3A). Lys236 in the CTD is conserved in all intrinsic and acquired enzymes, whereas the remaining residues tested, His54 in the N1 domain and Arg211, Arg241, and Met245, were previously identified in RmtB (16). Finally, because Arg241 and Met245 were previously tested only as part of a variant in which the Loop237–246 was replaced by four Ala residues (16), we prepared each individual residue substitution as well as the equivalent loop alteration. All variant RmtC proteins were expressed and could be purified as for WT RmtC. As a further quality control to ensure that residue substitutions did not substantially impact protein folding and stability, the unfolding inflection temperature (Ti) was determined for all purified proteins (Fig. S1 and Table S1). Almost all Ti values for both apparent unfolding transitions were <2.5 °C different from WT RmtC, indicative of retained structural integrity. The only exception was for the RmtC-K20E/R50E double variant, which exhibited slightly larger ΔTi values (4.0 and 4.5 °C).
Table 2.
Conservation of putative residues for 30S binding
NC, not conserved within the larger groups of sequences indicated.
| Residue (in RmtC) | Conservation (residue) |
Tested in | References | ||
|---|---|---|---|---|---|
| All m7G1405a | Intrinsic | Acquired | |||
| % | |||||
| Lys20 | 88 (Lys/Arg) | 100 (Arg) | 91 (Lys) | RmtC | This work |
| Arg50 | 100 (Arg/Lys) | 100 (Lys) | 100 (Arg/Lys) | Sgm | Ref. 19 |
| His54 | 96 (His) | 100 (His) | 100 (His) | RmtB | Ref. 16 |
| Arg68b | NC | NC | NC | RmtC | This work |
| Lys72b | NC | NC | NC | RmtC | This work |
| Arg211 | 40 (Arg)/40 (Gln) | NC | 73 (Arg)/18 (Gln) | RmtB | Ref. 16 |
| Lys236 | 72 (Arg/Lys) | 100 (Lys) | 100 (Arg/Lys) | RmtC | This work |
| Arg241 | 96 (Arg/Lys) | 100 (Arg) | 100 (Arg/Lys) | Sgm, RmtB | Refs. 16, 18, and 19 |
| Met245 | 96 (Met) | 100 (Met) | 100 (Met) | RmtB | Ref. 16c |
a Including sequences from aminoglycoside-producing bacteria (Intrinsic), pathogen-acquired enzymes (Acquired) and uncharacterized homologs in the chloroflexi.
b Residues are conserved only in within the RmtC subclade (seven of eight sequences).
c Residues previously only tested indirectly as part of a loop deletion variant in RmtB (residues 237–246).
As described in the following sections, each RmtC variant was assessed for 30S binding using the established FP assay and resistance (MIC) against kanamycin and gentamicin in bacteria expressing the enzymes (Table 3). Consistent expression of each RmtC variant was assessed under the culture conditions used for MIC measurements by immunoblotting using an anti-His6 antibody (Fig. S2). Thus, differences in resistance conferred and 30S binding affinity can be directly used to ascertain the role of each substituted residue in RmtC activity.
Table 3.
RmtC variant activity
NB, no binding.
| RmtC | Antibiotic MIC |
30S binding, Kia | |
|---|---|---|---|
| Kanamycin | Gentamicin | ||
| μg/ml | nm | ||
| Wildtype | >1024 | 1024 | 89.5 [72, 112] |
| K20E | <2 | <2 | NB |
| R50E | <2 | <2 | 977 [651, 1497] |
| K20E/R50E | <2 | <2 | NB |
| H54A | <2 | <2 | 75 [28, 203] |
| H54E | <2 | <2 | 90 [47, 169] |
| R68E | 256–512 | <2 | 1163 [545, 2969] |
| K72E | 256–1024 | 64–256 | 469 [225, 1005] |
| R68E/K72E | <2 | < 2 | NB |
| R211A | 1024 | 128 | 75 [28, 196] |
| R211E | 4 | < 2 | 62 [21, 188] |
| K236A | >1024 | 256–512 | 85 [47, 156] |
| K236E | 8 | < 2 | 76 [40, 146] |
| R241A | 1024 | 128 | 104 [79, 137] |
| R241E | 8 | <2 | 99 [39, 252] |
| M245A | <2 | <2 | 55 [31, 99] |
| Loop237–246-A4 | <2 | <2 | 63 [35, 114] |
a The values in parentheses are 95% confidence interval for the fit Ki after averaging replicate measurements in each experiment (see Table S2 for further details of Ki determination).
Residues in N1 and N2 primarily contribute to RmtC-30S binding affinity
Single substitutions with Glu of each basic residue in either the N1 (K20E and R50E) or N2 (R68E or K72E) domain reduces 30S binding affinity of the protein in FP assays (Fig. 4, A and B, and Table 3). The extent of the reduction in binding affinities range from ∼5-fold for K72E to ∼11–13-fold for R50E and R68E, whereas no binding was measurable for K20E. Consistent with these observations, double substitutions of each pair of residues in N1 or N2 also resulted in affinities below the detectable limit in the assay (Fig. 4, A and B, and Table 3), revealing the collective contributions of the two N2 residues (Arg68 and Lys72) to binding in addition to the N1 domain (Lys20 and Arg50).
Figure 4.

Functionally critical residues in the NTD contribute primarily to 30S binding affinity. A and B, competition FP binding experiments with unlabeled RmtC proteins with single or double substitutions of basic (Arg/Lys) residues with Glu in the N1 subdomain (K20E and R50E, A) or N2 subdomain (R68E and K72E, B). C, competition FP binding experiment with RmtC-H54E. In all panels, the WT RmtC fit shown for comparison (dotted black line) is the same as that shown in Fig. 1 (C and D). Error bars represent the S.E. Binding affinity (Ki) for each variant protein derived from these data are shown in Table 3.
The RmtC proteins with N2 substitutions R68E, K72E, or R68E/K72E were next tested for their ability to confer resistance to kanamycin or gentamicin. Intermediate MICs were determined for the single substitutions, indicating a partial loss of conferred resistance, whereas resistance was completely abolished in the double variant (Table 3). The activities of these RmtC variants in bacteria thus correlated well with the measured in vitro binding affinities. The effects of substitutions in the N1 domain were also largely consistent in their impact on binding and activity (MIC), although it is noteworthy that the R50E substitution completely restored susceptibility to both antibiotics despite only partially reducing the enzyme's 30S affinity (Table 3). This distinction may reflect a more complex role for Arg50 involving both a contribution to 30S binding affinity and a functionally critical conformational change in enzyme or substrate. For example, Arg50 might promote or stabilize a movement of the N1 subdomain relative to the CTD, as suggested by structural comparisons between RmtC and other enzymes (as noted above).
Finally, among the N1 subdomain variants, substitutions at His54 (to either Ala or Glu) produce the most striking results. For both variants, the enzyme is completely inactive, with MICs for both antibiotics at the same level as in the absence of enzyme, and yet neither substitution impacts 30S binding affinity (Fig. 4C and Table 3). Thus, although clearly critical for RmtC activity, His54 does not directly contribute to 30S binding but instead must play a distinct, critical role within the substrate recognition mechanism. This observation, along with the impacts of K20E and R50E, also further points to the primary importance of the N1 subdomain in specific 30S recognition.
Conserved CTD residues surrounding the SAM-binding pocket are functionally critical but do not contribute to 30S binding affinity
The RmtC CTD contains several residues and a structurally disordered loop region (Loop237–246) that are potentially critical for 30S binding. These residues line the protein surface adjacent to His54 of the N1 domain and surrounding the opening to the SAM-binding pocket (Table 2 and Fig. 3B). Consistent with prior analyses of RmtB (16), replacement of the RmtC loop with four Ala residues (Loop237–246 → A4) ablated the enzyme's ability to confer resistance to kanamycin and gentamicin, with the same result also observed for the single substitution M245A within the loop (Table 3). Single substitutions to either Ala to Glu were also made for three basic residues: one within Loop237–246 (Arg241), one immediately preceding the loop (Lys236), and a third more distant in primary sequence but on the adjacent protein surface (Arg211). Each substitution had the same impact on protein activity in all three cases. Substitution with Ala resulted in a partial reduction in resistance conferred by the RmtC variant to kanamycin and/or gentamicin (intermediate MICs), whereas substitution with Glu fully ablated resistance for all three variant enzymes (Table 3).
These results confirm the functional importance of the four tested residues, which line a continuous surface with His54 and the other critical residues of the N1 domain (Fig. 3B). The relative effects of Ala and Glu substitutions for each of the three basic residues, Arg211, Lys236, and Arg241, further suggest direct contact with the negative phosphate backbone of 16S rRNA given the greater defect with the charge reversal. Remarkably, however, none of the substitutions nor the loop swap (Loop237–246 → A4) resulted in a measurable change in 30S binding affinity (Fig. 5 and Table 3). Thus, like the N1 residue His54, these residues do not directly contribute to 30S binding affinity and instead must play a distinct but critical role in substrate recognition, such as promoting or stabilizing a conformationally altered state of the enzyme and/or substrate necessary for catalysis of m7G1405 modification.
Figure 5.
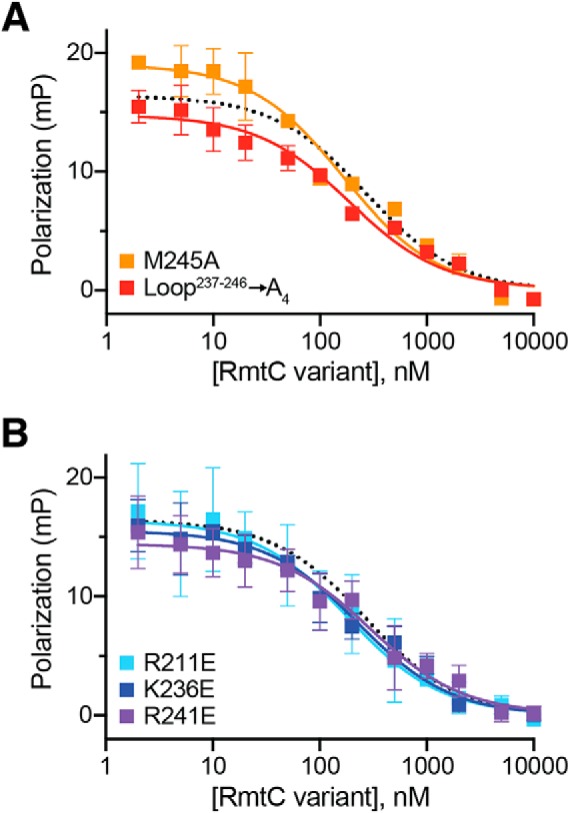
Functionally critical CTD residues do not contribute to 30S binding affinity. Competition FP binding experiments with unlabeled RmtC CTD variant proteins. A, analysis of RmtC with Loop237–246 → A4 (red) or M245A single substitution with the loop. B, analysis of RmtC proteins with single substitutions of basic (Arg/Lys) residues with Glu within the CTD. In both panels, the WT RmtC fit shown for comparison (dotted black line) is the same as that shown in Fig. 1 (C and D). Error bars represent the S.E. Binding affinity (Ki) values for all variant proteins derived from these data are shown in Table 3.
To gain direct insight into whether RmtC and other enzymes of this family disrupt the 30S structure upon binding, we screened a number of 30S–Rmt complexes for their suitability for single-particle cryo-EM analysis. Although strong preferred particle orientation currently precludes high-resolution 3D reconstruction, 2D class averages generated from images of a 30S–RmtG complex stabilized using the SAM analog sinefungin clearly show disordering of the subunit head domain in the presence of the methyltransferase (Fig. 6A). Thus, consistent with our interpretation of the biochemical analysis described above, m7G405 methyltransferase binding near the top of h44 causes significant disruption of the surrounding 16S rRNA structure, presumably allowing access to the relatively buried G1405 nucleotide for modification (Fig. 6B).
Figure 6.
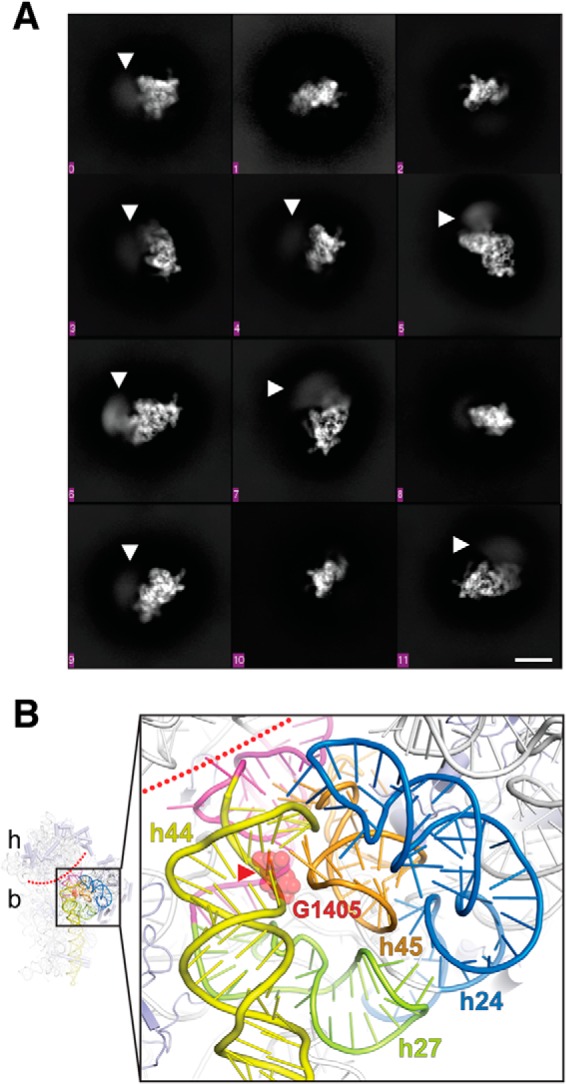
Cryo-EM analysis of a 30S–Rmt complex. A, 2D class averages showing different orientations of the 30S subunit. Disorder (blurring) of the 30S head is readily apparent in multiple averages (white arrowheads). The scale bar (white line) shown at the bottom right is 10 nm and applies to all class averages. B, zoomed view of the proposed 30S methyltransferase-binding site architecture (generated using E. coli 30S; PDB code 4V4Q). The conserved 16S rRNA tertiary surface formed by helices h24 (blue), h27 (green), and h45 (orange) is adjacent to h44 (yellow) containing the G1405 target nucleotide (red spheres). The buried location of the modified N7 atom is marked with a red arrowhead, and the approximate boundary between the 30S head (h) and body (b) is marked with a dotted red line. Nucleotides following h27 and preceding h44, which base pair at the head–body boundary, are colored in magenta.
Discussion
The bacterial ribosome is a major target for antibiotics such as aminoglycosides, which typically interfere with the fidelity of mRNA decoding (20, 21). Although side effects have limited aminoglycoside use to treatment of serious infections, increasing resistance to other widely used antibiotics has led to a reevaluation of their use in clinical practice (21–23). Additionally, progress in mitochondrial ribosome structural biology (24, 25) and semisynthesis of novel aminoglycosides (26, 27) can support future efforts to design new aminoglycosides with fewer side effects. As such, this important class of antimicrobials has the potential to be exceptionally useful in the treatment of serious hospital-based infections, especially those caused by Gram-negative pathogens. Unfortunately, however, the clinical emergence over the last decade of aminoglycoside-resistance 16S rRNA (m7G1405) methyltransferases (ArmA or RmtA-H) (2, 5) poses a new threat to the efficacy of both current and new aminoglycosides, such as plazomicin (6, 7). Detailed studies, such as those described here, of the resistance methyltransferases that incorporate these rRNA modification are thus needed to support development of strategies to counter the effects of these resistance determinants.
Previous studies of m7G1405 methyltransferases of pathogenic (RmtB) or aminoglycoside-producer (Sgm) bacterial origin have begun to reveal some details of 30S substrate recognition by this enzyme family (16–19). However, prior studies have typically relied on enzyme activity (e.g. MIC) measurements to indirectly infer the importance of specific residues in 30S binding. Without direct analysis of specific contributions of key residues to 30S binding affinity or other distinct roles in the process of specific substrate recognition, our understanding of the mechanism of 30S recognition and modification by the m7G1405 methyltransferases remained incomplete. We therefore adapted a previously developed FP assay (13) and used it here to more fully define substrate recognition by the m7G1405 methyltransferase enzymes.
The applicability of our FP assay using a probe based on the m1A1408 methyltransferase NpmA to the analysis of m7G1405 methyltransferase–30S interaction clearly demonstrates that the 30S binding site of these two groups of enzymes must substantially overlap. Both the m1A1408 and m7G1405 methyltransferases require the intact 30S subunit as their minimal substrate and the molecular basis for this requirement was revealed for the former enzyme subfamily by the structure of the 30S–NpmA complex. NpmA interacts exclusively with 16S rRNA and docks onto a conserved rRNA tertiary surface comprising helices 24, 27, and 45, adjacent to the h44 target site (10). This surface is bound by a group of positively charged residues, Lys66, Lys67, Lys70, and Lys71, that line a single helical region on the β2/β3 linker of the core methyltransferase fold (13). Our results with RmtC suggest that the m7G1405 methyltransferases likely exploit the same conserved rRNA tertiary surface for specific substrate recognition and that this is accomplished via interactions made by residues of the N1 and N2 domains. Specifically, a group of basic residues, Lys20, Arg50, Arg68, and Lys72, form a single positively charged surface, and each contributes directly to 30S binding affinity. Lys20 and Arg50 in the N1 subdomain are highly conserved in all m7G1405 methyltransferases, further underscoring their importance in 30S binding. In contrast, Arg68 and Lys72 in the first α-helix of the N2 domain are conserved only within the subclade comprising RmtC enzymes. However, in other m7G1405 methyltransferases, alternative basic residues positioned on the same surface of the protein may provide equivalent interactions with 16S rRNA, such as Lys76/Lys85 of the second α-helix of the RmtB N2 domain or Arg97/Arg106 of the second and third α-helices of the Sgm N2 domain. Thus, whereas some specific details may vary among different representatives of the m7G1405 methyltransferase subfamily, the extended positive surface created by residues of the N1/N2 domain is likely a critical first step in enzyme–substrate interaction.
Our results also reveal that multiple residues on the adjacent protein surface that surrounds the SAM-binding pocket, including His54 of the N1 subdomain and several others in the CTD, do not contribute to 30S binding affinity despite being critical for RmtC activity. These residues play no obvious direct role in RmtC protein structure and do not interact with SAM; in fact, despite their functional importance, Lys236, Arg241, and Met245 are in or adjacent to Loop237–246, which is disordered in the free protein. These observations and our findings that alteration of these residues abrogates activity but has no effect on 30S binding affinity suggest that they must play a distinct but essential role in substrate recognition. In NpmA, a single residue, Arg207, exhibits similar properties. Despite making no contribution to 30S binding affinity, Arg207 is nonetheless critical because it directly stabilizes the rRNA backbone of the flipped A1408 nucleotide. Our results suggest similar roles for these conserved residues in RmtC. The three basic residues, Arg211, Lys236, and Arg241, likely contact the 16S rRNA backbone to stabilize a binding-induced change in its structure. These residues and His54 and Met245 may also directly contact the G1405 nucleotide to position it for catalysis of methyltransfer.
Why multiple residues are required in this enzyme family compared with the single residue used by NpmA is unclear. However, our initial evidence from 2D cryo-EM class averages suggests that distortion of the 16S rRNA is large enough to cause disorder of the 30S head and body. It is noteworthy that G1405 is much less directly accessible at the top of h44 than A1408 and may thus require greater distortion of h44 and the surrounding 16S rRNA structure to create a conformation compatible with G1405 methylation by the enzyme. Reducing head–body interactions as we observe would likely allow opening of h44 near the target site and make G1405 accessible for modification, most likely via “base flipping” into the enzyme active site. We also note that nucleotides following h27, which is part of the conserved tertiary 16S rRNA surface recognized by these enzymes, are buried behind h44 near G1405 and extend to the 30S head–body boundary where they interact with residues that precede h44 (Fig. 6B). Thus, a plausible mechanism is that binding to this region could relay distortion of the 16S rRNA to h44 and the 30S head–body interface. The need for this major reorganization of 16S rRNA for G1405 methylation also explains why previous attempts to dock Sgm on the 30S subunit resulted in no models with the target base within 15 Å of the SAM methyl group (17).
In summary, our model for m7G1405 methyltransferase action on the 30S parallels that previously developed for the m1A1408 methyltransferase NpmA: initial binding to 30S is mediated by multiple residues of the N1 and N2 subdomains (analogous to the NpmA β2/β3 linker), and an extended surface adjacent to this docking point is necessary to promote and/or stabilize a novel, binding-induced 16S rRNA conformation. In particular, we speculate that one or more of the functionally critical residues on this surface is likely essential for stabilizing G1405 in a flipped conformation for methylation, as commonly observed for other RNA-modifying enzymes (10, 28–30). A high-resolution structure of a 30S:m7G1405 methyltransferase will be necessary to define these specific molecular details. However, our findings suggest that multiple aspects of m7G1405 methyltransferase–substrate binding and specific recognition will emerge that may present suitable molecular targets to interfere with the action of these resistance determinants in pathogenic bacteria.
Experimental procedures
Sequence analysis
m7G1405 methyltransferases sequences were retrieved by BLAST search using RmtB (UniProt code Q76G15) as the query sequence. Sequence redundancy was removed using CD-HIT (31) with a cutoff of 98% sequence identity and aligned using CLUSTAL omega. A neighbor joining phylogenetic tree was constructed using MEGA 6.0 (32), and the residue propensities were calculated using BioEdit (33).
Protein expression and purification
Constructs for expression of RmtA (UniProt code Q8GRA1), RmtB, and RmtC (UniProt code Q33DX5) from a modified pET44 plasmid (“pET44-HT”) were generated using synthetic E. coli codon-optimized genes (GenScript) as described previously (34). Equivalent expression constructs for RmtD (UniProt code B0F9V0) and RmtD2 (UniProt code A0A0U3JA93) were previously reported (35). Variants of RmtC were prepared using the megaprimer whole-plasmid PCR method (13, 36) and confirmed by automated DNA sequencing. Expression of all WT methyltransferases and variant RmtC proteins from the modified pET44 vector produced proteins with an N-terminal His6 tag and thrombin protease recognition sequence. For all experiments other than structural studies, proteins were used directly because the presence of the N-terminal sequence did not affect methyltransferase activity. For crystallization of RmtC, a construct for expression of tag-free WT RmtC (pET44-RmtC) was also generated, essentially as described previously (34).
Recombinant proteins were expressed and purified to near homogeneity using Ni2+-affinity and gel filtration chromatographies, as described previously (35). Purified proteins were concentrated to ∼1 mg/ml and flash-frozen for storage at −80 °C before use. Tag-free WT RmtC was expressed similarly except that terrific broth was used as the bacterial growth medium. Purification was accomplished using heparin-affinity chromatography in 20 mm HEPES buffer, pH 7.6, containing 150 mm NaCl, 10% glycerol, and 6 mm β-mercaptoethanol. After washing with eight column volumes of buffer, the protein was eluted using a 0.15–1 m NaCl gradient in the same buffer. Fractions containing RmtC were pooled and concentrated, and the protein was further purified by gel filtration chromatography on a Superdex 75 16/60 gel filtration column preequilibrated with the same buffer but containing no glycerol. Tag-free WT RmtC was stored as noted above or used directly for crystallization experiments (see below).
Ti measurements
The thermal stability of WT and variant RmtC proteins was assessed using a Tycho NT.6 instrument (NanoTemper) to ensure protein quality between different preparations of proteins and before/after freezing. In this assay, protein unfolding over a 35- 95 °C temperature ramp is monitored via intrinsic fluorescence at 350 and 330 nm and the Ti determined for each apparent unfolding transition from the temperature-dependent change in the ratio of these fluorescence measurements. All RmtC proteins unfolded in two similar apparent transitions (Fig. S1); Ti values reported in Table S1 are the averages of two measurements, and replicates were typically the same within 0.5 °C.
FP assay (Ki determination)
Preparation of 30S ribosomal subunits from E. coli (MRE600), generation of the fluorescein-labeled NpmA probe (NpmA*), and measurement of 30S–Rmt binding were accomplished essentially as previously described (13). Briefly, FP measurements were made using a Biotek Synergy Neo2 instrument with each 100-μl binding reaction containing 30S (50 nm), NpmA* (50 nm), and Rmt protein (2 nm to 10 μm) in 20 mm HEPES buffer, pH 7.0, containing 75 mm KCl, 5 mm Mg(OAc)2, 2 mm NH4Cl, and 3 mm β-mercaptoethanol. Solutions containing 30S and NpmA* were mixed first, incubated for 10 min at room temperature, aliquoted into the 96-well plate, and FP-measured to ensure uniform and stable FP signal prior to addition of the competing protein and final FP measurement. Initial experiments indicated that equilibration was reached quickly after the addition of competing protein and was stable for at least 20 min. Therefore, for subsequent assays, FP was measured immediately, and then each minute over the following ∼5-min period of incubation at 25 °C to ensure the system was at equilibrium with no major variations in the readings. These replicate readings were then averaged. Data handling, curve fitting to determine Ki values, and error calculations were performed in GraphPad Prism8. All binding measurements were made in at least three independent assays for WT Rmt enzymes and at least two independent assays for all RmtC variants. Each assay comprised three or four replicate experiments that were separately prepared and measured but that used the same preparations of protein, 30S, etc. These replicates were averaged prior to fitting in GraphPad Prism to yield the Ki values reported in Table 3. Ki values from fits performed on the individual values from each independent experiment were within ∼2-fold agreement or better for all variants except K236A (Table S2). The data were fit in GraphPad Prism 8 using the “one site–fit Ki” competition binding model,
| (Eq. 1) |
| (Eq. 2) |
where [NpmA*] and KdNpmA* are the concentration (in nm) and equilibrium dissociation of the labeled probe (NpmA*). Control experiments with the established competitor NpmA (13) or WT RmtC were included in all experiments to measure binding of the different Rmt enzymes and RmtC variants, respectively.
Crystallization, X-ray data collection, and structural refinement of the RmtC–SAH complex
Tag-free WT RmtC was concentrated to 12 mg/ml in the final purification buffer and mixed with a 2-fold molar excess of SAH for 10 min at room temperature prior to screening for crystallization conditions on a Crystal Phoenix (Art Robbins Instruments). Initial crystals were obtained at 20 °C using a 1:1 mixture of protein solution and 0.1 m HEPES pH 7.0 buffer containing 2 m ammonium sulfate. An additive screen was used to further optimize crystal size and diffraction with the best diffracting crystal coming from a condition containing 3 mm mellitic acid. X-ray data were collected remotely at the Southeast Regional Collaborative Access Team 22-ID Beamline at the Advanced Photon Source of the Argonne National Laboratory. The data were processed and scaled using X-ray detector software (XDS) (37) in space group P61. The structure was determined by molecular replacement in Phenix (38) using a structure of apo RmtC (PDB code 6CN0) that was deposited into the PDB during this study. The ligand docking and model optimization was accomplished using multiple rounds of refinement and model adjustment in Phenix (38) and Coot (39), respectively. PDB-Redo (40) was also used to optimize the quality of the final model. Complete X-ray data collection and refinement statistics are provided in Table 1.
Kanamycin and gentamicin MIC assays
Fresh lysogeny broth (5 ml) containing 100 μg/ml ampicillin was inoculated (1:100 dilution) with saturated overnight culture of E. coli BL21(DE3) harboring pET-HT plasmid encoding WT or variant RmtC. The cells were grown to A600 ∼0.1 at 37 °C with vigorous shaking. Cells from 1 ml of this culture were collected by centrifugation, washed twice with PBS solution (0.5 ml), and resuspended in cation-adjusted Mueller–Hinton (CA-MHB) medium to an A600 of 0.1 (5 × 107 cfu/ml). The cells were further diluted 50-fold with CA-MHB, and 100 μl was used to inoculate (1 × 105 cfu/well) an equal volume of CA-MHB medium containing 10 μm isopropyl β-d-thiogalactopyranoside and 4-2048 μg/ml antibiotic that was predispensed on a 96-well plate. For each RmtC protein, four to six individual colonies were tested from at least two independent transformations of bacterial cells with plasmid. Wells with no antibiotic or no cells served as controls in each replicate. The plates were incubated at 37 °C with shaking and A600 measurements taken after 24 h. The MIC was defined as the lowest concentration of antibiotic that inhibited growth (A600 of <0.05 above background).
To ensure all variant proteins were comparably expressed in the MIC assay, cultures were grown on microplates under identical conditions but without antibiotic. After confirming that all cultures had similar final cell densities (A600 = ∼0.45–0.5) at 24 h of growth, pelleted cells were resuspended in 100 μl of 2× SDS loading dye, and 5 μl was loaded per lane after boiling to lyse cells and denature the proteins. His-tagged RmtC proteins were detected by immunoblotting with a rabbit anti-His6 antibody (α-His6; Proteintech; 10001-0-AP) overnight at 4 °C. The blots were probed for 1 h at room temperature with a horseradish peroxidase–conjugated goat anti-rabbit secondary antibody (Sigma–Aldrich; A0545) treated with enhanced chemiluminescence reagent (Thermo Fisher) and imaged on a Bio-Rad ChemiDocTM imager.
Cryo-EM
30S–RmtG complex was cross-linked by addition of 25 μl of glutaraldehyde (0.04%) and incubated for 20 min on ice before quenching by addition of 10 μl of glycine (0.16 mm), pH 7.4. Sample (3.5 μl) was applied to glow discharged holey-carbon quantifoil grids (R 2/2, Cu 200) and blotted for 3.5 s at 95% humidity and 8 °C before vitrification by plunging into liquid ethane using a FEI Vitrobot Mark IV. A FEI Titan Krios microscope operating at 300 kV and equipped with a K3 direct detector camera (Gatan) was used to collect 1191 movie frames with Leginon (41). Sixty frames per movie were collected at the total dose of 60.13 e−/Å2 on the sample. The magnification was 81,000×, and the super-resolution frames were 2×-binned corresponding to a pixel size of 1.11 Å.
All preprocessing steps were performed in Appion (42). MotionCor2 (43) was used to align the frames of each micrograph, correct for global and local (5 × 5 patches) beam-induced motion and to dose weight individual frames. Defocus values were determined using CTFFIND4 (44). An initial 3000 particles were picked from using the reference-free particle picker DoG (45) and subjected to 2D classification in cryoSPARC (46). To generate an initial template, e2proc2d.py of EMAN2 (47) was used to compute the rotational average of the 10 best resolved classes. The resulting template was low pass filtered to 15 Å and used to extract particles from the entire set of frame-aligned micrographs using the FindEM template picking software (48). Mask diameter used for template picking was 300 Å. A total of 215,554 particles were extracted with 432 × 432 pixel boxes from full-size images, and the stack and metadata file were exported to cisTEM (49) and used to generate the 2D classes shown in Fig. 6.
Author contributions
M. N., D. D., S. E. S., and G. L. C. formal analysis; M. N., D. D., A. M., S. E. S., N. Z., E. D. H., S. M. S., and C. M. D. investigation; M. N. and G. L. C. methodology; M. N. and G. L. C. writing-original draft; M. N., D. D., A. M., S. E. S., N. Z., E. D. H., S. M. S., C. M. D., and G. L. C. writing-review and editing; N. Z. and G. L. C. project administration; C. M. D. and G. L. C. supervision; G. L. C. conceptualization; G. L. C. data curation; G. L. C. funding acquisition.
Supplementary Material
Acknowledgments
We thank members of the Conn and Dunham labs at Emory University for helpful discussions throughout the course of this work. The Southeast Regional Collaborative Access Team is supported by its member institutions and Equipment Grants S10 RR25528 and S10 RR028976 from the National Institutes of Health. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract W-31-109-Eng-38.
This work was supported by National Institutes of Health Grants R01-AI088025 (to G. L. C.) and T32-GM008367 and Postdoctoral Fellowship DEY18F0 from the Cystic Fibrosis Foundation (to D. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2 and Figs. S1 and S2.
The atomic coordinates and structure factors (code 6PQB) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- h44
- (16S rRNA) helix 44
- CA-MHB
- cation-adjusted Mueller–Hinton broth
- CTD
- C-terminal domain
- FP
- fluorescence polarization
- MIC
- minimum inhibitory concentration
- NTD
- N-terminal domain
- Rmt
- (aminoglycoside) resistance methyltransferase
- SAH
- S-adenosylhomocysteine
- SAM
- S-adenosyl-l-methionine
- RMSD
- root mean square deviation
- PDB
- Protein Data Bank.
References
- 1. Cundliffe E. (1989) How antibiotic-producing organisms avoid suicide. Annu. Rev. Microbiol. 43, 207–233 10.1146/annurev.mi.43.100189.001231 [DOI] [PubMed] [Google Scholar]
- 2. Wachino J., and Arakawa Y. (2012) Exogenously acquired 16S rRNA methyltransferases found in aminoglycoside-resistant pathogenic Gram-negative bacteria: an update. Drug Resist. Updat. 15, 133–148 10.1016/j.drup.2012.05.001 [DOI] [PubMed] [Google Scholar]
- 3. Rahman M., Shukla S. K., Prasad K. N., Ovejero C. M., Pati B. K., Tripathi A., Singh A., Srivastava A. K., and Gonzalez-Zorn B. (2014) Prevalence and molecular characterisation of New Delhi metallo-β-lactamases NDM-1, NDM-5, NDM-6 and NDM-7 in multidrug-resistant Enterobacteriaceae from India. Int. J. Antimicrob. Agents 44, 30–37 10.1016/j.ijantimicag.2014.03.003 [DOI] [PubMed] [Google Scholar]
- 4. Long H., Feng Y., Ma K., Liu L., McNally A., and Zong Z. (2019) The co-transfer of plasmid-borne colistin-resistant genes mcr-1 and mcr-3.5, the carbapenemase gene blaNDM-5 and the 16S methylase gene rmtB from Escherichia coli. Sci. Rep. 9, 696 10.1038/s41598-018-37125-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Doi Y., Wachino J. I., and Arakawa Y. (2016) Aminoglycoside resistance: the emergence of acquired 16S ribosomal RNA methyltransferases. Infect. Dis. Clin. North Am. 30, 523–537 10.1016/j.idc.2016.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aggen J. B., Armstrong E. S., Goldblum A. A., Dozzo P., Linsell M. S., Gliedt M. J., Hildebrandt D. J., Feeney L. A., Kubo A., Matias R. D., Lopez S., Gomez M., Wlasichuk K. B., Diokno R., Miller G. H., et al. (2010) Synthesis and spectrum of the neoglycoside ACHN-490. Antimicrob. Agents Chemother. 54, 4636–4642 10.1128/AAC.00572-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Livermore D. M., Mushtaq S., Warner M., Zhang J. C., Maharjan S., Doumith M., and Woodford N. (2011) Activity of aminoglycosides, including ACHN-490, against carbapenem-resistant Enterobacteriaceae isolates. J. Antimicrob. Chemother. 66, 48–53 10.1093/jac/dkq408 [DOI] [PubMed] [Google Scholar]
- 8. Garneau-Tsodikova S., and Labby K. J. (2016) Mechanisms of resistance to aminoglycoside antibiotics: overview and perspectives. Medchemcomm 7, 11–27 10.1039/C5MD00344J [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wachino J., Shibayama K., Kurokawa H., Kimura K., Yamane K., Suzuki S., Shibata N., Ike Y., and Arakawa Y. (2007) Novel plasmid-mediated 16S rRNA m1A1408 methyltransferase, NpmA, found in a clinically isolated Escherichia coli strain resistant to structurally diverse aminoglycosides. Antimicrob. Agents Chemother. 51, 4401–4409 10.1128/AAC.00926-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dunkle J. A., Vinal K., Desai P. M., Zelinskaya N., Savic M., West D. M., Conn G. L., and Dunham C. M. (2014) Molecular recognition and modification of the 30S ribosome by the aminoglycoside-resistance methyltransferase NpmA. Proc. Natl. Acad. Sci. U.S.A. 111, 6275–6280 10.1073/pnas.1402789111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Husain N., Obranic S., Koscinski L., Seetharaman J., Babic F., Bujnicki J. M., Maravic-Vlahovicek G., and Sivaraman J. (2011) Structural basis for the methylation of A1408 in 16S rRNA by a panaminoglycoside resistance methyltransferase NpmA from a clinical isolate and analysis of the NpmA interactions with the 30S ribosomal subunit. Nucleic Acids Res. 39, 1903–1918 10.1093/nar/gkq1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Macmaster R., Zelinskaya N., Savic M., Rankin C. R., and Conn G. L. (2010) Structural insights into the function of aminoglycoside-resistance A1408 16S rRNA methyltransferases from antibiotic-producing and human pathogenic bacteria. Nucleic Acids Res. 38, 7791–7799 10.1093/nar/gkq627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vinal K., and Conn G. L. (2017) Substrate recognition and modification by a pathogen-associated aminoglycoside resistance 16S rRNA methyltransferase. Antimicrob. Agents Chemother. 61, pii: e00077–17 10.1128/AAC.00077-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Savic M., Sunita S., Zelinskaya N., Desai P. M., Macmaster R., Vinal K., and Conn G. L. (2015) 30S Subunit-dependent activation of the Sorangium cellulosum So ce56 aminoglycoside resistance-conferring 16S rRNA methyltransferase Kmr. Antimicrob. Agents Chemother. 59, 2807–2816 10.1128/AAC.00056-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Witek M. A., and Conn G. L. (2016) Functional dichotomy in the 16S rRNA (m1A1408) methyltransferase family and control of catalytic activity via a novel tryptophan mediated loop reorganization. Nucleic Acids Res. 44, 342–353 10.1093/nar/gkv1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schmitt E., Galimand M., Panvert M., Courvalin P., and Mechulam Y. (2009) Structural bases for 16 S rRNA methylation catalyzed by ArmA and RmtB methyltransferases. J. Mol. Biol. 388, 570–582 10.1016/j.jmb.2009.03.034 [DOI] [PubMed] [Google Scholar]
- 17. Husain N., Tkaczuk K. L., Tulsidas S. R., Kaminska K. H., Cubrilo S., Maravić-Vlahovicek G., Bujnicki J. M., and Sivaraman J. (2010) Structural basis for the methylation of G1405 in 16S rRNA by aminoglycoside resistance methyltransferase Sgm from an antibiotic producer: a diversity of active sites in m7G methyltransferases. Nucleic Acids Res. 38, 4120–4132 10.1093/nar/gkq122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Savic M., Ilic-Tomic T., Macmaster R., Vasiljevic B., and Conn G. L. (2008) Critical residues for cofactor binding and catalytic activity in the aminoglycoside resistance methyltransferase Sgm. J. Bacteriol. 190, 5855–5861 10.1128/JB.00076-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maravić Vlahovicek G., Cubrilo S., Tkaczuk K. L., and Bujnicki J. M. (2008) Modeling and experimental analyses reveal a two-domain structure and amino acids important for the activity of aminoglycoside resistance methyltransferase Sgm. Biochim. Biophys. Acta 1784, 582–590 10.1016/j.bbapap.2007.09.009 [DOI] [PubMed] [Google Scholar]
- 20. Lin J., Zhou D., Steitz T. A., Polikanov Y. S., and Gagnon M. G. (2018) Ribosome-targeting antibiotics: modes of action, mechanisms of resistance, and implications for drug design. Annu. Rev. Biochem. 87, 451–478 10.1146/annurev-biochem-062917-011942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Becker B., and Cooper M. A. (2013) Aminoglycoside antibiotics in the 21st century. ACS Chem. Biol. 8, 105–115 10.1021/cb3005116 [DOI] [PubMed] [Google Scholar]
- 22. Takahashi Y., and Igarashi M. (2017) Destination of aminoglycoside antibiotics in the “post-antibiotic era.” J. Antibiot. (Tokyo), in press 10.1038/ja.2017.117 [DOI] [PubMed] [Google Scholar]
- 23. Durante-Mangoni E., Grammatikos A., Utili R., and Falagas M. E. (2009) Do we still need the aminoglycosides? Int. J. Antimicrob. Agents 33, 201–205 10.1016/j.ijantimicag.2008.09.001 [DOI] [PubMed] [Google Scholar]
- 24. Amunts A., Brown A., Toots J., Scheres S. H. W., and Ramakrishnan V. (2015) Ribosome: the structure of the human mitochondrial ribosome. Science 348, 95–98 10.1126/science.aaa1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Greber B. J., Bieri P., Leibundgut M., Leitner A., Aebersold R., Boehringer D., and Ban N. (2015) Ribosome: the complete structure of the 55S mammalian mitochondrial ribosome. Science 348, 303–308 10.1126/science.aaa3872 [DOI] [PubMed] [Google Scholar]
- 26. Sonousi A., Sarpe V. A., Brilkova M., Schacht J., Vasella A., Böttger E. C., and Crich D. (2018) Effects of the 1-N-(4-amino-2 S-hydroxybutyryl) and 6′-N-(2-hydroxyethyl) substituents on ribosomal selectivity, cochleotoxicity, and antibacterial activity in the sisomicin class of aminoglycoside antibiotics. ACS Infect. Dis. 4, 1114–1120 10.1021/acsinfecdis.8b00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matsushita T., Sati G. C., Kondasinghe N., Pirrone M. G., Kato T., Waduge P., Kumar H. S., Sanchon A. C., Dobosz-Bartoszek M., Shcherbakov D., Juhas M., Hobbie S. N., Schrepfer T., Chow C. S., Polikanov Y. S., et al. (2019) Design, multigram synthesis, and in vitro and in vivo evaluation of propylamycin: a semisynthetic 4,5-deoxystreptamine class aminoglycoside for the treatment of drug-resistant enterobacteriaceae and other Gram-negative pathogens. J. Am. Chem. Soc. 141, 5051–5061 10.1021/jacs.9b01693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee T. T., Agarwalla S., and Stroud R. M. (2005) A unique RNA fold in the RumA-RNA-cofactor ternary complex contributes to substrate selectivity and enzymatic function. Cell 120, 599–611 10.1016/j.cell.2004.12.037 [DOI] [PubMed] [Google Scholar]
- 29. Alian A., Lee T. T., Griner S. L., Stroud R. M., and Finer-Moore J. (2008) Structure of a TrmA-RNA complex: a consensus RNA fold contributes to substrate selectivity and catalysis in m5U methyltransferases. Proc. Natl. Acad. Sci. U.S.A. 105, 6876–6881 10.1073/pnas.0802247105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang C., Jia Q., Zeng J., Chen R., and Xie W. (2017) Structural insight into the methyltransfer mechanism of the bifunctional Trm5. Sci. Adv. 3, e1700195 10.1126/sciadv.1700195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li W., and Godzik A. (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 10.1093/bioinformatics/btl158 [DOI] [PubMed] [Google Scholar]
- 32. Tamura K., Stecher G., Peterson D., Filipski A., and Kumar S. (2013) MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hall T. (2011) BioEdit: an important software for molecular biology. GERF Bull. Biosci. 2, 60–61 [Google Scholar]
- 34. Witek M. A., and Conn G. L. (2014) Expansion of the aminoglycoside-resistance 16S rRNA (m1A1408) methyltransferase family: expression and functional characterization of four hypothetical enzymes of diverse bacterial origin. Biochim. Biophys. Acta 1844, 1648–1655 10.1016/j.bbapap.2014.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Corrêa L. L., Witek M. A., Zelinskaya N., Picão R. C., and Conn G. L. (2016) Heterologous expression and functional characterization of the exogenously acquired aminoglycoside resistance methyltransferases RmtD, RmtD2, and RmtG. Antimicrob. Agents Chemother. 60, 699–702 10.1128/AAC.02482-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Witek M. A., Kuiper E. G., Minten E., Crispell E. K., and Conn G. L. (2017) A novel motif for S-adenosyl-l-methionine binding by the ribosomal RNA methyltransferase TlyA from Mycobacterium tuberculosis. J. Biol. Chem. 292, 1977–1987 10.1074/jbc.M116.752659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kabsch W. (2010) XDS. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Joosten R. P., Long F., Murshudov G. N., and Perrakis A. (2014) The PDB_REDO server for macromolecular structure model optimization. IUCrJ 1, 213–220 10.1107/S2052252514009324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Suloway C., Pulokas J., Fellmann D., Cheng A., Guerra F., Quispe J., Stagg S., Potter C. S., and Carragher B. (2005) Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60 10.1016/j.jsb.2005.03.010 [DOI] [PubMed] [Google Scholar]
- 42. Lander G. C., Stagg S. M., Voss N. R., Cheng A., Fellmann D., Pulokas J., Yoshioka C., Irving C., Mulder A., Lau P. W., Lyumkis D., Potter C. S., and Carragher B. (2009) Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol. 166, 95–102 10.1016/j.jsb.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zheng S. Q., Palovcak E., Armache J. P., Verba K. A., Cheng Y., and Agard D. A. (2017) MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 10.1038/nmeth.4193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rohou A., and Grigorieff N. (2015) CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 10.1016/j.jsb.2015.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Voss N. R., Yoshioka C. K., Radermacher M., Potter C. S., and Carragher B. (2009) DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol. 166, 205–213 10.1016/j.jsb.2009.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Punjani A., Rubinstein J. L., Fleet D. J., and Brubaker M. A. (2017) cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 10.1038/nmeth.4169 [DOI] [PubMed] [Google Scholar]
- 47. Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., and Ludtke S. J. (2007) EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 10.1016/j.jsb.2006.05.009 [DOI] [PubMed] [Google Scholar]
- 48. Roseman A. M. (2004) FindEM–a fast, efficient program for automatic selection of particles from electron micrographs. J. Struct. Biol. 145, 91–99 10.1016/j.jsb.2003.11.007 [DOI] [PubMed] [Google Scholar]
- 49. Grant T., Rohou A., and Grigorieff N. (2018) cisTEM, user-friendly software for single-particle image processing. Elife 7, e35383 10.7554/eLife.35383 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.