

Abstract
Purpose:
Our goal was to evaluate the safety and toxicity of combining a PARP inhibitor, olaparib, with cetuximab and fractionated intensity-modulated radiotherapy for patients with locally advanced head and neck cancer and heavy smoking histories.
Patients and Methods:
Patients with ≥10 packs/year history of smoking were treated with olaparib at doses ranging from 25–200 mg orally twice daily beginning approximately 10 days prior to initiation of and with concurrent radiation (69.3 Gy in 33 fractions) using a time-to-event continual reassessment method model. Cetuximab was administered starting approximately 5 days prior to radiation per standard of care.
Results:
A total of 16 patients were entered onto the study, with 15 evaluable for acute toxicity. The most common treatment-related grade 3–4 side effects were radiation dermatitis and mucositis (38% and 69%, respectively). The MTD was determined to be 50 mg orally twice daily, but the recommended phase II dose was deemed to be 25 mg orally twice daily. At a median follow-up of 26 months, the actuarial median overall survival was 37 months, but was not reached for other endpoints. Two-year overall survival, progression-free survival, local control, and distant control rates were 72%, 63%, 72%, and 79%, respectively. Patients who continued to smoke during therapy experienced higher recurrence rates. MYC and KMT2A were identified as potential correlatives of response on gene amplification and mutational analysis.
Conclusions:
Olaparib at 25 mg orally twice daily with concurrent cetuximab and radiation was well tolerated with reduced dermatitis within the radiation field. Response rates were promising for this high-risk population.
Introduction
There are significant differences in the prognosis of patients with head and neck squamous cell carcinoma (HNSCC) depending on smoking history and human papillomavirus (HPV) status (1). Unfortunately, while the 2- to 5-year overall survival (OS) for HPV-positive (HPV+) patients ranges from 95% to 80% (2), the 5-year survival for patients with HPV-negative (HPV−) HNSCC, often affecting the oral cavity, larynx, or hypopharynx, and cancer associated with heavy smoking remains unacceptably low at around 45%-50% despite very aggressive chemoradiation regimens (1). Moreover, the risk of death significantly increased with each additional pack-year of tobacco smoking (1, 3). New therapies are needed for these high-risk populations that follow a more rational precision-based approach to improve survival.
Smoking-related and HPV− (p16 negative) HNSCC often express increased surface EGFR (1, 4). Increased surface EGFR correlates with poor outcomes and resistance to radiation, in part, due to association with enhanced DNA repair-associated genes (5, 6). Cetuximab is an FDA-approved anti-EGFR antibody foruse with radiation in locally advanced HNSCC that may be more effective in HPV−/smoking-related cancers (7). An additional cause of radiation and chemotherapy resistance in HPV-smoking-related tumors is the high frequency of TP53 mutations (8). Importantly, emerging data suggest resistance to cisplatin, seen in patients with high-risk TP53 mutations, may be abrogated with agents that attack DNA damage repair pathways (9, 10). We hypothesized that these smoking-related cancers have the capability for rapid DNA repair of single- and double-strand breaks resulting from radiation-related damage. Once DNA damage occurs, a multitude of critical DNA damage response enzymes are activated (11, 12). A therapy that could prevent DNA repair may therefore be effective for smoking-related HNSCC improving local-regional control and survival.
PARP is a 116-kDa nuclear protein that uses NAD+ to polymerize ADP-ribose and bind to single-strand DNA breaks (13). PARP inhibitors (PARPi) prevent the synthesis of pADPr, which prevents the downstream repair process from occurring. These agents can induce “synthetic lethality” in cancers with underlying DNA damage repair abnormalities, such as BRCA1/2-mutated ovarian cancer (11, 12). Olaparib (AZD2281) is one of several orally bioavailable inhibitors of both PARP-1 and PARP-2. It is now approved as maintenance therapy with women with BRCA1/2-mutated ovarian cancer on the basis of an improvement in progression-free survival (PFS; ref. 14). PARP inhibition's benefits may extend beyond BRCA1/2-mutated tumors. In preclinical data, synergistic activity between cetuximab, an anti-EGFR antibody, and PARP inhibition has also been demonstrated in several HNSCC cell lines (15). Moreover, PARP inhibition may synergize with the single- and double-stranded DNA breaks induced by radiotherapy (11, 12, 16).
In this study, we aimed to capitalize on DNA repair abnormalities in smoking-related cancers. Given the concerns about hematologic toxicities with combinations of PARPis and cisplatin-based chemotherapy regimens (17) and owing to possible synergy with cetuximab and radiation, we performed a phase I study of standard of care, curative-intent radiation plus cetuximab combined with olaparib. This article represents the first report from a phase I clinical trial examining the safety and tolerability of a PARPi, olaparib, in patients with HNSCC undergoing concurrent cetuximab-radiation therapy.
Patients and Methods
Patients
Inclusion criteria included patients with: histologically/cytologically confirmed AJCC 7th ed. III-IVB oropharynx, larynx, hypopharynx, and inoperable oral cavity; measurable disease per RECIST 1.1; age ≥18 years, life expectancy >12 weeks; adequate hepatic, hematologic, and renal function; Eastern Cooperative Oncology Group Performance Status (ECOG PS) of ≤2. Owing to the inferior survival of smoking-related HNSCC regardless of HPV status, both heavy smoker (≥10 pack-years) HPV+ and HPV− patients were eligible. Upto two prior cycles of induction chemotherapy were allowed. The primary goal of this study was to determine the MTD and the recommended phase II dose (RP2D) of olaparib in combination with concurrent cetuximab-radiation therapy. The protocol was approved by the institutional review board (COMIRB Protocol # 11–1658) and written informed consent was obtained from all patients before performing study-related procedures in accordance with federal and institutional guidelines. All studies were conducted in accordance with the International Ethical Guidelines for Biomedical Research Involving Human Subjects (CIOMS).
Design
Radiation.
Patients were simulated using CT-based imaging and treatment planning with mandatory custom aquaplast mask immobilization. Typically, 3-mm slice thickness was utilized. The treatment plan used for each patient was based on an analysis of the volumetric dose, including dose-volume histogram (DVH) analyses of the PTVs and critical normal structures. Inverse planning with computerized optimization was regularly employed. Radiotherapy was delivered in 33 fractions over 6.5 weeks using intensity-modulated radiation therapy delivered in a once daily fractionation regimen. Three PTVs were utilized with field within-field planning for simultaneous integrated boost: PTV1 to 69.3 Gy (2.1 Gy per fraction), PTV2 to 59.4 Gy (1.8 Gy/fraction), and PTV3 to 54 Gy (at 1.64 Gy per fraction). All plans were normalized such that 95% of the volume of PTV1 was covered with the prescription dose of 69.3 Gy. In addition, no more than 20% of PTV1 was allowed to receive ≥110% of the prescribed dose. No more than 1% of any PTV was allowed to receive ≤ 93% of the prescribed dose to that PTV. Daily cone beam image guidance was performed on all patients. Adaptive replanning was not formally written into the radiation guidelines but was permitted as necessary due to significant weight loss or major tumor changes during initial phases of treatment.
Cetuximab.
Patients received cetuximab according to the FDA package insert. Briefly, a loading dose of cetuximab 400 mg/m2 was administered intravenously over 120 minutes beginning approximately 5–7 days before initiation of radiation. Premedications included diphenhydramine with/without an H2 blocker or dexamethasone. Cetuximab 250 mg/m2 was subsequently administered weekly for a least 6 additional doses, although the number of doses could be extended should radiation be prolonged. Cetuximab rash was managed with standard supportive medications such as topical steroids, oral doxycycline, and topical clindamycin.
Olaparib dose escalation.
Olaparib was administered orally twice daily at the assigned dose starting 5 days prior to their first cetuximab infusion (cycle 1, day -5). The starting olaparib dose for each patient, taken orally in noncrushable 25 mg tablets, was selected on the basis of a time-to-event continual reassessment method (TITE-CRM) described below.
Patients continued taking olaparib throughout the radiation course, 7 days per week, without any planned interruptions. Olaparib was discontinued after the radiotherapy course had been completed.
Clinical evaluation and safety assessment.
Patients were monitored during therapy with serial physical, laryngoscopic, and laboratory exam by a combination of radiation oncologist, medical oncologists, and ENT physicians. Efficacy at the end of therapy was evaluated using RECIST 1.1 through preversus posttherapy CT or MRI scan imaging and by PET imaging when feasible. Patients were then followed per protocol at 1-month posttreatment, 2 months posttreatment, and then at 3-month intervals for the initial 2 years to assess local control, progression-free survival, overall survival, and long-term toxicity.
Determining dose-limiting toxicity assessment
Rather than use a standard 3+3 dose escalation design to assess toxicity and determine the MTD of olaparib, we incorporated a TITE-CRM (18). This design is an adaptive approach in the sense that the dose administered to a subject is based on the accumulated information of how long each prior subject has been on treatment and whether or not they have experienced a dose-limiting toxicity (DLT). A potential advantage of this design is the ability to enroll patients as they become available, as opposed to traditional phase I designs for which enrollment must wait until previous cohorts have finished their period of observation and a determination of whether or not a DLT has occurred is made. For locally advanced disease with radiation combinations and novel drugs, searching for new ways to safely and efficiently determine confidently the MTD and RP2D can be helpful to advance quickly into phase II—III trials. Thus, a TITE-CRM selects dose levels and estimates the probability of a patient experiencing a DLT, defined as any grade ≥3 toxicity attributable to olaparib excluding skin reaction or mucositis. For the latter two, only grade 4 mucositis or skin toxicity qualified as DLT. The trial design is shown in Supplementary Fig. S1. The TITE-CRM algorithm assigned patients to the dose with an estimated probability of DLT closest to the target DLT rate of 15%. Subjects were observed for the occurrence of a DLT for approximately 10 weeks (~70 days), which spanned the entire treatment period and included approximately 4 weeks of observation following treatment.
The trial was designed to select an acceptable MTD and RP2D from among the following 4 doses of olaparib: 25, 50, 100, and 200 mg twice daily continuously during radiation. At the time of study design, the accepted MTD of olaparib monotherapy was 400 mg twice daily although this changed with new tablet formations to 300 mg twice daily. Because of anticipated synergistic toxicity of radiation and cetuximab (19, 20), we set our highest dose at 200 mg. The relationship between dose and toxicity was modeled using a single-parameter (α) logistic regression function with the prior distribution of α set to be N (1, σ2), using σ = 0.3. The posterior distribution of the probability that a future patient would experience a DLT at a given dose was calculated at the time of enrollment for each patient using the prior distribution and available data from all patients at that time. Patients who had not experienced a DLT and had not yet completed the observation period were given a partial weight using a convex weighting function to down-weight the early observation period for which the probability of a DLT is lower than in later follow-up. To maximize safety, the starting dose was initiated at dose level 2 (50 mg, compared with the FDA-approved single agent dose of 400 mg twice daily at the time) to allow for a conservative progression through dose levels while allowing for the possibility of decreasing the dose should one of the early patients experience a DLT. The trial used a run-in period of 5 patients, and dose escalation was restricted to not escalate more than 1 dose level at a time. The TITE-CRM was implemented using version 8 of the TITE-CRM macro developed by the Biostatistics Unit of the University of Michigan Comprehensive Cancer Center (Ann Arbor, MI). This macro was run using SAS software, versions 9.3 and 9.4 (SAS Institute). At the conclusion of the trial, the dose level that has the probability of a DLT closest to the target rate of 15% without exceeding 20% was declared to be the MTD.
Correlative mutational analyses
Our exploratory, translational hypothesis was that patients with heavy smoking history are more sensitive to DNA damage. To characterize the mutational status of these patients, unstained paraffin slides were used for microdissection. Paraffin sections were thoroughly deparaffinized in xylene, hydrated through graded alcohols to water, and stained with Gill hematoxylin. Slides were manually microdissected under a dissecting microscope using a scalpel point into ethanol. The scraped material was washed in PBS and digested in proteinase K overnight at 37° C in ATL Buffer (Qiagen Inc.). DNA was then isolated using QIAamp DSP DNAFFPE extraction kit (catalog no. 60404) according to the manufacturer's instructions.
Mutational and gene amplification analysis was performed using the Illumina TruSightTumor 170 panel according to the manufacturer's instructions. For mutational analysis, FASTQ files were uploaded on Illumina BaseSpace software for variant interpretation. Only variants in coding regions or splice variants were retained. In addition, only variants that were present in <1% of the population according to ExACand 1000 genomes and which were present in >10% of reads with a minimum read depth of 30 variants were retained. For gene amplification analysis, only genes with greater than 2-fold change relative to reported amplification level were considered amplified.
We also performed total nucleic acid (TNA; for the purpose of obtaining RNA), but we prioritized DNA extraction for samples with limited tissue. We successfully extracted TNA from 9 patients. Seven passed quality checks and were analyzed for fusion using the ArcherDx FusionPlex Solid Tumor assay that covers fusions involving 53 genes (21).
The EGFR expression was assessed by visual estimation of predominant intensity of IHC labeling (i.e., none - 0, weak - 1, moderate - 2, and strong - 3) and percentage of positive cells. For this publication, EGFR levels of 2 or 3 were counted as positive. HPV positivity was assessed by p16 IHC.
Statistical analysis
Proportions are reported with exact 95% confidence intervals (CI). Event time distributions were estimated with the method of Kaplan and Meier (22) and compared using the log-rank statistic (23) and the proportional hazards regression model (24). Correlation of genomic differences with treatment outcome was performed using two-tailed Fisher exact test. All statistical analyses were performed using SAS V (SAS Inc.).
Results
Patients
A total of 17 patients were entered into this study with 16 evaluable for acute toxicity and 15 patients evaluable for survival outcomes. The 16 patients were enrolled in the study between November 5, 2012 and August 8, 2016 at the University of Colorado Hospital (Aurora, CO) and Denver Veterans Affairs Medical Center/Eastern Colorado Health Care System. The patient characteristics are shown in Table 1. Of the 16 patients, 14 were men and 2 were women with a median age of 60 years (range, 46–75). Of the 7 oropharyngeal patients, 5 (71%) were HPV-positive by p16, while the other 2 patients (29%) were p16-negative. All patients were heavy smokers with a 51 median pack-year history of smoking (range, 12–90). All were Caucasian, 10 patients (62%) had an ECOG of 1, most (88%) had EGFR expression. Six patients (38%) continued to smoke during treatment. Seven patients (44%) had oropharyngeal primaries and 8 (50%) were of laryngeal origin. Most (81%) presented with stage IVA as per the AJCC 7th edition.
Table 1.
Baseline characteristics of all treated patients
| Characteristics | Patient rates n = 16 |
|---|---|
| Age, y | |
| Median | 60.81 |
| Range | 46.13–75.48 |
| Sex, n (%) | |
| Male | 14 (87) |
| Female | 2 (13) |
| ECOG PS, n (%) | |
| 0 | 6 (38) |
| 1 | 10 (62) |
| Primary site of disease, n (%) | |
| Tonsil | 3 (19) |
| Base of tongue | 4 (25) |
| Supraglottic larynx | 6 (38) |
| Soft palate | 1 (6) |
| Larynx other | 2 (12) |
| P16-Positive oropharyngeal, n (%) | |
| Positive | 5 (71) |
| Negative | 2 (29) |
| EGFR Status | |
| Positive | 14 (87) |
| Negative | 1 (6) |
| Unknown | 1 (6) |
| Tobacco pack-year history | |
| Median | 51 |
| Range | 12–90 |
| Active tobacco use during treatment, n (%) | |
| Yes | 6 (38) |
| No | 10 (62) |
| Disease stage, AJCC 7th, n (%) | |
| III | 3 (19) |
| IVa | 13 (81) |
| T Stage, n (%) | |
| T1 | 1 (6) |
| T2 | 4 (25) |
| T3 | 5 (31) |
| T4a | 3 (19) |
| T4b | 2 (12) |
| N Stage, n (%) | |
| N0 | 3 (19) |
| N1 | 3 (19) |
| N2a | 0 (0) |
| N2b | 5 (31) |
| N2c | 4 (25) |
| N3 | 1 (06) |
Treatment
Cetuximab dosing.
All 16 evaluable patients received the cetuximab loading dose and started weekly cetuximab infusions. One patient received only 4 weeks of cetuximab due to diabetes complications unrelated to study therapy. The total number of infusions and cumulative cetuximab dosing is shown in Table 2. All but one patient who withdrew from treatment after 5,040 cGy received their full dose of radiotherapy for total of 6,930 cGy completed in a median period of 46 days (range 32–52;Table 2). One patient had a short treatment break (#12) due to poor compliance (#12) and another (#10) was given a treatment break of 4 days due to G4 dermatitis.
Table 2.
Patient dosing and DLT assessment
| Patient number |
HPV (p16) |
Induction chemo |
Olaparib dose (mg) |
Olaparib compliance (%) |
Cetuximab cycles |
XRT (cGy) |
XRT Days elapsed |
DLT |
|---|---|---|---|---|---|---|---|---|
| 1 | Y | N | 50 | 97 | 7 | 6,930 | 47 | N |
| 2 | N | Y | 50 | 100 | 7 | 6,930 | 49 | N |
| 3 | N | N | 50 | 97 | 7 | 6,930 | 69 | N |
| 4 | Y | N | 50 | 92 | 7 | 6,930 | 47 | N |
| 5 | Y | Y | 50 | 81 | 7 | 6,930 | 48 | N |
| 6 | Y | Y | 100 | 100 | 7 | 6,930 | 49 | Y G4 dermatitis |
| 7 | Y | N | 100 | 83 | 6 | 6,930 | 48 | N |
| 8 | Y | N | 200 | 70 | 7 | 6,930 | 45 | N |
| 9 | N | N | 200 | 93 | 7 | 6,930 | 48 | Y Nausea/vomiting |
| 10 | N | N | 100 | 93 | 8 | 6,930 | 52 | Y G4 dermatitis |
| 11 withdrew | N withdrew | N | 100 | < 50 | 4 | 5,040 | 24 | N |
| 12 | N | N | 25 | 97 | 7 | 6,930 | 52 | N |
| 13 | Y | N | 25 | 95 | 6 | 6,930 | 46 | N |
| 14 | N | N | 25 | 87 | 4 | 6,930 | 46 | N |
| 15 | Y | N | 25 | 97 | 6 | 6,930 | 46 | N |
| 16 | N | N | 25 | 92 | 7 | 6,930 | 46 | N |
Abbreviation: XRT, radiotherapy.
Olaparib dose escalation.
Olaparib dosing began at 50 mg twice daily with a 5-patient run in. Patient olaparib dosing and compliance are shown in Table 2. A total of 2,4, 5, and 5 patients were treated at dosing of 200,100,50, and 25 mg, respectively. With the exception of patient #11 who withdrew from treatment after 24 radiation fractions, treatment compliance ranged between 70% and 100%. To clarify, olaparib was distributed in an extended release tablet formation that was not crushable and was distributed at 25-mg strength thus necessitating more tablets with higher dosing to be ingested orally. Therefore, we were interested in tracking the overall compliance of the patients entered in terms of actually taking the allotted dose prescription of olaparib throughout the course of radiation or if there were difficulties in swallowing the tablets during therapy. If there were obvious deviations in terms of compliance, we felt this might be important to know in terms of any correlation with outcomes (or perhaps lack of toxicity). We identified no obvious correlations with outcomes to compliance or olaparib dosing in patients requiring to ingest more tablets, again understanding the limitations in a phase I study.
Safety.
Following the TITE-CRM model, olaparib was sequentially delivered at doses of 50, 200, 100, and 25 mg orally twice daily. The most common treatment-related AEs of all grades across all dose levels included dermatitis (88%), mucositis (88%), nausea (51%), acneiform rash (50%), dysphagia (44%), fatigue (44%), vomiting (44%), dysgeusia (40%), and hypomagnesemia (32%; Table 3). There were very few adverse hematologic toxicities with 1 patient (6%) developing leukopenia and 3 (19%) developing lymphopenia. The most common treatment-related grade 3–4 side effects were radiation dermatitis and mucositis (38% and 69%, respectively). There was no correlation between dermatitis and hotspots on radiation planning. Rather the dermatitis appeared like a diffused rash throughout the neck similar to what was seen with the cetuximab rash—some small areas related to skin breakdown and small areas of bleeding.
Table 3.
Adverse events possibly, probably, or definitely attributable to protocol therapy
| Event | Level 1 (25 mg twice daily); n (%) n = 5 |
Level 2 (50 mg twice daily); n (%) n = 5 |
Level 3 (100 mg twice daily); n (%) n = 4 |
Level 2 (200 mg twice daily); n (%) n = 2 |
All dose levels; n (%) n = 16 |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gr 1/2 | Gr 3/4 | Gr 1/2 | Gr 3/4 | Gr 1/2 | Gr 3/4 | Gr 1/2 | Gr 3/4 | Gr 1/2 | Gr 3/4 | |
| Acneiform rash | — | — | 5 (100) | — | 2(50) | — | 1(50) | — | 8(50) | — |
| Anorexia | — | — | 1 (20) | — | 1 (25) | — | — | — | 2 (13) | — |
| Chills | — | — | 2 (40) | — | — | — | — | — | 2 (13) | — |
| Constipation | — | — | — | — | — | — | 1 (50) | — | 1 (06) | — |
| Dehydration | 3 (60) | 1 (20) | 1 (20) | — | — | 1 (25) | — | — | 4 (25) | 2 (13) |
| Dermatitis | 3 (60) | 2 (40) | 3 (60) | 2 (40) | 2 (50) | 2 (50) | — | — | 8 (50) | 6 (38) |
| Diarrhea | — | — | 2 (40) | — | — | — | — | — | 2 (13) | — |
| Dysgeusia | 2(40) | — | 4 (80) | — | 1 (25) | — | 1 (50) | — | 8 (50) | — |
| Dysphagia | — | 2 (40) | 1 (20) | 1 (20) | 1 (25) | 2 (50) | — | — | 2 (13) | 5 (31) |
| Elevated TSH | — | — | — | — | 1 (25) | — | — | — | 1 (06) | — |
| Erythema (of ear) | 4 (80) | — | — | — | 2 (50) | — | — | — | 6 (38) | — |
| Facial swelling | — | — | 1 (20) | — | — | — | — | — | 1 (06) | — |
| Fatigue | — | — | 3 (60) | — | 2 (50) | — | 1 (50) | 1 (50) | 6 (38) | 1 (06) |
| Flatulence | 1(20) | — | — | — | — | — | — | — | 1 (06) | — |
| Headache | 1(20) | — | 2 (40) | — | — | — | — | — | 3(19) | — |
| Hemoptysis | — | — | 1 (20) | — | — | — | — | — | 1 (06) | — |
| Hoarseness | — | — | 1 (20) | — | 1 (25) | — | 1 (50) | — | 3 (19) | — |
| Hypermagnesemia | 2 (40) | — | — | — | — | — | — | — | 2 (13) | — |
| Hypokalemia | 1 (20) | — | — | — | — | — | — | — | 1 (06) | — |
| Hypomagnesemia | — | 1 (20) | 1 (20) | 1 (20) | 2 (50) | — | — | — | 3 (19) | 2 (13) |
| Hyponatremia | — | — | — | — | 1 (25) | — | — | — | 1 (06) | — |
| Infection of G-tube site | 1 (20) | — | — | — | — | — | — | — | 1 (06) | — |
| Insomnia | 1 (20) | — | — | — | — | — | — | — | 1 (06) | — |
| Intermittent hypocalcemia | 1 (20) | — | — | — | — | — | — | — | 1 (06) | — |
| Laryngeal edema | 1 (20) | — | — | — | — | — | — | — | 1 (06) | — |
| Leukopenia | — | — | 1 (20) | — | — | — | — | — | 1 (06) | — |
| Low albumin | — | — | 1 (20) | — | — | — | — | — | 1 (06) | — |
| Low T4 | 1 (20) | — | — | — | — | — | — | — | 1 (06) | — |
| Lymphopenia | — | — | — | 1(20) | — | 1 (25) | — | 1 (50) | — | 3 (19) |
| Malnutrition | 2 (40) | — | — | 1(20) | — | 1 (25) | — | — | 2 (13) | 2 (13) |
| Mucositis | 1 (20) | 3 (60) | 1 (20) | 4 (80) | 1 (25) | 3 (75) | — | 1 (50) | 3 (19) | 11 (69) |
| Nausea | 3 (60) | 1 (20) | 3 (60) | — | — | — | — | 1 (50) | 6 (38) | 2 (13) |
| Neck pain | 1 (20) | — | 2 (40) | — | 1 (25) | — | — | — | 4(25) | — |
| Neck/face skin infection | — | — | — | — | 1 (25) | — | — | — | 1 (06) | — |
| Nonhealing wound | 1 (20) | — | — | — | — | — | — | — | 1 (06) | — |
| Odynophagia | 2 (40) | — | 2 (40) | — | 3(75) | — | 1 (50) | — | 8 (50) | — |
| Oral pain | — | — | 1 (20) | 1 (20) | 1 (25) | — | — | — | 2 (13) | 1 (06) |
| Otalgia | 1 (20) | — | 1 (20) | — | — | — | — | — | 2 (13) | — |
| Hearing loss | — | — | — | — | 1 (25) | — | — | — | 1 (06) | — |
| Pharynx ulceration | — | — | — | — | 1 (25) | — | — | — | 1 (06) | — |
| Pruritus | 3 (60) | — | — | — | — | — | — | — | 3 (19) | — |
| Reflux | 1 (20) | — | 1 (20) | — | — | — | — | — | 2 (13) | — |
| Sinus disorder | 2 (40) | — | — | — | 1 (25) | — | — | — | 3 (19) | — |
| Sore throat | — | — | 2 (20) | — | 1 (25) | — | — | — | 3 (19) | — |
| Thrush | — | — | — | — | — | — | 1 (50) | — | 1 (06) | — |
| Tinnitus | 1 (20) | — | — | — | — | — | — | — | 1 (06) | — |
| Vomiting | 2 (40) | 1 (20) | 3 (60) | — | — | — | — | 1 (50) | 5 (31) | 2 (13) |
| Weight loss | 1 (20) | 1 (20) | 4 (80) | — | 2 (50) | — | 1 (50) | — | 8 (50) | 1 (06) |
| Xerosis/dry skin | 2 (40) | — | 1 (20) | — | 1 (25) | — | — | — | 4(25) | — |
| Xerostomia | 2 (40) | — | 4 (80) | — | 2 (50) | — | 1 (50) | — | 9 (56) | — |
There were three DLTs: two grade 4 dermatitis and one grade 3 nausea and vomiting. Patient #10, who was on the 100mgdoseof olaparib, developed grade 4 dermatitis compounded with skin candidiasis. He received supported care and a 4-day treatment break. Upon resolution to grade 2 dermatitis, he resumed cetuximab-radiation therapy with olaparib at 50 mg twice daily. Patient #6 developed grade 4 dermatitis on 100 mg twice-daily olaparib, 2 weeks after his treatment was completed. In an effort to be conservative in our toxicity evaluations, we coded this patient as a grade 4 dermatitis based on a small area of bleeding induced by removing the dressings after conclusion of radiation. Finally, patient #9 developed grade 3 nausea and vomiting at 200 mg of olaparib requiring hospitalization. He recovered well and his dose of olaparib was reduced to 100 mg twice daily. Although of note was the fact that he had been tolerating the 200 mg dose of olaparib for at least 4 weeks prior to the experience of nausea and vomiting so it was difficult to assign this as strictly study drug-based toxicity.
A summary of all adverse effects by grade is found in Table 3. Two patients were treated at 200 mg with one patient (50%) developing grade 3 nausea/vomiting (DLT) and grade 3 mucositis, fatigue, and lymphopenia (Table 3). Four patients were treated at 100 mg. Two patients (50%) experienced grade 3/4 dermatitis (DLT), 2 with grade 3 dysphagia, 3 (75%) experienced grade 3 mucositis, and 1 (25%) experienced grade 3/4 lymphopenia. None experienced severe nausea or vomiting. Five patients were treated at 50 mg. Two (40%) experienced grade 3 dermatitis and 4 (80%) experienced grade 3 mucositis, whereas only 1 patient (20%) experienced grade 3 lymphopenia and dysphagia and none experienced nausea or vomiting. Finally, 5 patients were treated 25 mg twice daily. Two (40%) patients experienced grade 3 dermatitis and dysphagia, and 3 (60%) experienced grade 3 mucositis. One patient also developed grade 3 nausea, vomiting, and hypomagnesemia requiring hospitalization. However, these complications were established to be related to complications to her diabetes and poor compliance with her insulin regimen and were not attributed to the study drug.
Subacute and late side effects of the treatments included pharyngeal wall necrosis at 6 months posttreatment requiring a replacement of a PEG tube for nutritional support due to dysphagia. This area was biopsied several times with no evidence of recurrent cancer. The changes subsequently resolved at approximately 9 months and the patient was able to return to oral intake. He subsequently experienced what was considered osteoradionecrosis (ORN) at around 1 year (patient #5, 50 mg) again without direct evidence of disease recurrence. His symptoms were treated with antibiotic, steroid treatment, pentoxifylline and vitamin E, and enteral nutritional treatment. He died at 19 months from exsanguination of unknown cause. He had received two cycles of induction chemotherapy prior to study treatment initiation. Another patient (#8,200 mg) who continued to smoke developed a fistula in the palate at 6 months posttreatment. It was not clear whether that was related to tumor regression at the site of the primary or treatment-related toxicity. Patient #10 developed telangiectasia and ORN and was resolved with conservative treatment with pentoxifylline and vitamin E.
DLTs attributable to the study drug were established to be at the 100-mg (2 patients, 50%) and the 200-mg (1 patient, 50%) dose levels (Table 2). However, given that the treatment at baseline is considerably toxic and given some delayed late-term toxicity, the MTD and RP2D was considered to be somewhere between 25 and 50 mg twice-daily range. The 50-mg twice-daily dose was declared to be the MTD as the posterior probability of a DLT was 13.3%, closest to the target of 15%. However, the RP2D, which takes into consideration all information published combining PARPi with alternative agents such as cisplatin (25), was deemed to be 25 mg orally daily twice daily of olaparib when given in combination with cetuximab and radiation.
Clinical activity.
Of the 16 patients, one patient withdrew from the trial (#11) and was lost to follow-up after 5,040 cGy, leaving 15 evaluable patients for disease response. Of the 5 patients who developed persistent or relapse HNSCC, almost all had continued to smoke during treatment despite smoking cessation encouragement (Table 4). Of the 6 patients that died, 3 died of reasons unrelated to HNSCC disease progression. Patient #2 who continued to smoke during and after treatment died of a cardiopulmonary event 6 months posttreatment that was felt to be attributable to a mucous plug and patient #3 died of progression of a second primary bladder cancer (Table 4). Patient #16 had developed severe supraglottic edema at 6 months posttreatment on nasopharyngoscopic exam as well as CT and PET imaging. It was unclear whether the cause of the edema could be attributed to disease progression or inflammation. He was placed on antibiotics and steroids and a biopsy was scheduled. However, he died before obtaining tissue biopsy. Two patients who relapsed (#12, #15) are still alive (Fig. 3; Table 4). Median PFS and OS were calculated from date of end of treatment to date of progressive disease/death. At a median follow-up of 26 months, the actuarial median OS was 37 months, but was not reached for other endpoints. Two-year OS, PFS, local control, and distant control rates were 72%, 63%, 72%, and 79%, respectively (Figs. 1 and 2).
Table 4.
Patients with any events
| Patient identifier |
Primary site of disease |
Primary disease stage |
Age | Olaparib dose (mg) |
Olaparib compliance (%) |
Smoked through treatment? |
PFS (d) |
Location of failure |
OS (d) |
Cause of death |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | Supraglottic larynx | IVa | 52 | 50 | 100 | N | NP | 180 | Cardiopulmonary arrest - suspected mucous pluga | |
| 3 | Base of tongue | IVa | 62 | 50 | 97 | N | NP | 930 | Bladder cancer | |
| 5 | Base of tongue | IVa | 55 | 50 | 81 | N | NP | 597 | Exsanguination (unknown etiology)a | |
| 7 | Supraglottic larynx | III | 69 | 100 | 83 | Y | 244 | Local and distant metastasis | 1098 | Disease progression |
| 8 | Tonsil | IVa | 56 | 200 | 70 | Y | 187 | Local and distant | 342 | Died of disease |
| 12 | Supraglottic larynx | IVa | 46 | 25 | 97 | Y | 303 | Local | Alive | |
| 15 | Tonsil | IVa | 59 | 25 | 97 | Y | 215 | Distant then local and distant after | Alive | |
| 16 | Supraglottic larynx | IVa | 69 | 25 | 92 | Questionable History of heavy smoking/drinking | 239 | Local | 259 | Likely persistent disease |
Patients NED at time of death.
Figure 1.

Survival outcomes. Kaplan-Meier survival curves of overall Survival (OS; A), progression-free interval (PFS; B); local control (C), and distant control (D).
Figure 2.

PFS for evaluable patients.
Correlative genomics profile and mutational testing results.
Mutational analysis is shown in Table 5A and genomic amplification data based on TruSight Tumor 170 assay are shown in Table 5B. The most commonly mutated genes are TP53 (n = 6), ERBB4 (n = 4), MSH3 (n = 4), and RAD51 (n = 4) among the 10 tested samples. Three of the samples were from patients who relapsed (patient #s 7, 15, and 16). Patients who progressed on this study had greater than 2-fold increase in MYC (mean: 2.24; range: 2.11–2.38) compared with patients who responded to treatment (mean: 1.43, range: 1.06–1.79; P = 0.0083). No other genes were found to be amplified. In 2 of 3 “nonresponders” KMT2A mutations were noticed, whereas none of the 7 other responder patients had KMT2A mutations (P = 0.0667). Of the seven TNA samples analyzed, none contained any evidence of fusion.
Table 5A.
Mutational analysis on 10 patients treated with olaparib and cetuximab–radiation therapy
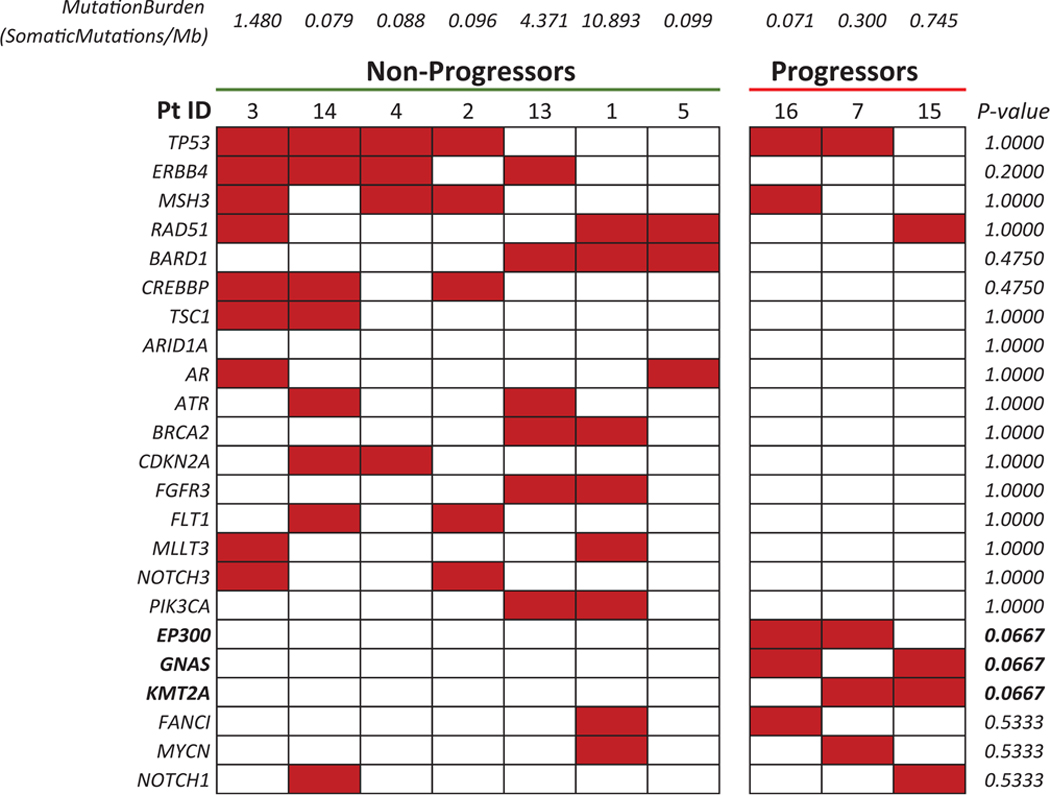 |
Patients 7, 15, and 16 are considered nonresponders. Boldface indicates a trend toward statistical significance.
Table 5B.
Correlative analysis of genomic amplification based on TruSight Tumor 170 assay
| Gene | Average relative expression | |
|---|---|---|
| Responsive | Resistant | |
| MYC | 1.42 | 2.24 |
| NBN | 1.08 | 1.56 |
| XRCC2 | 0.98 | 1.53 |
| SMARCB1 | 1.10 | 1.49 |
| MET | 0.95 | 1.32 |
| BRAF | 0.86 | 1.22 |
| ATR | 1.29 | 0.94 |
| INPP4B | 0.93 | 0.72 |
| BTK | 0.97 | 0.71 |
Discussion
Patients with HNSCC and heavy smoking histories have poor prognoses regardless of HPV status (3). This study demonstrates that olaparib, an oral PARPi, maybe combined with standard-of-care cetuximab and radiotherapy in patients with locally advanced HNSCC with high-risk, smoking-related tumors.
PARPs are critical regulators of DNA damage repair, regulation of cellular replication and differentiation, and other cellular processes (7). PARPis, such as olaparib, may attenuate the nuclear translocation of EGFR normally induced by DNA damages potentially leading to lower nuclear interaction between EGFR and DNA PK, and consequently lower DNA repair by c-NHEJ (15, 26). Combining PARPis with EGFR inhibitors yields synergistic increase in HNSCC cytotoxicity both in vitro and in vivo (27). These data formed the scientific justification to hypothesize that smoking-related HNSCC have the capability for rapid DNA repair of single- and double-strand breaks resulting form radiation; therefore, the addition of PARPis with cetuximab radiation will yield improved outcomes in HNSCC.
The clinical development of PARP-1 inhibitors in combination with radiotherapy is less advanced than monotherapy strategies (11). Safety and tolerability of such combinations are currently tested in several clinical phase 1 trials (11,12). To our knowledge, this is the first clinical trial examining the addition of a PARPi to cetuximab radiation in locally advanced HNSCC with heavy smoking history and EGFR expression (11). We believe our findings warrant further consideration of a phase II/III clinical trial design. For cisplatin-ineligible patients or as a way to move beyond cisplatin-radiation regimens, we envision additional options in the phase II-III setting that might look to combine radiation and olaparib with alternative agents as well. In this regard, the foundation of DNA damage repair inhibitor and radiation could be explored in the context of additional biologic or immunologic-based agents such as anti-PD-1 or PD-L1 drugs.
In our trial using a TITE-CRM approach, the MTD is based on a mathematical model that only considers DLTs. The RP2D takes into consideration all information published combining PARPis with alternative agents such as cisplatin or with radiation (ongoing study in the UK in LAHNC, NCT02308072). Here we declared the MTD to be 50 mg and the RP2D is 25 mg when combined with cetuximab and radiation, a much lower dose than described previously in olaparib monotherapy trials (11). Interestingly, this is consistent with preclinical data demonstrating that olaparib radiosensitizes HNSCC in vitro at much lower doses that for monotherapy dosing (16, 25). In a treatment regimen (cetuximab-radiotherapy) that is normally associated with significant acute toxicity that includes dermatitis and mucositis (28), it was difficult for us at times to establish how much of this toxicity was attributed to olaparib. However, it appeared that increased mucositis and increased dermatitis rates were observed primarily at doses above 25 mg twice daily, with 62% experiencing grade 3 mucositis, 1 patient experiencing grade 4 mucositis; dermatitis rates included 31% experiencing grade 3, and 1 grade 4 reaction experienced 14 days posttreatment with some minor bleeding of the skin of the anterior neck. Confounding toxicity assessment is the use of cetuximab in this phase I trial (29). It may be that adding on cetuximab, a potent cause of dermatitis, as an alternative to cisplatin, contributed to the increased skin toxicity observed at the higher doses of olaparib; however, at the 25 mg twice-daily level, the severity was reduced. It is important to point out that all analyzable patients healed appropriately as expected post radiation. Two patients died for reasons unrelated to active HNSCC, one of which appeared to be due to a cardiopulmonary arrest from a suspected mucous plug 6 months posttreatment in a patient (patient #2 at 50 mg twice-daily olaparib level) and one who reportedly exsanguinated (patient #5 with large BOT/pharyngeal wall involvement at 50 mg orally twice-daily olaparib level). The family refused an autopsy to determine exact cause of death. This speaks about the importance of careful follow up after resolution of acute side effects within the 60- to 90-day window from the start of therapy in patients with locally advanced disease in phase I clinical trials with radiation and novel therapeutics.
Preclinically, the extent of radiosensitization by the PARPi, olaparib, has also been shown to depend on the homologous recombination status of the cells (16, 30). We further showed that olaparib radiosensitizes at much lower doses than needed for single-agent activity. Our exploratory analysis identified MYC to be amplified in nonresponders. MYC is considered as a candidate for synthetic lethality gene partner for PARPis (31). Its overexpression accelerates the DNA replication stress, accumulates DNA DSBs, and associates with synthetic lethality to PARP1is (31–35). Although this is an attractive and sensible target, it is important to emphasize that the analysis is underpowered and would require confirmation in larger scale trials.
We had hypothesized that PARPis would also be a novel synthetic lethal therapeutic approach for HNSCC harboring activating mutations and overexpressed genes. In our mutational analysis, the most commonly mutated genes included TP53, EBB4, MSH3, and Rad51 among the 10 tested patients. Some of these have been reported as candidate genes for synthetic lethality gene partners for PARP interactions (36–39). It is interesting to note that 2 of 3 patients who relapsed (#7, #15) had KMT2A mutation. KMT2A is a mixed lineage leukemia (MLL) transcriptional coactivator, also known as histone-lysine N-methyltransferase 2A (40). Leukemia-driven KMT2A fusions with dominant transactivation ability are reportedly proficient in DNA damage repair and insensitive to PARP inhibition (41, 42). This suggests that the KMT2A signaling pathway may be a potential therapeutic target for locally advanced head and neck cancer. However, these data are limited by the small sample size in this analysis and by the limitations of the T170 illumina panel, which does not capture fusions found to be olaparib-sensitive markers in other cancer types, or large deletions of other genetic changes. Therefore, these data only serve as correlatives and remain hypothesis generating.
Any conclusions that we draw on efficacy are limited by our small sample size and by the fact that this is not a randomized controlled trial. The overall survival in this trial, albeit a small study, is worth commentary. In HNSCC with heavy smoking history, the expected OS rate at 2 years is approximately 60%, based on multiple studies (1). Our small study with a heterogeneous group of tumor types had a 2-year OS rate of 72%. We believe this combination warrants further study in the phase II setting as an alternative to cisplatin-based regimens where 3-year PFS remains below 45% in heavy smoker patients with T3–4 primaries (RTOG 0129; ref. 1). Of interest are several ongoing phase I trials currently investigating weekly cisplatin combined with olaparib given 3 days per week during radiotherapy for locally advanced HNSCC as well as a study evaluating the effects of olaparib alone with radiation in patients with locally advanced head and neck cancer (NCT02308072, NCT02229656) with an emphasis on HPV− laryngeal and oropharyngeal cancers and we await their assessments of acute toxicities. Newer approaches are indicated for this poor prognosis patient population that selectively attack the DNA damage repair pathways. Although this combination of cetuximab, olaparib, and radiation appeared quite effective and safe at doses in the range of 25 mg orally twice daily, we continue to search for ways to enhance immune-regulated anticancer approaches that might reduce acute toxicity even further while improving outcomes. To this end, with the recent approval of checkpoint inhibitors in advanced head and neck cancer, consideration of combining DNA damage repair inhibitors with immune-enabling drugs and radiation is gaining traction in the locally advanced setting and may offer further reduction of acute and chronic toxicity over traditional chemotherapy or anti-EGFR strategies. Recent preclinical studies indicate a cooperative effect between PARPi and anti-PD-L1 in syngeneic mouse models. Mechanistically, PARPi enhanced PARPi-mediated PD-L1 upregulation and blockade of PD-L1 resensitized PARPi-treated cancer cells to T-cell killing (43).
Finally, it is important to note that nearly all patients who progressed on treatment continued smoking during radiotherapy. This is consistent with the combined analysis of RTOG 0129 and RTOG 9003 (3). It has been long documented that smoking during radiotherapy reduced response rates and 2- and 5-year survival for patients with head and neck cancer compared with those who quit (44–46). Several possible explanations have been proposed for why active smoking during radiation might reduce effectiveness of therapy. Exacerbation of tissue hypoxia by continued smoking has been reported in patients and animal models (47, 48). Smoking exposure has also been reported to enhance resistance to DNA damage-induced cell death. Other mechanisms including nicotine's interaction with survival pathways such as MAPK and akt pathways have also been proposed for reducing with radiation's cytotoxic effects in active smokers (49–51).
In conclusion, we have demonstrated that olaparib may be safely combined with cetuximab and radiotherapy for patients with smoking-related HNSCC. It shows promising signs of activity and merits further investigation.
Supplementary Material
Translational Relevance.
In this phase I single-institutional trial, we pursued a novel combinatorial strategy of an orally bioavailable DNA-damaging agent, the PARP inhibitor olaparib, with cetuximab and conventionally fractionated radiation in patients with head and neck squamous cell carcinoma (HNSCC) with heavy smoking histories. Our primary endpoint was assessment of toxicity of this novel dual-biologic regimen. Secondary endpoints included progression-free survival, overall survival, local control, and distant control. As an exploratory measure, gene amplification profile and mutational analysis were evaluated. We hypothesized that our regimen might result in a tolerable combination and provide clinical benefit to patients with locally advanced HNSCC with heavy smoking history with resultant DNA damage repair defects and poor survival outcomes. To the best of our knowledge, this is the first study that combines the PARP inhibitor olaparib in patients with HNSCC undergoing cetuximab-radiation therapy.
Acknowledgments
Study drug only was provided by AstraZeneca. The company had no involvement in the study design or data interpretation. The authors thank the Wehling family for donations to support this clinical trial effort as well as the Marsico family for their endowment toward head and neck cancer research. This project was supported by Population Health Shared Resource, Molecular Pathology Shared Resource, and Genomics and Microarray Shared Resource, University of Colorado Cancer Center (P30CA046934).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
D.L. Aisner is a consultant/advisory board member for AbbVie and Genentech, and reports receiving commercial research grants from Genentech.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed by the other authors.
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med 2010;363:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Sullivan B, Huang SH, Su J, Garden AS, Sturgis EM, Dahlstrom K, et al. Development and validation of a staging system for HPV-related oropharyngeal cancer by the International Collaboration on Oropharyngeal cancer Network for Staging (ICON-S): a multicentre cohort study. Lancet Oncol 2016;17:440–51. [DOI] [PubMed] [Google Scholar]
- 3.Gillison ML, Zhang Q, Jordan R, Xiao W, Westra WH, Trotti A, et al. Tobacco smoking and increased risk of death and progression for patients with p16-positive and p16-negative oropharyngeal cancer. J Clin Oncol 2012; 30:2102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar B, Cordell KG, Lee JS, Worden FP, Prince ME, Tran HH, et al. EGFR, p16, HPVTiter, Bcl-xL and p53, sex, and smoking as indicators of response to therapy and survival in oropharyngeal cancer. J Clin Oncol 2008;26: 3128–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akimoto T, Hunter NR, Buchmiller L, Mason K, Ang KK, Milas L. Inverse relationship between epidermal growth factor receptor expression and radiocurability of murine carcinomas. Clin Cancer Res 1999;5:2884–90. [PubMed] [Google Scholar]
- 6.Huang SM, Harari PM. Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas: inhibition of damage repair, cell cycle kinetics, and tumor angiogenesis. Clin Cancer Res 2000;6:2166–74. [PubMed] [Google Scholar]
- 7.Dziadkowiec KN, Gąsiorowska E, Nowak-Markwitz E, Jankowska A. PARP inhibitors: review of mechanisms of action and BRCA1/2 mutation targeting. Prz Menopauzalny 2016;15:215–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Westra WH, Taube JM, Poeta ML, Begum S, Sidransky D, Koch WM. Inverse relationship between human papillomavirus-16 infection and disruptive p53 gene mutations in squamous cell carcinoma of the head and neck. Clin Cancer Res 2008;14:366–9. [DOI] [PubMed] [Google Scholar]
- 9.Osman AA, Monroe MM, Ortega Alves MV, Patel AA, Katsonis P, Fitzgerald AL, et al. Wee-1 kinase inhibition overcomes cisplatin resistance associated with high-risk “TP53” mutations in head and neck cancer through mitotic arrest followed by senescence. Mol Cancer Ther 2015;14:608–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gadhikar MA, Sciuto MR, Alves MVO, Pickering CR, Osman AA, Neskey DM, et al. Chk1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53. Mol Cancer Ther 2013;12:1860–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lesueur P, Chevalier F, Austry J-B, Waissi W, Burckel H, Noël G, et al. Poly-(ADP-ribose)-polymerase inhibitors as radiosensitizers: a systematic review of pre-clinical and clinical human studies. Oncotarget 2017;8: 69105–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bao Z, Cao C, Geng X, Tian B, Wu Y, Zhang C, et al. Effectiveness and safety of poly (ADP-ribose) polymerase inhibitors in cancer therapy: A systematic review and meta-analysis. Oncotarget 2016;7:7629–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Durkacz BW, Omidiji O, Gray DA, Shall S. (ADP-ribose)n participates in DNA excision repair. Nature 1980;283:593–6. [DOI] [PubMed] [Google Scholar]
- 14.Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1274–84. [DOI] [PubMed] [Google Scholar]
- 15.Nowsheen S, Bonner JA, Lobuglio AF, Trummell H, Whitley AC, Dobelbower MC, et al. Cetuximab augments cytotoxicity with poly (adp-ribose) polymerase inhibition in head and neck cancer. PLoS One 2011;6:e24148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verhagen CV, de Haan R, Hageman F, Oostendorp TP, Carli AL, O'Connor MJ, et al. Extent of radiosensitization by the PARP inhibitor olaparib depends on its dose, the radiation dose and the integrity of the homologous recombination pathway of tumor cells. Radiother Oncol 2015;116: 358–65. [DOI] [PubMed] [Google Scholar]
- 17.Zhou JX, Feng LJ, Zhang X. Risk of severe hematologic toxicities in cancer patients treated with PARP inhibitors: a meta-analysis of randomized controlled trials. Drug Des Devel Ther 2017;11:3009–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheung YK, Chappell R. Sequential designs for phase I clinical trials with lateonset toxicities. Biometrics 2000;56:1177–82. [DOI] [PubMed] [Google Scholar]
- 19.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med 2006;354:567–78. [DOI] [PubMed] [Google Scholar]
- 20.Magrini SM, Buglione M, Corvò R, Pirtoli L, Paiar F, Ponticelli P, et al. Cetuximab and radiotherapy versus cisplatin and radiotherapy for locally advanced head and neck cancer: a randomized phase II trial. J Clin Oncol 2016;34:427–35. [DOI] [PubMed] [Google Scholar]
- 21.Davies KD, Ng TL, Estrada-Bernal A, Le AT, Ennever PR, Camidge DR, et al. Dramatic response to crizotinib in a patient with lung cancer positive for an HLA-DRB1-MET gene fusion. JCO Precis Oncol 2017;2017:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Statist Assoc 1958;53:457–81. [Google Scholar]
- 23.Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 1959;22:719–48. [PubMed] [Google Scholar]
- 24.Cox DR. Regression models and life-tables. J Roy Stat Soc Ser B (Methodological) 1972;34:187–220. [Google Scholar]
- 25.Lourenco LM, Jiang Y, Drobnitzky N, Green M, Cahill F, Patel A, et al. PARP inhibition combined with thoracic irradiation exacerbates esophageal and skin toxicity in C57BL6 mice. Int J Radiat Oncol Biol Phys 2018;100: 767–75. [DOI] [PubMed] [Google Scholar]
- 26.Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Raju U, Milas L, et al. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J Biol Chem 2005;280:31182–9. [DOI] [PubMed] [Google Scholar]
- 27.Schulz D, Wirth M, Piontek G, Buchberger AM, Schlegel J, Reiter R, et al. HNSCC cells resistant to EGFR pathway inhibitors are hypermutated and sensitive to DNA damaging substances. Am J Cancer Res 2016;6: 1963–75. [PMC free article] [PubMed] [Google Scholar]
- 28.Magrini SM, Buglione M, Corvo R, Pirtoli L, Paiar F, Ponticelli P, et al. Cetuximab and radiotherapy versus cisplatin and radiotherapy for locally advanced head and neck cancer: a randomized phase II trial. J Clin Oncol 2016;34:427–35. [DOI] [PubMed] [Google Scholar]
- 29.Pinto C, Barone CA, Girolomoni G, Russi EG, Merlano MC, Ferrari D, et al. Management of skin reactions during cetuximab treatment in association with chemotherapy or radiotherapy: update of the italian expert recommendations. Am J Clin Oncol 2016;39:407–15. [DOI] [PubMed] [Google Scholar]
- 30.Wurster S, Hennes F, Parplys AC, Seelbach JI, Mansour WY, Zielinski A,et al. PARP1 inhibition radiosensitizes HNSCC cells deficient in homologous recombination by disabling the DNA replication fork elongation response. Oncotarget 2016;7:9732–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maifrede S, Martin K, Podszywalow-Bartnicka P, Sullivan-Reed K, Langer SK, Nejati R, et al. IGH/MYC translocation associates with BRCA2 deficiency and synthetic lethality to PARP1 inhibitors. Mol Cancer Res 2017;15:967–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reid RJ, Du X, Sunjevaric I, Rayannavar V, Dittmar J, Bryant E, et al. A synthetic dosage lethal genetic interaction between CKS1B and PLK1 is conserved in yeast and human cancer cells. Genetics 2016;204: 807–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shigetomi H, Sudo T, Shimada K, Uekuri C, Tsuji Y, Kanayama S, et al. Inhibition of cell death and induction of G2 arrest accumulation in human ovarian clear cells by HNF-1beta transcription factor: chemosensitivity is regulated by checkpoint kinase CHK1. Int J Gynecol Cancer 2014;24:838–43. [DOI] [PubMed] [Google Scholar]
- 34.Weterings E, Gallegos AC, Dominick LN, Cooke LS, Bartels TN, Vagner J, et al. A novel small molecule inhibitor of the DNA repair protein Ku70/80. DNA Repair 2016;43:98–106. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi Y, Sheridan P, Niida A, Sawada G, Uchi R, Mizuno H, et al. The AURKA/TPX2 axis drives colon tumorigenesis cooperatively with MYC. Ann Oncol 2015;26:935–42. [DOI] [PubMed] [Google Scholar]
- 36.Kawahara N, Ogawa K, Nagayasu M, Kimura M, Sasaki Y, Kobayashi H. Candidate synthetic lethality partners to PARP inhibitors in the treatment of ovarian clear cell cancer. Biomed Rep 2017;7:391–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takahashi M, Koi M, Balaguer F, Boland CR, Goel A. MSH3 mediates sensitization of colorectal cancer cells to cisplatin, oxaliplatin, and a poly (ADP-ribose) polymerase inhibitor. J Biol Chem 2011;286:12157–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Canfield K, Li J, Wilkins OM, Morrison MM, Ung M, Wells W, et al. Receptor tyrosine kinase ERBB4 mediates acquired resistance to ERBB2 inhibitors in breast cancer cells. Cell Cycle 2015;14:648–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Burness ML, Martin-Trevino R, Guy J, Bai S, Harouaka R, et al. RAD51 mediates resistance of cancer stem cells to PARP inhibition in triplenegative breast cancer. Clin Cancer Res 2017;23:514–22. [DOI] [PubMed] [Google Scholar]
- 40.Maifrede S, Martinez E, Nieborowska-Skorska M, Di Marcantonio D, Hulse M, Le BV, et al. MLL-AF9 leukemias are sensitive to PARP1 inhibitors combined with cytotoxic drugs. Blood Advances 2017;1:1467–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohmoto A, Yachida S. Current status of poly(ADP-ribose) polymerase inhibitors and future directions. OncoTargets Thera 2017;10:5195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Esposito MT, Zhao L, Fung TK, Rane JK, Wilson A, Martin N, et al. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat Med 2015;21:1481. [DOI] [PubMed] [Google Scholar]
- 43.Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res 2017;23:3711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Browman GP, Mohide EA, Willan A, Hodson I, Wong G, Grimard L, et al. Association between smoking during radiotherapy and prognosis in head and neck cancer: a follow-up study. Head Neck 2002;24:1031–7. [DOI] [PubMed] [Google Scholar]
- 45.Browman GP, Wong G, Hodson I, Sathya J, Russell R, McAlpine L, et al. Influence of cigarette smoking on the efficacy of radiation therapy in head and neck cancer. N Engl J Med 1993;328:159–63. [DOI] [PubMed] [Google Scholar]
- 46.Chen AM, Chen LM, Vaughan A, Sreeraman R, Farwell DG, Luu Q, et al. Tobacco smoking during radiation therapy for head-and-neck cancer is associated with unfavorable outcome. Int J Radiat Oncol Biol Phys 2011;79:414–9. [DOI] [PubMed] [Google Scholar]
- 47.Jensen JA, Goodson WH, Hopf HW, Hunt TK. Cigarette smoking decreases tissue oxygen. Arch Surg 1991;126:1131–4. [DOI] [PubMed] [Google Scholar]
- 48.Grau C, Nordsmark M, Khalil AA, Horsman MR, Overgaard J. Effect of carbon monoxide breathing on hypoxia and radiation response in the SCCVII tumor in vivo. Int J Radiat Oncol Biol Phys 1994;29:449–54. [DOI] [PubMed] [Google Scholar]
- 49.Heusch WL, Maneckjee R. Signalling pathways involved in nicotine regulation of apoptosis of human lung cancer cells. Carcinogenesis 1998;19:551–6. [DOI] [PubMed] [Google Scholar]
- 50.West KA, Brognard J, Clark AS, Linnoila IR, Yang X, Swain SM, et al. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J Clin Invest 2003;111: 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Onoda N, Nehmi A, Weiner D, Mujumdar S, Christen R, Los G. Nicotine affects the signaling of the death pathway, reducing the response of head and neck cancer cell lines to DNA damaging agents. Head Neck 2001; 23:860–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.