

Abstract
Objective:
Prader-Willi syndrome (PWS) is a rare genetic neuroendocrine disorder characterized by hypotonia, obesity, short stature, and mental retardation. Incomplete or delayed pubertal development as well as premature adrenarche are usually found in PWS, whereas central precocious puberty is rarely seen.
Methods:
This study reports the clinical, biochemical, and histologic findings in 2 boys with PWS who developed central precocious puberty.
Results:
Both boys were started on growth hormone therapy during the first years of life according to the PWS indication. They had both bilateral cryptorchidism at birth and had orchidopexy in early childhood. Retrospective histologic analysis of testicular biopsies demonstrated largely normal tissue architecture and germ cell maturation, but severely decreased number of prespermatogonia in one of the patients. Both boys had premature adrenarche around the age of 6. Precocious puberty was diagnosed in both boys with enlargement of testicular volume (>3 mL), signs of virilization and a pubertal response to a gonadotropin-releasing hormone (GnRH) test and they were both treated with GnRH analog.
Conclusion:
The cases described here displayed typical characteristics for PWS, a considerable heterogeneity of the hypothalamic-pituitary function, as well as testicular histology. Central precocious puberty is extremely rare in PWS boys, but growth hormone treatment may play a role in the pubertal timing.
INTRODUCTION
Prader-Willi syndrome (PWS) is a rare genetic disorder caused by the absence of paternal expression of several imprinted genes located on chromosome 15 (15q11-q13) most commonly due to deletion or uniparental disomy.
One of the major clinical findings in boys with PWS is cryptorchidism, hypoplastic external genitalia, and delayed pubertal development, suggesting a dysfunction of the hypothalamic-pituitary-gonadal axis (1,2). Some children with PWS experience premature adrenarche which is not linked to obesity (3), but central precocious puberty is extremely rare among boys with PWS (4–6). The concomitant presence of dysfunction of the hypothalamic-pituitary-gonadal axis with cryptorchidism and precocious puberty in PWS patients is complex and difficult to explain.
Growth hormone (GH) treatment is approved for children with PWS and has been found to improve growth and body composition. Both our cases were treated with GH despite of normal GH secretion. GH therapy has been proposed to accelerate the rate of sexual maturation during puberty, but only a few studies have examined this, and the results have been divergent (7).
METHODS
For this retrospective study, immunohistochemistry (IHC) staining was performed to characterize the presence and maturation of different testicular cell types. The following markers were used: OCT3/4, AP2γ, MAGE-A4 (to assess stages of germ cell differentiation and to detect the possible presence of germ cell neoplasia in situ [GCNIS]), SOX9 (Sertoli cell marker), COUP-TFII (marker of peritubular myoid and Leydig cells) and CYP11A1 (Leydig cell marker). The IHC staining was performed using a standard indirect immunoperoxidase method, according to previously published protocols (8). Tissues from specimens known to express the selected markers were used as positive controls, while for negative controls the antibodies were replaced by dilution buffer.
CASE REPORT
Informed consent was obtained from the patients' legal guardians for publishing this report. The boys were diagnosed with PWS early in life and both had a deletion in the q11–13 region of chromosome 15. Both boys had bilateral cryptorchidism at birth, premature adrenarche, and later developed central precocious puberty.
Patient 1
Patient 1 was born at term; birth weight was 3,115 g (−1.3 standard deviation score [SDS]) and birth length was 50 cm (−0.6 SDS). PWS was diagnosed during the first months of life. The patient was started on GH therapy at 8 months of age; he had a normal response to a growth hormone stimulation test.
At 6.9 years of age he developed pubic hair (PH) stage 2 (PH2) without testicular enlargement (2 mL). Measurements of 17-hydroxy progesterone were normal (0.4 nmol/L, −0.2 SDS), DHEAS was a little elevated (3,812 nmol/L, 2.3 SDS) and androstenedione was normal (0.78 nmol/L, 0.9 SDS), which suggested premature adrenarche. Bone age (BA) was 7.6 years at a chronologic age (CA) of 7.2 years, growth velocity was increased (7.1 cm/year, 1.3 SDS) and body mass index (BMI) SDS was 1.9. The serum concentration of FSH was 1.6 U/L, and LH and testosterone were undetectable (<0.05 U/L and <0.23 nmol/L, respectively). The boy had further virilization (PH2, genital development was G2), but testicular volume was unchanged (2 mL) and a gonadotropin-releasing hormone (GnRH) test showed a prepubertal response (peak LH 3.2 U/L and FSH 4.0 U/L).
At 8.2 years of age an increase in FSH concentration to 5.2 U/L and in LH concentration to 1.1 U/L was found. A GnRH test showed a pubertal response with stimulated FSH and LH concentrations of 7.0 and 11.1 U/L, respectively. Testicular volume increased to 3 mL and BA was advanced (BA 9.8 years at CA 8.5 years), and there was increased growth velocity of 7.5 cm/year (2.4 SDS) (Fig. 1 A). Insulin-like growth factor 1 concentrations were increased (253 μg/L, 2 SDS), androgen levels were elevated (androstenedione 0.78 nmol/L, 0.9 SDS; dehydroepiandrosterone sulfate [DHEAS] 3,812 nmol/L, 2.3 SDS) and testosterone was detectable (0.45 nmol/L, 2.5 SDS). Patient 1 was started on GnRH analog treatment (leuprorelin 11.25 mg subcutaneously every 12 weeks). The patient received treatment with GnRH analog until he was 12.3 years of age, and he is now 12.8 years of age and still prepubertal.
Fig. 1.
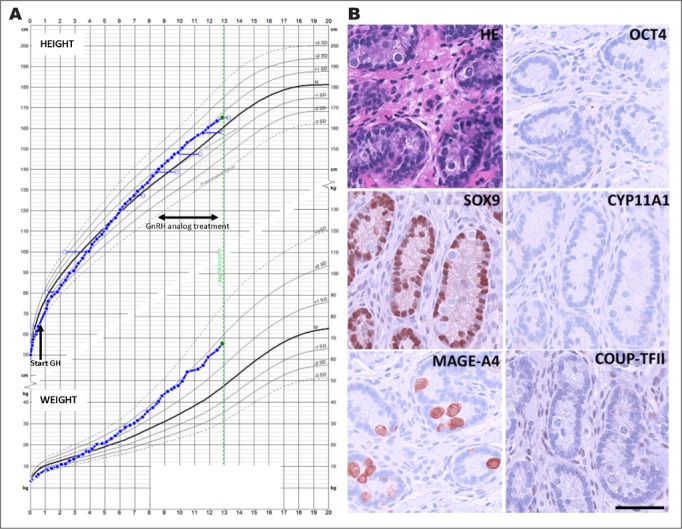
A, Growth chart for patient 1; height in the upper panel and weight in the lower panel. Age at start of growth hormone is marked by an arrow and the time span of GnRH analog treatment is marked by a double arrow. B, Evaluation of morphology and testicular cell lineage markers in patient 1 (1.2 years of age at time of biopsy). Hematoxylin eosin (HE) staining and immunohistochemical (IHC) staining of OCT4 (marker of gonocytes), MAGE-A4 (marker of spermatogonia), SOX9 (marker of Sertoli cells), CYP11A1 (marker of Leydig cells), and COUP-TFII (marker of peritubular myoid and Leydig cells). On all IHC images counterstaining is with Mayer's hematoxylin, scale bar corresponds to 100 μm. GH = growth hormone; GnRH = gonadotropin-releasing hormone.
Testicular Morphology Patient 1
Patient 1 had bilateral cryptorchidism at birth and required orchidopexy. The initial pathology evaluation of the biopsies at 1.2 years showed largely normal prepubertal testis structure with normal Sertoli and Leydig cells. A normal number of germ cells were present, without signs of malignancy. The IHC largely confirmed the morphologic findings. No OCT3/4- or AP2γ-positive gonocytes or premalignant GCNIS cells were detected. The germ cells present at this age were prespermatogonia (MAGE-A4 positive) and there were no multinucleated germ cells. The interstitial cells expressed COUP transcription factor 2 (COUP-TFII), while cytochrome P450 family 11 subfamily A member 1 (CYP11A1) expression could not be detected in Leydig cells, which is in accordance with observations in prepubertal testes at this age (9) (Fig. 1 B).
Patient 2
Patient 2 was born at 34 + 1 weeks of gestation; birth weight was 1,760 g (−2.6 SDS). PWS was diagnosed at 6 weeks. At 2 years the patient started GH therapy on PWS indication; he had a normal response to a growth hormone stimulation test.
At 6 years of age, pubic hair development (PH3) without testicular enlargement (2 mL bilaterally) was detected. BA was 6.9 years at CA of 6.1 years. Biochemistry showed normal 17-hydroxy progesterone but elevated DHEAS. An ACTH stimulation test was performed with a normal response.
At 7.2 years, the boy had PH3 and testicular volume had increased to 4 mL. A GnRH test showed a pubertal response with stimulated FSH and LH concentrations of 3.0 U/L and 8.5 U/L, respectively. Testosterone was 1.25 nmol/L and inhibin B was 198 pg/mL. BMI (SDS) was 2.9. GnRH analog treatment (leuprorelin 3.75 mg every 28 days) was started at 7.4 years of age and the patient was treated until 11.5 years of age. After cessation of treatment there was a normal progression in growth and puberty and at 14 years of age he was fully virilized with testicular volume of 10 mL (Fig. 2 A).
Fig. 2.
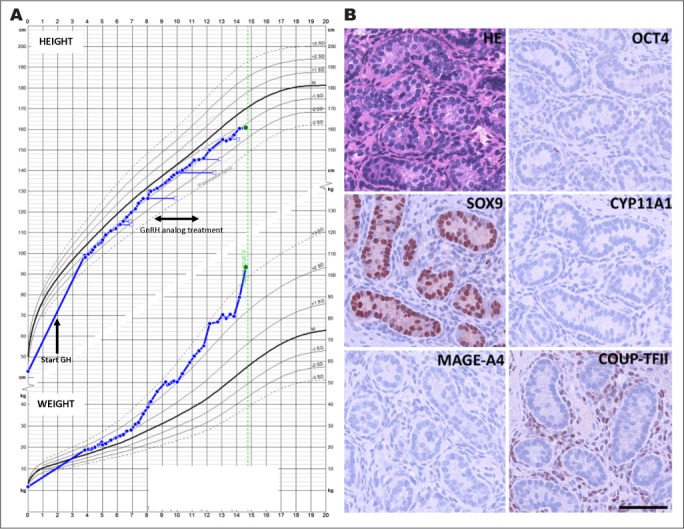
A, Growth chart for patient 2; height in the upper panel and weight in the lower panel. Age at start of growth hormone is marked by an arrow and the time span of GnRH analog treatment is marked by a double arrow. B, Evaluation of morphology and testicular cell lineage markers in patient 2 (3.5 years of age at time of biopsy). Hematoxylin eosin (HE) staining and immunohistochemical (IHC) staining of OCT4 (marker of gonocytes), MAGE-A4 (marker of spermatogonia), SOX9 (marker of Sertoli cells), CYP11A1 (marker of Leydig cells), and COUP-TFII (marker of peritubular myoid- and Leydig cells). On all IHC images, counterstaining is with Mayer's hematoxylin, scale bar corresponds to 100 μm. GH = growth hormone; GnRH = gonadotropin-releasing hormone.
Testicular Morphology Patient 2
Patient 2 had bilateral congenital cryptorchidism and required orchidopexy. The initial pathology evaluation of the biopsies at 3.5 years of age showed a reduced tubule diameter, but both Sertoli and Leydig cells appeared normal. In the right biopsy, only a few germ cells were detected, while no germ cells were observed in the left biopsy. The additional IHC analysis largely confirmed the initial morphologic findings, although no germ cells were detected (based on evaluation of OCT3/4, AP2γ, and MAGE-A4). In both biopsies, normal SOX9 expression in Sertoli cells and COUP-TFII expression in interstitial cells was observed, while no CYP11A1 expression in Leydig cells was detected as expected at this age (Fig. 2 B).
DISCUSSION
We report 2 cases of boys with PWS who developed central precocious puberty. Precocious puberty is rarely seen in children with PWS, but cases have been reported previously (4,6,10,11). More commonly delayed or incomplete puberty is seen due to the hypothalamic dysfunction and/or testicular dysfunction following cryptorchidism (2,12) and progression of hypogonadism over time has been described. Reproductive hormone production in PWS males was found to be normal during infancy, but most of the adults have a combined hypogonadism and are infertile (1,12).
Most boys with PWS have congenital bilateral cryptorchidism due to hypogonadotropic hypogonadism. This may contribute to primary testis dysgenesis due to loss of germ cells which may influence fertility later in life. Both our cases had congenital bilateral cryptorchidism and the testicular biopsies were re-evaluated. In one of the patients, germ cell loss was found; this patient had a normal progression through puberty after cessation of therapy.
The testicular biopsies in our 2 PWS cases revealed some heterogeneity, with largely normal histology in one patient, but absence of germ cells in the left testis and only few germ cells present in the right testis in the second patient. This is in agreement with previous histologic findings in PWS boys that ranged from normal morphology to reduced numbers or complete absence of spermatogonia (13,14). Reported histologic alterations in adult men with PWS include diffuse tubular hyalinization, presence of Sertoli-cell-only nodules and presence of vacuolized Leydig cells (13). These findings suggest a progressive testicular failure resulting in hypogonadism in almost all adult PWS patients. However, it is not completely clear to which extent this is due to cryptorchidism or the underlying genetic defects. Testicular dysgenesis is associated with an increased risk of germ cell neoplasia and some cases of testicular neoplasia have been reported among adult PWS patients (15–17).
Treatment with GH is approved for children with PWS independently of GH secretion. Former studies suggested that GH therapy in patients with normal GH secretion may accelerate timing of puberty and the duration of the growth spurt (7,18). The former case reports on precocious puberty in children with PWS included some patients on GH treatment (6), but not all of the reported cases were treated with GH (4,5). There is a strong interaction between GH and sex steroids and an appropriate secretion of growth hormone is essential for sexual maturation (19). In animal models injections of GH can induce a transcription factor involved in the GH signaling pathway (pSTAT5) in several brain nuclei involved in the regulation of the hypothalamic-pituitary-gonadal axis (20). Thus, GH treatment in prepubertal children may influence pubertal timing.
CONCLUSION
In conclusion, boys with PWS have a considerable heterogeneity of hypothalamic-pituitary function as well as primary testicular dysfunction. However, here we present 2 PWS boys with precocious puberty and it may be suggested that GH treatment played a role in the pubertal timing.
Abbreviations
- BA
bone age
- CA
chronologic age
- GH
growth hormone
- GnRH
gonadotropin-releasing hormone
- IHC
immunohistochemistry
- PH
pubic hair
- PWS
Prader-Willi syndrome
- SDS
standard deviation score
Footnotes
DISCLOSURE
The authors have no multiplicity of interest to disclose.
REFERENCES
- 1.Crinò A, Schiaffini R, Ciampalini P et al. Hypogonadism and pubertal development in Prader-Willi syndrome. Eur J Pediatr. 2003;162:327–333. doi: 10.1007/s00431-002-1132-4. [DOI] [PubMed] [Google Scholar]
- 2.Hirsch HJ, Eldar-Geva T, Benarroch F, Rubinstein O, Gross-Tsur V. Primary testicular dysfunction is a major contributor to abnormal pubertal development in males with Prader-Willi syndrome. J Clin Endocrinol Metab. 2009;94:2262–2268. doi: 10.1210/jc.2008-2760. [DOI] [PubMed] [Google Scholar]
- 3.Siemensma EP, de Lind van Wijngaarden RF, Otten BJ, de Jong FH, Hokken-Koelega AC. Pubarche and serum dehydroepiandrosterone sulphate levels in children with Prader-Willi syndrome. Clin Endocrinol. 2011;75:83–89. doi: 10.1111/j.1365-2265.2011.03989.x. [DOI] [PubMed] [Google Scholar]
- 4.Linnemann K, Schröder C, Mix M, Krüger G, Fusch C. Prader-Labhart-Willi syndrome with central precocious puberty and empty sella syndrome. Acta Paediatr. 1999;88:1295–1297. doi: 10.1080/080352599750030482. [DOI] [PubMed] [Google Scholar]
- 5.Crinò A, Di Giorgio G, Schiaffini R et al. Central precocious puberty and growth hormone deficiency in a boy with Prader-Willi syndrome. Eur J Pediatr. 2008;167:1455–1458. doi: 10.1007/s00431-008-0679-0. [DOI] [PubMed] [Google Scholar]
- 6.Ludwig NG, Radaeli RF, Silva MM et al. A boy with Prader-Willi syndrome: unmasking precocious puberty during growth hormone replacement therapy. Arch Endocrinol Metab. 2016;60:596–600. doi: 10.1590/2359-3997000000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crowe BJ, Rekers-Mombarg LT, Robling K et al. Effect of growth hormone dose on bone maturation and puberty in children with idiopathic short stature. J Clin Endocrinol Metab. 2006;91:169–175. doi: 10.1210/jc.2005-0891. [DOI] [PubMed] [Google Scholar]
- 8.Jørgensen A, Macdonald J, Nielsen JE et al. Nodal signaling regulates germ cell development and establishment of seminiferous cords in the human fetal testis. Cell Rep. 2018;25:1924–1937. doi: 10.1016/j.celrep.2018.10.064. [DOI] [PubMed] [Google Scholar]
- 9.Lottrup G, Nielsen JE, Maroun LL et al. Expression patterns of DLK1 and INSL3 identify stages of Leydig cell differentiation during normal development and in testicular pathologies, including testicular cancer and Klinefelter syndrome. Hum Reprod. 2014;29:1637–1650. doi: 10.1093/humrep/deu124. [DOI] [PubMed] [Google Scholar]
- 10.Lee HS, Hwang JS. Central precocious puberty in a girl with Prader-Willi syndrome. J Pediatr Endocrinol Metab. 2013;26:1201–1204. doi: 10.1515/jpem-2013-0040. [DOI] [PubMed] [Google Scholar]
- 11.Pusz ER, Rotenstein D. Treatment of precocious puberty in a female with Prader-Willi syndrome. J Pediatr Endocrinol Metab. 2008;21:495–500. doi: 10.1515/JPEM.2008.21.5.495. [DOI] [PubMed] [Google Scholar]
- 12.Radicioni AF, Di Giorgio G, Grugni G et al. Multiple forms of hypogonadism of central, peripheral or combined origin in males with Prader-Willi syndrome. Clin Endocrinol. 2012;76:72–77. doi: 10.1111/j.1365-2265.2011.04161.x. [DOI] [PubMed] [Google Scholar]
- 13.Vogels A, Moerman P, Frijns JP, Bogaert GA. Testicular histology in boys with Prader-Willi syndrome: fertile or infertile? J Urol. 2008;180:1800–1804. doi: 10.1016/j.juro.2008.03.113. [DOI] [PubMed] [Google Scholar]
- 14.Bakker NE, Wolffenbuttel KP, Looijenga LH, Hokken-Koelega AC. Testes in infants with Prader-Willi syndrome: human chorionic gonadotropin treatment, surgery and histology. J Urol. 2015;193:291–298. doi: 10.1016/j.juro.2014.07.113. [DOI] [PubMed] [Google Scholar]
- 15.Robinson AC, Jones WG. Prader Willi syndrome and testicular tumour. Clin Oncol. 1990;2:117. doi: 10.1016/s0936-6555(05)80799-x. [DOI] [PubMed] [Google Scholar]
- 16.Jaffray B, Moore L, Dickson AP. Prader-Willi syndrome and intratubular germ cell neoplasia. Med Pediatr Oncol. 1999;32:73–74. doi: 10.1002/(sici)1096-911x(199901)32:1<73::aid-mpo19>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 17.Kvist K, Clasen-Linde E, Cortes D, Petersen BL, Thorup J. Adult immunohistochemical markers fail to detect intratubular germ cell neoplasia in prepubertal boys with cryptorchidism. J Urol. 2014;191:1084–1089. doi: 10.1016/j.juro.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Clayton PE, Kamp GA, Waelkens JJ, de Muinck Keizer-Schrama SM et al. High dose growth hormone treatment induces acceleration of skeletal maturation and an earlier onset of puberty in children with idiopathic short stature. Arch Dis Child. 2002;87:215–220. doi: 10.1136/adc.87.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Juul A, Skakkebæk NE. Why do normal children have acromegalic levels of IGF-I during puberty? J Clin Endocrinol Metab. 2019;104:2770–2776. doi: 10.1210/jc.2018-02099. [DOI] [PubMed] [Google Scholar]
- 20.Furigo IC, Metzger M, Teixeira PD, Soares CR, Donato J., Jr. Distribution of growth hormone-responsive cells in the mouse brain. Brain Struct Funct. 2017;222:341–363. doi: 10.1007/s00429-016-1221-1. [DOI] [PubMed] [Google Scholar]