

Abstract
Objective:
DNA ligase IV syndrome is a rare genetic disorder characterized by pronounced radiosensitivity, growth failure, pancytopenia, hypogonadism, and immunodeficiency. Here, we describe a unique case of DNA ligase IV syndrome diagnosed in adulthood and review the endocrine manifestations of this rare disorder.
Methods:
We present detailed clinical, laboratory, and exam findings and review the relevant literature.
Results:
This patient initially presented in childhood with microcephaly, growth failure, and mild pancytopenia. At age 18, she developed secondary amenorrhea, with labs revealing hypergonadotropic hypogonadism. She was initially suspected to have Turner syndrome, but karyotype testing was normal. At age 34, genetic testing ultimately confirmed the diagnosis of DNA ligase IV syndrome.
Conclusion:
Severe growth failure and hypogonadism are important endocrine clues to the diagnosis of DNA ligase IV syndrome. The increased availability of genetic testing and whole-exome sequencing may allow for definitive diagnosis in patients that previously went unrecognized.
INTRODUCTION
DNA ligase IV (LIG4) deficiency (LIG4 syndrome) is a rare disorder associated with impaired response to DNA damage (1). The syndrome results from pathogenic variants in the gene encoding DNA ligase IV, an enzyme essential for nonhomologous end joining (NHEJ). NHEJ is required for the repair of DNA double strand breaks, which can arise from exposure to ionizing radiation (2). NHEJ is also involved in V(D)J recombination and class switch recombination (3). Patients with LIG4 syndrome have pronounced radiosensitivity (4). Clinical manifestations are heterogeneous and include microcephaly, growth failure, pancytopenia, hypogonadism, and severe combined immunodeficiency (5,6).
Here, we present a case of LIG4 syndrome diagnosed in adulthood. We propose that the endocrine manifestations of this disorder represent an important clinical clue that should prompt genetic evaluation.
CASE REPORT
Our patient was born from a full-term uncomplicated pregnancy weighing 4 pounds 3 ounces, with a length of 16 inches and head circumference of 12 inches (all <1st percentile). After delivery, she was noted to be small for gestational age, with dysmorphic facial features (rotated and posteriorly pointed ears, thin lower lip, beaked nose), but evaluation including infectious work-up and chromosome analysis did not reveal an etiology. At the age of 4, she was started on somatotropin injections for growth failure (see Fig. 1 for growth chart). She was concurrently treated with levothyroxine for borderline low thyroid studies. Her first year of treatment resulted in a steady rate of catch-up growth. However, in the subsequent years, she remained below the 1st percentile for height-for-age with normal growth velocity but exhibited no further catch-up growth. She continued on somatotropin until epiphyseal fusion at age 16. Levothyroxine was discontinued around this time, with normal thyroid studies thereafter. She achieved a final adult height of 4 feet 7 inches, corresponding to <1st percentile.
Fig. 1.
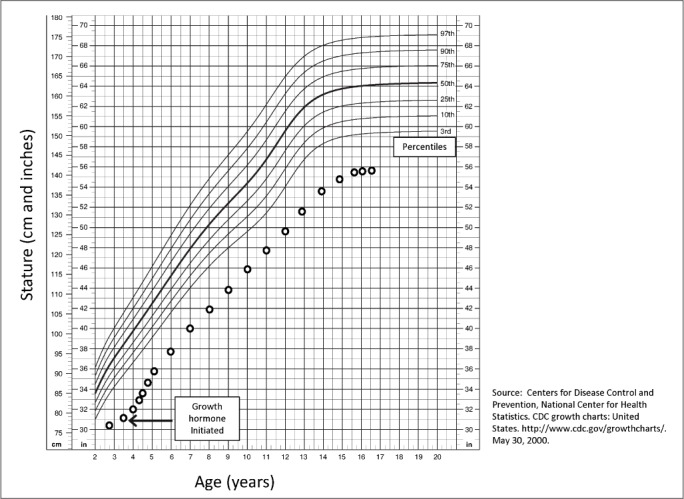
Stature-for-age growth chart.
The patient was diagnosed with secondary amenorrhea at age 15, after having 3 periods but none thereafter. Amenorrhea was initially attributed to low body weight (<1st percentile) and gymnastics training. Hormonal evaluation showed a follicle-stimulating hormone (FSH) of 14.8 mIU/mL. At the age of 18, repeat FSH was elevated at 59.8 mIU/mL, and luteinizing hormone was 21.3 mIU/mL, with low estradiol of 16 pg/mL. Pelvic magnetic resonance imaging (MRI) revealed a hypoplastic uterus, and ovaries were not well visualized. Karyotype was 46,XX. She was diagnosed with premature ovarian insufficiency. Conjugated equine estrogens were initiated at a small dose and then gradually increased with cyclic progesterone added.
Mild pancytopenia was first noted at age 16. A bone marrow biopsy demonstrated cellularity ranging from virtually acellular to approximately 30%, trilineage hematopoietic maturation, and neutropenia. A complete hematologic evaluation excluded infectious, inflammatory, or autoimmune processes. Blood counts remained borderline low, with white blood cells count ranging from 2.5 to 5.5 K/μL, absolute neutrophil count 0.5 to 4.5 K/μL, hemoglobin 10 to 12 g/dL, and platelets 90 to 130 K/μL.
The patient had her first malignancy at the age of 28, when she had a low-grade cystic mucoepidermoid carcinoma of the parotid gland resected.
The patient was diagnosed with diabetes at the age of 30, with hemoglobin A1c (HbA1c) 11.8% (105 mmol/mol). Glutamic acid decarboxylase 65 antibody was negative and C-peptide was 9.6 ng/mL when glucose was 164 mg/dL, thought to be most consistent with type 2 diabetes. However, she had no acanthosis, and her body mass index was just 19 kg/m2. Her diabetes has been managed with basal-bolus insulin therapy. Interestingly, she had a history of prediabetic and diabetic-range HbA1c measurements beginning around age 4 and peaking at 7.3% (56 mmol/mol) at age 6 before normalizing. At the time, this was attributed to her growth hormone (GH) treatments, and she was not formally diagnosed with diabetes until adulthood.
The patient had learning difficulties as a child (especially speech). For her undergraduate studies, she qualified for an Individualized Education Program. She has earned an associate's degree and is working on her bachelor's degree. She has a successful career as a paraprofessional educator.
The patient was evaluated by genetics initially at birth and again in her 30s. Work-up included a normal 46,XX peripheral karyotype with a total of 40 cells analyzed to rule out mosaicism. Additional work-up included normal chromosomal breakage analysis, ruling out Fanconi anemia and normal fragile X analysis. A chromosomal microarray analysis was negative for pathogenic chromosomal deletions and/or duplications. For further evaluation, trio-based whole-exome sequencing was performed and identified compound heterozygosity for two pathogenic variants inherited in trans in the LIG4 gene, c.2440C>T (p.R814*) and c.613delT (p.S205Lfs*29). The c.2440C>T variant was shown to be maternally inherited, with the c.613delT shown to be paternally inherited. Both variants were classified as pathogenic and have been previously reported in patients affected with LIG4 syndrome (4,7,8).
Following her diagnosis of LIG4 syndrome, she was found to have hepatocellular carcinoma. She had initially been diagnosed with hepatic steatosis at the age of 30 during work-up for mildly abnormal transaminases and alkaline phosphatase (aspartate aminotransferase, 69 IU/L [normal, 8 to 20 IU/L]; alanine transaminase, 162 IU/L [normal, 7 to 35 IU/L]; alkaline phosphatase, 254 IU/L [normal, 30 to 130 IU/L]). She later developed hepatic lesions suspicious for focal nodular hyperplasia versus hepatic adenomatosis. Due to interval growth and radiologic changes concerning for malignant transformation of a 6.0 × 5.4 cm liver lesion, she underwent laparoscopic left lateral lobectomy at age 35. Pathology confirmed well-differentiated hepatocellular carcinoma on the steatohepatitis background. Serial imaging studies showed stable hepatic adenomatosis and no recurrent cancer over the last 17 months. Figure 2 provides a timeline of the patient's clinical history.
Fig. 2.
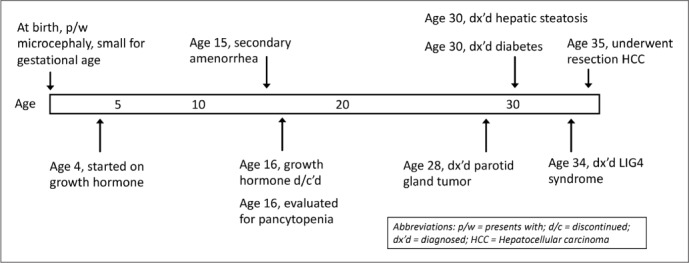
Timeline of clinical manifestations of patient's DNA ligase IV syndrome.
DISCUSSION
Approximately 28 cases of LIG4 syndrome have been described (6). Key clinical features include microcephaly, severe growth failure, distinctive facial features, severe combined immunodeficiency, and pancytopenia (3,5,6). Due to their impaired ability to repair DNA double strand breaks, patients with LIG4 syndrome are sensitive to ionizing radiation and predisposed to malignancy, particularly lymphoid malignancies. The first patient reported in the literature with LIG4 syndrome died of complications related to radiation therapy he received as part of his acute lymphoblastic leukemia treatment, and since then, at least 5 other patients with LIG4 syndrome have had malignancy as part of their clinical presentation (6). Thus, avoidance of unnecessary radiation exposure is essential (6,9). Whenever possible, clinicians should select imaging modalities without ionizing radiation, such as ultrasound and MRI, rather than X-ray, computed tomography, positron emission tomography, fluoroscopy, or mammography.
The phenotype of LIG4 syndrome is clinically heterogeneous, and patients with late truncating pathogenic variants typically have a milder phenotype than those with early truncating pathogenic variants (3). Importantly, the coding sequence for LIG4 resides solely in the terminal exon and therefore escapes nonsense-mediated decay (3,5). Biallelic null pathogenic variants though are thought to result in embryonic lethality (5). The majority of LIG4 syndrome cases to date have been diagnosed in childhood, so less is known about some of the later manifestations of the disease. Our patient, who was born several years before the first case of LIG4 syndrome was described in the literature, has many features consistent with this syndrome (Table 1). Diabetes mellitus is not an established feature of this syndrome, but our case would be the third reported patient with LIG4 syndrome who developed diabetes (4,7). LIG4 syndrome overlaps clinically with several other disorders of DNA damage response, including Nijmegen breakage syndrome, radiosensitive severe combined immunodeficiency, ataxia-telangiectasia, Bloom syndrome, and Seckel syndrome (1,8,10).
Table 1.
Clinical Features of DNA Ligase IV Syndrome
| Clinical manifestations | Feature present in current case? |
|---|---|
| Microcephaly | Yes - head circumference below 1st percentile |
| Short stature | Yes - final adult height 4 feet 7 inches |
| Pancytopenia | Yes - mild |
| Immunodeficiency | None clinically apparent |
| Hypogonadism | Yes - hypergonadotropic amenorrhea |
| Malignancy | Yes - cystic mucoepidermoid carcinoma of the parotid gland at age 28 and hepatocellular carcinoma at age 35 |
| Neurodevelopment | Attended speech therapy; had Individualized Education Program in college |
| Bone/dental abnormalities | Yes - selective tooth agenesis; no syndactyly or polydactyly |
Other diagnoses: asthma, type 2 diabetes, osteoporosis, hepatic steatosis
While LIG4 syndrome is clinically quite heterogeneous, significant growth failure is present in the vast majority of patients and is often one of the earliest manifestations of this disorder (3). Hence, this disease may be of interest to pediatric and adult endocrinologists who treat patients with growth failure. Our patient was treated with somatotropin for >10 years, despite concerns about inadequate response. It is unclear whether our patient's final adult height benefited from her somatotropin therapy. Given the high rates of malignancy in this population, the possible GH benefits should be weighed against the theoretical concerns about GH and cancer (11).
Similarly, hypergonadotropic hypogonadism is another common reason for referral to endocrinology. Hypogonadism has been described in several patients with LIG4 syndrome; however, exact estimates are difficult, as the majority of the patients described in the literature are prepubertal (6,8). A referral to genetics should be considered if syndromic causes are suspected and more common causes of premature ovarian insufficiency, such as fragile X and Turner syndrome, have been excluded.
CONCLUSION
LIG4 syndrome is a rare genetic disorder caused by pathogenic variants in the gene encoding DNA ligase IV. This enzyme is necessary for repair of double strand DNA breaks via NHEJ. Patients with LIG4 syndrome are sensitive to ionizing radiation and prone to malignancy. The disorder is clinically heterogeneous, and patients with milder phenotypes may go undetected. Early diagnosis can help patients avoid unnecessary radiation exposure, limit environmental and hepatotoxic exposures, and monitor for potential complications. Here, we presented a case of a patient with LIG4 syndrome that went undiagnosed until adulthood. Given the continued advances in genetic testing, as well as increased awareness of genetic instability syndromes, we anticipate that more patients with this disorder will be recognized and appropriately diagnosed.
Abbreviations:
- GH
growth hormone
- LIG4
DNA ligase IV
- NHEJ
nonhomologous end joining
Footnotes
DISCLOSURE
The authors have no multiplicity of interest to disclose.
REFERENCES
- 1.O'Driscoll M, Gennery AR, Seidel J, Concannon P, Jeggo PA. An overview of three new disorders associated with genetic instability: LIG4 syndrome, RS-SCID and ATR-Seckel syndrome. DNA Repair (Amst) 2004;3:1227–1235. doi: 10.1016/j.dnarep.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 2.Riballo E, Critchlow SE, Teo SH et al. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr Biol. 1999;9:699–702. doi: 10.1016/s0960-9822(99)80311-x. [DOI] [PubMed] [Google Scholar]
- 3.Murray JE, Bicknell LS, Yigit G et al. Extreme growth failure is a common presentation of ligase IV deficiency. Hum Mutat. 2014;35:76–85. doi: 10.1002/humu.22461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Driscoll M, Cerosaletti KM, Girard PM et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol Cell. 2001;8:1175–1185. doi: 10.1016/s1097-2765(01)00408-7. [DOI] [PubMed] [Google Scholar]
- 5.Chistiakov DA, Voronova NV, Chistiakov AP. Ligase IV syndrome. Eur J Med Genet. 2009;52:373–378. doi: 10.1016/j.ejmg.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 6.Altmann T, Gennery AR. DNA ligase IV syndrome; a review. Orphanet J Rare Dis. 2016;11:137. doi: 10.1186/s13023-016-0520-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stewart DR, Pemov A, Johnston JJ et al. Dubowitz syndrome is a complex comprised of multiple, genetically distinct and phenotypically overlapping disorders. PLoS One. 2014;9 doi: 10.1371/journal.pone.0098686. e98686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gruhn B, Seidel J, Zintl F et al. Successful bone marrow transplantation in a patient with DNA ligase IV deficiency and bone marrow failure. Orphanet J Rare Dis. 2007;2:5. doi: 10.1186/1750-1172-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plowman PN, Bridges BA, Arlett CF, Hinney A, Kingston JE. An instance of clinical radiation morbidity and cellular radiosensitivity, not associated with ataxia-telangiectasia. Br J Radiol. 1990;63:624–628. doi: 10.1259/0007-1285-63-752-624. [DOI] [PubMed] [Google Scholar]
- 10.Ben-Omran TI, Cerosaletti K, Concannon P, Weitzman S, Nezarati MM. A patient with mutations in DNA ligase IV: clinical features and overlap with Nijmegen breakage syndrome. Am J Med Genet A. 2005;137A:283–287. doi: 10.1002/ajmg.a.30869. [DOI] [PubMed] [Google Scholar]
- 11.Chae HW, Kim DH, Kim HS. Growth hormone treatment and risk of malignancy. Korean J Pediatr. 2015;58:41–46. doi: 10.3345/kjp.2015.58.2.41. [DOI] [PMC free article] [PubMed] [Google Scholar]