

Abstract
Objective:
Myotonic dystrophy (DM) is a monogenic disorder. It is caused by expansion of a cytosine-thymineguanine triplet in the DMPK gene which encodes for myotonic dystrophy protein kinase (DMPK).
Methods:
A 24-year-old man with DM and the DMPK mutation presented with elevated adrenocorticotropic hormone (ACTH) levels twice (152 and 185 pg/mL; normal value is 10 to 52 pg/mL) with normal cortisol levels (134.6 and 113.0 ng/mL, or 371.3 and 311.7 nmol/L; normal values are 67 to 226 ng/mL or 184.8 to 623.5 nmol/L). ACTH, corticotropin-releasing hormone (CRH) and insulin tolerance test (ITT) demonstrated normal cortisol response to ACTH and partial response to CRH and ITT tests, and ACTH hyperresponse to CRH and ITT. We suspected ACTH and/or ACTH receptor (ACTHR) mutations and evaluated the genetic profile for pro-opiomelanocortin (POMC), melanocortin 2 receptor (MC2R) and follicle-stimulating hormone receptor (FSHR) genes.
Results:
No mutations were found in either the MC2R or FSHR genes. The patient was heterozygous for the c.614A>G mutation corresponding to a p.53D>G substitution with a glycine instead of an aspartate in position 53 in POMC gene. This mutation was outside the sequence for ACTH (which spans amino acids 138 to 176) but was included in the part originating the N-terminal peptide of pro-opiomelanocortin (also called pro-γ-melanocyte stimulating hormone) which spans amino acids 27 to 102 and is involved in the regulation of adrenal steroidogenesis.
Conclusion:
The pathologic expansion of the cytosine-thymine-guanine triplet repeat in the 3' noncoding region of DMPK could explain the hyperresponse of ACTH typical of DM. The mutation of pro-γ-melanocyte-stimulating hormone could be associated with the abnormal response of cortisol, compatible with a partial adrenal insufficiency. Other studies are necessary to demonstrate this hypothesis.
INTRODUCTION
Myotonic dystrophy (DM), or Steinert myotonic dystrophy, is one of the most common lethal monogenic disorders. It is caused by expansion of a cytosine-thymineguanine (CTG) triplet repeat in the 3' noncoding region of the myotonic dystrophy protein kinase gene (DMPK). In the general population, the number of CTG repeats in this gene is between 5 to 37 while individuals with DM type 1 have at least 50 and in some cases >3,000 CTG repeats. A second form of the disease known as DM type 2 has been recently recognized. This second form results in a CCTG repeat in the first intron of zinc finger 9 (1).
The function of DMPK, the effects of CTG expansion on DMPK levels or structure, and the mechanisms leading to abnormalities in DM remain unclear. The multiple protein isoforms arising from the DMPK gene derive from alternative splicing (2). Moreover, the number of CTG repeats varies from tissue to tissue due to somatic mosaicism, thus explaining the wide phenotypic variation seen in DM (3).
The spectrum of DM type 1 symptoms extends from lethal effects in infancy to mild, late-onset symptoms. The most frequent symptom is myotonia, usually more pronounced after rest, which improves after muscle activity (the so-called “warm-up phenomenon”). Myotonia selectively involves specific muscle groups of the forearm, hand, tongue, and jaw. Most frequent systemic symptoms are cardiac dysrhythmia (heart block particularly) (4), ocular symptoms (the most frequent is cataract), and neurological symptoms (daytime hypersomnolence and behavioral and cognitive changes like anxiety, avoidant behavior, and apathy) (5). Other systemic features are gastrointestinal symptoms (cholelithiasis and intestinal dysmotility) (6), higher risk of cancer (7,8), insulin resistance and dyslipidemia, and abnormal liver function (9). Primary hypogonadism is the most common endocrine alteration present in DM type 1, characterized by testicular atrophy, reduced fertility, erectile dysfunction, and low testosterone levels (10).
Some studies have reported that some DM type 1 patients have altered hypothalamic-pituitary-adrenal axes, even in the absence of adrenal insufficiency symptoms, like increased adrenocorticotropic hormone (ACTH) response to corticotropin-releasing hormone (CRH) or insulin tolerance test (ITT) (11,12), or hyperresponse of ACTH after fenfluramine or naloxone test (13,14).
Here, we report the case of a young man affected by DM type 1 with elevated ACTH levels in the absence of adrenal insufficiency symptoms with a variant in his pro-opiomelanocortin (POMC) gene coding for pro-γmelanocyte-stimulating hormone (MSH).
CASE REPORT
Search for Genetic Alterations
A blood sample was obtained after informed consent in accordance with local ethical committee guidelines. Genomic DNA was extracted from peripheral blood leukocytes using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). DNA concentration was assessed with NanoDrop One (Thermo Fisher Scientific, Waltham, MA). The entire coding regions of POMC, melanocortin 2 receptor (MC2R, also known as ACTH receptor (ACTHR)), and follicle-stimulating hormone receptor (FSHR) genes were analyzed using end-point polymerase chain reaction followed by denaturing high-performance liquid chromatography and direct sequencing. Primer sequences and polymerase chain reaction conditions are available upon request.
Biochemical Tests
Serum thyroid-stimulating hormone, free triiodothyronine, and free thyroxine were measured by chemiluminescent immunometric assay (Immulite 2000; Siemens Medical Solutions Diagnostics, Los Angeles, CA). Cortisol levels were measured by immunoassay method using the Access Cortisol kit (Beckman Coulter, Inc., Brea, CA) with Beckman Coulter UniCel DxI 800 automatic analyzer. ACTH levels were measured by the immunometric chemiluminescence method with the Immulite 2000 XPI (Siemens AG, Munich, Germany) automatic analyzer using the ACTH kit (ACT Immulite 2000 systems; Siemens). Follicle-stimulating hormone, luteinizing hormone, and testosterone were evaluated using chemiluminescence performed with a commercial kit (Beckman Coulter).
Dynamic Tests
Insulin Tolerance
Intravenous insulin was administered (0.1 units/kg) with the intention of inducing hypoglycemia. Symptomatic hypoglycemia, with blood glucose values below 40 mg/dL, is required to evoke a reliable central stress response with activation of the hypothalamic-pituitary-adrenal axis. Measurements were obtained at 0, 30, 45, 60, 90 and 120 minutes with serum cortisol levels >200 ng/mL (>550 nmol/L) indicating a normal response.
Adrenocorticotropic Hormone
This test evaluates the ability of the adrenal cortex to produce cortisol after stimulation by synthetic ACTH. The test was performed in the morning and 250 μg of Synacthen (which contains tetracosactide, a synthetic ACTH) was administered intravenously. Measurements were obtained at 0, 30, and 60 minutes with serum cortisol levels >200 ng/mL (>550 nmol/L) indicating a normal response.
Corticotropin-Releasing Hormone
Human CRH at a dose of 1.0 μg/kg body weight was injected intravenously as a bolus over 30 seconds. Measurements were obtained at 0, 30, 45, 60, 90, and 120 minutes. Baseline ACTH increases 2 to 4-fold within 30 to 60 minutes of CRH administration. Plasma cortisol typically peaks at >200 ng/mL (>550 nmol/L) within the same period.
Results
A 24-year-old man received the diagnosis of DM type 1 in 2014, after the appearance of myotonic myopathy. DMPK mutation was identified with a CTG amplification between 170 and 500 repetitions. Cardiac function was normal except for sinus bradycardia.
The patient came to our attention because the evidence of elevated follicle-stimulating hormone levels (29.8 mU/mL; normal value is 0.7 to 11.0 mU/mL) with normal testosterone levels (6.35 ng/mL; normal value is 2.7 to 10.9 ng/mL). His thyroid function, glycemia, insulin, hemoglobin A1c, lipid levels, and liver function were all normal (Table 1). Based on the literature, we also measured ACTH and cortisol levels at 8:00 am. ACTH levels, in 2 different samples, were 152 and 185 pg/mL (normal value is 10 to 52 pg/mL) with normal cortisol levels of 134.6 and 113 ng/mL, equal to 371.3 and 311.7 nmol/L (normal value is 67 to 226 ng/mL, or 184.8 to 623.5 nmol/L). His aldosterone and plasma renin activity levels were also normal. As described above, measurements of ACTH and CRH and an ITT were performed. The patient demonstrated a normal cortisol response to ACTH, partial responses to CRH and ITT, and ACTH hyperresponse to CRH and ITT (Table 2).
Table 1.
Hormonal Results
| Patient's value | Reference range | |
|---|---|---|
| Aldosterone | 86 pg/mL | 70–295 pg/mL |
| Follicle-stimulating hormone | 29.9 mU/mL | 0.7–11 mU/mL |
| Free thyroxine | 11.4 pg/mL | 5.80–16.40 pg/mL |
| Free triiodothyronine | 3.8 pg/mL | 2.5–3.9 pg/mL |
| Insulin | 5.3 IU/mL | 2.6–34.9 IU/mL |
| Luteinizing hormone | 7.5 mU/mL | 0.8–8.0 mU/mL |
| Parathyroid hormone | 23 pg/mL | 10–60 pg/mL |
| Renin | 3.2 ng/mL/hour | 0.4–3.2 ng/mL/hour |
| Testosterone | 6.63 ng/mL | 2.7–10.9 ng/mL |
| Thyroid-stimulating hormone | 1.43 μU/mL | 0.4–4.0 μU/mL |
Table 2.
Test Results
| Time (minutes) | ACTH test (patient) | Corticotropin-releasing hormone test | Insulin tolerance test (patient) | |||||
|---|---|---|---|---|---|---|---|---|
| Cortisol (ng/mL) | Cortisol (ng/mL) | ACTH (pg/mL) | Cortisol (ng/mL) | ACTH (pg/mL) | Glycemia (mg/dL) | |||
| Patient | Father | Patient | Father | |||||
| 0 | 130.3 | 105.6 | 75.4 | 174.0 | 23.3 | 147.3 | 87.3 | 81 |
| 15 | - | 119.3 | 98.0 | 492.0 | 38.9 | - | - | - |
| 30 | 158.2 | 126.6 | 137.7 | 383.0 | 53.4 | 117.5 | 62.1 | 8 |
| 45 | - | - | - | - | - | 163.6 | 617.0 | 33 |
| 60 | 197.2 | 140.1 | 134.8 | 321.0 | 43.2 | 164.5 | 566.0 | 32 |
| 90 | - | - | - | - | - | 160.7 | 447.0 | 34 |
| 120 | - | 144.9 | 76.2 | 177.0 | 22.0 | 188.6 | 451.0 | 42 |
Abbreviation: ACTH = adrenocorticotropic hormone.
Due to the elevated basal and stimulated ACTH levels associated with normal cortisol response, we suspected ACTH and/or ACTHR mutations and evaluated the genetic profiles of POMC, MC2R, and FSHR genes. No mutations were found in either the MC2R or FSHR genes. However, the patient was heterozygous for the c.614A>G (rs28932470) mutation corresponding to a p.53D>G substitution with a glycine instead of an aspartate in position 53 in POMC gene (Fig. 1 A). The reported minor allele frequency is 0.2 to 0.3% in the general population in the Ensembl and Genecard databases (Fig. 1 B).
Fig. 1.
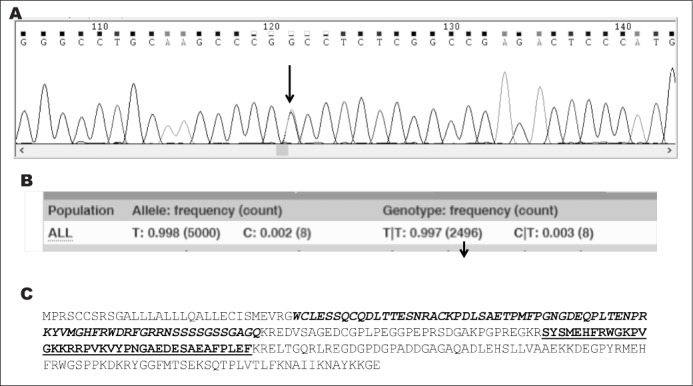
A, electropherogram showing the site of mutation (arrow). As indicated in the picture, the patient is heterozygous for c.614A>G. B, minor allele and genotype frequencies reported for the general population (Ensembl database). C, Sequence of pro-opiomelanocortin protein. In bold italic is shown the N-terminal peptide of pro-opiomelanocortin sequence with the arrow indicating the point of mutation. The sequence for adrenocorticotropic hormone is in bold.
Given that POMC is the precursor of several peptides including ACTH, we checked whether the p.53D>G modification could affect the ACTH protein sequence. As shown in Figure 1 C, this mutation was outside the sequence for ACTH (amino acids 138 to 176) but was included in the part originating the N-terminal peptide of pro-opiomelanocortin, also called pro-γ-MSH (amino acids 27 to 102). Modeling of the D53G mutation was performed using the Swiss model server (http://swissmodel.expasy.org/) and the molecular visualization system Pymol (https://pymol.org/2/) from the crystal structure of the normal protein (Fig. 2). The predicted phenotype for the variant was benign (PolyPhen software) and no splice sites were found to be associated (Human Splicing Finder version 3.1).
Fig. 2.
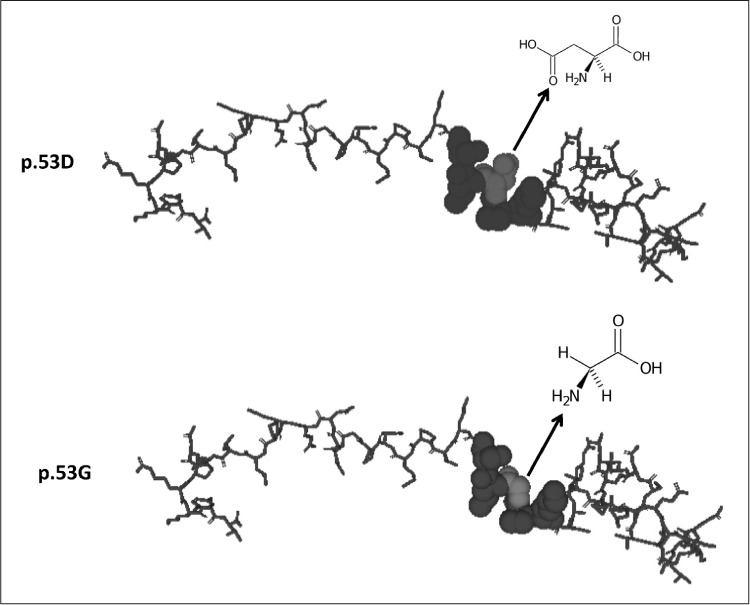
Modelling of the protein with aspartate (D; wild type) or glycine (G) at position 53. Aspartate, glycine, and surrounding amino acids are represented as spheres to show their interactions.
The parents were also analyzed for the c.614A>G mutation. The patient's mother was found to be wild type, whereas the father carried the same alteration. The parents had normal basal levels of cortisol and ACTH; the father's cortisol was 130.4 ng/mL (359.7 nmol/L) and ACTH was 33.2 pg/mL, the mother's cortisol 140 ng/mL (386.2 nmol/L) and ACTH was 24 pg/mL. His father was also submitted to a CRH-stimulation test which showed partial response of cortisol without ACTH hyperresponse (Table 2).
DISCUSSION
Diabetes mellitus and primary hypogonadism are endocrine manifestations typically seen in patients with DM type 1. In a few patients, abnormalities of the hypothalamic-pituitary axis have also been reported (11–14). Earlier studies in DM type 1 patients showed an increased ACTH response to exogenous CRH and endogenous CRH induced by naloxone, fenfluramine, or insulin-induced hypoglycemia, concluding that the ACTH hypersecretion is due to a defect in the interaction between CRH and pituitary corticotropin receptor or subsequent transduction.
Our patient had elevated ACTH levels in the absence of adrenal insufficiency symptoms and a mutation (p.D53G) in pro-γ-MSH (amino acids 27 to 102). It is known that γ-MSH has synergistic properties on the action of ACTH both in vitro and in vivo (15,16). During the stress response, ACTH and γ-MSH appear to coordinate the steroidogenic response of the adrenal gland. The γ-MSH peptide potentiates the response of ACTH by increasing the activity of the enzyme lipase, breaking down cholesterol esters to free cholesterol (17) and acting on adrenal mitogenesis and growth (18,19). Effects of γ-MSH have also been reported on the cardiovascular system, a pressure effect concomitant with an increase of heart rhythm and natriuresis.
Given the effects of γ-MSH, a morphological defect in this peptide might have a role in abnormalities of the pituitary-adrenal axis and cardiovascular system. Although the predicted phenotype for the p.D53G variant was benign, we can hypothesized from the 3-dimensional model of the protein that the substitution of aspartate with glycine may destabilize the protein. Aspartate is an α-amino acid with an acidic side chain (CH2COOH) which can react with other amino acids or enzymes. On the other hand, glycine is the is the simplest amino acid with only a hydrogen atom as its side chain. Due to their structural differences, we can hypothesize that the 53G variant differently interacts with other amino acids in the tridimensional structure and with other proteins, enzymes, or receptors compared to the wild type. Thus, the protein carrying the 53G variant may have a partial or diminished effect on its receptors, contributing to the clinical phenotype of DM characterized by endocrine dysfunctions and cardiac anomalies (including conduction block, tachyarrhythmia, sudden cardiac mortality, myocardial disease, or ischemic heart disease).
However, the ACTH hyperresponse is probably linked also to the pathologic expansion of the CTG triplet in the 3' noncoding region of DMPK. In normal individuals, CRH initiates ACTH release by binding to specific receptors on pituitary corticotropes, where cAMP-activated protein kinase-A is the essential link for the subsequent response cascade. Thus, it is possible to speculate that the DM mutation impacts the kinase pathway, leading to defective CRH-mediated signal transduction. Hockings et al (11) speculated on the abnormality of dihydropyridine-insensitive calcium transport in the corticotropes through experiments using nifedipine and naloxone, suggesting an association between DMPK and the regulation of voltage-dependent calcium channels.
Our patient and his father have different ACTH secretion responses (hypersecretion and normal secretion, respectively) but they have the same partial cortisol response to CRH. The absence of hyperresponse in the father, who shared the same mutation in pro-γ-MSH, remains difficult to explain. One hypothesis is that the p.53D>G substitution only partly affects the response of an adrenal gland to CRH. Another hypothesis could be because of the effect on adrenal growth, which could cause a primary insufficiency (19). The combination of p.53D>G in pro-γ-MSH, together with a pathologic expansion of the CTG triplet in DMPK (which was absent in the father), could be responsible for the abnormal response of the hypothalamic-pituitary-adrenal axis observed in our patient, characterized by ACTH hyperresponse and cortisol partial response to CRH.
One study on the pituitary-adrenal axis in DM patients showed differing results; some patients had normal ACTH and cortisol responses while others had hyperresponsive ACTH but normal cortisol while still others showed inappropriately low cortisol responses independently of normal or supranormal ACTH responses (20). It would be interesting to determine the genetic profile in these subgroups of DM patients to better understand the different hormonal patterns.
It has been demonstrated that endocrine abnormalities increase over time in patients with DM (21). Thus, we have suggested our patient undergo annual monitoring of hormonal status. In particular, because of the partial cortisol response to the CRH and ITT tests, we recommended to immediately report any high-stress incidents (such as illness, infection, fever, accidents, operations, or dental or medical procedures) and to keep hydrocortisone on hand for emergencies.
CONCLUSION
The pathologic expansion of the CTG triplet repeat in the 3' noncoding region of DMPK could explain the hyperresponse of ACTH typical of DM patients. The resulting mutation in pro-γ-MSH could be associated with the abnormal response of cortisol, compatible with partial adrenal insufficiency. Other studies are necessary to demonstrate this hypothesis in DM patients.
Abbreviations:
- ACTH
adrenocorticotropic hormone
- CRH
corticotropin-releasing hormone
- DM
myotonic dystrophy
- DMPK
myotonic dystrophy protein kinase
- FSHR
follicle-stimulating hormone receptor
- ITT
insulin tolerance test
- MC2R
melanocortin 2 receptor
- MSH
melanocyte-stimulating hormone
- POMC
pro-opiomelanocortin
Footnotes
DISCLOSURE
The authors have no multiplicity of interest to disclose.
REFERENCES
- 1.Turner C, Hilton-Jones D. Myotonic dystrophy: diagnosis, management and new therapies. Curr Opin Neurol. 2014;27:599–606. doi: 10.1097/WCO.0000000000000128. [DOI] [PubMed] [Google Scholar]
- 2.Forner F, Furlan S, Salvatori S. Mass spectrometry analysis of complexes formed by myotonic dystrophy protein kinase (DMPK) Biochim Biophys Acta. 2010;1804:1334–1341. doi: 10.1016/j.bbapap.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 3.Yum K, Wang ET, Kalsotra A. Myotonic dystrophy: disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes. Curr Opin Genet Dev. 2017;44:30–37. doi: 10.1016/j.gde.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McNally EM, Sparano D. Mechanisms and management of the heart in myotonic dystrophy. Heart. 2011;97:1094–1100. doi: 10.1136/hrt.2010.214197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serra L, Silvestri G, Petrucci A et al. Abnormal functional brain connectivity and personality traits in myotonic dystrophy type 1. JAMA Neurol. 2014;71:603–611. doi: 10.1001/jamaneurol.2014.130. [DOI] [PubMed] [Google Scholar]
- 6.Cardani R, Mancinelli E, Saino G, Bonavina L, Meola G. A putative role of ribonuclear inclusions and MBNL1 in impairment of gallbladder smooth contractility with cholelithiasis in myotonic dystrophy type 1. Neuromuscul Disord. 2008;18:641–645. doi: 10.1016/j.nmd.2008.06.366. [DOI] [PubMed] [Google Scholar]
- 7.Gadalla SM, Pfeiffer RM, Kristinsson SY et al. Quantifying cancer absolute risk and cancer mortality in the presence of competing events after a myotonic dystrophy diagnosis. PLoS One. 2013;8 doi: 10.1371/journal.pone.0079851. e79851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gadalla SM, Hilbert JE, Martens WB et al. Pigmentation phenotype, photosensitivity and skin neoplasms in patients with myotonic dystrophy. Eur J Neurol. 2017;24:713–718. doi: 10.1111/ene.13276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalafateli M, Triantos C, Tsamandas A, Kounadis G, Labropoulou-Karatza C. Abnormal liver function tests in a patient with myotonic dystrophy type 1. Ann Hepatol. 2012;11:130–133. [PubMed] [Google Scholar]
- 10.Peric S, Nisic T, Milicev M et al. Hypogonadism and erectile dysfunction in myotonic dystrophy type 1. Acta Myol. 2013;32:106–109. [PMC free article] [PubMed] [Google Scholar]
- 11.Hockings GI, Grice JE, Crosbie GV, Walters MM, Jackson RV. Altered hypothalamic-pituitary-adrenal axis responsiveness in myotonic dystrophy: in vivo evidence for abnormal dihydropyridine-insensitive calcium transport. J Clin Endocrinol Metab. 1993;76:1433–1438. doi: 10.1210/jcem.76.6.8388880. [DOI] [PubMed] [Google Scholar]
- 12.Johansson A, Andrew R, Forsberg H, Cederquist K, Walker BR, Olsson T. Glucocorticoid metabolism and adrenocortical reactivity to ACTH in myotonic dystrophy. J Clin Endocrinol Metab. 2001;86:4276–4283. doi: 10.1210/jcem.86.9.7865. [DOI] [PubMed] [Google Scholar]
- 13.Grice JE, Jackson J, Penfold PJ, Jackson RV. Adrenocorticotropin hyperresponsiveness in myotonic dystrophy following oral fenfluramine administration. J Neuroendocrinol. 1991;3:69–73. doi: 10.1111/j.1365-2826.1991.tb00241.x. [DOI] [PubMed] [Google Scholar]
- 14.Joyner JM, Grice JE, Hockings GI et al. Inhibition of naloxone-stimulated adrenocorticotropin release by alprazolam in myotonic dystrophy patients. J Neuroendocrinol. 1998;10:391–395. doi: 10.1046/j.1365-2826.1998.00220.x. [DOI] [PubMed] [Google Scholar]
- 15.Lowry P. 60 years of POMC: purification and biological characterisation of melanotrophins and corticotrophins. J Mol Endocrinol. 2016;56:T1–T12. doi: 10.1530/JME-15-0260. [DOI] [PubMed] [Google Scholar]
- 16.Samuels ME, Gallo-Payet N, Pinard S et al. Bioinactive ACTH causing glucocorticoid deficiency. J Clin Endocrinol Metab. 2013;98:736–742. doi: 10.1210/jc.2012-3199. [DOI] [PubMed] [Google Scholar]
- 17.Privalle CT, McNamara BC, Dhariwal MS, Jefcoate CR. ACTH control of cholesterol side-chain cleavage at adrenal mitochondrial cytochrome P-450scc. Regulation of intramitochondrial cholesterol transfer. Mol Cell Endocrinol. 1987;53:87–101. doi: 10.1016/0303-7207(87)90195-x. [DOI] [PubMed] [Google Scholar]
- 18.Estivariz FE, Carino M, Lowry PJ, Jackson S. Further evidence that N-terminal pro-opiomelanocortin peptides are involved in adrenal mitogenesis. J Endocrinol. 1988;116:201–206. doi: 10.1677/joe.0.1160201. [DOI] [PubMed] [Google Scholar]
- 19.Bicknell AB, Lomthaisong K, Woods RJ et al. Characterization of a serine protease that cleaves pro-gamma-melanotropin at the adrenal to stimulate growth. Cell. 2001;105:903–912. doi: 10.1016/s0092-8674(01)00403-2. [DOI] [PubMed] [Google Scholar]
- 20.Grice JE, Jackson RV, Hockings GI, Torpy DJ, Walters MM, Crosbie GV. Adrenocorticotropin hyperresponse to the corticotropin-releasing hormone-mediated stimulus of naloxone in patients with myotonic dystrophy. J Clin Endocrinol Metab. 1995;80:179–184. doi: 10.1210/jcem.80.1.7829609. [DOI] [PubMed] [Google Scholar]
- 21.Dahlqvist JR, Ørngreen MC, Witting N, Vissing J. Endocrine function over time in patients with myotonic dystrophy type 1. Eur J Neurol. 2015;22:116–122. doi: 10.1111/ene.12542. [DOI] [PubMed] [Google Scholar]