

Abstract
Objective:
Clinical evidence has linked low HDL cholesterol levels with high cardiovascular disease risk; however, its significance as a therapeutic target remains unestablished. We hypothesize that HDLs functional heterogeneity is comprised of metabolically distinct proteins, each on distinct HDL sizes and that are affected by diet.
Approach and Results:
Twelve participants were placed on two healthful diets high in monounsaturated fat or carbohydrate. After 4 weeks on each diet, participants completed a metabolic tracer study. HDL was isolated by apolipoprotein (Apo) A1 immunopurification and separated into 5 sizes. Tracer enrichment and metabolic rates for 8 HDL proteins – ApoA1, ApoA2, ApoC3, ApoE, ApoJ, ApoL1, ApoM, LCAT – were determined by parallel reaction monitoring and compartmental modeling, respectively. Each protein had a unique, size-specific distribution that was not altered by diet. However, carbohydrate, when replacing fat, increased the fractional catabolic rate of ApoA1 and ApoA2 on alpha3 HDL; ApoE on alpha3 and alpha1 HDL; and ApoM on alpha2 HDL. Additionally, carbohydrate increased the production of ApoC3 on alpha3 HDL and ApoJ and ApoL1 on the largest alpha0 HDL. LCAT was the only protein studied that diet did not affect. Finally, global proteomics showed that diet did not alter the distribution of the HDL proteome across HDL sizes.
Conclusions:
This study demonstrates that HDL in humans is composed of a complex system of proteins, each with its own unique size distribution, metabolism, and diet regulation. The carbohydrate-induced hypercatabolic state of HDL proteins may represent mechanisms by which carbohydrate alters the cardioprotective properties of HDL.
Keywords: high-density lipoprotein, apolipoprotein, metabolism, proteome, diet
Subject Terms: metabolism, proteomics, diet and nutrition, lipids and cholesterol, clinical studies
Graphical Abstract
HDL size and protien-based metabolic structure of the HDL particle system and the metabolic pathways increased when dietary carbohydrate replaces monounsaturated fat.
INTRODUCTION
Epidemiologic studies have established that high HDL-cholesterol (HDL-C) is associated with decreased risk of coronary heart disease (CHD).1, 2 Drug treatments that raise HDL-C, however, have failed to show beneficial effects in CHD prevention.3-5 The usefulness of HDL-C as a target for treatment has thus been put into question. We and others have demonstrated that HDL is a complex population of lipoproteins that vary in lipid and protein content as well as in particle size.6-12 Raising total HDL levels may thus not necessarily indicate that drugs enhance its beneficial effects. HDL contains over 100 proteins with diverse biological functions, such as those pertaining to cholesterol transport, immunity, antioxidation, hemostasis, and protease inhibition.6-9 HDL particles range in size from very small discoidal, cholesterol ester-poor prebeta to larger, spherical, cholesterol ester-rich alpha particles.8, 10, 11 Each of these sizes has its own distinct proteome, and proteins with similar functions tend to localize on the same HDL size particles.7-9
HDL metabolism is studied by labeling ApoA1 using radioisotopes or the stable isotope tracer trideuterated leucine (D3-Leu). The kinetic parameters, fractional catabolic rate (FCR) and production rate, are typically determined for HDL using plasma total ApoA1. Only a few studies have analyzed ApoA1 HDL metabolism by size, primarily large and small HDL13-15, or analyzed the metabolism of other HDL proteins in different HDL sizes.8 We recently developed a novel mass spectrometry (MS) strategy based on parallel reaction monitoring (PRM) to study the metabolism not only of ApoA1 across 5 HDL sizes but also of several additional HDL proteins simultaneously.8, 16, 17 Our compartmental modeling confirmed the findings of Mendivil et al.10, showing that the majority of ApoA1 on each alpha HDL size originates from the liver, which in turn is cleared from that size. A similar metabolic structure was seen for the other HDL proteins monitored.8 In the present study, we further leveraged this platform to determine the metabolism of several additional HDL proteins and the impact of an intervention, dietary fat and carbohydrate, on the HDL system’s metabolic structure.
Diets high in unsaturated fat, and low in saturated fat and carbohydrate, are continually being shown to improve lipoprotein risk factors, and reduce CHD risk.18 Carbohydrate, when replacing saturated fat, decreases plasma HDL-C and ApoA1 by decreasing total plasma ApoA1 production rate19-21; however, how carbohydrate, when replacing unsaturated fat, alters HDL metabolism is not clear.22, 23 Additionally, a diet high in carbohydrate and low in saturated fat decreases the proportion in plasma of large and increases small HDL,24 but no study has analyzed how dietary fat and carbohydrate alter the metabolism to cause these changes.
The primary goal of this study was to further elucidate the protein and size-based metabolic structure of the HDL particle system and how two healthful diets high in monounsaturated fat or carbohydrate alter this structure. The secondary goal was to determine how diet alters the composition and relative distribution of the entire HDL proteome across HDL size. Thus, we determined the metabolism of 8 HDL proteins as well as the proteome composition and distribution over 5 HDL sizes under two dietary conditions. The 8 HDL proteins included ApoJ, ApoL1, and lecithin-cholesterol acyltransferase (LCAT), whose metabolism has not been previously determined, as well as ApoA1, ApoA2, ApoC3, ApoE, and ApoM across the 5 HDL sizes. Overall, studying HDL protein metabolism and distribution across HDL size may increase our understanding of how these proteins modulate HDL function, and show how an intervention may affect these functions.
MATERIALS AND METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Participant recruitment
For this study, we analyzed plasma samples from 12 participants with low HDL-C (≤55 mg/dl for females, ≤45 mg/dl for men), and who were overweight or obese (body mass index (BMI) >25 kg/m2) (Supplemental Table I) (Clinicaltrials.gov ). Exclusion criteria included high LDL-cholesterol (LDL-C) (>190 mg/dl), very low HDL-C (<20 mg/dl), very high fasting triglyceride (TG) (>500 mg/dl), ApoE genotypes E2E2, E2E4, or E4E4, use of medications that can alter lipid levels, secondary hyperlipidemia, hepatic or renal complications, diabetes mellitus, and pregnancy.
Participants were screened for eligibility at Brigham and Women’s Hospital Center for Clinical Investigation (BWH CCI) nutrition research unit. Those who were eligible and agreed to participate in the study gave written informed consent. This study was approved by the Institutional Review Board of BWH and Harvard T.H. Chan School of Public Health.
Diet intervention
The 12 participants consumed two healthy diets, one high in fat, mainly monounsaturated derived from vegetable oil and nuts, and one high in carbohydrate, mainly complex derived from whole grains, in a randomized crossover design (Figure 1). Each diet was consumed for 32 days with a 3-week washout period between diets where participants consumed their own self-selected foods. Each diet adhered to the Institute of Medicine Dietary Reference Intake guidelines for healthy nutrient intake (http://ods.od.nih.gov/Health_Information/Dietary_Reference_Intake.aspx) and was formulated by BWH CCI. All food and beverages were provided for the duration of the study. Intake of alcoholic beverages was not permitted. Calories were adjusted to compensate for any complaints of hunger or satiety or changes in body weight.
Figure 1. Overview of the diet and HDL study design.

Twelve participants were placed on two controlled diets in a randomized crossover design (A). At the end of each diet period, each participant was infused with D3-Leu tracer and blood was collected for 70 hours (B). HDL was isolated and separated by size (C). For participants #1-6, HDL was analyzed by PRM mass spectrometry to detect D3-Leu tracer, and tracer quantification was automated by extracted PRM peak intensity (XPI) software (D, top panel). Compartmental modeling was then used to analyze the metabolism of HDL proteins on each HDL size (D, top panel). For participants #1-12, the HDL proteome was determined for each HDL size by data-dependent acquisition (DDA) mass spectrometry (D, bottom panel). IAC, immunoaffinity column chromatography; ND-PAGE, non-denaturing polyacrylamide gel electrophoresis.
Tracer infusion protocol
The 12 participants consumed each diet for 28 days prior to the kinetic study, and 4 days during the kinetic study. On the morning of day 28, participants were admitted to BWH CCI where they received an intravenous bolus injection of D3-Leu at a concentration of 10 mg/kg over 10 minutes. Blood was sampled before the injection (time 0 hour), and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 14, 16, 18, 22, 46, and 70 hours post-infusion. After the 22 hour sample was collected, participants were discharged. The 46 and 70 hour blood samples were collected at the ambulatory CCI.
Total plasma leucine tracer enrichment quantification
Total plasma leucine (D3-Leu labeled and endogenous) was isolated from 0.2 ml of plasma from time points 0, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 14, 16, 18, 22, 46, and 70 hours post-infusion using an AG 50W-X8 cation exchange resin (Bio-Rad, USA). Amino acids were dried under nitrogen, derivatized to heptafluorobutiric acid esters, and measured using gas chromatography MS (Agilent 6890 GC/5973 MS, USA). Total plasma leucine tracer enrichment was quantified for participants #1-6 on both diets by taking the area under the curve (AUC) of the tracer (D3-Leu) divided by the AUC of total plasma leucine (D3-Leu tracer + D0-Leu tracee).
HDL preparation, native gel electrophoresis, and proteolysis
Immediately after each blood sample collection, plasma was isolated by refrigerated centrifugation, aliquoted, and stored at −80°C. HDL was isolated from 1-15 time points per participant per diet. Underlined values indicate time points isolated only on the high fat diet, and values in parenthesis indicated time points isolated only on the high carbohydrate diet.
Participant 1: 0.5, 2, 4, 6, 12, 18, 22, 70 hours
Participant 2: (0.5), 1.5, 2, 3, 4, 6, (8), 10, 12, 14, 16, (18), 22, 70 hours
Participant 3: 0, 2, 4, 6, 12, 18, 22, 70 hours
Participant 4: 0, 0.5, 1, 1.5, 2, 4, 6, 8, 10, 12, 14, (18), 22, 70 hours
Participant 5: (0), 0.5, 2, 4, 6, 12, 18, 22, 70 hours
Participant 6: 0.5, 1, 1.5, 2, 3, 4, 6, 10, 12, 14, 16, 18, 22, 70 hours
Participants 7-12: 2 hour
HDL was purified from 1 ml of plasma by overnight incubation with anti-ApoA1 immunoglobulin (Academy Biomedical) bound to sepharose 4B resin (Academy Biomedical). The unbound fraction was collected by gravity flow and the bound fraction was eluted using 3M NaSCN (Sigma-Aldrich). Immediately after isolation, ApoA1-HDL was separated by size using non-denaturing polyacrylamide gel electrophoresis (ND-PAGE) on a 4-30% gradient gel (Jule INC) run at 15 mA for 16 hours. A molecular weight standard (GE/Amersham, 17-0445-01) was run alongside the samples.
After completion of the run, the gel was stained for 1-2 hours in Coomassie Blue (Invitrogen). Portions of the gel corresponding to each HDL size were excised: above 12.2 nm, the largest HDL size, alpha0; between 12.2 and 9.5 nm, alpha1; between 9.5 and 8.2 nm, alpha2; between 8.2 and 7.2 nm, alpha3; and band at 7.1 nm, prebeta8.
The excised gel pieces were trypsin digested for 4 hours at 37°C using a standard protocol25, with the exception that the alkylation step was omitted to increase throughput, and the final wash step (before digestion) comprising 2:1 ammonium bicarbonate:acetonitrile was done overnight to ensure complete removal of Coomassie stain. Peptides were re-suspended in 5% acetonitrile and 0.5% formic acid dissolved in MS-grade water.
HDL peptides samples from participants #1-6 were prepared for a single participant on one diet at a time. For participants #7-12, HDL samples from the same participant on both diets were isolated, run on the same ND-PAGE gel, and trypsin digested at the same time.
Lipid and protein concentrations in plasma and HDL
Fasting plasma levels of TG, total cholesterol (total-C), LDL-C, and HDL-C were measured in the 12 participants on both diets using standard cholesterol and TG assay kits (Thermo Scientific). Levels of ApoA1, ApoA2, ApoC3, ApoE, ApoJ, and ApoL1 in plasma and on HDL were measured in participants #1-12 and participants #1-6, respectively, on both diets by enzyme-linked immunosorbent assay (ELISA) (see Major Resources Table at the end of Supplemental Materials).26 Paired samples (same participant on both diets) were analyzed on the same plate, and HDL samples from participants #1-6 on both diets were isolated at the same time and analyzed by ELISA the same day.
Data-dependent acquisition (DDA) MS
Peptide samples were analyzed with the Q Exactive Quadrupole Orbitrap mass spectrometer fronted with a Nanospray FLEX ion source, and coupled to an Easy-nLC1000 HPLC pump (Thermo Scientific). The peptides were subjected to a dual column set-up: an Acclaim PepMap RSLC C18 trap column, 75 μm X 20 mm; and an Acclaim PepMap RSLC C18 analytical column 75 μm X 150 mm (Thermo Scientific). The analytical gradient was run at 250 nl/min from 5 to 18% Solvent B (acetonitrile/0.1% formic acid) for 10 or 30 minutes, followed by five minutes of 95% Solvent B. Solvent A was 0.1% formic acid.
For absolute quantification experiments, the Q Exactive was set to selected ion monitoring for the scan range of 400-1000 m/z.
For DDA of the HDL proteome, the Q Exactive was set to 140 K resolution, and the top 10 precursor ions (within a scan range of 380-1500 m/z) were subjected to higher energy collision induced dissociation (HCD), collision energy 25% (stepped CE +/− 10%), isolation width 3 m/z, and 17.5 K resolution for MS/MS scans. This portion of the study was performed once all peptide samples for the 12 participants on both diets were prepared (see methods, “HDL preparation”). The proteome was analyzed using the 2-hour time point for each participant on both diets. To ensure consistency across samples, participant #1’s sample was analyzed in triplicate, inter-spaced across the 3-week MS acquisition period.
PRM mass spectrometry
The Q Exactive was operated in the data-independent acquisition (DIA) mode.8 We monitored enrichment in 1 to 3 peptides per protein per HDL size (Supplemental Table II). Each peptide’s MS1 isolation window was centered on the average m/z of M0 and M3 peaks, +/− 5 m/z. The AGC was set to 5e5 or 1e6; the lower AGC was sometimes needed to avoid mass drift due to space charging. The HCD was set to 25 +/− 10% and the MS/MS ions were scanned at 140 K resolution. PRM was completed within ~1 month or less after peptide isolation for the majority of participant samples.
Mass spectral data analysis
The DDA data were queried against the Human UniProt database (downloaded August 1, 2014) using the HT-SEQUEST search algorithm via Proteome Discoverer (PD) (version 2.1, Thermo Scientific), using a 10 ppm tolerance window in the MS1 search space, and a 0.02 Da fragment tolerance window for HCD data. Methionine oxidation was set as a variable modification. The ‘precursor ions area detector’ module was used for quantification using the AUC of the extracted ion chromatograms of the MS1 peaks (top 3 most abundant peptides per protein). The peptide false discovery rate (1%) was calculated using PD Percolator. Proteins with 3 or more unique peptides were included for global proteome profiling.
The relative distribution of each protein across the 5 HDL sizes was determined by taking the AUC PD output of a given protein on a given size fraction divided by the total AUC for that same protein across the 5 HDL sizes (x100). This analysis was repeated for all identified proteins in each participant on each diet, generating a total of 24 proteome datasets.
Absolute quantification of protein pool sizes
In vitro synthesized peptide standards were used to quantify the pool size of 8 HDL proteins across the 5 HDL sizes in participants #1-6 on both diets. The following proteins were quantified using 1-2 peptides per protein: ApoA1 (THLAPYSDEL[R-labeled], ATEHLSTLSE[K-labeled]), ApoA2 (SPELQAEA[K-label]), ApoC3 (DALSSVQESQVAQQA[R-label]), ApoE (LGPLVEQG[R-label]), ApoJ (ELDESLQVAE[R-labeled]), ApoL1 (VTEPISAESGEQVE[R-labeled]), ApoM (FLLYN[R-label]), and LCAT (SSGLVSNAPGVQI[R-labeled]) (New England Peptides, NEP). Proteins were quantified by the absolute amino acid method (NEP). Arginines were labeled with 13C8,15N2 and lysines with 13C6,15N2. The peptide standards were chosen based on the following: 1) fully cleaved, 2) devoid of methionines and cysteines, 3) highest ionization / signal intensity relative to other observed peptides passing criteria 1 and 2, and 4) not reported to be post-translationally modified (Uniprot.org). Only the ApoL1 standard peptide could not meet the last criterion since it was recently show to be phosphorylated.27 However, since the MS-based measurements were adjusted to the ELISA-based measurements (see below), and the same peptide was used for all samples, any potential underestimation of absolute ApoL1 due to phosphorylation is consistent across samples.
We established the appropriate spike-in amount for the peptides by determining the linear range of ionization (AUC of M0) for both the standard and sample-derived peptides.8 Due to the large dynamic range of the sample peptides across the HDL sizes, we used two spiking mixtures. The first mixture contained a final on-column amount of 100 fmol of each ApoA1 peptide, 10 fmol of ApoA2 peptide, and 1 fmol for the remaining peptide standards. The second mixture contained the same peptides at a 10-fold lower on-column concentration. Peptides abundance was quantified for the 2 and 4 hour time points (4 quantification replicates). Skyline (https://skyline.gs.washington.edu) was used for quantification of the AUCs.
We calculated pool size (mg of protein in a given plasma pool) for each protein in the 5 HDL sizes. The fmol on-column (average of 4 replicates) of each protein per size fraction were converted to mg of protein per 1 ml of plasma. To determine the amount of protein loss during preparation, the mg/ml of each protein per size fraction were summed to get an estimated total protein concentration. This estimated total protein concentration was then compared to the concentration of ApoA1, ApoA2, ApoC3, ApoE, ApoJ, and ApoL1 on HDL, as determined by ELISA (see methods, “Lipid and protein concentrations in plasma and HDL”). The sample loss “correction factor” for each protein was calculated by dividing the ELISA total protein concentration by the MS-estimated total protein concentration. The mean (±SD) correction factor for ApoA1, ApoA2, ApoC3, ApoE, ApoJ, and ApoL1 were 43 (±28), 147 (±220), 847 (±601), 32 (±17), 49 (±23), and 1.2 (±0.4), respectively. The difference in correction factors likely reflect the variations in efficiency of peptide trypsin digest, peptide extraction from the ND-gel, and ELISA antibody binding to protein epitopes. Assuming that sample loss was similar for all sizes, the mg/ml estimated protein concentrations for each size was multiplied by the correction factor to determine the mg/ml concentration of each protein in each HDL size. The ApoA1 correction factor was used to adjust ApoM and LCAT sample loss. The mg/ml protein concentrations per size were then multiplied by the total plasma volume to determine the protein pool size per HDL size. Plasma volume per participant was calculated by the following: Plasma volume (dl) = Ideal body weight (kg) * 0.44 + excess weight (kg) * 0.1.28
Tracer enrichment quantification
We performed PRM to determine D3-Leu tracer enrichment in ApoA1, ApoA2, ApoC3, ApoE, ApoJ, ApoL1, ApoM, and LCAT across the 5 HDL sizes in participants #1-6 on both diets (see methods, “PRM mass spectrometry”). We next used our recently developed extracted PRM peak intensity (XPI) software that automates detection and extraction of tracee (M0) and tracer (2H M3) PRM peaks for enrichment calculations.16 Enrichment was calculated by taking the sum of the D3-Leu tracer PRM peak intensities divided by the total tracer + tracee PRM peak intensities per peptide fragment ion. Only fragment ions detected within a mass range of +/−0.01 of the theoretical mass of each peptide fragment ion were quantified. The XPI-generated peak assignments used to calculate enrichment were checked for each fragment ion, across all samples to ensure correct peak assignment. Median enrichment values per time point for all fragment ions per peptide were calculated and used to build compartmental models that describe the metabolism of each protein on each HDL size (see methods, “Compartmental modeling” below).
Compartmental modeling
Compartmental modeling was performed using SAAM II software (SAAM Institute, Seattle, Washington). Although enrichment was monitored for the 8 proteins across the 5 HDL sizes, only HDL sizes in which D3-Leu label was consistently detected in participants #1-6 on both diets were included in the compartmental model for each protein: ApoA1, alpha0, 1, 2, 3, prebeta; ApoA2, alpha1, 2, 3; ApoC3, alpha3; ApoE, alpha0, 1, 2, 3; ApoJ, alpha0; ApoL1, alpha0, 1; ApoM, alpha1, 2; and LCAT, alpha3. The ApoA1, ApoA2, ApoC3, ApoE, and ApoM models used in this study were developed previously.8, 10 This is the first study to determine the metabolism of ApoJ, ApoL1, and LCAT on HDL.
Each model contains an input, source, and HDL size compartments. The input compartment is the plasma amino acid precursor pool (D3-Leu tracer enrichment in plasma) expressed as a forcing function that drives the appearance of D3-Leu tracer in the model. Each participant’s plasma D3-Leu tracer enrichment curve per diet was used for all 8 protein models (Supplemental Figure IA). The source compartment accounts for the time necessary for D3-Leu-labeled protein to appear on each HDL size in plasma. One to five compartments were added to each model that represent the HDL sizes. The pool size and enrichment data were assigned to each HDL size compartment (example model fits are shown in Supplemental Figure IB,C). Two additional compartments were included in the ApoA1 model: 1) delay compartment connecting the source and prebeta. This may represent an extravascular delay (EVD) processing compartment that includes ApoA1 prebeta that has been secreted but is outside systemic circulation.8, 10 2) Compartment connecting alpha3 and prebeta. This compartment may represent lipidated ApoA1 (LA1) that has been released from alpha3 and is used to generate prebeta.8, 10 For all models, a direct secretion pathway into each HDL size was required for satisfactory fitting. Pathways among the sizes were included in the final model when flux through that pathway improved model fits for participants in which the pathway was detected.
The following kinetic parameters were calculated for each participant on both diets in the 8 protein models: 1) FCR, the fraction of a given plasma protein pool turned over per day, was determined for each protein in each HDL size by taking the sum of the rate constants exiting that compartment. 2) Production rate, the amount (mg) of protein produced or transferred into each HDL size per day per kg of body weight: production rate = FCR (pools/day) × pool size (mg) / body weight (kg). 3) System FCR, fraction of protein in the model system turned over per day (pools/day), was calculated by taking the system production rate divided by the system pool size (mg) * body weight (kg). 4) System production rate, amount of protein (mg) entering the model system per day per kg of body weight, was determined by taking the sum of all fluxes (mg/day) exiting the source compartment divided by body weight (kg). Steady-state kinetics were assumed for all proteins.
Data representation
Data graphs were constructed in Prism 7, Excel, and RStudio and figures were compiled in Microsoft PowerPoint and Adobe Photoshop. Heat maps and hierarchical clustering were executed using the Qlucore (version 3.4) software package (www.qlucore.com).
Statistics
Results are presented as mean (+/−SD) unless otherwise specified. Statistical differences between the high fat and high carbohydrate groups were determined by two-tailed, paired t-test.
RESULTS
Study design
Twelve participants (Supplemental Table I) completed a diet intervention (Figure 1A) and metabolic tracer study (Figure 1B). HDL was prepared by anti-ApoA1 immunopurification, separated into 5 sizes, and prepared for MS analysis (Figure 1C). For participants #1-6, tracer enrichment and metabolic rates for ApoA1, ApoA2, ApoC3, ApoE, ApoJ, ApoL1, ApoM, and LCAT were determined by PRM and compartmental modeling, respectively (Figure 1D, top panel). For participants #1-12, the HDL proteome was characterized by DDA (Figure 1D, bottom panel). BMI, and plasma levels of TG, total-C, LDL-C, and HDL-C were not significantly altered by diet (Figure 2). Relative to baseline, however, both diets decreased BMI, TG, and LDL-C; and did not significantly affect HDL-C (Figure 2).
Figure 2. BMI and plasma lipid concentrations before and after the diet intervention.

BMI and fasting plasma lipid levels at baseline and after 4 weeks on the high fat and high carbohydrate diets (participants #1-12). Values were compared by two-tailed, paired t-test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Non-significant P-values are shown above each comparison group. BMI, body mass index; TG, triglyceride; total-C, total cholesterol; LDL-C, LDL cholesterol; HDL-C, HDL cholesterol; Base, baseline; HF, high fat; HC, high carbohydrate.
Diet alters the abundance of proteins on total HDL and on specific HDL sizes
To determine the effect of diet on the abundance of HDL proteins, plasma HDL pool sizes were quantified for 8 proteins – ApoA1, ApoA2, ApoC3, ApoE, ApoJ, ApoL1, ApoM, and LCAT – across the 5 HDL sizes and in total HDL (sum of 5 HDL size pools) in participants #1-6 on both diets (Figure 3, Supplemental Table III). Overall, most proteins were detected in all HDL sizes, but with unique, size-specific distributions: ApoJ dominated on the largest alpha0 HDL; ApoL1 on alpha1; ApoM on alpha1 and 2; ApoE on alpha1, 2, and 3; ApoA1 and ApoA2 on alpha2 and 3; ApoC3 on alpha2, 3, and prebeta; and LCAT on alpha3 (Figure 3, Supplemental Table III). These size-specific distributions were maintained between diets.
Figure 3. Effect of diet on the pool size of 8 HDL proteins across 5 HDL sizes.

Pool size (mg of protein) of 8 HDL proteins across the 5 HDL sizes and in total HDL (sum of 5 HDL sizes) were determined for participants #1-6 on both diets. Pool size values per participant are shown as open circles; mean (±SD) pool sizes are shown as a bar graph. Pool sizes between diets were compared for each HDL size and in total HDL by two-tailed, paired t-test. Significant differences between diets are highlighted (*, P < 0.05; **, P < 0.01), and the P-values for each protein on total HDL are shown in the upper corner of each protein graph (“total HDL P=X”). The average pool size values for the 6 participants across the 5 HDL sizes on both diets are tabulated in Supplemental Table III. #, HDL sizes included in the compartmental model for a given protein.
Diet altered the abundance of ApoA1, ApoJ, ApoA2, and ApoC3 on specific HDL sizes. ApoA1 on alpha1 HDL decreased on the high carbohydrate diet by 37% (P=0.03) (Figure 3, Supplemental Table III). This result is consistent with previous reports showing increased carbohydrate consumption (and decreased fat) decreases the proportion of large HDL in plasma.24 ApoJ on alpha0 HDL increased on the high carbohydrate diet by 42% (P=0.007) (Figure 3, Supplemental Table III). Carbohydrate also tended to increase ApoA2 pool size on alpha1 and alpha2 HDL and ApoC3 pool size across all HDL sizes. These size-specific increases led to a 12% and 26% increase in total HDL ApoA2 and ApoC3 pool sizes, respectively (P=0.012; P=0.056) (Figure 3, Supplemental Table III). Plasma total levels of ApoA2 and ApoC3 were also significantly increased by carbohydrate (P=0.02; P=0.03) (Supplemental Figure II).
Effect of diet on the metabolism of 8 HDL proteins across 5 HDL sizes
We next used compartmental modeling to determined how dietary fat and carbohydrate affect the metabolism of 8 HDL proteins – ApoA1, ApoA2, ApoC3, ApoE, ApoJ, ApoL1, ApoM, and LCAT – in up to 5 HDL sizes in participants #1-6. The compartmental model for each protein contained an input, source, and 1 to 5 HDL size compartments (see methods, “Compartmental Modeling”). The FCR and production rate for each protein on a given HDL size per diet were determined by taking the sum of all removal pathways out of that size (pools/day), or the sum of all flux pathways entering that size (mg/kg/day), respectively, and are detailed in the following sections (Figures 4-6; Table 1; Supplemental Table IV).
Figure 4. ApoA1 metabolism on the high fat and high carbohydrate diets.

(A) ApoA1 enrichment curve fits generated by the compartmental model shown in B for participant #6. (B) Compartmental model used to determine the FCR (pools/day) and production rate (mg/kg/day) of ApoA1 on each HDL size. The input compartment represents the plasma amino acid precursor pool (D3-Leu tracer enrichment in plasma) expressed as a forcing function. The source compartment represents the sight of ApoA1 synthesis and secretion, likely the liver and/or small intestine. Grey, italic numbers next to arrows going out of a given HDL size represent the average ApoA1 FCR for that pathway on the high fat and high carbohydrate diet. Black, non-italic numbers next to arrows going into a given HDL size represent the average ApoA1 flux (mg/kg/day) for that pathway on the high fat and high carbohydrate diet. Pathways whose FCR and/or flux were significantly (P < 0.05) increased, or tended to increase (0.07 > P > 0.05) on the carbohydrate diet are highlighted by a solid and dashed red arrow, respectively. (C) ApoA1 FCR on each HDL size and in the total model system. The ApoA1 FCR on each HDL size was determined by taking the sum of all FCRs exiting that size. The system FCR represents the rate of total ApoA1 removal from the HDL system per day. (D) ApoA1 production rate on each HDL size and in the total model system. ApoA1 production rate on each size was determined by taking the sum of all fluxes entering that size. The system production rate represents the total amount of ApoA1 entering the HDL system per day. (B-D) Values between diets were compared by two-tailed, paired t-test in participants #1-6. (C,D) P-values are shown above each size fraction. *, P < 0.05. EVD, extravascular delay; LA1, lipidated ApoA1; PR, production rate.
Figure 6. Carbohydrate increases the production rate of ApoC3, ApoJ, and ApoL1 on specific HDL sizes.

(A) Compartmental models used to determine the FCR (pools/day) and production rate (mg/kg/day) of ApoC3, ApoJ, ApoL1, and LCAT on each HDL size. The input compartment represents the plasma amino acid precursor pool (D3-Leu tracer enrichment in plasma) expressed as a forcing function. The source compartment represents the sight of protein synthesis and secretion. Grey, italic numbers next to arrows going out of a given HDL size represent the average FCR for that pathway on the high fat and high carbohydrate diet. Black, non-italic numbers next to arrows going into a given HDL size represent the average flux (mg/kg/day) for that pathway on the high fat and high carbohydrate diet. Pathways whose FCR and/or flux were significantly (P < 0.05) increased, or tended to increase (0.07 > P > 0.05) on the high carbohydrate diet are highlighted by a solid and dashed red arrow, respectively. (B) FCR on each HDL size and in the total model system. The FCR on each size was determined by taking the sum of all FCRs exiting that size. The system FCR represents the rate of total protein removal from the HDL system per day. (C) Production rate on each size and in the total model system. Production rate on each size was determined by taking the sum of all fluxes entering that size. The system production rate represents the total amount of protein entering the HDL system per day. (A-C) Values between diets were compared by two-tailed, paired t-test in participants #1-6. (B,C) P-values are shown above each size fraction. *, P < 0.05; **, P < 0.01. PR, production rate.
Table 1. Percent change of kinetic parameters when carbohydrate replaces fat.
Summary of average percent change of a given kinetic parameter – FCR, production rate, and pool size – when carbohydrate replaces fat (HighCarbavg - HighFatavg / HighFatavg *100).
| FCR (pools/day) |
Production Rate (mg/kg/day) |
Pool Size (mg) |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| α0 | α1 | α2 | α3 | pβ | system | α0 | α1 | α2 | α3 | pβ | system | α0 | α1 | α2 | α3 | pβ | system | |
| ApoA1 | 90.1 | 208 | 56.7 | 38.5 | 82.6 | 37.4 | 69.2 | 47.6 | 40.3 | 64.4 | 28.6 | 38.2 | −14.3 | −37.1 | −6.2 | 6.7 | −38.3 | −5.0 |
| ApoA2 | - | 39.0 | 50.7 | 34.2 | - | 24.5 | - | 93.9 | 105 | 17.3 | - | 44.4 | - | 68.5 | 39.0 | −20.4 | - | 12.0 |
| ApoE | −4.8 | 45.9 | 43.0 | 28.9 | - | 33.9 | 70.2 | 73.3 | 71.8 | 49.8 | - | 54.2 | 38.9 | 16.1 | 17.2 | −0.8 | - | 14.2 |
| ApoJ | 22.5 | - | - | - | - | - | 88.1 | - | - | - | - | - | 41.7 | - | - | - | - | - |
| ApoL1 | 83.5 | 10.3 | - | - | - | 12.2 | 129 | 31.6 | - | - | - | 35.4 | 9.9 | 14.3 | - | - | - | 13.9 |
| ApoM | - | 42.4 | 56.9 | - | - | 50.8 | - | −29.6 | −13.0 | - | - | −20.5 | −46.5 | −43.2 | −44.6 | |||
| ApoC3 | - | - | - | 13.4 | - | - | - | - | - | 52.8 | - | - | - | - | - | 15.8 | - | - |
| LCAT | - | - | - | −6.9 | - | - | - | - | - | −41.9 | - | - | - | - | - | −12.8 | - | - |
Dark red boxes, parameters significantly (P < 0.05) increased on the high carbohydrate diet
Light red boxes, parameters that tended to increase (0.1 > P > 0.05) on the high carbohydrate diet
Blue boxes, parameters significantly decreased on the high carbohydrate diet
White boxes, parameters not altered by diet
Box with dash, size fractions not included in the compartmental model for a given protein
Carbohydrate increased the FCR and production rate of ApoA1 on specific HDL sizes
We first looked at the effect of diet on the metabolism of ApoA1 across the 5 HDL sizes. Carbohydrate, when replacing fat, tended to increase the ApoA1 enrichment curve ascending and descending slopes across the HDL sizes (Figure 4A), suggesting that carbohydrate increases ApoA1 FCR. We tested this using our ApoA1 compartmental model, which contains pathways from the source into each HDL size, transfer pathways from alpha3 to alpha2 and prebeta, prebeta to alpha2, 1, and 0, as well as two additional compartments, EVD and LA1 (see methods, “Compartmental Modeling”) (Figure 4B). Indeed, we found that carbohydrate tended to increase the removal rate of ApoA1 out of its major size fraction alpha3 by increasing ApoA1 transfer to alpha2, as well as to prebeta via LA1 (Figure 4B, arrows from alpha3 to alpha2 and LA1; Supplemental Figure IIIA). These increases led to a significant 39% increase in ApoA1 alpha3 FCR (P=0.02) (Figure 4C, Table 1, Supplemental Table IV). In addition, carbohydrate tended to increase the ApoA1 FCR on alpha0 by 90% (P=0.06) (Figure 4B, red dashed arrow out of alpha0, 4C; Table 1; Supplemental Table IV). Overall, when considering all HDL sizes, the total rate of ApoA1 catabolism out of the entire modeled system was 0.42 pools/day on the high fat, and 37% faster on the high carbohydrate diet at 0.58 pools/day (P=0.02) (Figure 4C, system; Table 1; Supplemental Table IV).
Similar to FCR, carbohydrate also increased the production rate of ApoA1 on specific HDL sizes. Carbohydrate significantly increased the production rate of ApoA1 on alpha3 by 64% (P=0.01) (Figure 4B, red arrow into alpha3; 4D) and tended to increase ApoA1 on alpha2 by 40% (Figure 4D; Table 1; Supplemental Figure IIIB; Supplemental Table IV). These size-specific production increases led to a 38% increase in total ApoA1 production into the modeled system per day, from 22 to 30 mg/kg/day (P=0.054) (Figure 4D, system; Table 1; Supplemental Table IV).
Carbohydrate increased the FCR and production rate of ApoA2 and ApoE, and the FCR of ApoM on HDL
Similar to ApoA1, carbohydrate increased the FCR and production rate of ApoA2 on specific HDL sizes. The ApoA2 compartmental model contains pathways from the source to alpha1, 2, and 3, as well as a transfer pathway from alpha3 to alpha2 (Figure 5A). Carbohydrate significantly increased the FCR of ApoA2 on alpha3 HDL by 34% (P=0.02;) (Figure 5B). The increase in ApoA2 alpha3 FCR was due to an increase in ApoA2 transfer from alpha3 to alpha2 (P=0.015), and not due to an increase in clearance out of the system (Figure 5A, red versus black arrow out of alpha3; Supplemental Figure IVA). This increase led to a concomitant increase in ApoA2 alpha3 to alpha2 flux (P=0.03) (Figure 5A, red arrow from alpha3 to alpha2; Supplemental Figure IVB). This increase in ApoA2 alpha3 to alpha2 flux, along with an increase in ApoA2 flux into alpha2 from the source (P=0.06) (Figure 5A, red dashed arrow into alpha2; Supplemental Figure IVB), doubled the production rate of ApoA2 on alpha2 HDL (P=0.014) (Figure 5C; Table 1; Supplemental Table IV). Overall, the carbohydrate-induced increase in ApoA2 alpha3 FCR and ApoA2 alpha2 production rate led to a 25% (P=0.08) and 44% (P=0.019) increase in the system FCR and production rate, respectively (Figure 5B, C, system; Table 1; Supplemental Table IV).
Figure 5. Carbohydrate increases the FCR of ApoA2, ApoE, and ApoM on specific HDL sizes.

(A) ApoA2 compartmental model used to determine the FCR (pools/day) (B) and production rate (mg/kg/day) (C) of ApoA2 on each HDL size and in the total model system. (D) ApoE compartmental model used to determine the FCR (E) and production rate (F) of ApoE on each HDL size and in the total model system. (G) ApoM compartmental model used to determine the FCR (H) and production rate (I) of ApoM on each HDL size and in the model system. (A, D, G) In each model, the input compartment represents the plasma amino acid precursor pool (D3-Leu tracer enrichment in plasma) expressed as a forcing function. The source compartment represents the sight of protein synthesis and secretion. Grey, italic numbers next to arrows going out of a given HDL size represent the average FCR for that pathway on the high fat and high carbohydrate diet. Black, non-italic numbers next to arrows going into a given HDL size represent the average flux (mg/kg/day) for that pathway on the high fat and high carbohydrate diet. Pathways whose FCR and/or flux were significantly (P < 0.05) increased, or tended to increase (0.07 > P > 0.05) on the high carbohydrate diet are highlighted by a solid and dashed red arrow, respectively. (B, E, H) The FCR on each size was determined by taking the sum of all FCRs exiting that size. The system FCR represents the rate of total protein removal from the modeled HDL system per day. (C, F, I) Production rate on each size was determined by taking the sum of all fluxes entering that size. The system production rate represents the total amount of protein entering the HDL modeled system per day. (A-I) Values between diets were compared by two-tailed, paired t-test in participants #1-6. In all bar graphs, P-values are shown above each size fraction. *, P < 0.05; **, P < 0.01. PR, production rate.
Also similar to ApoA1, carbohydrate increased the FCR and production rate of ApoE on specific HDL sizes. The ApoE compartmental model contains pathways from the source into each HDL size, and transfer pathways from alpha3 to alpha0, 1, and 2, and alpha2 to alpha0 (Figure 5D). Carbohydrate increased the FCR of ApoE on alpha1 and alpha3 by 46% (P=0.04) and 29% (P=0.036), respectively, and the FCR of the ApoE in the HDL modeled system by 34% (P=0.007), from 5.4 to 7.3 pools/day. (Figure 5E; Table 1; Supplemental Figure IVC; Supplemental Table IV). Carbohydrate also increased the source production of ApoE on alpha2 and alpha3 by 69% (P=0.04) and 50% (P=0.011), respectively (Figure 5D, red arrows out of source; 5F; Supplemental Figure IVD). Due to these size-specific production increases, the total ApoE production into the HDL system went from 16.5 to 25.5 mg/kg/day (P=0.009) (Figure 5F, system; Table 1; Supplemental Table IV).
Additionally, carbohydrate also increased the FCR of ApoM. The ApoM compartmental model includes pathways from the source into each HDL size (Figure 5G). The ApoM FCR tended to increase on alpha1 and increased significantly on alpha2 HDL by 42% and 57%, respectively (P=0.07; P=0.045) (Figure 5G, red arrow out of alpha2; 5H; Table 1; Supplemental Table IV). This led to an overall 51% increase in ApoM catabolism in the ApoM model system, from 0.37 to 0.56 pools/day (P=0.055) (Figure 5H, system; Table 1; Supplemental Table IV). ApoM production rate was not altered by diet (Figure 5I; Table 1; Supplemental Table IV).
ApoC3, ApoJ, and ApoL1 production rates on specific HDL sizes are increased by carbohydrate
The compartmental models for ApoC3, ApoJ, ApoL1, and LCAT contain pathways from the source into each HDL size, and the ApoL1 model also contains a transfer pathway from alpha1 to alpha0 (Figure 6A). Unlike ApoA1, ApoA2, ApoE, and ApoM, the FCRs of ApoC3, ApoJ, ApoL1, and LCAT were not significantly altered by diet (Figure 6B; Supplemental Figure VA). However, carbohydrate significantly increased the production rate of ApoC3 on alpha3, ApoJ on alpha0, and ApoL1 on alpha0 HDL by 53%, 88%, and 129%, respectively (P=0.018; P=0.011; P=0.004, respectively) (Figure 6A, red arrows out of source compartment, 6C; Table 1; Supplemental Figure VB). Despite the increase in ApoL1 alpha0 production rate, the system production rate of ApoL1 was not significantly altered by diet (Figure 6C, ApoL1 system; Table 1; Supplemental Table IV). LCAT production rate on alpha3 was not altered by diet (Figure 6A, C, LCAT; Table 1; Supplemental Table IV).
HDL Proteome Distribution Across HDL Size is Conserved Between Diets
After determining that diet alters the metabolism of several HDL proteins, we next wanted to delineate the effect of diet on the composition of the entire HDL proteome and how these proteins are distributed across HDL size. The HDL proteome across 5 HDL sizes was determined for participants #1-12 on both diets, generating a total of 24 proteome datasets. We limited our quantification analysis to stable, reliably detected proteins that were identified by 3 or more unique peptides per participant’s combined high fat and high carbohydrate proteome dataset resulting in 145 total proteins: 53 of these proteins were identified in all 24 proteome datasets (Protein Set 1), 44 were found in 12-23 datasets (Protein Set 2), and 48 were found in 1-11 datasets (Protein Set 3) (Figure 7A, Supplemental Table V). The proteome composition was remarkably conserved between diets (Figure 7B-D, Supplemental Table V). All proteins detected in more than 2 datasets were detected on both diets, and 97% of proteins identified in a given participant on the high fat diet were identified in that same participant on the high carbohydrate diet (Figure 7B-D, Supplemental Table V). No proteins unique to the high fat or high carbohydrate diet were identified (Figure 7B-D, Supplemental Table V).
Figure 7. Relative distribution of the HDL proteome across HDL size is conserved between diets.

(A) 145 HDL proteins identified in the global proteome analysis. These proteins were identified in 24 datasets (all 12 participants on both diets, Protein Set 1), in 50-99% of datasets (Protein Set 2), or in <50% of datasets (Protein Set 3). Heat maps of sum-normalized area under the curve (AUC) values of Set 1 (B), 2 (C), and 3 (D) proteins were clustered based on the similarity of their protein distribution profiles across the 5 HDL sizes. Color scale indicates the relative % of total protein in each size fraction from 100% (red) to 0% (white). Size-specific clusters are indicated by Groups I-V.
Each protein’s relative distribution across the 5 HDL sizes was then determined by calculating the protein-specific sum-normalized AUC measurements (per proteome dataset). Hierarchical clustering was used to compare the proteome of the 12 participants on both diets for Protein Sets 1, 2, and 3, and the cluster matrix for each Protein Set was sorted by decreasing HDL size (Figure 7B-D, Supplemental Table V). Protein Set 1 formed 5 distinct groups based on their relative distribution across the 5 HDL sizes (Figure 7B, Supplemental Table V). These HDL size-specific protein groups were conserved across the 12 participants and between diets (Figure 7B, Supplemental Table V). Proteins enriched in the largest alpha0 HDL (Group I) included ApoJ; alpha1 and/or alpha2-dominant proteins (Group II) included ApoL1, ApoM, ApoE, and ApoA2. Proteins primarily associated with alpha2, alpha3, and/or prebeta (Group III) contained ApoA1 and ApoC3. Alpha3 (Group IV) and prebeta (Group V) proteins included LCAT and ApoA4, respectively (Figure 7B, Supplemental Table V). Additionally, these HDL size-specific protein distributions were consistent with the distributions in our pool size data (Figure 3).
This distinct size-specific protein distribution profile, defined by Protein Set 1, was readily visualized due to the inclusion of proteins identified in all participant datasets (Figure 7B). Nonetheless, despite fewer observations across the participants for Protein Set 2, the HDL proteome size-specific architecture was discernable (Figure 7C, Supplemental Table V). Alpha0, and alpha0 and alpha1-dominant proteins (Group I and II) consisted of several immunoglobulins and complement factors as well as haptoglobin-related protein (HPR). Proteins enriched in alpha1, 2, and 3 HDL (Group III), alpha3 (Group IV), and alpha3/prebeta (Group V) included ApoC1, ceruloplasmin (CP), and serum paraoxonase 3 (PON3), respectively (Figure 7C, Supplemental Table V).
Unlike Protein Set 1 and 2, Protein Set 3 did not form distinct protein clusters across the HDL sizes; proteins instead were scattered across the HDL sizes (Figure 7D, Supplemental Table V). This is likely due to the fact that these proteins were only identified in a few participants. This protein set included ApoA5, apolipoprotein(a) (LPA), ApoF, and ApoC4 (Figure 7D, Supplemental Table V), all of which were previously identified on HDL (HDL Proteome Watch, http://homepages.uc.edu/~davidswm/HDLproteome.html). However, similar to Protein Sets 1 and 2, each participant’s Protein Set 3 proteome composition was highly conserved between diets (i.e. a protein found in a given participant on the high fat diet was typically detected in that same participant on the high carbohydrate diet), suggesting that these proteins are not nonspecific contaminants, but potentially low abundant HDL proteins at the limit of instrument detection, and may only be detectable in certain participants (Figure 7D, Supplemental Table V).
DISCUSSION
This study further elucidates the protein and size-based metabolic structure of the HDL particle system in humans recently introduced by Mendivil et al. and Singh et al.8, 10 These studies demonstrated that the majority of ApoA1, as well as other HDL proteins, on different sizes of alpha HDL are secreted directly into plasma from the source compartment and remain mainly within their specific size until they are removed from circulation; protein transfer between HDL sizes only provided a minor contribution to the metabolism of these proteins.8, 10 In the present study, we expanded upon these findings and showed that several additional HDL proteins are secreted and metabolized in plasma within a given HDL size range (Figure 8). These findings suggest that the majority of these HDL proteins are incorporated onto the surface of a given HDL size during HDL synthesis in the liver (or small intestine), or are picked up in the extravascular space before entering the systemic circulation (Figure 8). The structural and compositional properties of each HDL size – such as the size-specific surface curvature, lipid packing density, or the lipid and protein content – may dictate which proteins can bind to each HDL size.12, 29, 30 Additionally, the metabolism of HDL proteins within a given HDL size, taken together with the unique proteome composition of a given HDL size, suggest that HDL’s multiple biological functions may be orchestrated by several distinct size and protein-based subspecies.
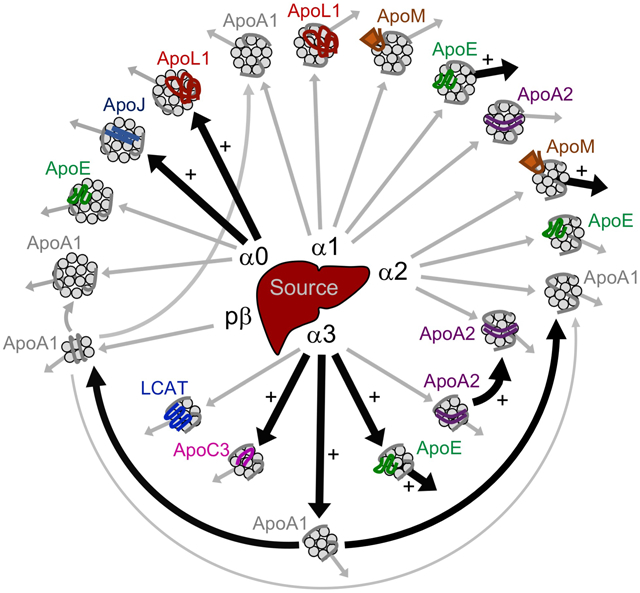
Figure 8. Model of the HDL system when dietary carbohydrate replaces monounsaturated fat.

This model highlights the general metabolic structure of the HDL particle system as well as the metabolic pathways altered when dietary carbohydrate replaces fat. ApoA1-HDL particles of all sizes (alpha0, 1, 2, 3, prebeta) are secreted by the source compartment, representative of the liver primarily and small intestine secondarily (arrows out of the source into each HDL size). In addition to ApoA1 (shown in grey on each HDL particle), additional proteins are incorporated onto the surface of specific HDL sizes – ApoJ on alpha0; ApoL1 on alpha0 and alpha1; ApoE on alpha0, 1, 2, 3; ApoM on alpha1 and alpha2; ApoA2 on alpha1, 2, 3; ApoC3 on alpha3; and LCAT on alpha3. These proteins are likely incorporated onto specific HDL sizes during particle synthesis, or are picked up in an extravascular compartment prior to entering systemic circulation. An arrow into each HDL size represents the production rate of a given protein onto that size fraction, and an arrow out of each HDL size represents the FCR of that protein out of that size fraction. Carbohydrate and fat did not alter the general metabolic structure of the HDL particle system; all pathways identified on the high fat diet were also identified on the high carbohydrate diet. However, carbohydrate, when replacing fat, significantly increased the production rate of ApoJ and ApoL1 on alpha0, ApoA2 on alpha2, and ApoE, ApoA1, and ApoC3 on alpha3 (large black arrows into these size particles), and increased the FCR of ApoE on alpha1, ApoM on alpha2, and ApoA2, ApoE, and ApoA1 on alpha3 (large black arrows out of these size particles). Carbohydrate increased ApoA2 alpha2 production rate and ApoA2 alpha3 FCR by increasing the conversion of ApoA2 alpha3 to alpha2 (large black arrow from ApoA2 alpha3 to alpha2), and increased the FCR of ApoA1 alpha3 by increasing the conversion of ApoA1 alpha3 to prebeta and alpha2 (large black arrows out of ApoA1 alpha3). Grey arrows indicate metabolic pathways that were not significantly altered by carbohydrate. For simplicity, ApoE and ApoL1 conversion pathways between size fractions were not included in this model, as these pathways were not altered by carbohydrate. FCR, fractional catabolic rate.
Furthermore, we investigated the effect of an intervention, dietary fat and carbohydrate, on the HDL system’s protein and size-based metabolic structure. We found that our study diets did not alter the HDL system’s metabolic structure; the pathways identified in a given protein’s compartmental model across the HDL sizes were similar between diets (Figure 8). Additionally, we found that the composition and relative distribution of the HDL proteome was conserved between diets. Taken together, these findings illustrate the remarkable conservation of the HDL proteome’s size-specific architecture and metabolic structure (Figure 7,8). Although the metabolic structure of the HDL system was conserved between diets, carbohydrate, when replacing mainly monounsaturated fat, increased the FCR and production rate of several of these HDL proteins on specific HDL sizes (Figure 8). These carbohydrate-induced metabolic changes suggest that diet may be an important regulator of HDL function in humans.
Carbohydrate significantly increased the FCR of ApoA1, ApoA2, and ApoE on alpha3 HDL (Figure 4, 5, 8; Table 1). Interestingly, the increase in FCR for ApoA1 and ApoA2 on alpha3 HDL was not due to an increase in removal from the HDL system, but instead due to an increase in transfer to alpha2 HDL, and in the case of ApoA1, also an increase in transfer to prebeta HDL. The alpha3 to alpha2 pathway detected in the ApoA1 and ApoA2 models may be representative of the same particles, as 60% or more of ApoA1 particles contain ApoA2.26 This alpha3 to alpha2 pathway may also reflect in vivo particle fusion and size expansion, as alpha3 HDL are more unstable and participate more readily in particle fusion compared to larger HDL (such as alpha1, 2).29, 31 Additionally, the carbohydrate-induced increase in ApoE FCR on alpha3 HDL is likely due to an increase in the removal of ApoE itself from the particle surface, and not due to the increase in removal of ApoE-containing alpha3 HDL particles, as the clearance rate of these particles is not altered by increasing dietary carbohydrate at the expense of dietary fat.32 Carbohydrate may enhance the ApoA1 and ApoA2 alpha3 to alpha2 conversion and ApoE detachment from alpha3 by further destabilizing the alpha3 HDL particle, potentially by altering alpha3 lipid composition, which has a major effect on the stability and protein binding affinity of HDL.12, 29 Although ApoA2 and ApoE can form a heterodimer complex on HDL33, the differential effect of carbohydrate on ApoA2 and ApoE metabolism suggest that ApoA2 and ApoE may be on different alpha3 particles. If all ApoE and ApoA2 formed a heterodimer on alpha3, we would expect them to be catabolized together at the same rate. However, the FCR of ApoE on alpha3 is 20-times faster than that of ApoA2 on alpha3 (Supplemental Table IV). Additionally, only 30% of total ApoE on HDL forms a heterodimer with ApoA2, while the remaining 70% exists as either a homodimer or monomer33, and ApoE bound to ApoA2 has a slower catabolism relative to the ApoE homodimer and monomer.34 If a subset of ApoE is bound to ApoA2 in our alpha3 samples, its slower metabolism may be masked by the more abundant and rapidly catabolized ApoE monomer and homodimer. Follow-up studies are needed to delineate the specific effects of carbohydrate on HDL subspecies containing ApoE only, ApoA2 only, and ApoE and ApoA2.
In vitro studies have suggested that ApoM is anti-atherogenic, as ApoM on HDL inhibits LDL oxidation and stimulates cholesterol efflux from macrophage foam cells.35-37 Studies in mice have shown that hepatocyte-specific ApoM overexpression facilitates the formation of larger, ApoM and cholesterol ester-rich plasma HDL particles.38 ApoM’s role as an anti-atherogenic entity in humans, however, is less clear, as plasma levels of ApoM are not associated with incidence of CHD in humans.39 Consistent with our previous study also in humans8, we found that ApoM is primarily associated with large alpha1 and alpha2 HDL, and that dietary fat and carbohydrate do not alter this distribution (Figure 3). However, carbohydrate, when replacing fat, significantly increased the FCR of ApoM on alpha2, and tended to increase the FCR of ApoM on alpha1 HDL (Figure 5, 8; Table 1). This increase in ApoM FCR suggests that ApoM itself may be removed from the HDL particle, or potentially that ApoM-containing HDL particles are removed from circulation more rapidly on the high carbohydrate diet.
Carbohydrate increased ApoC3 levels on total HDL, and this increase is likely due to an increase in ApoC3 production, as shown for ApoC3 on alpha3 HDL, ApoC3’s major size fraction (Figure 3, 6, 8; Table 1). Glucose, through the transcription factors carbohydrate response element-binding protein (ChREBP) and hepatocyte nuclear factor-4alpha (HNF-4alpha), induces ApoC3 gene expression in primary rat and immortalized human hepatocytes.40 Total plasma levels of ApoC3 are also positively correlated with fasting glucose in overweight and obese subjects, suggesting glucose levels may mediate ApoC3 gene expression and protein levels in humans.40 An increase in HDL levels of ApoC3 is likely unfavorable, as levels of HDL particles containing ApoC3 independently predict increased risk for CHD.41, 42 ApoC3 on HDL may exert its atherogenic function by blocking liver uptake of ApoE-containing HDL particles, and thus inhibiting HDL’s ability to remove cholesterol from circulation.42 Thus, the overall increase in ApoC3 on HDL by carbohydrate may decrease clearance of ApoE-containing HDL particles32, and increase risk of CHD.
ApoJ on HDL may be cardioprotective due to its ability to activate cholesterol efflux43 and protect LDL from oxidation.44, 45 In addition to these functions, ApoJ-containing HDL is enriched in proteins involved in hemostasis and acute phase response, suggesting ApoJ may also play a role in these functions.26 We found that ApoJ is enriched on the largest alpha0 HDL, with smaller amounts on alpha1, 2, 3, and prebeta HDL (Figure 3). Carbohydrate significantly increased ApoJ pool size on alpha0 HDL by increasing its production (Figure 6, 8), shifting the distribution of ApoJ from 43% on alpha0 and 57% on the other HDL sizes, to 56% and 44%, respectively (P=0.015). This shift in ApoJ distribution from small to large HDL suggests that carbohydrate may promote transfer of ApoJ from the smaller HDL to the largest alpha0 HDL, or increase ApoJ alpha0 production from the source or transfer from another non-sampled pool.
ApoL1 is a component of trypanosome lytic factor (TLF) and protects against Trypanosoma brucei, the protozoan that causes African sleeping sickness.46-48 ApoL1 forms 2 distinct lipoproteins complexes, TLF1 and TLF2, that are ~12 and 20 nm in size, respectively, and each of which also contain ApoA1 and haptoglobin-related protein (HPR).46-49 Consistent with these reports, we found that ApoL1 is enriched primarily on alpha1 HDL (similar to TLF1 size), and to a smaller extent on the largest alpha0 HDL (similar to TLF2 size) (Figure 3), and that HPR had a similar HDL size distribution to that of ApoL1 (Figure 7). These similarities suggest that ApoL1 on alpha1 and alpha0 may be ApoL1 on TLF1 and TLF2, respectively. Within a given diet, the catabolic rates of ApoL1 on alpha0 and alpha1 were similar, however, carbohydrate significantly increased ApoL1 production on its minor alpha0 fraction but not on alpha1 HDL, where most ApoL1 resides (Figure 6, 8, Table 1). If TLF1 and TLF2 have different functions or mechanisms of action, as suggested by differences in their proteome47, 48 and different pathways of uptake by the trypanosome50, 51, these data suggest that diet could be a potential regulator of ApoL1 function in humans in vivo, and this regulation could have implications in immune function and the ability of ApoL1 to fight infection.
LCAT esterifies free cholesterol and alters the shape (discoidal to spherical) and increases the size of HDL.52, 53 Thus, LCAT activity likely accounts at least partially for the conversion of ApoA1 on small prebeta and alpha3 to larger alpha HDL particles identified in our ApoA1 kinetic model (Figure 4). Similar to ApoA1, we expected to detect LCAT transfer from small to large HDL. However, due to the low abundance of LCAT on alpha0, 1, 2, and prebeta (<2 mg per size), we were only able to quantify D3-Leu tracer in LCAT on alpha3, its major size (~8 mg) (Figure 3). Enrichment of LCAT-containing HDL may be necessary to determine the metabolism of LCAT on these additional HDL sizes. Additionally, the higher affinity of LCAT for small alpha3 HDL may be due to its size-specific lipid composition. Relative to alpha3, the larger alpha particles contain higher amounts of sphingomyelin,12 which inhibits LCAT binding.54 We also found that, unlike ApoA1, ApoA2, ApoE, ApoC3 on alpha3, LCAT metabolism on alpha3 was not altered by diet (Figure 6, 8; Table 1). This suggests that LCAT may reside on a subset of alpha3 HDL particles whose metabolism is not affected by diet, or that the ability of LCAT to attach and detach from the HDL surface is not altered by diet.
In addition to HDL protein metabolism, we also determined the effect of dietary fat and carbohydrate on the composition and relative distribution of the HDL proteome across 5 HDL sizes in 12 participants with low HDL-C and who were overweight or obese. Our protein identification cutoff was stringent, as we only considered proteins identified by 3 or more unique peptides in each participant. We identified 145 HDL proteins, 97 of which were shared by 50-100% of participants (Figure 7). No proteins unique to the high fat or high carbohydrate diet were identified. The relative distribution of these 97 proteins across HDL size was remarkably conserved not only across participants, but also between diets. This degree of conservation was surprising, given the diversity of our study population - males and females with a large age range (Supplemental Table I). It would be interesting to compare this profile to high HDL-C, normal weight individuals to further assess the degree of conservation of this size-specific, proteome distribution.
In addition to the 97 proteins discussed above, we identified 48 proteins that were found in <50% of participants. Due to the identification of these proteins in only a few participants, we were not able to obtain a clear picture of their HDL size-specific distribution (Figure 7D). These proteins, however, still exhibited a high degree of conservation between diets, as most proteins identified in a given participant on the high fat diet were also identified in that same participant on the high carbohydrate diet (Supplemental Table V). For example, LPA and ApoA5, both of which have previously been identified on HDL (HDL Proteome Watch), were only identified in 1 out of 12 participants studied, but they were identified in that same participant on both diets. This consistent detection of a given protein in one participant on both diets decreases the likelihood that these proteins are merely nonspecific sample contaminants. It is possible this set of proteins was not detected in more participants because, 1) they are at the lower limit of the MS acquisition range, so we only identified them in participants in which they are the most abundant; 2) these proteins were identified in additional participants, but they did not meet our 3 or more unique peptide identification cutoff; and 3) the abundance of these proteins may be low in our study population. It is possible that these proteins may define or correspond to various clinical attributes.
Past studies of dietary fat and carbohydrate on total ApoA1 metabolism have yielded mixed results. Carbohydrate, when replacing saturated fat, decreased ApoA1 production rate 19-21, and increased19 or did not alter ApoA1 FCR20, 21. Carbohydrate, when replacing monounsaturated fat, decreased ApoA1 production rate and FCR22, or increased ApoA1 FCR without altering production rate.23 We found that carbohydrate, when replacing mainly monounsaturated fat, increased total ApoA1 production rate and FCR (Figure 4, 8; Table 1). The discrepancy in findings across these studies is likely due to differential effects of monounsaturated and saturated fat on ApoA1 metabolism, and differences in study populations. Our study is limited to individuals with low HDL-C and who are overweight or obese, and individuals with low HDL-C respond less to carbohydrate and fat compared to those with higher HDL-C.55 Future HDL metabolism studies would benefit from comparing the effect of carbohydrate and monounsaturated fat in individuals with low and high levels of HDL-C.
We determined the effect of diet on a given kinetic parameter – FCR, production rate, and pool size – for each of the 8 proteins on each HDL size by paired t-test, and reported all unadjusted P-values. In situations where multiple, independent comparisons are performed, adjusting the P-value to account for multiple testing may be necessary to avoid false positives. However, kinetic studies are unique in that the main outcomes – the kinetic parameters – are dependent relative to each other as well as across the HDL sizes for a given protein model: for instance, production rate is directly proportional to FCR and pool size, production rate = FCR × pool size / body weight (see methods, “Compartmental Modeling”), so an alteration in one parameter will affect the others. Within a given kinetic model, all compartments are metabolically connected, either directly by a given pathway or via another compartment, and changing a metabolic pathway into or out of one compartment can alter the pathways into and out of the other compartments. Because of this interdependence, we do not adjust for multiple comparisons when comparing two groups in our lipoprotein kinetic studies.32, 42, 56-58 Additionally, these comparisons were all preplanned and are not opportunistic, and our findings are consistent and support the same conclusion – that carbohydrate, when replacing fat, creates a hypermetabolic state of HDL proteins. Taken together, these considerations decrease the likelihood that the differences we detect between diets are due to chance alone.
The major strength of this study is the ability of our PRM-based MS platform to detect tracer in multiple HDL proteins simultaneously. This allowed us to determine the metabolism of 8 HDL proteins across 5 HDL sizes, and overall obtain the most comprehensive picture to-date of the metabolic structure of the HDL particle system. This is the first study to describe the metabolism of ApoJ, ApoL1, and LCAT on HDL. This is also the first study to determine the effect of carbohydrate and fat on the metabolism of ApoA1 and ApoA2 on different HDL sizes, and of ApoC3, ApoE, ApoJ, ApoL1, ApoM, and LCAT on HDL. In addition to HDL protein metabolism, we also determined the effect of diet on the proteome of 5 HDL sizes, which provides further insight on the protein and size-based structure of the HDL system. To determine protein pool sizes, we utilized both MS and ELISA-based quantification methods to determine the amount of protein in each HDL size and to correct for protein loss during preparation (HDL isolation through MS acquisition), respectively. The amount of loss differed across the proteins (see methods, “Absolute quantification of protein pool sizes”), emphasizing the importance of both quantification methods to more accurately estimate protein pool sizes. Additionally, our diet intervention study design has several strengths: this study was a randomized crossover study, so each participant served as his/her own control; both diets were completely controlled and did not depend on participants preparing and recording their own self-selected foods; study participants were diverse and included men and women over a large age range.
This study also has several limitations. First, due to the small sample size, the study was not powered to detect small differences between the metabolic parameters on the high fat and high carbohydrate diets. Second, relative to the high fat diet, the high carbohydrate diet contains not only a higher amount of carbohydrate, but also less saturated fat. Thus, any of the observed changes could be due to a reduction in saturated fat, and not necessarily due to an increase in carbohydrate. Third, both study diets were healthy, enriched in monounsaturated fat or complex carbohydrates, and do not represent the average American diet, which contains higher amounts of saturated fat and refined carbohydrates. Fourth, although we hypothesize that the diet-induced changes in HDL protein metabolism alter HDL function, we did not directly test the effect of diet on HDL function. Future studies on the effect of diet on HDL function, such as HDL cholesterol efflux capacity, would be of interest. Fifth, our study is limited to participants with low HDL-C and who are overweight or obese only, and does not contain a normal HDL-C, non-obese comparison group. Lastly, the accuracy of our protein pool size estimates is dependent on the assumption that protein loss is similar across the HDL sizes, and deviations from this assumption would lead to inaccurate estimates.
In closing, we have shown that HDL in blood is composed of metabolically distinct proteins that localize to distinct HDL size subspecies and that their metabolisms can be altered by diet. These findings underscore the capabilities of a MS-based platform to detect not only metabolic alterations in the total HDL system, but also in the flux and removal pathways in and out of, and between, any given HDL size(s), and how these pathways can be altered by an intervention. Carbohydrate, when replacing mainly monounsaturated fat, increased the flux and/or removal pathways of several HDL proteins into and out of specific HDL sizes. Given that diets high in carbohydrate, and low in unsaturated fat, negatively impact lipoprotein risk factors, and increase CHD risk18, this hypermetabolic state of HDL proteins may represent a mechanism by which carbohydrate decreases the cardioprotective properties of HDL. Future intervention studies or clinical trials aimed at treating dyslipidemia or other diseases via alterations in HDL will benefit from HDL protein kinetic studies, as they allow us to look beyond HDL-C and total HDL and gain a better understanding of the structure and function of the HDL particle system in humans.
Supplementary Material
HIGHLIGHTS.
HDL in humans is composed of a complex system of proteins, each protein with its own unique size distribution, metabolism, and diet regulation.
Dietary carbohydrate, when replacing fat, increases the production and catabolism of several proteins on specific HDL sizes, indicating that diet may be an important regulator of HDL function in humans.
The composition of the HDL proteome across the 5 HDL sizes is remarkably conserved across participants and between the high fat and high carbohydrate diets.
ACKNOWLEDGEMENTS
Sources of funding. This work was supported by research grants from the National Institutes of Health [R01HL095964 (F.M.S.); R01HL123917 (F.M.S.); R01HL107550 (M.A.); R01HL126901 (M.A.)], Kowa Company Ltd. [Nagoya, Japan (M.A.)], and the American Heart Association [17PRE32740000 (A.B.A.)].
ABBREVIATIONS
- HDL
high-density lipoprotein
- Apo
apolipoprotein
- LCAT
lecithin-cholesterol acyltransferase
- FCR
fractional catabolic rate
- MS
mass spectrometry
- PRM
parallel reaction monitoring
- D3-Leu
trideuterated leucine
- AUC
area under the curve
Footnotes
Disclosures. None
REFERENCES
- 1.Emerging Risk Factors C, Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, Collins R, Thompson SG, Danesh J. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson A, Danesh J. Associations between apolipoprotein B, apolipoprotein AI, the apolipoprotein B/AI ratio and coronary heart disease: a literature-based meta-analysis of prospective studies. J Intern Med. 2006;259:481–492. [DOI] [PubMed] [Google Scholar]
- 3.Investigators A-H, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267. [DOI] [PubMed] [Google Scholar]
- 4.Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–2122. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz GG, Olsson AG, Abt M, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–2099. [DOI] [PubMed] [Google Scholar]
- 6.Vaisar T, Pennathur S, Green PS, et al. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest. 2007;117:746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davidson WS, Silva RA, Chantepie S, Lagor WR, Chapman MJ, Kontush A. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler Thromb Vasc Biol. 2009;29:870–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh SA, Andraski AB, Pieper B, Goh W, Mendivil CO, Sacks FM, Aikawa M. Multiple apolipoprotein kinetics measured in human HDL by high-resolution/accurate mass parallel reaction monitoring. J Lipid Res. 2016;57:714–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gordon SM, Deng J, Lu LJ, Davidson WS. Proteomic characterization of human plasma high density lipoprotein fractionated by gel filtration chromatography. J Proteome Res. 2010;9:5239–5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mendivil CO, Furtado J, Morton AM, Wang L, Sacks FM. Novel Pathways of Apolipoprotein A-I Metabolism in High-Density Lipoprotein of Different Sizes in Humans. Arterioscler Thromb Vasc Biol. 2016;36:156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenson RS, Brewer HB Jr., Chapman MJ, Fazio S, Hussain MM, Kontush A, Krauss RM, Otvos JD, Remaley AT, Schaefer EJ. HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin Chem. 2011;57:392–410. [DOI] [PubMed] [Google Scholar]
- 12.Kontush A, Lhomme M, Chapman MJ. Unraveling the complexities of the HDL lipidome. J Lipid Res. 2013;54:2950–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walsh BW, Li H, Sacks FM. Effects of postmenopausal hormone replacement with oral and transdermal estrogen on high density lipoprotein metabolism. J Lipid Res. 1994;35:2083–2093. [PubMed] [Google Scholar]
- 14.Brinton EA. Oral estrogen replacement therapy in postmenopausal women selectively raises levels and production rates of lipoprotein A-I and lowers hepatic lipase activity without lowering the fractional catabolic rate. Arterioscler Thromb Vasc Biol. 1996;16:431–440. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Stolinski M, Umpleby AM. Development of a method to measure prebetaHDL and alphaHDL apoA-I enrichment for stable isotopic studies of HDL kinetics. Lipids. 2012;47:1011–1018. [DOI] [PubMed] [Google Scholar]
- 16.Lee LH, Andraski AB, Pieper B, Higashi H, Sacks FM, Aikawa M, Singh SA. Automation of PRM-dependent D3-Leu tracer enrichment in HDL to study the metabolism of apoA-I, LCAT and other apolipoproteins. Proteomics. 2017;17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh SA, Aikawa M. Unbiased and targeted mass spectrometry for the HDL proteome. Curr Opin Lipidol. 2017;28:68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sacks FM, Lichtenstein AH, Wu JHY, Appel LJ, Creager MA, Kris-Etherton PM, Miller M, Rimm EB, Rudel LL, Robinson JG, Stone NJ, Van Horn LV, American Heart A. Dietary Fats and Cardiovascular Disease: A Presidential Advisory From the American Heart Association. Circulation. 2017;136:e1–e23. [DOI] [PubMed] [Google Scholar]
- 19.Brinton EA, Eisenberg S, Breslow JL. A low-fat diet decreases high density lipoprotein (HDL) cholesterol levels by decreasing HDL apolipoprotein transport rates. J Clin Invest. 1990;85:144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Velez-Carrasco W, Lichtenstein AH, Barrett PH, Sun Z, Dolnikowski GG, Welty FK, Schaefer EJ. Human apolipoprotein A-I kinetics within triglyceride-rich lipoproteins and high density lipoproteins. J Lipid Res. 1999;40:1695–1700. [PubMed] [Google Scholar]
- 21.Ooi EM, Lichtenstein AH, Millar JS, Diffenderfer MR, Lamon-Fava S, Rasmussen H, Welty FK, Barrett PH, Schaefer EJ. Effects of Therapeutic Lifestyle Change diets high and low in dietary fish-derived FAs on lipoprotein metabolism in middle-aged and elderly subjects. J Lipid Res. 2012;53:1958–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desroches S, Paradis ME, Perusse M, Archer WR, Bergeron J, Couture P, Bergeron N, Lamarche B. Apolipoprotein A-I, A-II, and VLDL-B-100 metabolism in men: comparison of a low-fat diet and a high-monounsaturated fatty acid diet. J Lipid Res. 2004;45:2331–2338. [DOI] [PubMed] [Google Scholar]
- 23.Labonte ME, Jenkins DJ, Lewis GF, Chiavaroli L, Wong JM, Kendall CW, Hogue JC, Couture P, Lamarche B. Adding MUFA to a dietary portfolio of cholesterol-lowering foods reduces apoAI fractional catabolic rate in subjects with dyslipidaemia. Br J Nutr. 2013;110:426–436. [DOI] [PubMed] [Google Scholar]
- 24.Berglund L, Oliver EH, Fontanez N, Holleran S, Matthews K, Roheim PS, Ginsberg HN, Ramakrishnan R, Lefevre M. HDL-subpopulation patterns in response to reductions in dietary total and saturated fat intakes in healthy subjects. Am J Clin Nutr. 1999;70:992–1000. [DOI] [PubMed] [Google Scholar]
- 25.Singh S, Kirchner M, Steen JA, Steen H. A practical guide to the FLEXIQuant method. Methods Mol Biol. 2012;893:295–319. [DOI] [PubMed] [Google Scholar]
- 26.Furtado JD, Yamamoto R, Melchior JT, Andraski AB, Gamez-Guerrero M, Mulcahy P, He Z, Cai T, Davidson WS, Sacks FM. Distinct Proteomic Signatures in 16 HDL (High-Density Lipoprotein) Subspecies. Arterioscler Thromb Vasc Biol. 2018;38:2827–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tagliabracci VS, Wiley SE, Guo X, et al. A Single Kinase Generates the Majority of the Secreted Phosphoproteome. Cell. 2015;161:1619–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nikkila EA, Kekki M. Plasma triglyceride metabolism in thyroid disease. J Clin Invest. 1972;51:2103–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen D, Nickel M, Mizuguchi C, Saito H, Lund-Katz S, Phillips MC. Interactions of apolipoprotein A-I with high-density lipoprotein particles. Biochemistry. 2013;52:1963–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gordon SM, Deng J, Tomann AB, Shah AS, Lu LJ, Davidson WS. Multi-dimensional co-separation analysis reveals protein-protein interactions defining plasma lipoprotein subspecies. Mol Cell Proteomics. 2013;12:3123–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pownall HJ, Hosken BD, Gillard BK, Higgins CL, Lin HY, Massey JB. Speciation of human plasma high-density lipoprotein (HDL): HDL stability and apolipoprotein A-I partitioning. Biochemistry. 2007;46:7449–7459. [DOI] [PubMed] [Google Scholar]
- 32.Morton AM, Furtado JD, Mendivil CO, Sacks FM. Dietary unsaturated fat increases HDL metabolic pathways involving apoE favorable to reverse cholesterol transport. JCI Insight. 2019;4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weisgraber KH, Shinto LH. Identification of the disulfide-linked homodimer of apolipoprotein E3 in plasma. Impact on receptor binding activity. J Biol Chem. 1991;266:12029–12034. [PubMed] [Google Scholar]
- 34.Ikewaki K, Rader DJ, Zech LA, Brewer HB Jr. In vivo metabolism of apolipoproteins A-I and E in patients with abetalipoproteinemia: implications for the roles of apolipoproteins B and E in HDL metabolism. J Lipid Res. 1994;35:1809–1819. [PubMed] [Google Scholar]
- 35.Christoffersen C, Nielsen LB, Axler O, Andersson A, Johnsen AH, Dahlback B. Isolation and characterization of human apolipoprotein M-containing lipoproteins. J Lipid Res. 2006;47:1833–1843. [DOI] [PubMed] [Google Scholar]
- 36.Wolfrum C, Poy MN, Stoffel M. Apolipoprotein M is required for prebeta-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nat Med. 2005;11:418–422. [DOI] [PubMed] [Google Scholar]
- 37.Ren K, Tang ZL, Jiang Y, Tan YM, Yi GH. Apolipoprotein M. Clin Chim Acta. 2015;446:21–29. [DOI] [PubMed] [Google Scholar]
- 38.Liu M, Seo J, Allegood J, Bi X, Zhu X, Boudyguina E, Gebre AK, Avni D, Shah D, Sorci-Thomas MG, Thomas MJ, Shelness GS, Spiegel S, Parks JS. Hepatic apolipoprotein M (apoM) overexpression stimulates formation of larger apoM/sphingosine 1-phosphate-enriched plasma high density lipoprotein. J Biol Chem. 2014;289:2801–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahnstrom J, Axler O, Jauhiainen M, Salomaa V, Havulinna AS, Ehnholm C, Frikke-Schmidt R, Tybjaerg-Hansen A, Dahlback B. Levels of apolipoprotein M are not associated with the risk of coronary heart disease in two independent case-control studies. J Lipid Res. 2008;49:1912–1917. [DOI] [PubMed] [Google Scholar]
- 40.Caron S, Verrijken A, Mertens I, et al. Transcriptional activation of apolipoprotein CIII expression by glucose may contribute to diabetic dyslipidemia. Arterioscler Thromb Vasc Biol. 2011;31:513–519. [DOI] [PubMed] [Google Scholar]
- 41.Jensen MK, Aroner SA, Mukamal KJ, Furtado JD, Post WS, Tsai MY, Tjonneland A, Polak JF, Rimm EB, Overvad K, McClelland RL, Sacks FM. High-Density Lipoprotein Subspecies Defined by Presence of Apolipoprotein C-III and Incident Coronary Heart Disease in Four Cohorts. Circulation. 2018;137:1364–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morton AM, Koch M, Mendivil CO, Furtado JD, Tjonneland A, Overvad K, Wang L, Jensen MK, Sacks FM. Apolipoproteins E and CIII interact to regulate HDL metabolism and coronary heart disease risk. JCI Insight. 2018;3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gelissen IC, Hochgrebe T, Wilson MR, Easterbrook-Smith SB, Jessup W, Dean RT, Brown AJ. Apolipoprotein J (clusterin) induces cholesterol export from macrophage-foam cells: a potential anti-atherogenic function? Biochem J. 1998;331 (Pt 1):231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Navab M, Anantharamaiah GM, Reddy ST, Van Lenten BJ, Wagner AC, Hama S, Hough G, Bachini E, Garber DW, Mishra VK, Palgunachari MN, Fogelman AM. An oral apoJ peptide renders HDL antiinflammatory in mice and monkeys and dramatically reduces atherosclerosis in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2005;25:1932–1937. [DOI] [PubMed] [Google Scholar]
- 45.Swertfeger DK, Li H, Rebholz S, Zhu X, Shah AS, Davidson WS, Lu LJ. Mapping Atheroprotective Functions and Related Proteins/Lipoproteins in Size Fractionated Human Plasma. Mol Cell Proteomics. 2017;16:680–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shiflett AM, Bishop JR, Pahwa A, Hajduk SL. Human high density lipoproteins are platforms for the assembly of multi-component innate immune complexes. J Biol Chem. 2005;280:32578–32585. [DOI] [PubMed] [Google Scholar]
- 47.Raper J, Fung R, Ghiso J, Nussenzweig V, Tomlinson S. Characterization of a novel trypanosome lytic factor from human serum. Infect Immun. 1999;67:1910–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weckerle A, Snipes JA, Cheng D, Gebre AK, Reisz JA, Murea M, Shelness GS, Hawkins GA, Furdui CM, Freedman BI, Parks JS, Ma L. Characterization of circulating APOL1 protein complexes in African Americans. J Lipid Res. 2016;57:120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duchateau PN, Pullinger CR, Orellana RE, Kunitake ST, Naya-Vigne J, O’Connor PM, Malloy MJ, Kane JP. Apolipoprotein L, a new human high density lipoprotein apolipoprotein expressed by the pancreas. Identification, cloning, characterization, and plasma distribution of apolipoprotein L. J Biol Chem. 1997;272:25576–25582. [DOI] [PubMed] [Google Scholar]
- 50.Vanhollebeke B, De Muylder G, Nielsen MJ, Pays A, Tebabi P, Dieu M, Raes M, Moestrup SK, Pays E. A haptoglobin-hemoglobin receptor conveys innate immunity to Trypanosoma brucei in humans. Science. 2008;320:677–681. [DOI] [PubMed] [Google Scholar]
- 51.Vanhollebeke B, Pays E. The trypanolytic factor of human serum: many ways to enter the parasite, a single way to kill. Mol Microbiol. 2010;76:806–814. [DOI] [PubMed] [Google Scholar]
- 52.Glomset JA, Parker F, Tjaden M, Williams RH. The esterification in vitro of free cholesterol in human and rat plasma. Biochim Biophys Acta. 1962;58:398–406. [DOI] [PubMed] [Google Scholar]
- 53.Shamburek RD, Bakker-Arkema R, Auerbach BJ, Krause BR, Homan R, Amar MJ, Freeman LA, Remaley AT. Familial lecithin:cholesterol acyltransferase deficiency: First-in-human treatment with enzyme replacement. J Clin Lipidol. 2016;10:356–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bolin DJ, Jonas A. Sphingomyelin inhibits the lecithin-cholesterol acyltransferase reaction with reconstituted high density lipoproteins by decreasing enzyme binding. J Biol Chem. 1996;271:19152–19158. [DOI] [PubMed] [Google Scholar]
- 55.Asztalos B, Lefevre M, Wong L, Foster TA, Tulley R, Windhauser M, Zhang W, Roheim PS. Differential response to low-fat diet between low and normal HDL-cholesterol subjects. J Lipid Res. 2000;41:321–328. [PubMed] [Google Scholar]
- 56.Zheng C, Khoo C, Furtado J, Ikewaki K, Sacks FM. Dietary monounsaturated fat activates metabolic pathways for triglyceride-rich lipoproteins that involve apolipoproteins E and C-III. Am J Clin Nutr. 2008;88:272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng C, Khoo C, Furtado J, Sacks FM. Apolipoprotein C-III and the metabolic basis for hypertriglyceridemia and the dense low-density lipoprotein phenotype. Circulation. 2010;121:1722–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng C, Murdoch SJ, Brunzell JD, Sacks FM. Lipoprotein lipase bound to apolipoprotein B lipoproteins accelerates clearance of postprandial lipoproteins in humans. Arterioscler Thromb Vasc Biol. 2006;26:891–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.