

Abstract
Choroideremia (CHM) is an X-linked chorioretinal dystrophy characterized by progressive degeneration of the choroid, retinal pigment epithelium and retina. This disease is caused by mutations in the X-linked CHM gene encoding a Ras-related GTPase Rab escort protein (REP)-1, which is extremely important for the retinal function. Clinically, male-affected patients have a progressive reduction in visual acuity. This disease is formally considered incurable, although new promising treatments have been recently introduced. In this article, a review of the salient pathogenetic features of choroideremia, essential for the proper interpretation of therapeutic approaches, is followed by a discussion of the fundamental clinical features of this hereditary disease. Finally, relevant new therapeutic approaches in this disease will be discussed, including gene therapy, stem cells, small molecules, and retinal prosthesis.
Keywords: choroideremia, heredodystrophies, gene, therapy, clinical trials, stem cells, gene therapy, small molecules, retinal prosthesis
Introduction
Choroideremia (CHM) is an uncommon heredodystrophy with an estimated prevalence of 1 in 50,000 patients. This disorder mainly involves males because of its X-linked inheritance pattern1 and is dependent on mutations in the CHM gene. This gene is known to be related to membrane transportation protein in the retina and retinal pigment epithelium (RPE).1
Assuming that CHM is associated with a progressive chorioretinal atrophy, this dystrophy was named choroideremia since its first descriptions.2 In detail, choroideremia is characterized by a progressive deterioration of the outer retina, RPE and inner choroid, although there are still controversies regarding which of these structures is first and mainly affected.3,4
Affected subjects generally complain night blindness in their teenage years, and their visual symptoms slowly progress toward a severe peripheral vision loss during adulthood. Carrier female patients are generally asymptomatic; however, they may feature mild disease characteristics, including nyctalopia and pigmentary changes in the fundus oculi.5–7
Nowadays, the main problem in the management of patients with choroideremia is that there is lack of approved treatments available for patients with this disease.8,9 In this review, we will summarize the emerging treatments for choroideremia. To place the therapeutic options in proper context, this review begins with the two core components essential for proper interpretation of therapy: pathophysiology and clinical aspects of choroideremia.
Genetics
Choroideremia is an X-linked recessive inherited disorder due to mutation in the CHM gene (OMIM 303390), which is placed on chromosome X at position q21.2. CHM messenger RNA (mRNA) is responsible for the creation of the Rab escort protein (REP)-1 which is ubiquitously expressed. This protein has 653 amino acids and is involved in intracellular migration of organelles and molecules. Especially, REP-1 manages the process aimed at attaching the unprenylated Rab proteins to the geranyl-geranyl transferase 2 enzyme.10,11 As a consequence, in the deficiency of this process, Rabs proteins are not able to participate in intracellular trafficking. Assuming that REP-1 is extremely important in the dynamics of the RPE and photoreceptors, these cells are particularly affected by this impairment.12
The CHM phenotype is related to ~280 identified mutations, including sequence variations, translocations, point mutations, small deletions, insertions, nonsense, and frameshift mutations. Nonsense and frameshift mutations are the most prevalent and are responsible for ~70% of cases.13,14
It is important to know that genotype-phenotype correlations remain unclear as it was not possible to provide a definite correlation between different CHM mutations and disease severity. Thus, there can be variability in clinical characteristics and severity of this disease, even within a single family with the same mutation.15,16
Pathophysiology
Although the expanded knowledge about the mutational spectrum of CHM, it is still controversial the specific pathogenesis. There are histopathological evidences suggesting the choroid as primarily affected.17 On the contrary, other important studies demonstrated that RPE cells or photoreceptors are involved first.18,19
Furthermore, inflammation seems to be a relevant part of this disorder. In detail, in a post-mortem study, McDonald et al20 presented the ocular histopathological findings in a patient with choroideremia. This study displayed the presence of lymphocytic infiltration into the choroid and inflammation was therefore speculated to represent an early event in choroideremia.
Clinical Diagnosis
Detection of variants in the CHM gene was previously identified using a range of methods.21,22 Importantly, a recent paper has demonstrated the utility of cell‑free fetal DNA in combination with next‑generation sequencing (NGS) for diagnosing choroideremia and non-invasive prenatal testing for Y chromosome determination.23 This approach might improve the use of prenatal diagnosis, increase genetic counseling, and grant a personalization of the clinical management in choroideremia.
Clinically, male patients with choroideremia typically report nyctalopia in their first to second decade of life, and in their 20s they are usually aware of reduction in peripheral vision.15 Peripheral visual field loss progresses and it usually culminates in loss of central vision in their fifth or sixth decade of life.5
The phenotype variability in female carriers is probably related to random X-chromosome inactivation: severe choroideremia in female patients has been attributed to a skewed X-chromosome inactivation pattern.24
Although CHM is mainly considered as an isolated retinal disease, this disorder may be also characterized by syndromic phenotypes such as intellectual disability, clip lip and palate, skeletal deformities.25,26
Clinical evaluation of patients with choroideremia involves several imaging modalities and a visual assessment.
Fundus Examination
In the first phases of this dystrophy, peripheral pigmentary changes may characterize the retina of affected patients. At a later time, distinct regions of chorioretinal atrophy are usually visible. Of note, these degenerative alterations usually start at the equator and centripetally and progressively involve the posterior pole and the peripapillary region (Figure 1).25
Figure 1.
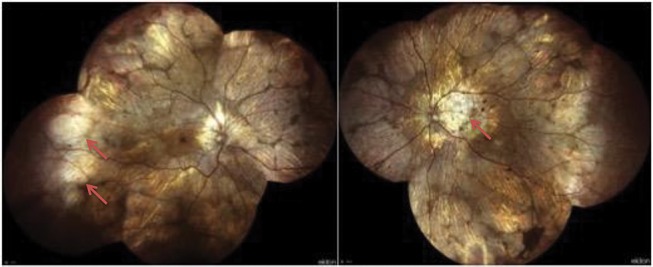
Widefield color fundus photography from an affected CHM male patient. Widefield images show areas of chorioretinal atrophy mainly localised in the periphery and peripapillary regions (red arrows). Images of this patient courtesy of Dr. Andrea Scupola, MD.
Female carriers are usually unaffected, even though minimal retinal signs such as pigmentary changes can be observed on their fundus examination.10
Moreover, a few articles have reported examples of female CHM carriers with areas of chorioretinal atrophy in the retinal periphery, highlighting that also carriers have clinical heterogenicity.27,28
Furthermore, posterior subcapsular cataract, macular edema and choroidal neovascularization may develop in these patients.
Fundus Autofluorescence
Fundus autofluorescence (FAF) is considered as a surrogate of lipofuscin distribution in RPE and has become an essential biomarker for disease progression in CHM. FAF reveals regions of chorioretinal atrophy in patients with choroideremia as hypoautofluorescent areas with sharp hyperautofluorescent edges because of remaining degenerating RPE and loss of photoreceptors (Figure 2).29 Therefore, FAF allows the evaluation of disease progression by estimating the rate of shrinkage autofluorescence.5,30
Figure 2.

Multimodal imaging from an affected CHM male patient. Multicolor images (left) show areas of RPE atrophy in the macula. Blue fundus autofluorescence (middle left) and fluorescein angiography (middle right) images display the presence of RPE and choroidal atrophy, respectively. Structural OCT images (right) demonstrate a thin choroid and an RPE and outer retinal atrophy. Images of this patient courtesy of Prof. Eric Souied, MD, PhD.
The larger reduction in autofluorescence is topographically located in the nasal retina in the region near to the optic disc, while the macular area is usually less involved likely because of different rod–cone density ratios in these regions. Jolly et al31 reported a 10-fold decrease in FAF every 25 years, corresponding to a rate of 7.7% reduction in residual retinal area every year.
Of note, female carrier patients may have a unique pattern of speckled autofluorescence as hyperautofluorescence dots alternate with hypoautofluorescence granular areas (Figure 3).32
Figure 3.

Multimodal imaging from a female CHM carrier. Multicolor images (left) show areas of RPE alteration and mottling in the macula. Blue fundus autofluorescence images (middle) show a characteristic pattern of speckled autofluorescence. Structural OCT images (right) demonstrate a normal appearance of the macular retina and choroid.
Optical Coherence Tomography
Several studies employing optical coherence tomography (OCT) have been performed in CHM patients. Using OCT, the central macular thickness (CMT) was displayed to be increased in the early childhood.30 Successively, they develop progressive CMT thinning as visual acuity declines.33
OCT studies also identified other variations, including areas of decreased RPE reflectance and alterations in the external limiting membrane (ELM) and ellipsoid zone (EZ).5 Furthermore, outer retinal tubulations were identified to occur with chorioretinal atrophy and they were speculated to represent remodeling of degenerating photoreceptors.3,34,35 Using OCT, important reports also displayed the presence of triangular outer nuclear layer (ONL) structures, which were showed in correspondence of the junction between atrophic and trophic retina and were suspected to represent hyperplastic Müller cells.34,36 Moreover, microcysts in the inner retinal layers were showed to occur in ~20% of CHM cases and they were associated with a negative prognosis.3,37
Optical Coherence Tomography Angiography
Patients with choroideremia have been recently investigated using optical coherence tomography angiography (OCTA).38 In a recent study on 2 carriers and 7 affected patients with choroideremia, the authors demonstrated a significant reduction in choriocapillaris (CC) perfusion in both patients and carriers.39 Notably, patients with choroideremia displayed definite transition between regions of moderately conserved and damaged CC, while carriers were characterized by patchy areas of CC loss. Interestingly, this paper also investigated the correlation between CC perfusion and outer retinal declining. The reduction in CC perfusion was demonstrated to be inferior in areas characterized by damaged EZ, which further demonstrated the strict interdependence between these two structures in this disorder. Moreover, the areaof conserved EZ evaluated using OCT was greater than the region of conserved RPE, the latter quantified using FAF. These evidences could suggest that CC and photoreceptors' damages follow the RPE death.40,41
Fluorescein Angiography
Fluorescein angiography (FA) is not commonly employed in patients with choroideremia, excluding cases with suspect of choroidal neovascularization. In CHM patients, FA may also display narrowing of the retinal vessels and retarded retinal and choroidal flow.42
Electroretinography
In the earlier stages of the disease, full-field electroretinogram (ERG) may show pathological changes in CHM patients. In detail, abnormal responses for the scotopic component may be first detected, which correlate symptomatically with a reduction in rod-mediated night vision.15,29 As the disease progresses, ERG findings may display an abnormal photopic component secondary to cone involvement.15,29 Importantly, the ERG examination has a high sensitivity in detecting functional reduction, assuming that in those patients with stable visual acuity, a reduction in ERG responses may be detected.15,29
Notably, while most female carriers have normal ERG responses, they can rarely have subtle alterations such as decreased scotopic waves.43
Therapeutic Options
Despite there are currently no treatment options available for patients with choroideremia, several attempts to managing this disease and its progression are under investigation.
Gene Therapy
Gene therapy is a promising therapeutic option, assuming that choroideremia is monogenic, its diagnosis is relatively precocious, and it is moderately homogenous in clinical findings and evolution, thereby providing a wide therapeutic window for intervention.4 Furthermore, the eye may be considered as an ideal target for gene therapy, assumed that there is a low risk of immune reaction and systemic penetration.
Several adeno-associated virus (AAV) vector-based gene therapies have been used in the CHM treatment. AAVs constitute tiny, single-stranded DNA viruses of the parvovirus family. Furthermore, they are the prevalent viral vector employed in retinal dystrophies because of a favorable immunologic, inflammatory and toxicity profile. In detail, specific AAV species (2, 5 and 7–9) have been employed for transducing RPE and photoreceptors.
In 2017, a multicentric nonrandomized open-label phase one-half clinical trial (NCT01461213) employing an AAV.REP1 vector was performed in CHM patients.9,44 Fourteen patients at different stages of the disease were included and underwent subretinal injection of AAV2.REP1 vector during pars plana vitrectomy.9,44 Two out of 14 patients experienced procedure-related complications and were treated off protocol, while the remaining patients demonstrated a significant improvement in vision in the follow-up visits, as compared with the untreated fellow eyes.9
In addition, Xue et al45 recently employed an adeno-associated viral vector to express RPE1 in patients with choroideremia. In the latter study, the authors evaluated visual changes in treated eyes as compared to the untreated fellow eyes. They demonstrated that retinal gene therapy can improve visual acuity in a cohort of predominantly late-stage choroideremia patients.
Even though gene therapy is a promising treatment, several challenges are still unaddressed, including alternative delivery methods, such as intravitreal injection. However, intravitreal injection of viral vectors has still several restrictions, including an inefficacious cell penetration and possible humoral response.
Stem Cells
Previous reports have evaluated stem cells as a potential treatment in patients with different advanced retinal dystrophies.46–48
These approaches might be used in patients with CHM. However, it would be first needed to define the most suitable tissue to be regenerated (RPE cells or photoreceptors) and the best stem cell category to use. Previous approaches with both fibroblast-derived49 and hESC-derived50 RPE cells have been used in a few patients with advanced age-related macular degeneration (AMD) and seemed to effect in a functional improvement. Future larger trials will shed further light on the utility of this treatment in CHM patients.
Duong et al51 recently promoted the use of induced pluripotent stem cells in non-human primates. They demonstrated that the delivery of these cells may restore normal prenylation, phagocytosis, and protein trafficking in the CHM cells.
Small Molecules
In-frame nonsense mutations cause a significant number of CHM cases (~30%) by premature stop codons. Small-molecule drugs may be helpful in these cases by promoting ribosomal read-through of premature stop codons and thus bypassing abnormal termination signals. This approach was demonstrated to be useful in other disorders, such as cystic fibrosis.52 In the nonsense-mediated zebrafish model of choroideremia, this approach was displayed to be effective in determining an increase in RPE1 expression.53
Retinal Prosthesis Systems
Retinal prosthesis systems provide long-term retinal stimulation in cases with advanced stages of outer retinal dystrophies. This treatment is mainly aimed at either preserve or reach essential vision.
In a prospective, multicenter trial using the Argus II system, 47 individuals (most patients were affected by retinitis pigmentosa, while 1 patient had choroideremia) with light perception vision had this treatment.54 The Argus II retinal prosthesis system (Second Sight Medical Products, Sylmar, California, USA) consists of a video camera mounted on glasses, a video processing unit and an epiretinal stimulating array. The array stimulates residual inner retinal layers and signal is transmitted to the visual cortex. This device granted the identification of letters and words, indicating reproducible spatial resolution. The latter study adds to the growing evidence that retinal prothesis systems may have an important role as a therapeutic option in patients with severe vision loss.
Conclusion
CHM represents a genetic eye disease that causes a profound vision loss in affected male subjects. For this reason, although this is a rare disease, its impact is enormous.
Although this review highlights the therapeutic options for choroideremia, these treatments are not mature, and remain in a state of fast progression. Gene therapy may be a crucial treatment option in these patients, assuming that choroideremia represents a perfect target disease for this kind of treatment.
In summary, new promising therapies are available in patients with choroideremia. Future clinical trials will shed further light on the different therapeutic options in these patients.
Disclosure
Professor Francesco Bandello reports personal fees from Allergan, Bayer, BoehringerIngelheim, Fidia Sooft, Hoffmann-La Roche, Novartis, NTC Pharma, SIFI, ThromboGenics and from Zeiss, outside the submitted work. The authors report no other conflicts of interest in this work.
References
- 1.Kalatzis V, Hamel CP, MacDonald IM. Choroideremia: towards a therapy. Am J Ophthalmol. 2013;156(3):433.e3–437.e3. doi: 10.1016/j.ajo.2013.05.009 [DOI] [PubMed] [Google Scholar]
- 2.L M. Original description of choroideremia as a retinal dystrophy. Ber Naturwissensch-Med Ver Inssbruck. 1872;2191–2197. [Google Scholar]
- 3.Xue K, Oldani M, Jolly JK, et al. Correlation of optical coherence tomography and autofluorescence in the outer retina and choroid of patients with choroideremia. Invest Ophthalmol Vis Sci. 2016;57(8):3674–3684. doi: 10.1167/iovs.15-18364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xue K, MacLaren RE. Ocular gene therapy for choroideremia: clinical trials and future perspectives. Expert Rev Ophthalmol. 2018;13(3):129–138. doi: 10.1080/17469899.2018.1475232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitsios A, Dubis AM, Moosajee M. Choroideremia: from genetic and clinical phenotyping to gene therapy and future treatments. Ther Adv Ophthalmol. 2018. 10 2515841418817490–2515841418817490. doi: 10.1177/2515841418817490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonilha VL, Trzupek KM, Li Y, et al. Choroideremia: analysis of the retina from a female symptomatic carrier. Ophthalmic Genet. 2008;29(3):99–110. doi: 10.1080/13816810802206499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edwards TL, Groppe M, Jolly JK, Downes SM, MacLaren RE. Correlation of retinal structure and function in choroideremia carriers. Ophthalmology. 2015;122(6):1274–1276. doi: 10.1016/j.ophtha.2014.12.036 [DOI] [PubMed] [Google Scholar]
- 8.Barnard AR, Groppe M, MacLaren RE. Gene therapy for choroideremia using an adeno-associated viral (AAV) vector. Cold Spring Harb Perspect Med. 2014;5(3):a017293–a017293. doi: 10.1101/cshperspect.a017293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacLaren RE, Groppe M, Barnard AR, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet (London, England). 2014;383(9923):1129–1137. doi: 10.1016/S0140-6736(13)62117-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanchez-Alcudia R, Garcia-Hoyos M, Lopez-Martinez MA, et al. A comprehensive analysis of choroideremia: from genetic characterization to clinical practice. PLoS One. 2016;11(4):e0151943. doi: 10.1371/journal.pone.0151943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seabra MC, Brown MS, Slaughter CA, Südhof TC, Goldstein JL. Purification of component A of rab geranylgeranyl transferase: possible identity with the choroideremia gene product. Cell. 1992;70(6):1049–1057. doi: 10.1016/0092-8674(92)90253-9 [DOI] [PubMed] [Google Scholar]
- 12.Preising M, Ayuso C. Rab escort protein 1 (REP1) in intracellular traffic: a functional and pathophysiological overview. Ophthalmic Genet. 2004;25(2):101–110. doi: 10.1080/13816810490514333 [DOI] [PubMed] [Google Scholar]
- 13.Furgoch MJB, Mewes-Arès J, Radziwon A, Macdonald IM. Molecular genetic diagnostic techniques in choroideremia. Mol Vis. 2014;20:535–544. [PMC free article] [PubMed] [Google Scholar]
- 14.Coussa RG, Traboulsi EI. Choroideremia: a review of general findings and pathogenesis. Ophthalmic Genet. 2012;33(2):57–65. doi: 10.3109/13816810.2011.620056 [DOI] [PubMed] [Google Scholar]
- 15.Pennesi ME, Birch DG, Duncan JL, Bennett J, Girach A. CHOROIDEREMIA: retinal degeneration with an unmet need. RETINA. 2019;39(11):2059–2069. doi: 10.1097/IAE.0000000000002553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simunovic MP, Jolly JK, Xue K, et al. The spectrum of CHM gene mutations in choroideremia and their relationship to clinical phenotype. Invest Ophthalmol Vis Sci. 2016;57(14):6033–6039. doi: 10.1167/iovs.16-20230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cameron JD, Fine BS, Shapiro I. Histopathologic observations in choroideremia with emphasis on vascular changes of the uveal tract. Ophthalmology. 1987;94(2):187–196. doi: 10.1016/S0161-6420(87)33479-7 [DOI] [PubMed] [Google Scholar]
- 18.Flannery JG, Bird AC, Farber DB, Weleber RG, Bok D. A histopathologic study of a choroideremia carrier. Investig Ophthalmol Vis Sci. 1990;31(2):229–236. [PubMed] [Google Scholar]
- 19.Mura M, Sereda C, Jablonski MM, MacDonald IM, Iannaccone A. Clinical and functional findings in choroideremia due to complete deletion of the CHM gene. JAMA Ophthalmol. 2007;125(8):1107–1113. doi: 10.1001/archopht.125.8.1107 [DOI] [PubMed] [Google Scholar]
- 20.MacDonald IM, Russell L, Chan -C-C. Choroideremia: new findings from ocular pathology and review of recent literature. Surv Ophthalmol. 2009;54(3):401–407. doi: 10.1016/j.survophthal.2009.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gregg AR, Van den Veyver IB, Gross SJ, Madankumar R, Rink BD, Norton ME. Noninvasive prenatal screening by next-generation sequencing. Annu Rev Genomics Hum Genet. 2014. doi: 10.1146/annurev-genom-090413-025341 [DOI] [PubMed] [Google Scholar]
- 22.Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010. doi: 10.1038/nmeth0810-575 [DOI] [PubMed] [Google Scholar]
- 23.Zhu L, Cheng J, Zhou B, et al. Diagnosis for choroideremia in a large Chinese pedigree by next-generation sequencing (NGS) and non-invasive prenatal testing (NIPT). Mol Med Rep. 2017. doi: 10.3892/mmr.2017.6119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang AS, Kim LA, Fawzi AA. Clinical characteristics of a large choroideremia pedigree carrying a novel CHM mutation. Arch Ophthalmol (Chicago, Ill 1960). 2012;130(9):1184–1189. doi: 10.1001/archophthalmol.2012.1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coussa RG, Kim J, Traboulsi EI. Choroideremia: effect of age on visual acuity in patients and female carriers. Ophthalmic Genet. 2012;33(2):66–73. doi: 10.3109/13816810.2011.623261 [DOI] [PubMed] [Google Scholar]
- 26.Rosenberg T, Schwartz M, Niebuhr E, et al. Choroideremia in interstitial deletion of the X chromosome. Ophthalmic Paediatr Genet. 1986;7(3):205–210. doi: 10.3109/13816818609004140 [DOI] [PubMed] [Google Scholar]
- 27.MacDonald IM, Hume S, Chan S, et al. Choroideremia In: Adam MP, Ardinger HH, Pagon RA, et al. editors. GeneReviews®. Seattle:University of Washington, Seattle; 1993–2019. 2003 (updated 2015). [Google Scholar]
- 28.Campos-Pavón J, Torres-Peña JL. Choroidal neovascularization secondary to choroideremia. Arch Soc Esp Oftalmol. 2015;90(6):289–291. doi: 10.1016/j.oftale201506008. [DOI] [PubMed] [Google Scholar]
- 29.Renner AB, Kellner U, Cropp E, et al. Choroideremia: variability of clinical and electrophysiological characteristics and first report of a negative electroretinogram. Ophthalmology. 2006. doi: 10.1016/j.ophtha.2006.05.045 [DOI] [PubMed] [Google Scholar]
- 30.Khan KN, Islam F, Moore AT, Michaelides M. Clinical and genetic features of choroideremia in childhood. Ophthalmology. 2016;123:2158–2165. doi: 10.1016/j.ophtha.2016.06.051 [DOI] [PubMed] [Google Scholar]
- 31.Jolly JK, Edwards TL, Moules J, Groppe M, Downes SM, MacLaren RE. A qualitative and quantitative assessment of fundus autofluorescence patterns in patients with choroideremia. Invest Ophthalmol Vis Sci. 2016;57(10):4498–4503. doi: 10.1167/iovs.15-18362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Preising MN, Wegscheider E, Friedburg C, Poloschek CM, Wabbels BK, Lorenz B. Fundus autofluorescence in carriers of choroideremia and correlation with electrophysiologic and psychophysical data. Ophthalmology. 2009;116(6):1201.e2–1209.e2. doi: 10.1016/j.ophtha.2009.01.016 [DOI] [PubMed] [Google Scholar]
- 33.Heon E, Alabduljalil T, McGuigan DB, et al. Visual function and central retinal structure in choroideremia. Investig Ophthalmol Vis Sci. 2016. doi: 10.1167/iovs.15-18421 [DOI] [PubMed] [Google Scholar]
- 34.Heon E, Alabduljalil T, McGuigan III DB, et al. Visual function and central retinal structure in choroideremianatural history of choroideremia. Invest Ophthalmol Vis Sci. 2016;57(9):OCT377–OCT387. doi: 10.1167/iovs.15-18421 [DOI] [PubMed] [Google Scholar]
- 35.Morgan JIW, Han G, Klinman E, et al. High-resolution adaptive optics retinal imaging of cellular structure in choroideremia. Invest Ophthalmol Vis Sci. 2014;55(10):6381–6397. doi: 10.1167/iovs.13-13454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abbouda A, Lim W, Sprogyte L, Webster AMM. Quantitative and qualitative features of spectral-domain optical coherence tomography provide prognostic indicators for visual acuity in patients with choroideremia. Ophthalmic Surg Lasers Imaging Retina. 2017;48(9):711–716. doi: 10.3928/23258160-20170829-05 [DOI] [PubMed] [Google Scholar]
- 37.Syed R, Sundquist SM, Ratnam K, et al. High-resolution images of retinal structure in patients with choroideremia. Invest Ophthalmol Vis Sci. 2013;54(2):950–961. doi: 10.1167/iovs.12-10707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borrelli E, Sarraf D, Freund KB, Sadda SR. OCT angiography and evaluation of the choroid and choroidal vascular disorders. Prog Retin Eye Res. 2018. doi: 10.1016/j.preteyeres.2018.07.002 [DOI] [PubMed] [Google Scholar]
- 39.Jain N, Jia Y, Gao SS, et al. Optical coherence tomography angiography in choroideremia: correlating choriocapillaris loss with overlying degeneration. JAMA Ophthalmol. 2016;134(6):697–702. doi: 10.1001/jamaophthalmol.2016.0874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patel RC, Gao SS, Zhang M, et al. Optical coherence tomography angiography of choroidal neovascularization in four inherited retinal dystrophies. Retina. 2016;36(12):2339–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abbouda A, Dubis AM, Webster AR, Moosajee M. Identifying characteristic features of the retinal and choroidal vasculature in choroideremia using optical coherence tomography angiography. Eye (Lond). 2018;32(3):563–571. doi: 10.1038/eye.2017.242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forsius H, Hyvärinen LEA, Nieminen H, Flower R. Fluorescein and indocyanine green fluorescence angiography in study of affected males and in female carriers with choroideremia. Acta Ophthalmol. 1977;55(3):459–470. doi: 10.1111/j.1755-3768.1977.tb06123.x [DOI] [PubMed] [Google Scholar]
- 43.Sieving PA, Niffenegger JH, Berson EL. Electroretinographic findings in selected pedigrees with choroideremia. Am J Ophthalmol. 1986;101(3):361–367. doi: 10.1016/0002-9394(86)90832-9 [DOI] [PubMed] [Google Scholar]
- 44.Xue K, Groppe M, Salvetti AP, MacLaren RE. Technique of retinal gene therapy: delivery of viral vector into the subretinal space. Eye (Lond). 2017;31(9):1308–1316. doi: 10.1038/eye.2017.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xue K, Jolly JK, Barnard AR, et al. Beneficial effects on vision in patients undergoing retinal gene therapy for choroideremia. Nat Med. 2018;24(10):1507–1512. doi: 10.1038/s41591-018-0185-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perin EC, Silva GV. Stem cell therapy for cardiac diseases. Curr Opin Hematol. 2004;11:399–403. doi: 10.1097/01.moh.0000143359.77689.aa [DOI] [PubMed] [Google Scholar]
- 47.Ramsden CM, Powner MB, Carr A-JF, Smart MJK, da Cruz L, Coffey PJ. Stem cells in retinal regeneration: past, present and future. Development. 2013;140:2576–2585. doi: 10.1242/dev.092270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwartz SD, Regillo CD, Lam BL, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet. 2015;385:509–516. doi: 10.1016/S0140-6736(14)61376-3 [DOI] [PubMed] [Google Scholar]
- 49.Mandai M, Watanabe A, Kurimoto Y, et al. Autologous induced stem-cell–derived retinal cells for macular degeneration. N Engl J Med. 2017;376(11):1038–1046. doi: 10.1056/NEJMoa1608368 [DOI] [PubMed] [Google Scholar]
- 50.da Cruz L, Fynes K, Georgiadis O, et al. Phase 1 clinical study of an embryonic stem cell–derived retinal pigment epithelium patch in age-related macular degeneration. Nat Biotechnol. 2018;36:328. doi: 10.1038/nbt.4114 [DOI] [PubMed] [Google Scholar]
- 51.Duong TT, Vasireddy V, Ramachandran P, et al. Use of induced pluripotent stem cell models to probe the pathogenesis of Choroideremia and to develop a potential treatment. Stem Cell Res. 2018;27:140–150. doi: 10.1016/j.scr.2018.01.009 [DOI] [PubMed] [Google Scholar]
- 52.Richardson R, Smart M, Tracey-White D, Webster AR, Moosajee M. Mechanism and evidence of nonsense suppression therapy for genetic eye disorders. Exp Eye Res. 2017;155:24–37. doi: 10.1016/j.exer.2017.01.001 [DOI] [PubMed] [Google Scholar]
- 53.Moosajee M, Tracey-White D, Smart M, et al. Functional rescue of REP1 following treatment with PTC124 and novel derivative PTC-414 in human choroideremia fibroblasts and the nonsense-mediated zebrafish model. Hum Mol Genet. 2016;25(16):3416–3431. doi: 10.1093/hmg/ddw184 [DOI] [PubMed] [Google Scholar]
- 54.Schaffrath K, Schellhase H, Walter P, et al. One-year safety and performance assessment of the argus ii retinal prosthesis: a postapproval study. JAMA Ophthalmol. 2019;137:896. doi: 10.1001/jamaophthalmol.2019.1476 [DOI] [PMC free article] [PubMed] [Google Scholar]