

SUMMARY
Compensation among paralogous transcription factors (TFs) confers genetic robustness of cellular processes, but how TFs dynamically respond to paralog depletion on a genome-wide scale in vivo remains incompletely understood. Using single and double conditional knockout of myocyte enhancer factor 2 (MEF2) family TFs in granule neurons of the mouse cerebellum, we find that MEF2A and MEF2D play functionally redundant roles in cerebellar-dependent motor learning. Although both TFs are highly expressed in granule neurons, transcriptomic analyses show MEF2D is the predominant genomic regulator of gene expression in vivo. Strikingly, genome-wide occupancy analyses reveal upon depletion of MEF2D, MEF2A occupancy robustly increases at a subset of sites normally bound to MEF2D. Importantly, sites experiencing compensatory MEF2A occupancy are concentrated within open chromatin and undergo functional compensation for genomic activation and gene expression. Finally, motor activity induces a switch from non-compensatory to compensatory MEF2-dependent gene regulation. These studies uncover genome-wide functional interdependency between paralogous TFs in the brain.
Graphical Abstract
In Brief
Majidi et al. study how transcription factors respond to paralog depletion by conditionally depleting MEF2A and MEF2D in mouse cerebellum. Depletion of MEF2D induces functionally compensatory genomic occupancy by MEF2A. Compensation occurs within accessible chromatin in a context-dependent manner. This study explores the interdependency between paralogous transcription factors.
INTRODUCTION
The development and function of the mammalian brain requires precise control of gene expression (Cholewa-Waclaw et al., 2016; de la Torre-Ubieta and Bonni, 2011; Ziats et al., 2015). Combinatorial interactions of DNA-binding transcription factors (TFs) regulate diverse gene programs that specify neuronal sub-types, develop and refine circuits, and link sensory experience to adaptive responses of the brain (Mazzoni et al., 2013; Molyneaux et al., 2007; Kawashima et al., 2013; Pulimood et al., 2017; Sharma et al., 2019). Additionally, deregulation of TFs contributes to the pathogenesis of neurological diseases (Porter et al., 2018; Ebert and Greenberg, 2013; Li et al., 2018). Although genome-wide patterns of TF cooperation are just beginning to be revealed in the nervous system, how TF family members cooperate to orchestrate gene expression in the mammalian brain remains poorly understood.
The majority of mammalian TFs are members of multigene families that have evolved by duplication events of a single TF (Teichmann and Babu, 2004; Levine and Tjian, 2003). Members of a multigene TF family, known as paralogous TFs, typically have highly conserved DNA-binding domains. Because paralogous TFs often bind virtually identical short DNA sequences, they are thought to participate in cooperative mechanisms distinct from their non-paralogous counterparts (Weirauch et al., 2014; Wei et al., 2010; Luna-Zurita et al., 2016). However, despite the prevalence of paralogous TFs, the nature and importance of the coordinated function of paralogous TFs at a genome-wide level remains unexplored.
Importantly, paralogous TFs are thought to confer genetic robustness to cellular processes through evolutionary retention of functionally redundant activities (Macneil and Walhout, 2011). Despite the prevalence of phenotypic redundancy, the underlying molecular mechanisms by which paralogous TFs regulate this widespread phenomenon are relatively unexplored. Individual overexpression studies of paralogous TFs in Saccharomyces cerevisiae have revealed similar DNA-binding specificities to exogenous DNA sequences (Fuxman Bass et al., 2015). Recently, individual transfection of Hox proteins followed by chromatin profiling yielded insights into their binding distribution in insect cells (Porcelli et al., 2019). Although similar studies have advanced our understanding of paralogous TF binding, how endogenous TFs dynamically respond within the chromatin context to paralog depletion remains unknown and will require the integrative study of co-expressed paralogous TFs. Regional and single cell analyses of gene expression in the developing and adult brain have revealed diverse expression patterns of paralogous TFs, suggesting that they may act in concert to impart genetic robustness during brain development and function (Lyons et al., 1995; Saunders et al., 2018). However, in vivo mechanisms of paralogous TF interplay, and their roles in neuronal gene expression and function are as of yet unknown.
The MEF2 (myocyte enhancer factor 2) proteins play fundamental roles in the development and function of the brain, and deregulation of MEF2 activity contributes to the pathogenesis of neurological diseases (Shalizi and Bonni, 2005; Yap and Greenberg, 2018; Lipton et al., 2009). However, the interdependency and functional output of paralogous MEF2 proteins on a genome-wide scale have not yet been explored. The four vertebrate MEF2 family members, MEF2A–D, share a highly conserved MADS domain that mediates DNA binding to the consensus MEF2 response element (MRE) YTAWWWWTAR (Flavell et al., 2008; Potthoff and Olson, 2007). Expression studies show different but overlapping patterns of MEF2A–D expression in the brain (Lyons et al., 1995; Potthoff and Olson, 2007), suggesting that distinct combinations of MEF2 family members coordinate gene expression (Estrella et al., 2015). MEF2 family members play key roles in neuronal survival, differentiation, and maturation (Gaudilliere et al., 2002; Flavell et al., 2006; Yamada et al., 2013), as well as neural plasticity (Rashid et al., 2014; Chang et al., 2017; Chen et al., 2012; Pulipparacharuvil et al., 2008). Importantly, MEF2 factors are thought to confer phenotypic robustness to these neuronal processes across multiple brain regions. Despite the significant and diverse roles of MEF2 proteins in the nervous system, mechanisms of combinatorial gene regulation by these factors remain to be elucidated.
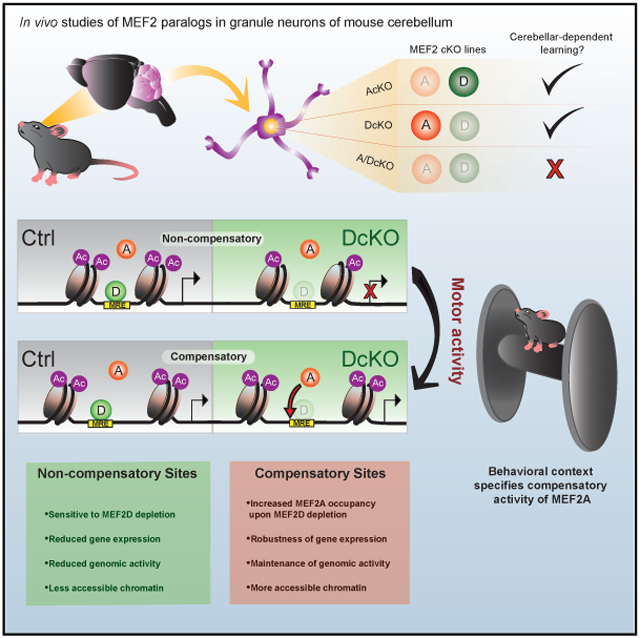
Here, we reveal an in vivo interdependent mechanism of gene regulation mediated by the paralogous TFs MEF2A and MEF2D in granule neurons of mouse cerebellum. Despite strong co-expression of MEF2A and MEF2D and high amino acid identity of their respective DNA-binding domains, genome-wide profiling shows that MEF2D appears to be the predominant regulator of gene expression in granule neurons in the mouse cerebellum. Strikingly, upon MEF2D depletion, the genomic occupancy of MEF2A robustly increases at a distinct subpopulation of formerly bound MEF2D sites, revealing differential compensation by MEF2A on a genome-wide level. Epigenome and transcriptome analyses reveal that sites experiencing compensatory MEF2A occupancy undergo functional compensation for genomic activation and gene expression. In contrast, a distinct population of sites without compensatory MEF2A activity undergo significant dysregulation upon loss of MEF2D. The two populations of MEF2 target sites are further stratified by relative chromatin accessibility, with compensatory MEF2A activity concentrated within more open chromatin. Behavioral context also plays a key role in specifying MEF2A compensatory activity, as revealed by a dynamic switch from non-compensatory to compensatory MEF2-dependent gene regulation in the context of motor activity. Collectively, our study defines a compensatory transcriptional regulatory scheme for MEF2A and MEF2D that imparts genetic robustness during mammalian brain development and function, hence providing insight into the functional interdependency between paralogous TFs.
RESULTS
MEF2A and MEF2D Regulate Cerebellar-Dependent Motor Learning in a Compensatory Manner
Granule neurons of the mouse cerebellum provide a uniquely robust model to study the interplay of MEF2 family members in the mammalian brain. Whereas other neuronal subtypes solely express one MEF2 or variable levels of three or four MEF2 family members, cerebellar granule neurons strongly co-express MEF2A and MEF2D (Lyons et al., 1995). Importantly, granule neurons vastly outnumber all other cells in the cerebellum, making these neurons a suitably homogeneous cell type for in vivo studies of the neuronal epigenome (Yamada et al., 2014; Yang et al., 2016; Frank et al., 2015).
In granule neurons of mouse cerebellum, the temporal expression of MEF2A and MEF2D coincides with the expression of the granule-neuron-enriched protein GABA(A)a6 receptor (G6R) (Lin and Bulleit, 1996). Therefore, to characterize the roles of MEF2A and MEF2D in granule neurons, we used a G6R-promoter-driven Cre transgenic line to conditionally knock out Mef2a (AcKO), Mef2d (DcKO), or both Mef2a and Mef2d (ADcKO) selectively in granule neurons (Figure 1A) (Fünfschilling and Reichardt, 2002; Andzelm et al., 2015, 2019). The expression of MEF2A and MEF2D proteins concurrently increased in the mouse cerebellum as granule neurons differentiate and mature (Roussel and Hatten, 2011; de la Torre-Ubieta and Bonni, 2011), reaching peak levels at postnatal day 15 (P15) and continuing into adulthood (Figure 1B). MEF2A and MEF2D proteins were downregulated specifically in the internal granule layer of the cerebellar cortex during the third postnatal week in AcKO and DcKO mice, respectively (Figures 1B and 1C). Importantly, conditional knockout of MEF2A failed to effectively alter the levels of MEF2D RNA or protein, and conversely conditional knockout of MEF2D failed to effectively alter the levels of MEF2A RNA or protein in the cerebellum (Figure 1B; Figure S1A).
Figure 1. MEF2A and MEF2D Regulate Cerebellar-Dependent Motor Learning in a Compensatory Manner.

(A) Schematic depiction of single- and double-conditional knockouts of MEF2A and MEF2D in cerebellar granule neurons in mice. Transgenic mice expressing the recombinase Cre downstream of the granule-neuron-enriched GABA(A)a6-receptor (G6R) gene promoter are crossed to mice harboring conditional alleles for Mef2a, Mef2d, or both Mef2a and Mef2d to generate Mef2afl/fl;G6RCre+/− (AcKO), Mef2dfl/fl;G6RCre+/− (DcKO), and Mef2a/dfl/fl;G6RCre+/− (ADcKO), respectively.
(B) Immunoblotting of MEF2A (top left) and MEF2D (bottom left) in lysates of cerebellum harvested from postnatal day 3 through 56 (P3–P56) AcKO (top) and DcKO (bottom) mice, respectively. Immunoblotting also performed for MEF2D in P22 AcKO mice (top right) and MEF2A in P22 DcKO mice (bottom right).
(C) Sagittal sections of P22 cerebellum from different MEF2 conditional knockout mouse lines were subjected to immunohistochemistry by using antibodies recognizing calbindin (first column), and brain-enriched MEF2 family members (second column), as well as the DNA dye bisbenzimide (Hoechst) (third column). Immunohistochemical analyses of MEF2A performed on control (top row) and AcKO (second row) mouse cerebellum, of MEF2D on control (third row) and DcKO (fourth row) mouse cerebellum, and of MEF2C on control (fifth row) mouse cerebellum. IGL, internal granule layer; PCL, Purkinje cell layer; ML, molecular layer. Scale bar: 100 μm, 20× magnification.
(D) Cerebellar-dependent eyeblink conditioning learning paradigm was performed on AcKO and control (n = 6 per genotype), DcKO and control (n = 7 and n = 8, respectively), and ADcKO and control (n = 11 and n = 9, respectively) mice. Percent conditioned response (CR) is shown as mean ± SEM for each session day. ****p < 10−4, **p < 10−2 repeated-measures ANOVA, Sidak’s multiple comparison test.
See also Figure S1.
In immunohistochemical analyses, MEF2D expression was predominantly restricted to granule neurons and Purkinje cells, whereas MEF2A was expressed in granule neurons and other neurons of the molecular and internal granule layers (Figure 1C). MEF2B was undetectable, and MEF2C was expressed predominantly in Purkinje cells (Figure 1C; Figure S1B; Mellén et al., 2012). In RNA sequencing analyses of the mouse cerebellum, mRNA copy numbers of MEF2A, C, and D were 37.42, 6.55, and 56.74 RPKM (reads per kilobase of transcript, per million mapped reads), respectively. Thus, MEF2A and MEF2D were in the top 10% of detected transcripts, whereas MEF2C was in the lower 50th percentile of detected transcripts. MEF2B transcript levels were undetectable by RNA sequencing of mouse cerebellum. These data show that MEF2A and MEF2D are robustly expressed in granule neurons of the developing mouse cerebellum.
To determine whether MEF2A and MEF2D are required for the proper function of granule neurons, we first subjected AcKO, DcKO, and ADcKO mice to the cerebellar-dependent eyeblink conditioning learning paradigm (Figure 1D; Heiney et al., 2014; Valnegri et al., 2017). During this associative task, mice learn to blink in response to an initially neutral conditioned stimulus (blue light) after repeated pairing with an eyeblink-eliciting unconditioned stimulus (periocular air puff). As expected, the learned eyelid blink conditioned response (CR) gradually increased each session day in control littermate mice. Strikingly, the rate of CRs was significantly reduced in ADcKO mice by day 3 of conditioning, which persisted for the remaining session days (Figure 1D). However, neither AcKO nor DcKO mice had significant learning deficits (Figure 1D). In other analyses, general motor coordination assessed by the accelerating rotarod and DigiGait assays (Puram et al., 2011; Hurlock et al., 2009) was not affected upon knockout of MEF2A, MEF2D, or both proteins (Figures S1C and S1D). Taken together, these data reveal that MEF2A and MEF2D are required redundantly in cerebellar-dependent learning, suggesting a potential compensatory mechanism of MEF2A and MEF2D in granule neurons.
Because MEF2 family members regulate neuronal survival and synapse formation and refinement in diverse brain regions (Shalizi et al., 2006; Flavell et al., 2006; Gaudilliere et al., 2002), we next characterized the effect of combined knockout of MEF2A and MEF2D on these fundamental developmental events. The architecture of the cerebellar cortex was not altered in ADcKO mice, nor was there a detectable change in neuronal survival (Figure S1E). In electron microscopy analyses, the density of granule neuron parallel fiber boutons synapses onto Purkinje neuron dendritic spines was not significantly altered in ADcKO mice (Figure S1F). In vivo electroporation of granule neurons in ADcKO mice followed by morphological analyses of their dendrites revealed no differences in dendrite length (Figure S1G).
MEF2A and MEF2D Exhibit Complex Patterns of Gene Regulation in the Cerebellum
Because of the redundant contribution of MEF2A and MEF2D to cerebellar dependent motor learning, we reasoned that the two paralogous TFs may exert compensatory mechanisms of gene regulation. To test this possibility, we first characterized the relative effects of individual and combined conditional knockouts of MEF2A and MEF2D on gene expression in granule neurons in vivo. We, therefore, performed RNA-seq in the cerebellum from P22 mice in four biological replicates each of AcKO, DcKO, ADcKO, and respective sex-matched control littermates.
We next characterized how genetic depletion of MEF2A or MEF2D individually contributes to gene dysregulation in the combined MEF2A and MEF2D knockout. Differential mRNA expression analysis of ADcKO and control littermates led to the identification of 130 “MEF2-repressed genes” that were significantly upregulated and 175 “MEF2-activated” genes that were significantly downregulated in the ADcKO mouse cerebellum. Principal component analysis of MEF2-regulated genes showed smaller variation between control littermates for each condition, with the majority caused by differences between the three conditional knockout conditions (Figure S2A). The MEF2-regulated genes were then organized into distinct clusters based on their expression in AcKO, DcKO, ADcKO, and respective control littermates by subjecting them to hierarchical clustering by using the dynamic tree cut algorithm (Figure 2A). This analysis yielded two major clusters each for MEF2-repressed (C1, C2) and activated genes (C3, C4), which respectively represent ~14.8%, 27.9%, 20.7%, and 36.7% of MEF2-regulated genes. Quantitative assessment of the relative behavior of single- and double-conditional knockouts on gene expression in each cluster revealed significantly stronger effects in DcKO mice compared to AcKO and control mice across all four identified clusters (Figure 2B). These data suggest that depletion of MEF2D predominantly affects gene expression in comparison to the modest effects of MEF2A depletion in granule neurons.
Figure 2. MEF2A and MEF2D Exhibit Complex Patterns of Gene Regulation in Cerebellum.

(A) Hierarchical clustering of gene expression of AcKO, DcKO, ADcKO, and respective control (Ctrl) P22 mouse cerebellum for genes detected as significantly dysregulated (false discovery rate [FDR], <0.05) in analysis of RNA-seq from Ctrl and ADcKO cerebellum (n = 4 biological replicates per genotype). Four clusters are indicated on the left side of the heatmap. Heat represents Z score of log2 cpm (counts per million) for a given gene.
(B) Box-whisker plots representing median and distribution of the Z score of log2 cpm for control, AcKO, DcKO, and ADcKO mice show distinct trends in gene expression for each of the four clusters (C1–C4) of genes identified in (A). ****p < 10−4, ***p < 10−3, one-way ANOVA, Tukey’s multiple comparison test; n.s., not significant.
(C) WashU Epigenome browser view of RNA-seq coverage from AcKO, DcKO, ADcKO, and respective control (Ctrl) mice, illustrating changes in gene expression for each of the four clusters (C1–C4).
See also Figure S2.
Two major patterns emerged upon closer examination of clusters of significantly altered genes between DcKO and ADcKO mice. First, the C1 and C3 clusters of differentially regulated genes in the mouse cerebellum displayed no significant differences between DcKO and ADcKO mice, suggesting that these groups of genes are primarily affected by the depletion of MEF2D in granule neurons of the mouse cerebellum. In contrast, the C2 and C4 clusters showed significantly stronger dysregulation in ADcKO relative to DcKO mice (Figure 2C; Figure S2B), suggesting compensatory regulation of C2 and C4 cluster genes by MEF2A and MEF2D in granule neurons.
MEF2A Displays Functionally Compensatory Binding Activity at a Distinct Subset of MEF2D-Bound Genomic Sites
The gene expression patterns in single- and double-conditional knockout mice suggested shared as well as distinct roles for MEF2A and MEF2D. To better understand the underlying basis of the relationships between these two paralogous TFs, we performed chromatin immunoprecipitation sequencing (ChIP-seq) of MEF2A and MEF2D in the cerebellum in control, AcKO, and DcKO mice. Among these three conditions, we identified 203 MEF2A-binding sites and 1388 MEF2D-binding sites (Figure 3A). Due to the strong conservation between paralogous TFs, we validated the specificity of MEF2A and MEF2D ChIP-seq signal in the cerebellum of AcKO and DcKO mice, respectively (Figures S3A and S3B). De novo motif discovery demonstrated the canonical MRE YTAWWWTAR as the most significantly enriched motif at >95% of peaks (Figure 3B), further strengthening the conclusion that the identified MEF2A and MEF2D ChIP-seq sites represent high confidence MEF2-binding sites. Analyses of promoters and enhancers, identified based on histone modifications in ChIP-seq of the mouse cerebellum in P22 mice (Figures 3A and 3C; Figure S3D; Yamada et al., 2014), revealed that MEF2A and MEF2D bound active intergenic enhancers at a frequency higher than the normal genomic distribution. However, MEF2A proportionally bound promoters to a greater extent than MEF2D (Figure 3C).
Figure 3. MEF2A Displays Functionally Compensatory Binding Activity at a Subset of MEF2D-Bound Genomic Sites.

(A) Aggregate plot and heatmap of ChIP-seq signal for MEF2A (red, n = 203) and MEF2D (green, n = 1388) genomic binding sites in Ctrl, AcKO, and DcKO P22 mouse cerebellum (n = 3–4 biological replicates per ChIP condition). ChIP-seq signal for H3K27ac and H3K4me3 (purple) from P22 mouse cerebellum centered on MEF2 genomic binding sites.
(B) Significantly enriched de novo binding motifs at MEF2A and MEF2D peaks. Below each position, weighted matrix is the E-value followed by the most significant match to a TF motif.
(C) Pie charts displaying regulatory element distribution of MEF2A (top) and MEF2D (bottom) peaks. Pro, promoter; Enh, enhancer. For regulatory element distribution of genomic background, refer to Figure S3D.
(D) MEF2D sites experiencing increased MEF2A occupancy in DcKO mouse cerebellum are termed compensatory. Aggregate plots are shown for MEF2A and MEF2D ChIP-seq performed in different control (Ctrl) or cKO mice. A schematic depicts outcome based on the ChIP-seq peak signal detected in each condition.
(E) Non-compensatory sites, defined as control MEF2D sites without increased MEF2A binding in DcKO mouse cerebellum, are shown. Aggregate plots are shown for MEF2A and MEF2D ChIP-seq performed in different control (Ctrl) or cKO mice. A schematic depicts outcome based on the ChIP-seq peak signal detected in each condition.
(F) Heatmap of ChIP-seq signal for MEF2A (left) and MEF2D (right) at compensatory sites, sorted in descending order based on MEF2A ChIP-seq signal.
(G) Scatterplot of MEF2A and MEF2D ChIP-seq signal (RPKM) at compensatory sites. Coefficient of determination using Pearson correlation.
(H) For compensatory genomic binding sites, box-whisker plots for log2-transformed fold change of H3K27ac in AcKO, DcKO, and ADcKO over respective control mice. ****p < 10−4, ANOVA followed by Tukey’s multiple comparison test. n.s., not significant.
(I) WashU Epigenome Browser view of a compensatory site (highlighted), showing MEF2A, MEF2D, and H3K27ac ChIP-seq coverage from AcKO, DcKO, AdcKO, and respective control (Ctrl) mice.
(J) Same format as (I) for non-compensatory sites.
See also Figure S3.
Strikingly, the vast majority of MEF2A peaks exclusively appeared in DcKO mice, in which 199 sites were statistically enriched above background (Figure 3A). The dynamic upregulation of MEF2A binding activity upon depletion of MEF2D was highly consistent, appearing in each of the four biological ChIP-seq replicates (Figure 3A; Figure S3C). In contrast, MEF2D was stably present at the majority of MEF2-bound sites in the control condition, with its occupancy mostly unaffected by conditional knockout of MEF2A. These data suggest that MEF2D may play the predominant role in regulating gene expression in granule neurons.
Because MEF2A and MEF2D both bind to the canonical MRE, we next determined the extent of overlap between DcKO-induced MEF2A peaks and sites normally occupied by MEF2D. Intersectional peak analysis revealed that conditional knockout of MEF2D induced a robust increase of MEF2A mainly at sites previously bound by MEF2D (Figure 3D). Specifically, 80% of MEF2A peaks that appeared in DcKO mice were occupied by MEF2D in control littermate mice. Heretofore, we refer to sites at which MEF2A increased in DcKO mice as compensatory (Figure 3D), whereas the subset of MEF2D sites that did not experience MEF2A binding statistically enriched above the background are termed non-compensatory (Figure 3E). As an example of a compensatory MEF2 binding site, the Inpp4b intragenic enhancer showed binding to MEF2D in control mice, whereas MEF2A occupancy was statistically undetectable (Figure 3I). However, the absence of MEF2D in DcKO mice led to significantly increased MEF2A occupancy at the Inpp4b intragenic enhancer. In contrast, an example of a non-compensatory binding site is a normally MEF2D-bound proximal enhancer for Gng7, at which no MEF2A occupancy was observed in DcKO mice (Figure 3J).
The finding that MEF2A displays compensatory binding at a subset of MEF2D sites raises the question of whether the strength of MEF2D occupancy at a given site dictates the extent of MEF2A compensatory binding. We found no correlation between MEF2A and MEF2D signal intensity at compensatory sites, suggesting that the strength of MEF2D occupancy is not predictive of the degree to which MEF2A binds a given site (Figures 3F and 3G).
To determine whether regulation of compensatory sites depends on MEF2A and MEF2D, we analyzed the levels of histone H3 lysine 27 acetylation (H3K27ac) at these sites upon single- or double-conditional knockout of the two TFs (Figure 3H). As expected, because compensatory sites were predominantly bound to MEF2D in control mice, conditional knockout of MEF2A minimally affected H3K27ac levels at these sites (Figures 3H and 3I). Conditional knockout of MEF2D also failed to significantly alter H3K27ac levels at these sites (Figures 3H and 3I). By contrast, H3K27ac levels were significantly reduced at compensatory sites upon conditional knockout of both MEF2A and MEF2D (Figures 3H and 3I), suggesting that either MEF2A or MEF2D is sufficient for activation of these regulatory sites. Just as for compensatory sites, conditional knockout of MEF2A had minimal effects on H3K27ac levels at non-compensatory sites. In contrast, however, conditional knockout of MEF2D significantly reduced H3K27ac levels at non-compensatory sites (Figure 3J; Figure S3E), suggesting that MEF2D is selectively required for activation of non-compensatory sites. Taken together, our data suggest that the dynamically increased occupancy of MEF2A at compensatory sites may confer on these targets a uniquely robust ability to maintain normal activation in the presence of a single MEF2 factor.
Compensatory Binding by MEF2A at a Subset of MEF2-Activated Target Genes Confers Genetic Robustness to MEF2D Depletion
To understand how MEF2A and MEF2D occupancy regulates gene expression, we first identified MEF2 target genes by performing an intersectional analysis of all MEF2 ChIP-seq and MEF2 activated/repressed genes. These analyses revealed that MEF2A and MEF2D bound in the vicinity of 49.1% of MEF2-activated genes. In contrast, only 9.2% of MEF2-repressed genes were associated with MEF2-binding sites (Figure 4A). Next, we analyzed patterns of MEF2 occupancy for each of the RNA-seq clusters, C1–C4. Remarkably, the two MEF2-activated clusters C3 and C4 exhibited distinct patterns of MEF2A and MEF2D target gene occupancy. The C3 cluster was solely enriched for MEF2D-occupied non-compensatory sites, consistent with the finding that these MEF2D-bound genes are primarily dysregulated upon depletion of MEF2D (Figure 4B). In contrast, C4 was the only cluster of genes with significant enrichment of compensatory MEF2A and MEF2D occupancy (Figure 4B). In other analyses, H3K27ac levels at compensatory direct target gene regulatory elements were most significantly reduced in ADcKO mice (Figure 4C). At non-compensatory direct target genes, DcKO and ADcKO mice showed similarly reduced levels of H3K27ac (Figure S4A), suggesting relatively stronger sensitivity to genetic perturbation of MEF2D. As an example of a compensatory MEF2-binding site, the stimulus-responsive gene Tll1 also exhibited compensatory MEF2A occupancy at an intragenic enhancer, at which H3K27ac levels were reduced most strongly in ADcKO mice. RNA-seq coverage at Tll1 showed substantial reduction in ADcKO mice (Figure 4D). In contrast, a loss of MEF2D at the intragenic enhancer of the Blc9l gene did not lead to compensatory MEF2A binding, which was associated with a significant reduction of H3K27ac levels at this regulatory element and reduced gene expression in both DcKO and ADcKO mice (Figure 4E). In summary, compensatory binding by MEF2A at a subset of MEF2-activated target genes diminishes the influence of MEF2D depletion on gene expression and associated regulatory element activation.
Figure 4. Compensatory Binding by MEF2A at a Subset of MEF2-Activated Target Genes Confers Robustness to MEF2D Depletion on Gene Expression.

(A) Pie chart representing proportion of MEF2-repressed (top) and MEF2-activated (bottom) genes associated with MEF2A and/or MEF2D genomic binding sites.
(B) Table represents significance of overlap of RNA-seq clusters with MEF2A and MEF2D peaks identified in various ChIP-seq conditions. Columns from left to right: MEF2A ChIP in control mice; MEF2A ChIP in DcKO mice (compensatory sites); MEF2D ChIP in control mice; and MEF2D ChIP in AcKO mice. Rows from top to bottom represent RNA-seq clusters 1, 2, 3, and 4 (C1, C2, C3, and C4), as depicted by a schematic of the trends in gene expression (schematization of Figure 2B): control (black circle), AcKO (red circle), DcKO (green circle), and ADcKO (blue circle) mice. Heat represents −log-10 p value significance of overlap determined by hypergeometric test, with significant values displayed.
(C) For compensatory direct target genes (defined as genes from compensatory C4 associated with MEF2-bound sites): box-whisker plots show median and distribution of log2-transformed fold change of H3K27ac in different conditional knockout mice over respective control mice. ***p < 10−3, ANOVA followed by Tukey’s multiple comparison test.
(D) WashU Epigenome Browser view of an intragenic enhancer site (highlighted) of a compensatory direct target gene, showing MEF2A, MEF2D, and H3K27ac ChIP-seq and RNA-seq coverage from AcKO, DcKO, AdcKO, and respective control (Ctrl) mice.
(E) Same format as (D) for intragenic enhancer site (highlighted) at non-compensatory direct target gene.
See also Figure S4.
Chromatin Accessibility and Cellular State Specify Compensatory Action of MEF2A
To investigate the basis for distinct MEF2A compensatory activities, we next compared genomic features at compensatory and non-compensatory MEF2-regulated sites. Because the MRE directly binds both MEF2A and MEF2D, we first characterized whether MREs at compensatory versus non-compensatory sites exhibit different levels of degeneracy. These analyses revealed no significant difference in the distribution of MRE degeneracy scores between compensatory and non-compensatory sites (Figure 5A; Figure S5A), suggesting factors beyond the MRE sequence direct compensatory action of MEF2A.
Figure 5. Chromatin Accessibility and Cellular State Specify Compensatory Action of MEF2A.

(A) Box-whisker plot of MRE degeneracy scores relative to a consensus MRE for compensatory and non-compensatory sites. n.s., not significant.
(B) Box-whisker plot of chromatin accessibility for compensatory and non-compensatory sites. Two-sided unpaired t test, ****p < 10−4.
(C) Plots represent bins of MEF2 sites sorted by increasing MEF2A peak read density in DcKO mice. Relative distribution of compensatory (light gray) and non-compensatory (black) MEF2 sites are shown as percentage (%) of regions comprising each bin.
(D) Box-whisker plots of DNaseI sequencing read density for bins of increasing MEF2A density in DcKO mice (classified in C). ANOVA followed by Tukey’s multiple comparison test. **p < 10−2; n.s., not significant.
(E) (Top) Relative motif enrichment for compensatory sites relative to non-compensatory sites. (Bottom) Relative motif enrichment at MEF2A ChIP peaks (some of which are overlap MEF2C) relative to MEF2C-only ChIP peaks in cortical neurons (Telese et al., 2015). Predicted binding factor followed in parentheses by number of significant motif occurrences and the top q-value representing significance of relative enrichment.
(F) ChIP-seq signal of MEF2A (red, left) and MEF2D (green, right) at stimulus responsive genes listed in the “Response to Stimulus” Gene Ontology term (GO 0050896).
(G) Schematic depicting accelerating rotarod paradigm (Yang et al., 2016; Yamada et al., 2019).
(H) Rotarod paradigm performed for control and ADcKO mice followed by qRT-PCR on cerebellar RNA for select rotarod-activated genes experiencing compensatory MEF2A binding. ****p < 10−4, ***p < 10−3, **p < 10−2, *p < 10−4, two-way repeated-measures ANOVA, Sidak’s multiple comparison test.
(I) Total RNA-seq analysis of rotarod-activated gene expression in cerebellum of AcKO, DcKO, ADcKO, and respective control mice subjected to the rotarod paradigm (n = 4 biological replicates per condition). Box-whisker plots of log2-transformed fold change (FC) of gene expression in AcKO, DcKO, and ADcKO over respective control mice for MEF2-occupied rotarod-activated genes at baseline (left) and after rotarod stimulation (middle), as well as unoccupied rotarod-activated genes after rotarod stimulation (right). ****p < 10−4, ANOVA followed by Tukey’s multiple comparison test. n.s., not significant.
See also Figure S5.
In addition to binding site affinity, the chromatin environment plays a critical role in regulating the permissibility of TF binding (Spitz and Furlong, 2012). To assess the relationship of chromatin accessibility and compensatory MEF2A activity, we compared DnaseI sequencing levels between compensatory and non-compensatory sites in P22 mouse cerebellum (Yamada et al., 2019). We found that compensatory sites displayed significantly higher chromatin accessibility than non-compensatory sites, as measured by DNaseI sequencing read density (Figure 5B). Further examination revealed that chromatin accessibility showed a graded relationship to compensatory occupancy by MEF2A. Sites of the highest compensatory occupancy by MEF2A were concentrated in more accessible chromatin, whereas sites in relatively less accessible chromatin were selective for MEF2D (Figures 5C and 5D). These data indicate that chromatin accessibility rather than MRE affinity may restrict target selection by MEF2A to a distinct subset of formerly bound MEF2D sites in conditional MEF2D knockout mice.
Because combinatorial TF occupancy serves as an energetically favorable mechanism to outcompete nucleosomes for a genomic binding site (Lambert et al., 2018; Grossman et al., 2018), we next characterized the presence of other TF-binding sites that may distinguish compensatory and non-compensatory sites. Compensatory MEF2 sites in the cerebellum showed differential motif enrichment for AP-1 complex components compared to non-compensatory MEF2 sites (Figure 5E; Figure S5B). The AP-1 motif is the binding site for the early response proteins FOS and JUN (Eferl and Wagner, 2003; Sheng and Greenberg, 1990). Our interrogation of ChIP-seq datasets of MEF2A and MEF2C binding in cortical neurons (Telese et al., 2015) revealed that sites co-regulated by MEF2A and MEF2C were also significantly enriched for the AP-1 motif compared to sites solely regulated by MEF2C (Figure 5E; Figure S5B). In accordance with higher accessibility at compensatory than non-compensatory sites, AP-1 is thought to increase chromatin accessibility at enhancers by recruitment of the BAF complex (Vierbuchen et al., 2017).
Neuronal stimuli significantly modify the chromatin landscape by increasing accessibility at stimulus-responsive enhancers (Su et al., 2017). Importantly, MEF2 TFs play key roles in neuronal stimulus-dependent gene expression (Assali et al., 2019; Flavell et al., 2008; Lyons et al., 2012). Furthermore, MEF2A and MEF2D display functional redundancy for cerebellar-dependent motor learning (Figure 1D), a process that likely requires stimulus-dependent gene expression to link sensory experiences to adaptive responses of the brain (Yamada et al., 2019). We asked whether neuronal state might influence the compensatory action of MEF2A (Assali et al., 2019; Malik et al., 2014; Ataman et al., 2016; Flavell et al., 2008). Analysis of MEF2A and MEF2D ChIP-seq peaks at stimulus-responsive genes revealed strongly increased MEF2A occupancy at MEF2D-bound sites upon depletion of MEF2D (Figure 5F). Although the expression of stimulus responsive genes is often relatively low in the mouse cerebellum, exposure of mice to forced locomotion in an accelerating rotarod paradigm triggers significant upregulation of canonical immediate early genes and other stimulus-responsive genes in the cerebellum (Figure 5G; Yang et al., 2016). Thus, we performed the accelerating rotarod paradigm followed by qRT-PCR in ADcKO mice on several of the 113 MEF2-bound rotarod-activated genes, including the canonical immediate early genes Fosb, Nr4a2, and Nr4a3. The expression of MEF2-bound rotarod-activated genes was significantly reduced in the cerebellum in ADcKO mice (Figure 5H; Figure S5C), suggesting a role for MEF2 TFs in rotarod-activated gene expression.
We next used an unbiased characterization of MEF2A- and MEF2D-dependent changes in rotarod-activated gene expression by performing the rotarod paradigm followed by RNA-seq in the cerebellum of AcKO, DcKO, or ADcKO mice and their respective control littermates (Figure S5D). Under baseline conditions, MEF2-bound rotarod-activated genes manifested similar dysregulation in both DcKO and ADcKO mice (Figure 5I), suggesting that the MEF2-target genes were regulated in a non-compensatory manner. In contrast, following rotarod stimulation, MEF2-bound rotarod-activated genes were robustly dysregulated in ADcKO mice compared to single-cKO mice (Figure 5I), revealing that rotarod activity induced a switch to compensatory MEF2-dependent regulation. As expected, control rotarod-activated genes with no MEF2 binding were minimally altered in the cerebellum in AcKO, DcKO, or ADcKO mice (Figure 5I). Together, these results reveal that motor-activity-induced changes in neuronal state induce a dynamic switch from non-compensatory to compensatory MEF2-dependent gene regulation, demonstrating the context-dependent nature of paralogous TF interdependency.
DISCUSSION
Redundancy is an inherently dynamic process that involves the substitution of one paralog upon the loss of the other (Macneil and Walhout, 2011). However, prior to our study, how paralogs respond to the absence of a family member at a genome-wide level remained unknown. Our study unveils robust compensatory binding activity of MEF2A at sites normally bound to the predominant genomic occupant MEF2D. This finding suggests competitive binding may operate among MEF2 family members, which may be a widespread phenomenon extending to some of the >20 other TF families comprised of paralogs with highly similar DNA-binding domains (Messina et al., 2004). In addition, due to the widespread expression of MEF2 family members, our findings may provide insights into redundant gene regulation in other cell types. For instance, studies in cardiomyocytes have revealed similar roles for MEF2A and MEF2D in the repression of cell cycle genes and the activation of a subset of sarcomeric markers (Desjardins and Naya, 2017). Moreover, both MEF2A and MEF2D may be redundant for neonatal cardiomyocyte survival, as the overexpression of either factor diminished programmed cell death in the context of MEF2A deficiency (Desjardins and Naya, 2017).
In view of the high amino acid identity in the DNA-binding domains of TF paralogs, what features may allow MEF2D to dominate occupancy at sites co-regulated by both MEF2D and MEF2A? Although a higher prevalence of MEF2D-binding sites might be attributed to technical differences in the ChIP-seq efficiency of MEF2A and MEF2D antibodies, recent studies suggest that paralogs may exhibit differential DNA-binding specificities (Shen et al., 2018). Although the DNA-binding domains of MEF2A and MEF2D share >95% amino acid identity, the few non-consensus amino acids may contribute to differential binding (Potthoff and Olson, 2007). Sampling frequency and target site occupancy are also sensitive to the local concentrations of available TFs, as recently demonstrated for the cooperative TFs Sox2 and Oct4 in embryonic stem cells (Chen et al., 2014). Although MEF2 proteins are expressed at variable concentrations in neuronal cell types, these differences probably do not apply to granule neurons, which fortuitously co-express high levels of both MEF2A and MEF2D (Lyons et al., 1995). Although our analyses of mRNA levels of MEF2A and MEF2D by RNA sequencing of mouse cerebellum reveal similarly high levels of these two MEF2 proteins, MEF2D transcripts are more abundant, which may translate into a higher concentration of the MEF2D protein. The disparity between MEF2A and MEF2D occupancies may arise from their highly divergent transactivation domains. The crystal structure of a MEF2A homodimer demonstrates that the highly divergent region beyond the DNA-binding domain may interact with the genome, possibly conferring distinct binding activities to different MEF2 family members (Wu et al., 2010). Beyond its potential interaction with DNA, the transactivation domain of each MEF2 family member may undergo unique post-translational modifications or bind distinct co-factors that stabilize the binding of one paralog over the other (Shalizi et al., 2006; Shalizi and Bonni, 2005). In addition to co-factor interactions, MEF2A and MEF2D interact to form homodimers and heterodimers (Potthoff and Olson, 2007), which may further influence the distribution and activity of these two proteins across the genome. Perhaps, compensatory sites are more frequently bound by MEF2A/D heterodimers under baseline conditions in comparison to non-compensatory sites, which may predominantly bind MEF2D homodimers.
Importantly, true redundancy is defined as little or no change in output following perturbation of one factor because another one masks the effect (Macneil and Walhout, 2011; Conant and Wagner, 2004). Surprisingly, however, the molecular consequences of paralog occupancy have not yet been adequately explored. The integration of unbiased epigenome and transcriptome analyses in our study reveals that increased MEF2A occupancy upon the loss of MEF2D is functionally compensatory. These findings provide a tangible explanation for a common observation in which TF-binding sites detected by ChIP appear to be nonfunctional due to the unchanged mRNA levels of target genes, following depletion of the assayed TF (Li et al., 2008; Cao et al., 2010; Andzelm et al., 2015).
Although we have discovered the compensatory genomic features of MEF2A and MEF2D, we also find a large number of non-compensatory MEF2D-bound sites. Following gene duplication, TF paralogs are thought to maintain a degree of ancestral function while also gaining new specificities termed “neo-functionalization” (Badis et al., 2009; Macneil and Walhout, 2011). The identification of both compensatory and non-compensatory MEF2 sites supports the occurrence of these dual evolutionary processes for MEF2A and MEF2D. Global analyses of paralog evolution suggest that non-compensatory sites arising from neo-functionalization of MEF2D may have emerged by the evolution of co-factor interactions. TF paralogs arising from local duplication events undergo rapid divergence of protein-protein interactions, with older paralogs acquiring relatively more protein interactions (Guan et al., 2007; Reece-Hoyes et al., 2013; Grove et al., 2009). As phylogenetic analysis indicates that MEF2D arose from an earlier local duplication event, it may have developed more co-factor interactions than MEF2A, thereby acquiring non-compensatory binding sites in granule neurons (Wu et al., 2011).
The chromatin environment plays a critical role in regulating the permissibility of TF binding (Spitz and Furlong, 2012). The concentration of MEF2A compensatory activity within more open chromatin suggests that chromatin accessibility may play a key role in directing the compensatory activity of paralogous TFs. Therefore, compensatory activity by MEF2A may be influenced by competition with nucleosomes at formerly bound MEF2D sites. Neuronal activity dynamically increases the accessibility at enhancers of stimulus-responsive genes (Su et al., 2017). State-dependent alterations in the chromatin environment may explain how motor activity increases MEF2A compensatory activity at formerly non-compensatory sites. Combinatorial TF occupancy serves as an energetically favorable mechanism to outcompete nucleosomes for a genomic binding site (Lambert et al., 2018; Grossman et al., 2017). We show that compensatory sites are differentially enriched for AP-1 motifs. Recent evidence reveals the importance of AP-1 for increasing chromatin accessibility by the recruitment of the BAF complex (Vierbuchen et al., 2017). Upon depletion of MEF2D, AP-1 may be sufficient to maintain adequate chromatin accessibility for incoming MEF2A. Collectively, these data suggest a model whereby collaborative TFs increase chromatin accessibility by recruitment of chromatin remodelers, thus allowing for compensatory regulation by multiple MEF2 family members.
Redundant mechanisms are thought to mediate the robustness for genes that are essential, such as ETS family co-occupancy at housekeeping genes and HOX factors at developmental patterning genes (Macneil and Walhout, 2011; Hollenhorst et al., 2007; Slattery et al., 2011). Because stimulus-responsive genes experience compensatory redundancy by MEF2A and MEF2D, this gene program may represent a shared feature of the MEF2 family. Interestingly, stimulus responsive genes are common targets of MEF2 family members in multiple cell types, including cardiac myocytes, T cells, fibroblasts, and neurons (Andzelm et al., 2015; Black and Olson, 1998). Furthermore, as stimulus-dependent gene expression links sensory experience to adaptive responses of the brain (West and Greenberg, 2011; Alberini and Kandel, 2014; Zovkic et al., 2014), the identification of compensatory regulation of motor-activity-induced gene expression may explain the redundancy of MEF2A and MEF2D in cerebellar-dependent learning.
Non-compensatory MEF2D sites may represent more specialized gene targets in granule neurons. Consistently, in photoreceptors, MEF2D is recruited away from stimulus responsive genes to retinal-specific genes by cooperativity with CRX, a photoreceptor-specific TF (Andzelm et al., 2015). Thus, it will be interesting to determine whether MEF2D plays a more specialized biological role in granule neurons.
Although we have focused on compensatory functions for MEF2A and MEF2D as well as MEF2D-predominant roles in the regulation of transcriptional activation, MEF2 proteins also play critical roles in transcriptional repression, as revealed by studies of sumoylated MEF2 (Grégoire and Yang, 2005; Shalizi et al., 2006, 2007; Yamada et al., 2013). In addition, in vivo knockdown and structure-function studies in rat pups during the first two postnatal weeks suggest that sumoylated MEF2A drives the formation of postsynaptic dendritic claw differentiation and the maturation of presynaptic sites in the rat cerebellum (Shalizi et al., 2006; Yamada et al., 2013). The absence of major changes in transcriptomic analyses of the cerebellum in P22 AcKO mice raises the question of whether sumoylated MEF2A operates at a distinct developmental temporal window to repress transcription and trigger consequent developmental effects.
Due to the diverse states a neuron undergoes during development and plasticity, the context-dependent nature of compensation by TF family members should advance our understanding of brain development and function. As we learn more about the interdependency of paralogous TFs, we should gain further insight into how paralogs respond to TF loss-of-function mutations in the context of disease (Ebert and Greenberg, 2013; Südhof, 2017; Li et al., 2018). Furthermore, identifying the genomic signatures of non-compensatory sites may allow us to predict regulatory elements that might be more susceptible to gene dysregulation upon perturbation of different TF paralogs in disease states.
STAR ★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Azad Bonni (bonni@wustl.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice were maintained in a pathogen-free environment. All procedures involving animals were performed according to protocols approved by the Animal Studies Committee of Washington University School of Medicine and in accordance with both the National Institute of Health Standings Committee on Animals as well as the National Institutes of Health guidelines. Mice used for behavioral experiments were housed individually in cages. MEF2A fl/fl, MEF2D fl/fl and GABAa(6)R-Cre have been described (Andzelm et al., 2015, 2019; Fünfschilling and Reichardt, 2002). For all experiments, biological replicates are individual mice. For each experimental mouse, the control mouse is a sex-matched double floxed littermate without the G6R-Cre transgene. Experimental mice were not involved in previous procedures or multiple types of experiments. Administration of anesthesia for surgical operations on mice is described under Method Details.
METHOD DETAILS
Antibodies
Antibodies to Calbindin (Millipore ab1778; IHC), Mef2a (Santa Cruz sc-313; ChIP/IP/IB), Mef2c (Protein-Tech 18290-1-AP; IB), 14-3-3 (Santa Cruz sc-1675; IB), Cre (Millipore 69050-3; IB), histone H3K27ac (Abcam ab4729; ChIP), cleaved caspase 3 (Cell Signaling Technology 9661S; IHC), GFP (Abcam ab13970; IHC) were purchased. Antibodies to Mef2a (IB) and Mef2d (ChIP/IP/IB) have been described (Flavell et al., 2008; Andzelm et al., 2015).
Immunohistochemistry
The cerebellum from mice was fixed with 4% PFA and 4% sucrose and subjected to cryo-sectioning on the Leica CM3050S Cryostat. Sections were blocked with blocking buffer (10% goat serum, 3% BSA, and 0.4% Triton X in PBS). Subsequently, sections were incubated overnight with relevant primary antibodies followed by a two-hour incubation with Alexa Fluor conjugated secondary antibodies. The DNA dye Bisbenzimide (Hoechst 33258) was used to label cell nuclei. Confocal images were acquired with a Zeiss LSM 880 II Airyscan FAST Confocal Microscope or an Olympus FV1200 Confocal Microscope.
Delay eye-blink conditioning
Delay eye-blink conditioning assay was adapted from the procedure used by Heiney et al. (2014). Sex-matched littermate conditional MEF2A, MEF2D, MEF2A/D cKO or control mice at five to eight weeks of age were used. Surgical procedures were performed as described (Yang et al., 2016). Head plates were implanted and stabilized with screws using Metabond cement (Parkell) over the Bregma skull landmark in mice anesthetized with ketamine/xylazine (100mg/kg; 10mg/kg). After five days of post-surgical recovery, head-fixed mice underwent two consecutive days of one hour habituation sessions on a cylindrical treadmill. After training, mice underwent experimental testing in the head-fixed eyeblink conditioning apparatus. Mice gradually associate a conditioned stimulus (CS; blue LED) with an eye-blink-eliciting unconditioned stimulus (US, 20psi periocular air puff through a 25-gauge needle; CS-US inter-stimulus interval, 150 msec). 100 trials of CS-US pairings were performed each day over six consecutive days. The learned eyelid conditioned response was recorded using a high-speed monochrome camera (Allied Vision). Fraction of eyelid closure, ranging from 0 (fully open) to 1 (fully closed), was calculated on each frame as described previously (Heiney et al., 2014). During the inter-stimulus period, eyelid closure > 0.1 was designated as a conditioned eyelid response (CR). Our measure for motor learning was the percentage of CR-positive trials on each session day (Percent CR). Investigators were blind to genotype during running of behavioral experiment and unblinded for analysis of results.
DigiGait analysis
The DigiGait imaging platform (Mouse Specifics Inc, Quincy, MA, USA) was employed to assess gait dynamics in sex-matched littermate five-week-old conditional knockout and control mice as described (Valnegri et al., 2017; Puram et al., 2011; Amende et al., 2005). During mouse ambulation on a transparent treadmill (20 cm/s), digital paw prints were captured by high-speed camera. Subsequently, gait-related variables were quantified and analyzed by software specialized for the DigiGait imaging system. Investigators were blinded to genotype during running of behavioral experiment and unblinded for analysis of results.
Accelerating rotarod behavior assay
The accelerating rotarod assay was performed using sex-matched littermate five-week-old MEF2A/D cKO and control mice. On the first day, mice underwent habituation on the rotarod apparatus (IITC) at a constant 5 rotations per minute (rpm) for 10 min. Following habituation, mice underwent three consecutive days of testing, with each session day consisting of 5 trials of forced ambulation at 5 to 40 rpm over a period of 3 minutes, with a 1 minute inter-trial interval (15 trials total). Latency to falling (sec) from the rod onto the platform below was recorded. Investigators were blinded to genotype during running of behavioral experiment and unblinded for analysis of results.
In vivo electroporation
In vivo electroporation of postnatal mouse pups was performed as described (Yamada et al., 2014; Konishi et al., 2004; Kim et al., 2009; Yang et al., 2009; Chen et al., 2019). P12-P14 littermate conditional MEF2A/D knockout and control mouse pups were injected with pCAG-GFP (Matsuda and Cepko, 2004), and subjected to four electric pulses of 135mV with 950ms intervals. Electroporated pups were returned to moms and examined in a blinded manner by immunofluorescence confocal microscopy eight days later. Investigators were blinded to genotype during running of experiment and unblinded for analysis of results.
Electron microscopy
P24-P28 mice were perfusion fixed with warmed (37°C) mammalian Ringer’s solution for 2 minutes followed by a mixture of 2.5% glutaraldehyde and 2% paraformaldehyde in 0.15 M cacodylate buffer containing 2mM CaCl2, pH 7.4 for 5 minutes. Mouse brains were carefully dissected and placed into excess fixative overnight. The following day, 100 μm vibratome sections were taken of the cerebellum. Tissue slices were then stained according the methods described by Deerinck et al. (2010). In brief, coverslips were rinsed in cacodylate buffer 3 times for 10 minutes each, and subjected to a secondary fixation for one hour in 2% osmium tetroxide/1.5% potassium ferrocyanide in cacodylate buffer for one hour, rinsed in ultrapure water 3 times for 10 minutes each, and stained in an aqueous solution of 1% thiocarbohydrazide for one hour. After this, the coverslips were once again stained in aqueous 2% osmium tetroxide for one hour, rinsed in ultrapure water 3 times for 10 minutes each, and stained overnight in 1% uranyl acetate at 4°C. The samples were then again washed in ultrapure water 3 times for 10 minutes each and en bloc stained for 30 minutes with 20 mM lead aspartate at 60°C. After staining was complete, coverslips were briefly washed in ultrapure water, dehydrated in a graded acetone series (50%, 70%, 90%, 100% ×2) for 10 minutes in each step, and infiltrated with microwave assistance (Pelco BioWave Pro, Redding, CA) into Durcupan resin, and flat embedded between two slides that had previously been coated with PTFE release agent (Miller-Stephenson #MS-143XD, Danbury, CT) and clamped with binder clips. Samples were cured in an oven at 60°C for 48 hours. Post resin curing, the slides were separated and regions containing central vermal lobules of the cerebellum were cut out by saw and mounted onto blank resin stubs before 70 nm thick sections were cut and placed onto silicon wafer chips. These chips were then adhered to SEM pins with carbon adhesive tabs and large areas (~200 × 200 μm) were then imaged at high resolution in a FE-SEM (Zeiss Merlin, Oberkochen, Germany) using the ATLAS (Fibics, Ottowa, Canada) scan engine to tile large regions of interest. High-resolution tiles were captured at 20,480 × 20,480 pixels at 10 nm/pixel with a 8 μs dwell time and line average of 2. The SEM was operated at 8 KeV and 900 pA using the solid-state backscatter detector. Tiles were aligned and exported using ATLAS 5. Investigators were blinded to genotype during running of experiment and synapse quantification, then unblinded for analysis of results.
qRT-PCR
Reverse transcription reactions were performed with Superscript III (Invitrogen) according to manufacturer’s protocol. Real-time PCR reactions using iTaq Universal SYBR Green Supermix (BioRad) were performed on the LightCycler 480 II (Roche).
RNA-sequencing
For RNA-seq, total RNA was extracted from the cerebellum of sex-matched littermate mice using Trizol (ThermoFisher) according to the manufacturer’s instructions. RNA was reverse-transcribed with oligo-dT priming and the cDNA was sequenced on an Illumina HiSeq 2500 (Genome Technology Access Center at Washington University). Four biological replicates were sequenced in all experiments.
Chromatin immunoprecipitation
ChIP-seq assays were performed with P22 mouse cerebella as described with modifications (Andzelm et al., 2015). For MEF2A and MEF2D ChIP-seq, prior to immunoprecipitation, the respective antibody was coupled with Dynabeads protein A (ThermoFisher). For histone H3K27ac ChIP-seq, prior to immunoprecipitation, the antibody was coupled to Dynabeads protein G (ThermoFisher). Following immunoprecipitation, MEF2 ChIP library prep and sequencing was performed at the Genome Technology Access Center at Washington University as described (Yang et al., 2016). H3K27ac ChIP libraries were prepared with Accel-NGS 2S Plus DNA Library Kit (Swift Biosciences) and sequenced on the Illumina NextSeq 500 platform at the Center for Genomic Sciences (Washington University in St. Louis School of Medicine). Three to four biological ChIP replicates were sequenced in all experiments.
Rotarod activation paradigm
Five- to eight-week-old sex-matched littermate AcKO, DcKO, ADcKO and control mice were trained on an accelerating rotarod on the first day for 30 min (6 trials for each of the following speeds: 5-10rpm, 5-15rpm, 5-20rpm; trial duration: 90 s; inter-trial interval: 10 s; ramp speed: 90 s). On the second day, mice were placed on the rotarod for 1 hour (36 trials of 5-20rpm; trial duration: 90 s; inter-trial interval 10 s; ramp speed: 90 s), immediately followed by extraction of total RNA from cerebellum using Trizol (Thermo Fisher Scientific) according to the manufacturer’s instructions. ~100 ng of RNA was treated with NEBNext rRNA Depletion Kit and libraries prepared with NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs) and sequenced on the Illumina NextSeq 500 platform at the Center for Genomic Sciences (Washington University in St. Louis School of Medicine) to obtain 75bp single-end reads. Four biological replicates were performed in all experiments. Investigators were blinded to genotype during running of behavioral experiment and unblinded for analysis of results.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Statistical analysis for each experiment is detailed in the figure legends. For analysis of genomic distribution, the distribution of enhancers and promoters bound by MEF2A and MEF2D were compared to the genomic distribution of all enhancers and promoters. Statistical significance was evaluated using a two-tailed Chi-square test. Box-whisker plots display median value with whiskers representing the 5th and 95th percentile. Significance testing for box-whisker plots were performed using two-tailed unpaired t test or ANOVA with Tukey multiple comparison test, when appropriate. MEF2-bound genes were defined as the single nearest gene based on distance to a MEF2 ChIP-seq peak. Significance for overlap of RNA-seq clusters with MEF2A and MEF2D peaks identified in various ChIP-seq conditions was evaluated by hypergeometric test followed by Bonferroni multiple comparison. For behavioral experiments, independent t test and repeated-measures ANOVA followed by Sidak’s multiple comparison correction were used when appropriate. Threshold for calling statistical significance for all analyses mentioned above was p < 0.05. Statistical analyses were performed using Graphpad PRISM 6.0.
ChIP-seq alignment and peak calling
Single-end reads of 50 or 75 base pairs were obtained for all datasets. Samples were sequenced to a minimum depth of 18.5 million reads and aligned to the mm10 genome using Bowtie2 with default parameters for Galaxy platform. Reads were then filtered for a map quality score greater than 10 (mapQuality > 10). Peaks were called using MACS2 on pooled data. Blacklist regions were subsequently removed prior to downstream analysis and visualization of ChIP-seq data.
Motif Analysis
MEME suite was used to perform de novo motif discovery for MEF2A and MEF2D peaks, with similarly sized flanking regions of MEF2A and MEF2D peaks serving as genomic background. MRE degeneracy was determined by scanning MEF2A and MEF2D peaks for a consensus MRE using FIMO software. Identification of motifs relatively enriched in compensatory sites compared to non-compensatory sites was performed using AME software, in which compensatory sites served as the experimental dataset and non-compensatory sites as the control dataset.
RNA-seq analysis
Differential mRNA-seq analysis was performed for RNA extracted from the cerebellum of P22 AcKO, DcKO, ADcKO, and control mice. Reduction of potential line-specific differences between conditional knockout lines was performed by overlapping genes identified by two types of differential mRNA analyses, one in which conditional knockout mice were compared to respective control littermates and another in which they were compared to controls from all conditional lines.
DNaseI-seq analysis
DNaseI-seq peaks were called using MACS2 at a q-value of less than 0.01 (-q 0.01) without model building (-nomodel), an extension of 200bp (-extsize 200), and a shift of −100bp (-shift −100). DNaseI-hypersensitivity performed in two biological replicates of cerebellum harvested from p22 mouse in Yamada et al., (2019).
DATA AND CODE AVAILABILITY
The accession number for the RNA-sequencing, anti-MEF2A, anti-MEF2D, and anti-H3K27ac ChIP-sequencing datasets reported in this paper is GEO repository: GSE138028
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Calbindin D-28K antibody | Millipore | Cat# AB1778; RRID: AB_2068336 |
| Rabbit anti-MEF2A | M.E. Greenberg lab | Andzelm et al., 2015 |
| Rabbit anti-MEF2D | M.E. Greenberg lab | Flavell et al., 2008 |
| Rabbit anti-MEF2A | Santa Cruz Biotechnology | Cat# sc-313; RRID: AB_631920 |
| Rabbit anti-MEF2C | Proteintech | Cat# 18290-1-AP; RRID:AB_2142849 |
| Mouse anti-14-3-3 (H-8) antibody | Santa Cruz Biotechnology | Cat# sc-1657, RRID: AB_626618 |
| Rabbit anti-Cre recombinase | Millipore | Cat# 69050-3; RRID: AB_10806983 |
| Rabbit anti-H3K27ac | Abcam | Cat# ab4729; RRID:AB_2118291 |
| Rabbit anti-Cleaved Caspase-3 (Asp175) | Cell Signaling Technology | Cat# 9661; RRID:AB_2341188 |
| Chicken anti-GFP | Abcam | Cat# ab13970; RRID:AB_300798 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dynabeads Protein G | Thermo Fisher | Cat#10003D |
| Dynabeads Protein A | Thermo Fisher | Cat#10001D |
| AMPure XP beads | Beckman Coulter | Cat# A63880 |
| Formaldehyde | Sigma Aldrich | Cat# 252549 |
| Hoechst 33258 Solution | Sigma Aldrich | Cat# 94403 |
| Critical Commercial Assays | ||
| Accel-NGS 2S Plus DNA Library Kit (24 rxns) | Swift Biosciences | Cat#21024 |
| 2S Indexing Kit (12 indices, Set A) | Swift Biosciences | Cat#26148 |
| 2S Indexing Kit (12 indices, Set B) | Swift Biosciences | Cat#26248 |
| NEBNext Ultra Directional RNA Library Prep Kits | New England Biolabs | Cat#E7760S |
| NEBNext Multiplex Oligos for Illumina | New England Biolabs | Cat#E7335S |
| NEBNext rRNA Depletion Kit | New England Biolabs | Cat#E6310L |
| Qubit dsDNA HS Assay Kit | Thermo Fisher | Cat#Q32854 |
| Deposited Data | ||
| RNA-sequencing and anti-MEF2A, anti-MEF2D, and anti-H3K27ac ChIP-sequencing data | This paper | GEO: GSE138028 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Mef2d conditional knockout mice: Mef2dfl/fl | M.E. Greenberg lab | Andzelm et al., 2015 |
| Mouse: Mef2a conditional knockout mice: Mef2afl/fl | M.E. Greenberg lab | Andzelm et al., 2019 |
| Mouse: G6R-Cre: GABA(A)α6 receptor promoter driven Cre-recombinase transgene | L.F. Reichardt lab | Fünfschilling and Reichardt, 2002 |
| Oligonucleotides | ||
| qPCR primers used in this study | Table S1 | |
| Recombinant DNA | ||
| pCAG-GFP | Matsuda and Cepko, 2004 | RRID:Addgene_11150 (gift from Connie Cepko) |
| Software and Algorithms | ||
| GraphPad Prism 6 | Open Source | SCR_002798 |
| EdgeR | Open Source | SCR_012802 |
| GREAT | Open Source | SCR_005807 |
| Adobe Photoshop | Software | SCR_014199 |
| Adobe Illustrator | Software | SCR_010279 |
| Bowtie 2 (mm10) | Open Source | SCR_005476 |
| Bedtools | Open Source | SCR_006646 |
| MACS2 | Open Source | SCR_013291 |
| Galaxy | Open Source | SCR_006281 |
| Imaris | Software | SCR_007370 |
| MEME Suite | Open Source | SCR_001783 |
| R | Open Source | SCR_001905 |
| Other | ||
| anti-MEF2 ChIP-seq in cortical neurons | Telese et al., 2015 | GEO: GSE66710 |
| anti-H3K4me3 ChIP-seq in p22 mouse cerebellum | Yamada et al., 2014 | GEO: GSE57758 |
| Rotarod activation RNA-seq | Yamada et al., 2019 | GEO: GSE127995 |
Highlights.
MEF2D is the predominant regulator of gene expression in the cerebellum
Depletion of MEF2D induces functionally compensatory occupancy by MEF2A
Compensatory MEF2A activity is concentrated within more accessible chromatin
Behavioral context plays a key role in specifying compensatory MEF2A activity
ACKNOWLEDGMENTS
We thank members of the Bonni, Greenberg, Gabel, Yang, and Yamada laboratories for helpful discussions and critical reading of the manuscript; and Matt Joens and Drs. Peter Bayguinov and James Fitzpatrick for electron microscopy (EM) studies and analyses at the Washington University Center for Cellular Imaging (WUCCI), which is supported by Washington University School of Medicine, Children’s Discovery Institute of Washington University, St. Louis Children’s Hospital (CDI-CORE-2015-505), and the Foundation for Barnes-Jewish Hospital (3770). We also thank Krikor Dikranian for insights in EM analyses and the Genome Technology Access Center (GTAC) and Center for Genomic Sciences (CGS) at Washington University in St. Louis for sequencing analyses. This work was supported by NIH grant NS041021 (A.B.), the Mathers Foundations (A.B.), and NIH grant R37 NS028829 (M.E.G.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no conflicts of interest.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.033.
REFERENCES
- Alberini CM, and Kandel ER (2014). The regulation of transcription in memory consolidation. Cold Spring Harb. Perspect. Biol 7, a021741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amende I, Kale A, McCue S, Glazier S, Morgan JP, and Hampton TG (2005). Gait dynamics in mouse models of Parkinson’s disease and Huntington’s disease. J. Neuroeng. Rehabil 2, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andzelm MM, Cherry TJ, Harmin DA, Boeke AC, Lee C, Hemberg M, Pawlyk B, Malik AN, Flavell SW, Sandberg MA, et al. (2015). MEF2D drives photoreceptor development through a genome-wide competition for tissue-specific enhancers. Neuron 86, 247–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andzelm MM, Vanness D, Greenberg ME, and Linden DJ (2019). A Late Phase of Long-Term Synaptic Depression in Cerebellar Purkinje Cells Requires Activation of MEF2. Cell Rep. 26, 1089–1097.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assali A, Harrington AJ, and Cowan CW (2019). Emerging roles for MEF2 in brain development and mental disorders. Curr. Opin. Neurobiol 59, 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataman B, Boulting GL, Harmin DA, Yang MG, Baker-Salisbury M, Yap EL, Malik AN, Mei K, Rubin AA, Spiegel I, et al. (2016). Evolution of Osteocrin as an activity-regulated factor in the primate brain. Nature 539, 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. (2009). Diversity and complexity in DNA recognition by transcription factors. Science 324, 1720–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BL, and Olson EN (1998). Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu. Rev. Cell Dev. Biol 14, 167–196. [DOI] [PubMed] [Google Scholar]
- Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, Mac-Quarrie KL, Davison J, Morgan MT, Ruzzo WL, et al. (2010). Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev. Cell 18, 662–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CW, Wilkerson JR, Hale CF, Gibson JR, and Huber KM (2017). Distinct stages of synapse elimination are induced by burst firing of CA1 neurons and differentially require MEF2A/D. eLife 6, e26278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SX, Cherry A, Tari PK, Podgorski K, Kwong YK, and Haas K (2012). The transcription factor MEF2 directs developmental visually driven functional and structural metaplasticity. Cell 151, 41–55. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhang Z, Li L, Chen BC, Revyakin A, Hajj B, Legant W, Dahan M, Lionnet T, Betzig E, et al. (2014). Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 156, 1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Chanda A, Ikeuchi Y, Zhang X, Goodman JV, Reddy NC, Majidi SP, Wu DY, Smith SE, Godec A, et al. (2019). The Transcriptional Regulator SnoN Promotes the Proliferation of Cerebellar Granule Neuron Precursors in the Postnatal Mouse Brain. J. Neurosci 39, 44–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholewa-Waclaw J, Bird A, von Schimmelmann M, Schaefer A, Yu H, Song H, Madabhushi R, and Tsai LH (2016). The Role of Epigenetic Mechanisms in the Regulation of Gene Expression in the Nervous System. J. Neurosci 36, 11427–11434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant GC, and Wagner A (2004). Duplicate genes and robustness to transient gene knock-downs in Caenorhabditis elegans. Proc. Biol. Sci 271, 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, and Bonni A (2011). Transcriptional regulation of neuronal polarity and morphogenesis in the mammalian brain. Neuron 72, 22–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deerinck TJ, Bushong EA, Lev-Ram V, Shu X, Tsien RY, and Ellisman MH (2010). Enhancing Serial Block-Face Scanning Electron Microscopy to Enable High Resolution 3-D Nanohistology of Cells and Tissues Microscopy and Microanalysis, 16 (Cambridge University Press; ), pp. 1138–1139. [Google Scholar]
- Desjardins CA, and Naya FJ (2017). Antagonistic regulation of cell-cycle and differentiation gene programs in neonatal cardiomyocytes by homologous MEF2 transcription factors. J. Biol. Chem 292, 10613–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, and Greenberg ME (2013). Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eferl R, and Wagner EF (2003). AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859–868. [DOI] [PubMed] [Google Scholar]
- Estrella NL, Desjardins CA, Nocco SE, Clark AL, Maksimenko Y, and Naya FJ (2015). MEF2 transcription factors regulate distinct gene programs in mammalian skeletal muscle differentiation. J. Biol. Chem 290, 1256–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, and Greenberg ME (2006).Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 311, 1008–1012. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, Markenscoff-Papadimitriou E, Bear DM, and Greenberg ME (2008). Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron 60, 1022–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CL, Liu F, Wijayatunge R, Song L, Biegler MT, Yang MG, Vockley CM, Safi A, Gersbach CA, Crawford GE, and West AE (2015). Regulation of chromatin accessibility and Zic binding at enhancers in the developing cerebellum. Nat. Neurosci 18, 647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fünfschilling U, and Reichardt LF (2002). Cre-mediated recombination in rhombic lip derivatives. Genesis 33, 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxman Bass JI, Sahni N, Shrestha S, Garcia-Gonzalez A, Mori A, Bhat N, Yi S, Hill DE, Vidal M, and Walhout AJM (2015). Human gene-centered transcription factor networks for enhancers and disease variants. Cell 161, 661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudilliere B, Shi Y, and Bonni A (2002). RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival. J. Biol. Chem 277, 46442–46446. [DOI] [PubMed] [Google Scholar]
- Grégoire S, and Yang XJ (2005). Association with class IIa histone deacetylases upregulates the sumoylation of MEF2 transcription factors. Mol. Cell. Biol 25, 2273–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SR, Engreitz J, Ray JP, Nguyen TH, Hacohen N, and Lander ES (2018). Positional specificity of different transcription factor classes within enhancers. Proc. Natl. Acad. Sci. USA 115, E7222–E7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove CA, De Masi F, Barrasa MI, Newburger DE, Alkema MJ, Bulyk ML, and Walhout AJ (2009). A multiparameter network reveals extensive divergence between C. elegans bHLH transcription factors. Cell 138, 314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Dunham MJ, and Troyanskaya OG (2007). Functional analysis of gene duplications in Saccharomyces cerevisiae. Genetics 175, 933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiney SA, Wohl MP, Chettih SN, Ruffolo LI, and Medina JF (2014). Cerebellar-dependent expression of motor learning during eyeblink conditioning in head-fixed mice. J. Neurosci 34, 14845–14853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenhorst PC, Shah AA, Hopkins C, and Graves BJ (2007). Genome-wide analyses reveal properties of redundant and specific promoter occupancy within the ETS gene family. Genes Dev. 21, 1882–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlock EC, Bose M, Pierce G, and Joho RH (2009). Rescue of motor coordination by Purkinje cell-targeted restoration of Kv3.3 channels in Kcnc3-null mice requires Kcnc1. J. Neurosci 29, 15735–15744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, Kitamura K, Suzuki K, Nonaka M, Kamijo S, Takemoto-Kimura S, Kano M, Okuno H, Ohki K, and Bito H (2013). Functional labeling of neurons and their projections using the synthetic activity-dependent promoter E-SARE. Nat. Methods 10, 889–895. [DOI] [PubMed] [Google Scholar]
- Kim AH, Puram SV, Bilimoria PM, Ikeuchi Y, Keough S, Wong M, Rowitch D, and Bonni A (2009). A centrosomal Cdc20-APC pathway controls dendrite morphogenesis in postmitotic neurons. Cell 136, 322–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Stegmüller J, Matsuda T, Bonni S, and Bonni A (2004). Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science 303, 1026–1030. [DOI] [PubMed] [Google Scholar]
- Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, and Weirauch MT (2018). The Human Transcription Factors. Cell 175, 598–599. [DOI] [PubMed] [Google Scholar]
- Levine M, and Tjian R (2003). Transcription regulation and animal diversity. Nature 424, 147–151. [DOI] [PubMed] [Google Scholar]
- Li XY, MacArthur S, Bourgon R, Nix D, Pollard DA, Iyer VN, Hechmer A, Simirenko L, Stapleton M, Luengo Hendriks CL, et al. (2008). Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLoS Biol. 6, e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, Sunkin SM, Li Z, Shin Y, Zhu Y, et al. ; BrainSpan Consortium; PsychENCODE Consortium; PsychENCODE Developmental Subgroup (2018). Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362, eaat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, and Bulleit RF (1996). Cell intrinsic mechanisms regulate mouse cerebellar granule neuron differentiation. Neurosci. Lett 220, 81–84. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Li H, Zaremba JD, McKercher SR, Cui J, Kang YJ, Nie Z, Soussou W, Talantova M, Okamoto S, and Nakanishi N (2009). Autistic phenotype from MEF2C knockout cells. Science 323, 208. [DOI] [PubMed] [Google Scholar]
- Luna-Zurita L, Stirnimann CU, Glatt S, Kaynak BL, Thomas S, Baudin F, Samee MA, He D, Small EM, Mileikovsky M, et al. (2016). Complex Interdependence Regulates Heterotypic Transcription Factor Distribution and Coordinates Cardiogenesis. Cell 164, 999–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons GE, Micales BK, Schwarz J, Martin JF, and Olson EN (1995). Expression of mef2 genes in the mouse central nervous system suggests a role in neuronal maturation. J. Neurosci 15, 5727–5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons MR, Schwarz CM, and West AE (2012). Members of the myocyte enhancer factor 2 transcription factor family differentially regulate Bdnf transcription in response to neuronal depolarization. J. Neurosci 32, 12780–12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macneil LT, and Walhout AJ (2011). Gene regulatory networks and the role of robustness and stochasticity in the control of gene expression. Genome Res. 21, 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik AN, Vierbuchen T, Hemberg M, Rubin AA, Ling E, Couch CH, Stroud H, Spiegel I, Farh KK, Harmin DA, and Greenberg ME (2014). Genome-wide identification and characterization of functional neuronal activity-dependent enhancers. Nat. Neurosci 17, 1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, and Cepko CL (2004). Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc. Natl. Acad. Sci. USA 101, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni EO, Mahony S, Closser M, Morrison CA, Nedelec S, Williams DJ, An D, Gifford DK, and Wichterle H (2013). Synergistic binding of transcription factors to cell-specific enhancers programs motor neuron identity. Nat. Neurosci 16, 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellén M, Ayata P, Dewell S, Kriaucionis S, and Heintz N (2012). MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 151, 1417–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina DN, Glasscock J, Gish W, and Lovett M (2004). An OR-Feome-based analysis of human transcription factor genes and the construction of a microarray to interrogate their expression. Genome Res. 14, 2041–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, and Macklis JD (2007). Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci 8, 427–437. [DOI] [PubMed] [Google Scholar]
- Porcelli D, Fischer B, Russell S, and White R (2019). Chromatin accessibility plays a key role in selective targeting of Hox proteins. Genome Biol. 20, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter RS, Jaamour F, and Iwase S (2018). Neuron-specific alternative splicing of transcriptional machineries: Implications for neurodevelopmental disorders. Mol. Cell. Neurosci 87, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potthoff MJ, and Olson EN (2007). MEF2: a central regulator of diverse developmental programs. Development 134, 4131–4140. [DOI] [PubMed] [Google Scholar]
- Pulimood NS, Rodrigues WDS, Atkinson DA, Mooney SM, and Medina AE (2017). The Role of CREB, SRF, and MEF2 in Activity-Dependent Neuronal Plasticity in the Visual Cortex. J. Neurosci 37, 6628–6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulipparacharuvil S, Renthal W, Hale CF, Taniguchi M, Xiao G, Kumar A, Russo SJ, Sikder D, Dewey CM, Davis MM, et al. (2008). Cocaine regulates MEF2 to control synaptic and behavioral plasticity. Neuron 59, 621–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puram SV, Riccio A, Koirala S, Ikeuchi Y, Kim AH, Corfas G, and Bonni A (2011). A TRPC5-regulated calcium signaling pathway controls dendrite patterning in the mammalian brain. Genes Dev. 25, 2659–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid AJ, Cole CJ, and Josselyn SA (2014). Emerging roles for MEF2 transcription factors in memory. Genes Brain Behav. 13, 118–125. [DOI] [PubMed] [Google Scholar]
- Reece-Hoyes JS, Pons C, Diallo A, Mori A, Shrestha S, Kadreppa S, Nelson J, Diprima S, Dricot A, Lajoie BR, et al. (2013). Extensive rewiring and complex evolutionary dynamics in a C. elegans multiparameter transcription factor network. Mol. Cell 51, 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel MF, and Hatten ME (2011). Cerebellum development and medulloblastoma. Curr. Top. Dev. Biol 94, 235–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174, 1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalizi AK, and Bonni A (2005). brawn for brains: the role of MEF2 proteins in the developing nervous system. Curr. Top. Dev. Biol 69, 239–266. [DOI] [PubMed] [Google Scholar]
- Shalizi A, Gaudillière B, Yuan Z, Stegmüller J, Shirogane T, Ge Q, Tan Y, Schulman B, Harper JW, and Bonni A (2006). A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science 311, 1012–1017. [DOI] [PubMed] [Google Scholar]
- Shalizi A, Bilimoria PM, Stegmüller J, Gaudillière B, Yang Y, Shuai K, and Bonni A (2007). PIASx is a MEF2 SUMO E3 ligase that promotes postsynaptic dendritic morphogenesis. J. Neurosci 27, 10037–10046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, Pollina EA, Nagy MA, Yap EL, Dibiase FA, Hrvatin S, Hu L, Lin C, and Greenberg ME (2019). ARNT2 Tunes Activity-Dependent Gene Expression through NCoR2-Mediated Repression and NPAS4-Mediated Activation. Neuron 102, 390–406.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen N, Zhao J, Schipper JL, Zhang Y, Bepler T, Leehr D, Bradley J, Horton J, Lapp H, and Gordan R (2018). Divergence in DNA Specificity among Paralogous Transcription Factors Contributes to Their Differential In Vivo Binding. Cell Syst. 6, 470–483.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, and Greenberg ME (1990). The regulation and function of cfos and other immediate early genes in the nervous system. Neuron 4, 477–485. [DOI] [PubMed] [Google Scholar]
- Slattery M, Riley T, Liu P, Abe N, Gomez-Alcala P, Dror I, Zhou T, Rohs R, Honig B, Bussemaker HJ, and Mann RS (2011). Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell 147, 1270–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, and Furlong EE (2012). Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet 13, 613–626. [DOI] [PubMed] [Google Scholar]
- Su Y, Shin J, Zhong C, Wang S, Roychowdhury P, Lim J, Kim D, Ming GL, and Song H (2017). Neuronal activity modifies the chromatin accessibility landscape in the adult brain. Nat. Neurosci. 20, 476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC (2017). Molecular Neuroscience in the 21st Century: A Personal Perspective. Neuron 96, 536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichmann SA, and Babu MM (2004). Gene regulatory network growth by duplication. Nat. Genet 36, 492–496. [DOI] [PubMed] [Google Scholar]
- Telese F, Ma Q, Perez PM, Notani D, Oh S, Li W, Comoletti D, Ohgi KA, Taylor H, and Rosenfeld MG (2015). LRP8-Reelin-Regulated Neuronal Enhancer Signature Underlying Learning and Memory Formation. Neuron 86, 696–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valnegri P, Huang J, Yamada T, Yang Y, Mejia LA, Cho HY, Oldenborg A, and Bonni A (2017). RNF8/UBC13 ubiquitin signaling suppresses synapse formation in the mammalian brain. Nat. Commun 8, 1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, and Greenberg ME (2017). AP-1 Transcription Factors and the BAF Complex Mediate Signal-Dependent Enhancer Selection. Mol. Cell 68, 1067–1082.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei GH, Badis G, Berger MF, Kivioja T, Palin K, Enge M, Bonke M, Jolma A, Varjosalo M, Gehrke AR, et al. (2010). Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 29, 2147–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, and Greenberg ME (2011). Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol 3, a005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Dey R, Han A, Jayathilaka N, Philips M, Ye J, and Chen L (2010). Structure of the MADS-box/MEF2 domain of MEF2A bound to DNA and its implication for myocardin recruitment. J. Mol. Biol 397, 520–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, de Folter S, Shen X, Zhang W, and Tao S (2011). Vertebrate paralogous MEF2 genes: origin, conservation, and evolution. PLoS One 6, e17334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Yang Y, Huang J, Coppola G, Geschwind DH, and Bonni A (2013). Sumoylated MEF2A coordinately eliminates orphan presynaptic sites and promotes maturation of presynaptic boutons. J. Neurosci 33, 4726–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Yang Y, Hemberg M, Yoshida T, Cho HY, Murphy JP, Fioravante D, Regehr WG, Gygi SP, Georgopoulos K, and Bonni A (2014). Promoter decommissioning by the NuRD chromatin remodeling complex triggers synaptic connectivity in the mammalian brain. Neuron 83, 122–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Yang Y, Valnegri P, Juric I, Abnousi A, Markwalter KH, Guthrie AN, Godec A, Oldenborg A, Hu M, et al. (2019). Sensory experience remodels genome architecture in neural circuit to drive motor learning. Nature 569,708–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Kim AH, Yamada T, Wu B, Bilimoria PM, Ikeuchi Y, de la Iglesia N, Shen J, and Bonni A (2009). A Cdc20-APC ubiquitin signaling pathway regulates presynaptic differentiation. Science 326, 575–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Yamada T, Hill KK, Hemberg M, Reddy NC, Cho HY, Guthrie AN, Oldenborg A, Heiney SA, Ohmae S, et al. (2016). Chromatin remodeling inactivates activity genes and regulates neural coding. Science 353, 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap EL, and Greenberg ME (2018). Activity-Regulated Transcription: Bridging the Gap between Neural Activity and Behavior. Neuron 100, 330–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziats MN, Grosvenor LP, and Rennert OM (2015). Functional genomics of human brain development and implications for autism spectrum disorders. Transl. Psychiatry 5, e665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovkic IB, Paulukaitis BS, Day JJ, Etikala DM, and Sweatt JD (2014). Histone H2A.Z subunit exchange controls consolidation of recent and remote memory. Nature 515, 582–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-sequencing, anti-MEF2A, anti-MEF2D, and anti-H3K27ac ChIP-sequencing datasets reported in this paper is GEO repository: GSE138028