

Abstract
We investigated the impact of time interval, primary vs. metastatic biopsy site, variant allele fraction (VAF) and histology on concordance of KRAS alterations in tissue vs. circulating tumor DNA (ctDNA), and association of concordance with survival. Blood and tissue were evaluated by next-generation sequencing in 433 patients with diverse cancers. Altogether, 101 patients (23.3%) had KRAS alterations: 56, ctDNA (12.9%); 81, tissue (18.7%); and 36, both (8.3%). The overall blood and tissue concordance rate for KRAS alterations was 85%, but was mainly driven by the large negative/negative subset. Therefore, specificity of one test for the other was high (88.1–94.3%), while sensitivity was not high (44.4–64.3%) and was lower still in patients with >6 vs. ≤2 months between blood and tissue sampling (31.0–40.9% vs. 51.2–84.0%; p = 0.14 time interval-dependent sensitivity of blood for tissue; p = 0.003, tissue for blood). Positive concordance rate for KRAS alterations was 57.1% vs. 27.4% (colorectal vs. noncolorectal cancer; p = 0.01), but site of biopsy (primary vs. metastatic) and VAF (%ctDNA) was not impactful. The presence of KRAS alterations in both tests was independently associated with shorter survival from diagnosis (hazard ratio, 1.72; 95% confidence interval, 1.04–2.86) and from recurrent/metastatic disease (1.70; 1.03–2.81). Positive concordance of KRAS alterations between ctDNA and tissue was negatively affected by a longer time period between blood and tissue sampling and was higher in colorectal cancer than in other malignancies. The presence of KRAS alterations in both tests was an independent prognostic factor for poor survival.
Keywords: KRAS, circulating tumor DNA, next-generation sequencing, survival
Introduction
KRAS is a protein in the mitogen-activated protein kinase (MAPK) pathway that is responsible for various aspects of cell growth and regulation, including cell proliferation, apoptosis, and differentiation. Alterations in the KRAS oncogene occur frequently and play a major role in many human cancers.1,2 As an example, in pancreatic ductal adenocarcinoma, oncogenic KRAS is present in more than 90% of cases.2,3 KRAS is also present in 30–40% of all colorectal cancer cases and is associated with decreased survival.1,2
Investigation of molecular alterations in cancerous tumors has created a new wave of personalized cancer medicine that uses targeted cancer therapies matched to a patient’s specific molecular alterations.4 Recent research has shown that advancements in genomic testing has led to higher response rates to treatment in patients with advanced cancers.5 With the availability of these multigene panels, malignant tissues have been found to harbor multiple alterations that may differ from patient to patient and, hence, require customized combination therapies.5 In the case of KRAS, mutations may also serve as a negative marker, indicating a contraindication to epidermal growth factor receptor (EGFR) antibody therapy in colorectal cancer.6,7
Interrogating blood-derived circulating tumor DNA (ctDNA) allows for the detection of genetic heterogeneity in cancer without the invasive nature of tissue biopsies. Solid tumors release ctDNA into the bloodstream. The amount present or absent in the blood may be an indicator of tumor burden.8–10 This ctDNA is collected through a liquid biopsy (i.e., a blood draw) and sequenced in order to identify characterized somatic alterations. Blood-derived ctDNA may provide valuable molecular data as an adjunct to the tumor biopsy for the following reasons: (i) tumor biopsies can be complicated procedures with morbidity; (ii) some tumors are not readily accessible for biopsy; (iii) tissue biopsies can be expensive; (iv) as time elapses, the tissue that was biopsied may become less representative of the tumor, since cancers evolve; (v) genomics performed on a tissue biopsy reflects the alterations that are in the small tissue specimen, while genomics performed on ctDNA may reflect alterations found in shed DNA from multiple metastatic sites; and (vi) dynamic changes in ctDNA can occur and reflect response to therapy or emergence of resistance. In addition, interrogating ctDNA before or after surgery may provide a predictive tool for risk of recurrence.11,12 There are also disadvantages to ctDNA as compared to tissue. For instance, ctDNA is found in only small amounts in the bloodstream, making it difficult to detect alterations. In addition, ctDNA carrying tumor-specific alterations may represent only a small fraction of the total genomic alterations in the tumor, since not all cancer-derived DNA may be shed into the bloodstream. Therefore, variability in concordance rates between blood-derived ctDNA samples and tissue samples can be due to temporal and spatial factors, as well as dynamic changes with therapy and disease evolution.
Several studies have investigated the concordance in molecular alterations between tissue and ctDNA samples. A study conducted in prostate cancer found that, in the majority of patients, a ctDNA assay was sufficient to identify all driver DNA alterations present in matched metastatic tissue.13 Other studies demonstrated that, among various cancer types, concordance was variable with a range of ~70–98%, depending on the gene(s) examined.8,14,15 Several previous studies evaluated the concordance of KRAS alterations between tissue and ctDNA and found overall concordance to range 67–96%.6,16–18 These studies did not evaluate the impact of time interval between tissue biopsy and blood draw, nor did they examine the correlation with site of biopsy or the association with outcome for discordant vs. concordant KRAS alterations.
For this present study, results from tumor biopsies and blood-derived ctDNA biopsies were used to investigate the level of concordance for altered KRAS among 433 patients with diverse cancer types in order to determine temporal and spatial effects on concordance across the spectrum of malignancies, as well as to ascertain the relationship between concordance and survival.
Methods
Patients
The molecular profiles of both liquid and tissue biopsies from 433 consecutive eligible patients seen at the UC San Diego Moores Cancer Center starting in June 2014 were reviewed (Supporting Information Fig. S1). Demographics of each of these patients were provided by chart review, including, but not limited to age, gender, cancer diagnosis, tumor origin, date of biopsy report or blood test, date of diagnosis and survival time. Patients included in the study were analyzed and consent obtained as appropriate in accordance with an internal review board-approved protocol ().
Next-generation sequencing
The ctDNA molecular profiles came from patients with diverse cancers and were provided by Guardant Health Inc. (https://www.guardanthealth.com/); tissue testing was performed by Foundation Medicine (https://www.foundationmedicine.com/genomic-testing#how-does-it-work). Both laboratories are Clinical Laboratory Improvement Amendment (CLIA)-accredited.
ctDNA testing.
As reported in Lanman et al.,19 5–30 ng of ctDNA was isolated from plasma (two 10 ml Streck tubes drawn for each patient) and sequencing libraries were made with custom in-line barcode molecular tagging and complete sequencing at 15,000× read depth. The panels use hybrid capture followed by next-generation sequencing (NGS) of the crucial exons in a panel of 54–73 genes and report all four major types of genomic alterations (indels, fusions, point mutations and copy number amplifications). Postsequencing bioinformatics matches the complementary strands of each barcoded DNA fragment to remove false-positive results.19 The variant allele fraction (VAF; %ctDNA) is calculated as the number of mutated DNA molecules divided by the total number (mutated plus wild type) of DNA fragments at that allele. We used the maximum %ctDNA if a patient had two different KRAS alterations, unless we referred to a specific KRAS alteration. The majority of cell-free DNA is wild type; hence, the median % ctDNA of somatic alterations is <0.5%. The analytic sensitivity reaches detection of one to two single-mutant fragments from a 10 ml blood sample (0.1% limit of detection), and the analytic specificity is greater than 99.9999%.19
Tissue NGS.
Tissue NGS was performed at Foundation Medicine with assay panels of 236 or 315 genes as previously described (Cambridge, MA, https://www.foundationmedicine.com).20 Average depth of sequencing was greater than 250×, with 100× at >99% of exons. This method of sequencing allows for detection of copy number alterations, gene rearrangements, and somatic mutations with 99% specificity and >99% sensitivity for base substitutions at ≥5 mutant allele frequency and >95% sensitivity for copy number alterations. A threshold of ≥8 copies for gene amplification was used.
Variants of unknown significance.
Synonymous alterations and other variants of unknown significance (VUS) were excluded and only characterized alterations were included in the analysis.21
Defining concordance
Concordant alterations between ctDNA blood-derived biopsy samples and tissue biopsy samples were defined as a KRAS alteration being detected in both samples. If patients had more than one tissue or blood sample, the samples closest together were counted. Concordance between tissue DNA and ctDNA was assessed with overall concordance rate and the Kappa value. Kappa values were interpreted by commonly used agreement categories: 1 (perfect agreement) to 0 (no agreement [the same as would be expected by chance]).
Outcome endpoints and statistical analysis
Difference in overall concordance rate between two groups was compared to Fisher’s exact test to assess statistical significance (p ≤ 0.05). Descriptive characteristics were reported for all patients and the most common characterized alterations found in both liquid and tissue biopsies were highlighted. Patients were then categorized into groups based on whether they were diagnosed with colorectal cancer or noncolorectal cancers. Survival was examined by the method of Kaplan–Meier; patients still alive at the last follow up were censored at that time. Survival time was calculated for three dates of interest; data of diagnosis, date of blood draw for ctDNA, and date of metastatic or recurrent disease. Data cutoff for survival analysis was February 22, 2018. Log-rank test was used to compare Kaplan–Meier curves. In terms of the investigation of factors associated with overall survival (OS), variables with p-value <0.15 in univariate analysis were included in the multivariate Cox-regression model. Statistical analyses were performed by coauthor RO using the IBM SPSS version 25.0 (SPSS Inc., Chicago, IL).
Data availability
The data that support the findings of our study are available upon request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Results
Patients and molecular alterations
Our study population consisted of 433 patients with diverse cancers who had both ctDNA and tissue NGS performed. Their median age at diagnosis was 59 years (range, 2–91 years) and 237 patients were women (54.7%; Table 1). The most common types of cancers were gastrointestinal, lung, brain and breast. All but 28 patients had advanced disease that was surgically unresectable, metastatic or both.
Table 1.
Patient characteristics among the 433 patients that had both blood-derived ctDNA and tissue testing done
| Patient characteristics | Total patients n = 433 |
|---|---|
| Age at diagnosis (years: median, range) | 59 (2–91) |
| Gender (n, %) | |
| Women | 237 (54.7%) |
| Men | 196 (45.3%) |
| Type of cancer (n, %) | |
| Gastrointestinal1 | 104 (24.0%) |
| Lung | 78 (18.0%) |
| Brain | 56 (12.9%) |
| Breast | 50 (11.5%) |
| Hepatic/pancreatic/biliary | 40 (9.2%) |
| Head and neck | 31 (7.2%) |
| Gynecologic | 22 (5.1%) |
| Others2 | 52 (12.0%) |
| Tissue biopsy site (n, %) | |
| Liver | 63 (14.5%) |
| Brain | 59 (13.6%) |
| Lung | 54 (12.5%) |
| Gastrointestinal | 51 (11.8%) |
| Lymph node | 18 (4.2%) |
| Breast | 13 (3.0%) |
| Gynecologic | 13 (3.0%) |
| Others | 162 (37.4%) |
| Time between tissue biopsy and blood draw for ctDNA | |
| Median months (interquartile range) | 5.0 (0.03–241) |
| Category (n, %) | |
| ≤2 months | 165 (38.1%) |
| >2 to 6 months | 69 (15.9%) |
| >6 months | 199 (46.0%) |
Includes colorectal cancer (n = 54), appendiceal cancer (n = 20), gastric cancer (n = 8), esophageal cancer (n = 7), gastrointestinal stromal tumor (n = 7), anal cancer (n = 3), small bowel cancer (n = 3), and neuroendocrine tumor (n = 2).
Includes genitourinary/prostate cancers (n = 16), cancers of unknown primary (n = 10), soft tissue sarcoma (n = 8), hematologic malignancies (n = 7), mesothelioma/peritoneal carcinoma (n = 5), skin carcinoma (n = 4), and thymoma (n = 2).
In the 433 patients, the most common non-VUS alterations found in both ctDNA and tissue biopsies were TP53 (ctDNA: 36.7% of patients, tissue: 49.7% of patients), KRAS (ctDNA: 12.9%, tissue: 18.7%), CDKN2A/B (ctDNA: 1.9%, tissue: 20.3%) and EGFR (ctDNA: 10.6%, tissue: 11.6%; Fig. 1).
Figure 1.
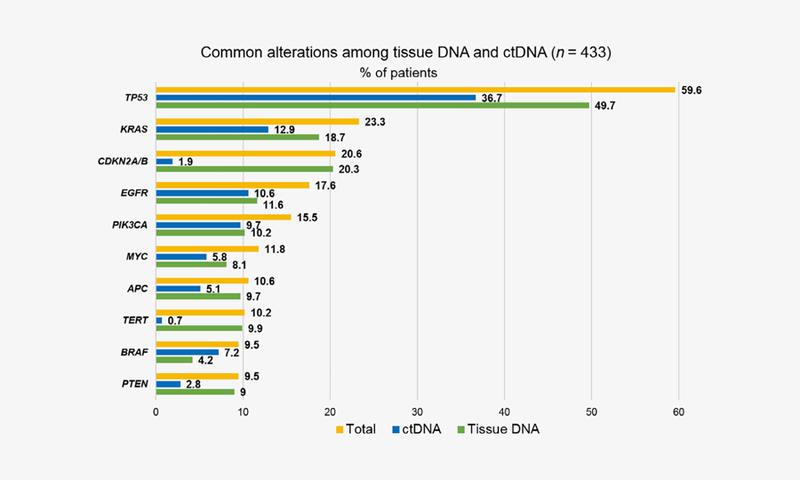
Frequent alterations among blood-derived ctDNA and tissue biopsy (n = 433). Percentage denotes percent of patients with an alteration. Most common alterations in both ctDNA and tissue in 433 patients with diverse cancers who had both ctDNA and tissue biopsy are shown. VUS alterations were excluded. If two different samples were collected for a patient, only the tissue and blood samples closest together timewise were counted.
The types of KRAS alterations seen were missense alterations in 70% and amplifications in 37% of patients (ctDNA) vs. 91% and 9%, respectively, in tissue (Supporting Information Fig. S2). Altogether, 15 kinds of KRAS alterations were seen in our study (in ctDNA or tissue DNA): KRAS amplification (n = 26); KRAS A59T (n = 1); KRAS D33E (n = 1); KRAS G12A (n = 6); KRAS G12C (n = 11); KRAS G12D (n = 19); KRAS G12R (n = 10); KRAS G12S (n = 5); KRAS G12 V (n = 15); KRAS G13D (n = 10); KRAS K117 N (n = 2); KRAS Q61H (n = 1); KRAS Q61K (n = 1); KRAS Q61R (n = 1); and KRAS V14I (n = 1).
Overall, 101 patients (23.3% of 433 tumors) harbored a KRAS alteration: 20 patients (4.6% of 433) had KRAS found only in ctDNA; 45 (10.3% of 433) had KRAS only in tissue; and 36 (8.3% of 433) in both (Supporting Information Fig. S1).
Overall concordance
Temporal effects and overall concordance.
Overall concordance for KRAS alterations in blood ctDNA vs. tissue was 85.0% (Table 2). When comparing the concordance rate between the 165 patients who had both ctDNA and tissue biopsy within 2 months of each other vs. the 199 patients with the time period being >6 months, the concordance rate was 85.5% vs. 83.4% (p = 0.67; Table 2). The concordance rate for subtypes of the more common KRAS alterations was consistently over 90% regardless of the time interval between tissue and ctDNA (Supporting Information Tables S1 and S2; keeping in mind that, because of the small numbers of patients with each KRAS alteration subtype, the ability to analyze statistical differences was limited).
Table 2.
Overall concordance of KRAS alterations in all patients (n = 433) and positive concordance in patients with KRAS alterations (n = 101), both stratified by time between blood draw and tissue biopsy (≤2, >2–6 and >6 months) and by tissue biopsy site (primary tumor vs. metastatic sites)
|
All patients (n = 433) | ||||||||||
|
Tissue DNA results |
Overall concordance rate | Kappa (SE) | Positive concordance rate1 | |||||||
| Positive | Negative | |||||||||
| ctDNA results | Positive | 36 | 20 | 85.0% | 0.44 (0.06) | 35.6% | ||||
| Negative | 45 | 332 | ||||||||
| Temporal and spatial effects on concordance | ||||||||||
|
Test results (ctDNA/tissue DNA) |
Overall concordance |
Positive concordance1 |
||||||||
| (+/+) | (+/− plus −/+) | (−/−) | Rate | Kappa (SE) | p-value | Rate | p-value | |||
| Time interval between blood draw and tissue biopsy (months) |
≤2 months (n = 165) |
21 | 24 | 120 | 85.5% | 0.55 (0.08) | 46.7% |
0.02 (≤2 vs. >6 months) |
||
| >2–6 months (n = 69) |
6 | 8 | 55 | 88.4% | 0.53 (0.15) | 0.67 (≤2 vs. >6) 0.42 (≤6 vs. >6) |
42.9% |
0.02 (≤6 vs. >6 months) |
||
| >6 months (n = 199) |
9 | 33 | 157 | 83.4% | 0.26 (0.09) | 21.4% | ||||
| Tissue biopsy site | Primary (n = 228) |
13 | 31 | 184 | 86.4% | 0.38 (0.09) |
0.42 |
29.5% |
0.30 |
|
| Metastatic (n = 205) |
23 | 34 | 148 | 83.4% | 0.48 (0.08) | 40.4% | ||||
.
Spatial effects and overall concordance.
Concordance between ctDNA and tissue for KRAS alterations was not dependent on whether the tissue biopsy site was primary or metastatic disease (86.4% vs. 83.4%; p = 0.42; Table 2).
Disease histology and overall concordance.
Concordance rates for KRAS alterations were not statistically different in patients with colorectal vs. noncolorectal cancers (77.8% vs. 86.0%; p = 0.15; Supporting Information Table S3).
%ctDNA (variant allele frequency) and overall concordance.
Concordance rate also did not differ when dichotomized by % ctDNA (using the median (1.55%) %ctDNA for KRAS alterations): 84.2% vs. 90%; p = 0.66 (Supporting Information Table S4; but only small numbers of patients were assessable).
Positive concordance (effects of temporal, spatial and histologic factors as well as VAF)
Figure 2 shows that positive concordance was 35.6%. Positive concordance decreased from 46.7% to 21.4% when the time period between blood draw and tissue biopsy was ≤2 months vs. over 6 months (p = 0.02; Table 2). Positive concordance did not differ depending on whether the tissue biopsy was from the primary or metastatic site (29.5% vs. 40.4%; p = 0.30; Table 2). The positive concordance rate for KRAS alterations in colorectal cancer was higher than in noncolorectal cancer (57.1% vs. 27.4%, p = 0.01; Fig. 2, Supporting Information Table S3 and Figure S3). %ctDNA was not correlated with positive concordance (Supporting Information Table S4); positive concordance was also similar (92.3% and 92.9%, respectively for %ctDNA <1.55 and ≥1.55%) when only the 27 patients with ≤6 months between tests were considered (data not shown).
Figure 2.

KRAS alterations among 433 patients, categorized by colorectal cancer and noncolorectal cancers for patients with ≤2 months, >2–6 months and >6 months between blood draw and tissue biopsy. If two different samples were collected for a patient, only the tissue and blood samples closest together timewise were counted. Numbers in parentheses represent number of patients.
Accuracy of ctDNA for tissue DNA for KRAS alterations and vice versa
As mentioned earlier, the prevalence of KRAS alterations in ctDNA was 12.9% while the prevalence of KRAS alterations in tissue was 18.7%. Supporting Information Table S5 shows that the positive predictive power of ctDNA for tissue DNA positivity (which is equivalent to the sensitivity of tissue for ctDNA. Supporting Information Table S6) for KRAS alterations was 64.3%. When comparing between the patients whose blood and tissue samples were ≤2 months apart and those with the time period being >6 months, the value was 84% and 40.9% (p = 0.003). Also, the positive predictive power of tissue for ctDNA positivity (which is equivalent to the sensitivity of ctDNA for tissue) for KRAS alterations was 44.4%. The value for the patients whose blood and tissue samples were ≤2 months apart vs. >6 months apart was 51.2% vs. 31%, respectively (p = 0.14).
In contrast, specificity and negative predictive power of ctDNA for tissue and vice versa were 94.3 and 88.1%, and did not differ regardless of the time interval between tests (Supporting Information Tables S5 and S6).
Survival analysis
Patients with discordant or absent KRAS alterations live longer when compared to patients with concordant KRAS alterations (n = 433 patients). Univariate and multivariate analyses were performed to determine factors associated with outcome. In multivariate analysis of all 433 patients, older age (≥60 years) and the presence of KRAS alterations in both ctDNA and tissue (vs. presence in one or the other or in neither) were associated with a shorter OS (Table 3). The multivariate hazard ratios (HRs; 95% confidence interval; p values) were 1.72 (1.04–2.86; p = 0.04), 1.52 (0.94–2.46; p = 0.09), and 1.70 (1.03–2.81; p = 0.04) when OS was measured from diagnosis, time of blood sample for ctDNA analysis, and time of recurrent/metastatic disease, respectively. Figure 3 further demonstrates that, regardless of whether survival is measured from diagnosis, from blood draw or from time of advanced/metastatic disease, patients with discordant KRAS alterations (KRAS alterations in blood or tissue but not both) live longer than those with concordant KRAS alterations (KRAS in both blood and tissue) (all p values significant or trend). Patients with no KRAS alterations also live longer compared to those with concordant KRAS alterations. However, patients with discordant KRAS alterations show no difference in survival as compared to those with no KRAS alterations. The observations were analogous when only the 234 patients with ≤6 months between blood draw and tissue biopsy were evaluated (Supporting Information Fig. S4).
Table 3.
Factors associated with survival from date of diagnosis, date of blood draw for ctDNA test and from date of metastatic/recurrent disease (n = 433 patients)
| Variables | Univariate |
Multivariate1 |
|||
|---|---|---|---|---|---|
| Median OS (months) |
HR (95% CI) | p-value | HR (95% CI) | p-value | |
| OS from date of diagnosis | |||||
| Age | |||||
| ≥60 years (n = 199) vs. <60 years (n = 234) | 57.6 vs. 103.8 | 1.84 (1.40–2.43) | <0.001 | 1.99 (1.50–2.64) | <0.001 |
| Gender | |||||
| Women (n = 237) vs. men (n = 196) | 78.1 vs. 68.0 | 0.95 (0.72–1.24) | 0.69 | – | – |
| Type of cancer | |||||
| GI or HPB (n = 144) vs. not (n = 289) | 53.2 vs. 88.8 | 1.65 (1.24–2.18) | <0.001 | 1.47 (1.08–1.99) | 0.01 |
| Time interval between tissue biopsy and blood draw | |||||
| ≤6 months (n = 234) vs. >6 months (n = 199) | 65.1 vs. 85.1 | 1.41 (1.08–1.84) | 0.01 | 1.43 (1.09–1.88) | 0.009 |
| KRAS alteration seen in ctDNA and/or tissue DNA | |||||
| Yes (n = 101) vs. no (n = 332) | 73.5 vs. 78.1 | 1.21 (0.89–1.66) | 0.23 | – | – |
| KRAS alteration seen in both ctDNA and tissue DNA | |||||
| Yes (n = 36) vs. no (n = 397) | 32.2 vs. 78.1 | 1.98 (1.26–3.12) | 0.003 | 1.72 (1.04–2.86) | 0.04 |
| OS from date of blood draw for ctDNA test | |||||
| Age | |||||
| ≥60 years (n = 199) vs. <60 years (n = 234) | 13.7 vs. 21.8 | 1.34 (1.03–1.75) | 0.03 | 1.37 (1.05–1.80) | 0.02 |
| Gender | |||||
| Women (n = 237) vs. men (n = 196) | 18.0 vs. 21.8 | 1.05 (0.81–1.38) | 0.71 | – | – |
| Type of cancer | |||||
| GI or HPB (n = 144) vs. not (n = 289) | 13.3 vs. 18.8 | 1.31 (0.99–1.72) | 0.06 | 1.21 (0.90–1.62) | 0.21 |
| Time interval between tissue biopsy and blood draw | - | - | |||
| ≤6 months (n = 234) vs. >6 months (n = 199) | 17.2 vs. 18.8 | 1.07 (0.81–1.39) | 0.65 | ||
| KRAS alteration seen in ctDNA and/or tissue DNA | |||||
| Yes (n = 101) vs. no (n = 332) | 15.0 vs. 18.2 | 1.19 (0.87–1.63) | 0.27 | – | – |
| KRAS alteration seen in both ctDNA and tissue DNA | |||||
| Yes (n = 36) vs. not (n = 397) | 9.0 vs. 18.8 | 1.61 (1.03–2.53) | 0.04 | 1.52 (0.94–2.46) | 0.09 |
| OS from date of metastatic or recurrent disease2 | |||||
| Age | |||||
| ≥60 years (n = 185) vs. <60 years (n = 220) | 30.0 vs. 44.2 | 1.54 (1.18–2.02) | 0.001 | 1.66 (1.26–2.18) | <0.001 |
| Gender | |||||
| Women (n = 222) vs. men (n = 183) | 42.1 vs. 35.1 | 0.82 (0.63–1.08) | 0.16 | – | – |
| Type of cancer | |||||
| GI or HPB (n = 137) vs. not (n = 268) | 32.2 vs. 43.6 | 1.56 (1.18–2.06) | 0.002 | 1.45 (1.07–1.96) | 0.02 |
| Time interval between tissue biopsy and blood draw | |||||
| ≤6 months (n = 221) vs. >6 months (n = 184) | 34.9 vs. 43.4 | 1.46 (1.12–1.92) | 0.006 | 1.48 (1.13–1.94) | 0.005 |
| KRAS alteration seen in ctDNA and/or tissue DNA | |||||
| Yes (n = 97) vs. no (n = 308) | 35.5 vs. 40.7 | 1.19 (0.86–1.62) | 0.29 | – | – |
| KRAS alteration seen in both ctDNA and tissue DNA | |||||
| Yes (n = 36) vs. not (n = 369) | 25.9 vs. 41.7 | 1.98 (1.26–3.13) | 0.003 | 1.70 (1.03–2.81) | 0.04 |
The bold values represent the statistically significant p-values.
Values of p < 0.15 in univariate were selected for multivariate analysis.
A total of 28 patients were excluded from the latter analysis because their disease was surgically resected.
Abbreviations: CI, confidence interval; GI, gastrointestinal; HPB, hepatic/pancreatic/biliary; HR, hazard ratio; NGS, next-generation sequencing; OS, overall survival.
Figure 3.
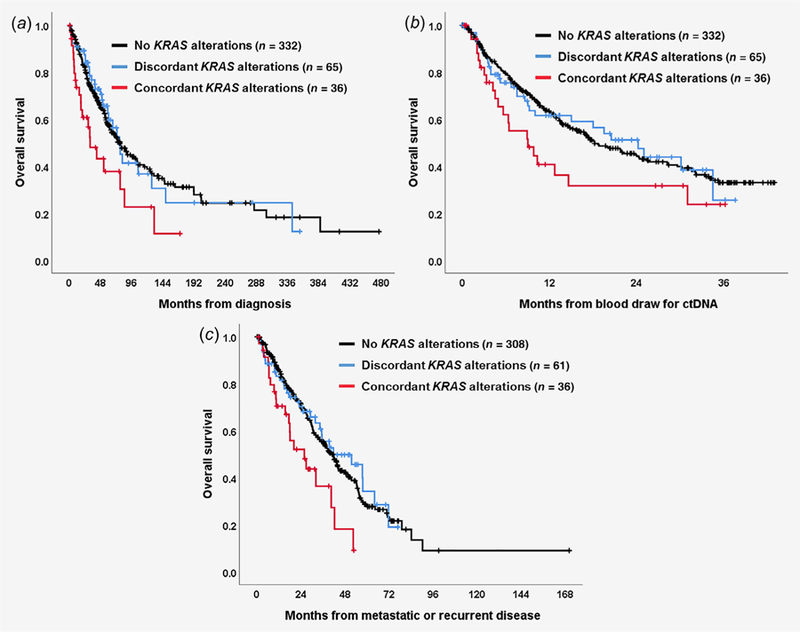
Survival analysis among patients with concordant KRAS alterations, those with discordant KRAS alterations and those with no KRAS alterations (n = 433 patients). (a) Overall survival from date of diagnosis (concordant vs. discordant, HR [95%CI] = 1.96 [1.11–3.45], p = 0.02; concordant vs. no KRAS, HR [95%CI] = 1.40 [1.12–1.76], p = 0.003; discordant vs. no KRAS, HR [95%CI] = 0.96 [0.65–1.41], p = 0.82); (b) Overall survival from date of blood draw for ctDNA test (concordant vs. discordant, HR [95%CI] = 1.53 [0.88–2.68], p = 0.13; concordant vs. no KRAS, HR [95%CI] = 1.28 [1.02–1.60], p = 0.03; discordant vs. no KRAS, HR [95%CI] = 1.01 [0.68–1.49], p = 0.98); (c) Overall survival from date of metastatic or recurrent disease (concordant vs. discordant, HR [95%CI] = 2.05 [1.14–3.68], p = 0.02; concordant vs. no KRAS, HR [95%CI] = 1.41 [1.12–1.77], p = 0.003; discordant vs. no KRAS, HR [95%CI] = 0.93 [0.63–1.37], p = 0.72). *Twenty-eight patients were excluded from this analysis because their disease was surgically resected.
In patients with KRAS alterations, those with discordant KRAS status live longer when compared to patients with concordant blood and tissue KRAS status (n = 101 patients). The OS was significantly shorter (or trended to be shorter; depending on the start point of survival analysis) in univariate and multi-variate analysis of factors affecting OS in the 101 patients with KRAS alterations, when comparing those with KRAS alterations in both blood and tissue vs. those with KRAS alterations in either blood or tissue, but not both (Fig. 3 and Supporting Information Table S7). Kaplan–Meier curves demonstrate that these trends persist when only the 59 patients with KRAS alterations and ≤6 months between blood draw and tissue sample were analyzed—patients with KRAS alterations in both blood and tissue vs. only blood or only tissue had a trend toward shorter survival regardless of whether survival was measured from time of diagnosis, time of ctDNA blood draw or time of recurrent/metastatic disease (Supporting Information Fig. S4: p = 0.06, 0.06 and 0.16, respectively).
Discussion
NGS has unveiled useful genomic biomarkers for predicting diagnosis, prognosis, and response to cancer treatment.22–27 NGS can be performed on either tissue or blood-derived ctDNA; these technologies may be complementary. Herein, by using the data from 433 patients with diverse cancer types, we assessed the effect of temporal (time interval between tissue and blood sample) and spatial (tissue from primary vs. meta-static site) factors on concordance of KRAS alterations in tissue and ctDNA, and their association with survival.
We found that 23.3% of diverse cancer patients had a KRAS alteration; 12.9% in ctDNA and 18.7% in tissue (8.3% in both). These percentages are consistent with a previous large-scale survey of over 78,000 tumor tissues showing KRAS alterations in 22% of cancers.28 In our dataset, genomic alterations (in either blood, tissue or both) were also commonly seen in TP53 (59.6% of patients), CDKN2A/B (20.6%) and EGFR (17.6%; Fig. 1). For the most part, alterations in ctDNA and tissue were found in a similar percentage of patients. However, CDKN2A/B loss was found in only 1.9% of patients in ctDNA, but in 20.3% of patients by tissue NGS; this discrepancy is most likely due to the failure to capture allelic loss in older panels of ctDNA sequencing.
In regard to pathogenic KRAS alterations, gene amplification and 14 kinds of mutation were seen. Further studies may be required to determine whether the impact of these alterations differs or not. Interestingly, a study in colorectal cancer showed that alterations that occurred in codons 59, 61, 117 and 146 had higher MAPK activity than those in codons 12 and 13 and that the mutations with higher MAPK activity were associated with a shorter survival.29
The overall concordance rate for KRAS alterations in ctDNA and tissue was 85.0% (Table 2). When assessed according to sub-type of KRAS alteration, the concordance rate was over 90% for each subtype (Supporting Information Tables S1 and S2). In terms of the temporal (≤2 months vs. >6 months between blood draw and tissue biopsy) and spatial (tissue biopsy site was primary tumor vs. metastatic sites) effects on the overall concordance rate, no statistical differences were observed (85.5% vs. 83.4% [p = 0.67] and 86.4% vs. 83.4% [p = 0.42], respectively). Furthermore, overall concordance did not vary by %ctDNA (Supporting Information Table S4). However, the overall concordance rates were largely impacted by the ctDNA-negative/tissue-negative population. In fact, when limited to the patients with KRAS alterations, the positive concordance rate (positive in both ctDNA and tissue divided by positive in ctDNA or positive in tissue or in both) was only 35.6% (Fig. 2); furthermore, KRAS alteration-positive concordance significantly decreased from 46.7% to 21.4% when the time period between blood draw and tissue biopsy was ≤2 months vs. >6 months (p = 0.02); there was however no statistical difference when tissue biopsy was from primary or metastatic site (Table 2). In comparison, a prior study in colorectal cancer also showed a high overall concordance rate (~90%) for RAS alterations in ctDNA vs. tissue;18 although positive concordance was not formally analyzed, that study did demonstrate that some patients had discordant results, albeit at a lesser frequency than in our study, perhaps because the population was limited to colorectal cancer or because the blood and tissue samples were taken at time points close together. Indeed, in our patients, the positive concordance rate for KRAS alterations in colorectal cancer was higher than in noncolorectal cancer (57.1% vs. 27.4%, p = 0.01; Fig. 2).
The positive predictive power of ctDNA for tissue DNA positivity (= the sensitivity of tissue for ctDNA) was 64.3% for detecting KRAS alterations (Supporting Information Tables S5 and S6). This means that, of the KRAS-altered ctDNA tests, 64.3% were positive by tissue. The value was substantially lower when blood and tissue samples were >6 months apart than when they were ≤2 months apart (40.9% vs. 84.0%; p = 0.003). The positive predictive power of tissue for ctDNA positivity (= the sensitivity of ctDNA for tissue) was 44.4%. This means that, of the KRAS-altered tissue tests, 44.4% were positive by ctDNA. This value also showed a trend to be lower when blood and tissue samples were >6 months apart than when they were ≤2 months apart (31.0% vs. 51.2%; p = 0.14). Hence, substantial numbers of the KRAS-altered ctDNA tests were not picked up by tissue testing and vice versa; further-more, in each case, this dichotomy increased with greater time interval between the blood sample and tissue biopsy dates (albeit the increase in discrepancy was only statistically significant for the ctDNA tests that were not picked up by tissue). In contrast, specificity and negative predictive power of ctDNA for tissue (= the negative predictive power and specificity of tissue for ctDNA, respectively) remained high (94.3 and 88.1%), regardless of the time interval between tests. These findings suggested that, if one test (ctDNA or tissue DNA) showed negative for KRAS alterations, the other would also show negative, regardless of the time interval.
In the multivariate analysis of all 433 patients, the presence of KRAS alterations in both ctDNA and tissue (vs. not [“not” means the presence of KRAS alterations in tissue alone, blood alone or neither]) was an independent poor prognostic factor for patients’ OS, even if the time interval between blood draw and tissue biopsy was considered as a confounder (Table 3). Interestingly, the survival of patients with no KRAS alterations did not differ from that of patients with discordant KRAS alterations, while the survival of patients with concordant KRAS alterations in ctDNA and tissue was significantly worse (Fig. 3). A prior systematic review analyzing ctDNA results in diverse cancer patients showed that KRAS alterations were associated with a poorer OS (HR = 2.02, 95%CI 1.63–2.51).30 However, our findings suggested that KRAS alterations in both tests were more meaningful as a prognostic factor. In addition, even among 101 patients with KRAS alterations, the presence of KRAS alterations in both ctDNA and tissue (i.e., concordant) still tended to be a strong prognostic factor for poor survival (the HRs ranged 1.81–2.30 [p values 0.02–0.13], Figure 3 and Supporting Information Table S7). It is unclear why concordant blood and tissue alterations are associated with poorer survival. Alterations were not more concordant with higher %ctDNA (Supporting Information Table S4), which has previously been shown to be associated with poorer survival.8 To the best of our knowledge, this phenomenon is not previously reported and, hence, deserves additional investigation.
There were several limitations in our study. First, prior treatment type at time of blood draw or tissue biopsy was not considered. It was previously reported that KRAS status in ctDNA could be affected by specific therapeutic pressure such as administration of EGFR antibodies.31,32 Indeed, 7 out of 20 patients who had KRAS alterations only in ctDNA received anti-EGFR therapies prior to their ctDNA analyses in this series. Moreover, three additional patients received systemic therapies with other targeted agents, which may lead to the emergence of KRAS alterations as a mechanism of acquired resistance. Second, the number of samples in each cancer type depended on the physician choice to examine ctDNA or tissue by NGS. Third, the tissue and ctDNA tests were performed by different vendors, though this could also be considered an advantage of the study in that it permitted a comprehensive comparison (which is a topic of interest to the liquid biopsy field). Finally, while %ctDNA was not associated with concordance, the number of patients evaluable was small (since, by definition, only patients positive for KRAS in ctDNA could be assessed) and may have precluded robust comparisons.
In conclusion, KRAS alterations were seen in 23.3% of pan-cancer patients and the overall concordance between ctDNA and tissue was 85.0%. Temporal and spatial effects did not impact the overall concordance of KRAS alterations. High overall concordance was mainly driven by the fact that, if either blood or tissue was negative for a KRAS alteration, the other was likely negative as well. In contrast, substantial numbers of the KRAS-altered ctDNA tests were not picked up by tissue testing and vice versa. Positive concordance was significantly higher when the time interval between blood draw and tissue biopsy was ≤2 months vs. >6 months. This result is consistent with prior studies that show that tumors evolve over time.33 Positive concordance was also higher in colorectal cancer vs. other tumors. Importantly, concordance for KRAS alteration positivity between blood and ctDNA was an independent factor associated with a significantly worse survival as compared to patients with no KRAS alterations or with discordant KRAS alterations. Further studies are warranted to evaluate the prognostic impact of concordance between blood and tissue sequencing for other molecular alterations.
Supplementary Material
What’s new?
Interrogating blood-derived circulating tumor DNA (ctDNA) allows for the detection of genetic heterogeneity in cancer without the invasive nature of tissue biopsies. However, the factors influencing concordance in molecular alterations between tissue and ctDNA samples remain largely understudied. Here, positive concordance between patient ctDNA and tissue DNA for KRAS alterations was inversely associated with time interval between blood and tissue sampling and was higher in colorectal versus non-colorectal cancer. Concordant KRAS alterations (versus discordant KRAS alterations or no KRAS alterations) in blood and tissue correlated with shorter survival, suggesting complementary clinical utility for ctDNA and tissue DNA sequencing in prognostication.
Acknowledgement
Funded in part by the Joan and Irwin Jacobs Fund and by National Cancer Institute grants P30 CA023100 (RK).
Grant sponsor: Joan and Irwin Jacobs Fund; Grant sponsor: National Cancer Institute; Grant number: P30 CA023100
Abbreviations:
- CLIA
Clinical Laboratory Improvement Amendment
- ctDNA
circulating tumor DNA
- EGFR antibody
epidermal growth factor receptor antibody
- HR
hazard ratio
- MAPK
mitogen-activated protein kinase
- NGS
next-generation sequencing
- OS
overall survival
- VAF
variant allele fraction
- VUS
variant of unknown significance
Footnotes
Conflict of interest: Shumei Kato serves as a consultant for Foundation Medicine. Razelle Kurzrock has research funding from Incyte, Genentech, Merck Serono, Pfizer, Sequenom, Foundation Medicine, Guardant Health, Grifols, and Konica Minolta, as well as consultant fees from LOXO, X-Biotech, Actuate Therapeutics, Genentech and NeoMed. She receives speaker fees from Roche, and has an ownership interest in IDbyDNA and Curematch, Inc.
References
- 1.Arrington AK, Heinrich EL, Lee W, et al. Prognostic and predictive roles of KRAS mutation in colorectal cancer. Int J Mol Sci 2012;13:12153–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garrido-Laguna I, Hong DS, Janku F, et al. KRASness and PIK3CAness in patients with advanced colorectal cancer: outcome after treatment with early-phase trials with targeted pathway inhibitors. PLoS One 2012;7:e38033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eser S, Schnieke A, Schneider G, et al. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 2014;111:817–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurzrock R, Giles FJ. Precision oncology for patients with advanced cancer: the challenges of malignant snowflakes. Cell Cycle 2015;14:2219–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wheler JJ, Janku F, Naing A, et al. Cancer therapy directed by comprehensive genomic profiling: a single center study. Cancer Res 2016;76:3690–701. [DOI] [PubMed] [Google Scholar]
- 6.Schmiegel W, Scott RJ, Dooley S, et al. Blood-based detection of RAS mutations to guide anti-EGFR therapy in colorectal cancer patients: concordance of results from circulating tumor DNA and tissue-based RAS testing. Mol Oncol 2017;11:208–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lievre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 2006;66:3992–5. [DOI] [PubMed] [Google Scholar]
- 8.Schwaederle M, Husain H, Fanta PT, et al. Use of liquid biopsies in clinical oncology: pilot experience in 168 patients. Clin Cancer Res 2016;22: 5497–505. [DOI] [PubMed] [Google Scholar]
- 9.Schwaederle M, Husain H, Fanta PT, et al. Detection rate of actionable mutations in diverse cancers using a biopsy-free (blood) circulating tumor cell DNA assay. Oncotarget 2016;7:9707–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kidess-Sigal E, Liu HE, Triboulet MM, et al. Enumeration and targeted analysis of KRAS, BRAF and PIK3CA mutations in CTCs captured by a label-free platform: comparison to ctDNA and tissue in metastatic colorectal cancer. Oncotarget 2016;7:85349–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sclafani F, Chau I, Cunningham D, et al. KRAS and BRAF mutations in circulating tumour DNA from locally advanced rectal cancer. Sci Rep 2018; 8:1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mok T, Wu YL, Lee JS, et al. Detection and dynamic changes of EGFR mutations from circulating tumor DNA as a predictor of survival out-comes in NSCLC patients treated with first-line intercalated Erlotinib and chemotherapy. Clin Cancer Res 2015;21:3196–203. [DOI] [PubMed] [Google Scholar]
- 13.Wyatt AW, Annala M, Aggarwal R, et al. Concordance of circulating tumor DNA and matched metastatic tissue biopsy in prostate cancer. J Natl Cancer Inst 2017;109:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Danese E, Minicozzi AM, Benati M, et al. Comparison of genetic and epigenetic alterations of primary tumors and matched plasma samples in patients with colorectal cancer. PLoS One 2015;10: e0126417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwaederle MC, Patel SP, Husain H, et al. Utility of genomic assessment of blood-derived circulating tumor DNA (ctDNA) in patients with advanced lung adenocarcinoma. Clin Cancer Res 2017;23:5101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernard V, Kim DU, San Lucas FA, et al. Circulating nucleic acids are associated with outcomes of patients with pancreatic cancer. Gastroenterology 2019;156:108–18.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato S, Okamura R, Baumgartner JM, et al. Analysis of circulating tumor DNA and clinical correlates in patients with Esophageal, Gastroesophageal junction, and gastric adenocarcinoma. Clin Cancer Res 2018;24:6248–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grasselli J, Elez E, Caratu G, et al. Concordance of blood- and tumor-based detection of RAS mutations to guide anti-EGFR therapy in meta-static colorectal cancer. Ann Oncol 2017;28: 1294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lanman RB, Mortimer SA, Zill OA, et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS One 2015;10:e0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31: 1023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwaederle M, Daniels GA, Piccioni DE, et al. On the road to precision cancer medicine: analysis of genomic biomarker actionability in 439 patients. Mol Cancer Ther 2015;14:1488–94. [DOI] [PubMed] [Google Scholar]
- 22.Khagi Y, Goodman AM, Daniels GA, et al. Hypermutated circulating tumor DNA: correlation with response to checkpoint inhibitor-based immunotherapy. Clin Cancer Res 2017;23: 5729–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ikeda S, Schwaederle M, Mohindra M, et al. MET alterations detected in blood-derived circulating tumor DNA correlate with bone metastases and poor prognosis. J Hematol Oncol 2018;11:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwaederle M, Zhao M, Lee JJ, et al. Association of biomarker-based treatment strategies with response rates and progression-free survival in refractory malignant neoplasms: a meta-analysis. JAMA Oncol 2016;2:1452–9. [DOI] [PubMed] [Google Scholar]
- 25.Kato S, Janku F. Cell-free DNA as a novel marker in cancer therapy. Biomark Med 2015;9:703–12. [DOI] [PubMed] [Google Scholar]
- 26.Schwaederle M, Chattopadhyay R, Kato S, et al. Genomic alterations in circulating tumor DNA from diverse cancer patients identified by next-generation sequencing. Cancer Res 2017;77:5419–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kato S, Krishnamurthy N, Banks KC, et al. Utility of genomic analysis in circulating tumor DNA from patients with carcinoma of unknown primary. Cancer Res 2017;77:4238–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res 2012;72:2457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loree JM, Miron B, Holla V, et al. Not all RAS mutations created equal: functional and clinical characterization of 80 different KRAS and NRAS mutations. J Clin Oncol 2017;35:3589. [Google Scholar]
- 30.Zhuang R, Li S, Li Q, et al. The prognostic value of KRAS mutation by cell-free DNA in cancer patients: a systematic review and meta-analysis. PLoS One 2017;12:e0182562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Del Re M, Tiseo M, Bordi P, et al. Contribution of KRAS mutations and c.2369C > T (p.T790M) EGFR to acquired resistance to EGFR-TKIs in EGFR mutant NSCLC: a study on circulating tumor DNA. Oncotarget 2017;8:13611–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vidal J, Muinelo L, Dalmases A, et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann Oncol 2017;28: 1325–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swanton C Intratumor heterogeneity: evolution through space and time. Cancer Res 2012;72: 4875–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of our study are available upon request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.