

Abstract
PARN encodes poly(A)-specific ribonuclease. Biallelic and monoallelic PARN variants are associated with Hoyeraal-Hreidarsson syndrome/dyskeratosis congenita and idiopathic pulmonary fibrosis (IPF), respectively. The molecular features associated with incomplete penetrance of PARN-associated IPF have not been described. We report a family with a rare missense, p.Y91C, and a novel insertion, p.(I274*), PARN variant. We found PARN p.Y91C had reduced deadenylase activity and the p.(I274*) transcript was depleted. Detailed analysis of the consequences of these variants revealed that, while PARN protein was lowest in the severely affected biallelic child who had the shortest telomeres, it was also reduced in his mother with the p.(I274*) variant but telomeres at the 50th percentile. Increased adenylation of telomerase RNA, hTR, and certain snoRNAs, and impaired rRNA maturation was observed in cells derived from the severely affected biallelic carrier, but not in the other, less affected biallelic carrier, who had less severely shortened telomeres, nor in the monoallelic carriers who were unaffected and had telomeres ranging from the 1st to the 50th percentiles. We identified hsa-miR-202–5p as a potential negative regulator of PARN. We propose one or more genetic modifiers influence the impact of PARN variants on its targets and this underlies incomplete penetrance of PARN-associated disease.
Keywords: PARN, poly(A)-specific ribonuclease, incomplete penetrance, telomere biology disorder, MIR202, idiopathic pulmonary fibrosis
Graphical Abstract
INTRODUCTION
Telomeres are nucleoprotein structures that cap the ends of linear chromosomes (Moyzis et al., 1988; Palm & de Lange, 2008). Telomeric DNA, which consists of tandem 5’TTAGGG/3’CCCTAA repeats spanning up to 10 kb in length at birth, shortens with each round of semi-conservative DNA replication due to the so-called end replication problem (Harley, Futcher, & Greider, 1990; Vaziri et al., 1994). Most cells lack a mechanism to counter this attrition; thus, telomere length shortens progressively with each cell division. Telomeres may ultimately reach a critical length that triggers a DNA damage checkpoint, inducing cellular senescence (Allsopp & Harley, 1995; d’Adda di Fagagna et al., 2003; Herbig, Jobling, Chen, Chen, & Sedivy, 2004). In contrast, a subset of cells, such as germ cells, stem cells, and activated lymphocytes, express the specialized reverse transcriptase telomerase, which catalyzes the de novo synthesis of telomeric repeats on to the chromosome terminus, thereby maintaining telomere length and replicative potential (Kim et al., 1994; Wright, Piatyszek, Rainey, Byrd, & Shay, 1996).
While telomeres naturally shorten as humans age, some individuals are endowed with abnormally short or dysfunctional telomeres due to germline pathogenic variants in telomere biology genes, which can result in a collection of pathologies referred to as the telomere biology disorders (TBDs) (Barbaro, Ziegler, & Reddel, 2016; Dodson & Bertuch, 2018; Savage, 2018). Depending on the gene, the inheritance pattern can be autosomal dominant, autosomal recessive, or X-linked recessive and the age of onset and the disease severity and manifestations vary substantially (Bertuch, 2016). The TBDs include adult-onset diseases, such as idiopathic pulmonary fibrosis (IPF), and pediatric-onset syndromes, including dyskeratosis congenita (DC) and Hoyeraal-Hreidarsson (HH) syndrome (MIM# 127550, 616553, 612199, 305000, 613987, 224230, 616353, 615190, 616371, 616373, 617341, 614743, 613989, 614742, 268130, 613990, and 613988). DC is classically defined by a mucocutaneous triad of oral leukoplakia, reticulated skin pigmentation, and nail dysplasia, and is also associated with increased risks of bone marrow failure, cancer, pulmonary fibrosis, cirrhosis, and gastrointestinal disease among others. HH is classically defined by intrauterine growth retardation, developmental delay, microcephaly, immunodeficiency, and bone marrow failure. The mucocutaneous triad and additional features of DC may also be present. More recently, clinicians have used cerebellar hypoplasia as a defining feature of HH in patients who have additional clinical features of DC.
Fourteen genes have been associated with the TBDs (Dodson & Bertuch, 2018). Among them are TERT and TERC, which encode the core components of telomerase (TERT and hTR), and PARN, which encodes a factor involved in telomerase biogenesis. PARN was first associated with IPF through identification of multiple probands bearing rare or novel monoallelic PARN variants (Stuart et al., 2015). Subsequently, patients with biallelic variants in PARN were identified in a cohort of patients with HH (Tummala et al., 2015). To date, greater than 50 cases with IPF, 8 cases with HH, and 3 cases with DC, have been reported with PARN as the underlying etiology (Alder et al., 2018; Benyelles et al., 2019; Burris et al., 2016; Dhanraj et al., 2015; Kropski et al., 2017; Moon et al., 2015; Newton et al., 2016; Petrovski et al., 2017; Tummala et al., 2015). The phenomenon of monoallelic variants causing adult-onset IPF and biallelic variants causing a pediatric TBD has also been observed for TERT and RTEL1 (Armanios et al., 2007; Ballew, Joseph, et al., 2013; Ballew, Yeager, et al., 2013; Deng et al., 2013; Gramatges, Qi, Sasa, Chen, & Bertuch, 2013; Kannengiesser et al., 2015; Le Guen et al., 2013; Marrone et al., 2007; Stuart et al., 2015; Tsakiri et al., 2007; Walne, Vulliamy, Kirwan, Plagnol, & Dokal, 2013). Notably, there is incomplete penetrance of IPF in individuals with monoallelic PARN variants (Stuart et al., 2015). The average age of diagnosis of IPF in PARN variant carriers is 64 +/− 8 years (Newton et al., 2016). In one study, of the 19 reported individuals aged 72 years or older known or inferred to carry a pathogenic PARN variant (Stuart et al., 2015) (Supp. Table S1), 5 did not have lung disease (74% penetrance). This penetrance was similar for the 7 women (71%) and 12 men (75%), suggesting that gender alone does not account for incomplete penetrance. The nine unaffected individuals had a splice site, stopgain, or missense variant, suggesting that the type of mutation does not determine penetrance either. These results raise the possibility of a relatively common genetic modifier.
PARN is 3’ exoribonuclease with a preference for adenosines. It acts on many RNA targets, including messenger, ribosomal, small nucleolar, small Cajal body-specific, microRNAs (mRNAs, rRNAs, snoRNAs, scaRNAs, miRNAs, respectively), Y RNAs and hTR, the integral RNA component of telomerase (Berndt et al., 2012; Dhanraj et al., 2015; Lee, Park, Park, & Shin, 2018; Montellese et al., 2017; Shukla & Parker, 2017; Virtanen, Henriksson, Nilsson, & Nissbeck, 2013; Yoda et al., 2013). A longer hTR precursor is post-transcriptionally adenylated, but PARN removes the terminal adenosines and facilitates its trimming to the mature length (Dhanraj et al., 2015). Deadenylation of hTR also prevents its degradation via the exosome (Nguyen et al., 2015; Tseng et al., 2015). Thus, PARN plays a critical role in regulating hTR levels.
Consistent with its role in hTR processing, fibroblasts, induced pluripotent stem cells, and lymphoblastoid cell lines (LCLs) derived from patients with biallelic PARN variants have increased hTR adenylation, reduced overall level of hTR, and reduced telomerase activity (Dhanraj et al., 2015; Moon et al., 2015; Tummala et al., 2015). Samples derived from biallelic variant carriers also have increased adenylation of some snoRNAs and scaRNAs and altered ribosome biogenesis (Dhanraj et al., 2015). Studies of adenylation and level of PARN RNA targets have only been performed on samples derived from one monoallelic variant carrier, who was the father of a patient with HH and had normal telomere length, and these were all similar to controls as expected (Tummala et al., 2015). However, in order to understand the pathogenesis of IPF, there is a need for functional studies performed on samples derived from monoallelic variant carriers with both normal and short telomeres.
The two most commonly used methods of measuring telomere length and determining age-adjusted percentiles in patient cohorts are telomere fluorescence in situ hybridization coupled with flow cytometry (telomere flow FISH) and quantitative PCR (qPCR). In addition to differences in the technical approach, qPCR is generally performed on DNA isolated from whole blood, whereas telomere flow FISH measurements may be determined on the granulocytes, pan-lymphocytes, and lymphocyte subsets. Studies that have compared telomere flow FISH and qPCR to the gold standard Southern blotting or to each other in cohorts with known pathogenic variants in TBD-related genes indicate that telomere flow FISH has greater sensitivity and specificity in the detection of patients with short telomeres and TBDs (Behrens et al., 2017; Gadalla et al., 2016; Gutierrez-Rodrigues, Santana-Lemos, Scheucher, Alves-Paiva, & Calado, 2014; Khincha et al., 2017). With respect to previous telomere length analyses of kindreds with PARN variants, the largest study reported measurements by qPCR and the lengths corresponding to affected versus unaffected family members were not delineated (Stuart et al., 2015). Four reports described families with biallelic PARN variant carriers, in which pan-lymphocyte telomere flow FISH was reported on five sets of parents carrying a single variant and seven affected children carrying both mutant alleles (Burris et al., 2016; Dhanraj et al., 2015; Moon et al., 2015; Tummala et al., 2015). The types of mutations included whole gene deletion, a promoter single nucleotide variant, a multi-exon deletion, a stopgain, four missense changes, and one yet-to-be described mutation that led to loss of expression from one of the parental alleles. Whereas each of the affected children had lymphocyte telomere lengths well below the 1st percentile, the lymphocyte telomere lengths of the parents varied from the 1st percentile to between the 50th to 90th percentiles. Telomere lengths in the lymphocyte subsets were not reported. Overall, the level of consistency with which monoallelic PARN variants shorten telomeres has not been shown. Moreover, the contribution of normal telomere length in monoallelic PARN variant carriers to incomplete penetrance of IPF has not been explored.
Here we describe a three-generation, 10-person pedigree with biallelic and monoallelic PARN variant carriers, each with telomere length measurements by telomere flow-FISH and cellular analyses of PARN protein levels, hTR and snoRNA adenylation and abundance, and rRNA processing in LCLs. Analysis of the missense and exonic insertion variants revealed both were pathogenic, yet there was variable impact of each of these in family members with respect to telomere length and in the cellular assays. In search of a genetic modifier we identified a microRNA, hsa-miR-202–5p, encoded by MIR202, as a potential regulator of PARN protein level. We investigated the possibility that a common single nucleotide polymorphism, rs12355840, which impacts the processing of the hsa-pre-mir202, may play a role in the variable impact of given PARN variants on telomere length, but do not conclude that it explains the phenotype. Instead, the comprehensive analyses underscore a complexity of PARN regulation and impact of PARN variants on telomere length that remains to be elucidated.
MATERIALS AND METHODS
Human subjects
This investigation was conducted according to Declaration of Helsinki Principles. Informed consent was received from participants prior to inclusion in the study according to protocol H-17698 Genetic and Biological Determinants of Bone Marrow Failure approved by the Institutional Review Board for Baylor College of Medicine and affiliated hospitals.
Telomere length analyses
Telomere flow FISH analysis was performed on peripheral blood leukocytes by Repeat Diagnostics (Vancouver, British Columbia) as previously described (33). Telomere restriction fragment length analysis of HCT116 PARN wild type (WT) and heterozygous cell lines was performed by Southern blotting as previously described (Nelson et al., 2018). Mean telomere length was determined using TeloTool V1.3 (Gohring, Fulcher, Jacak, & Riha, 2014). In addition, telomere length of the cell lines was analyzed by quantitative telomere FISH as previously described (Ourliac-Garnier & Londono-Vallejo, 2017). Metaphase chromosomes were prepared by treatment of cells with colcemid (Sigma-Aldrich) 0.1 μg/ml for 30 min at 37˚. Fluorescence in situ hybridization (FISH) was performed as previously described (Ourliac-Garnier & Londono-Vallejo, 2017) using TelG Alexa Fluor 647 (PNA Bio, catalog # F1014). DNA was counterstained with DAPI in SlowFade Diamond Antifade Mountant (Life Technologies, catalog # S36964). Images were captured using a GE Healthcare DVLive epifluorescence image restoration microscope with an Olympus PlanApo 100x/1.4 NA objective and a 1.9k x 1.9x sCMOS camera. Z stacks (0.24 μm) covering entire metaphases were acquired before applying a conservative restorative algorithm for quantitative image deconvolution. Exposures for DAPI and Alexa Fluor 647 were set on the control sample and maintained constant within each set of samples. Maximum intensity projections were generated and used for image analysis. Telomere length intensity was calculated using the NIH software ImageJ (https://imagej.nih.gov/ij/) with the plugin Telometer version 3 (http://demarzolab.pathology.jhmi.edu/telometer).
DNA sequencing and data analyses
Whole exome sequencing was conducted on proband and parents according to standard protocols at Baylor Genetics. Re-interpretation of exome data was conducted in the research setting using GATK software for calling variants and custom perl scripts for interpretation of variants that annotate and prioritize variants based on multiple sources of information including SIFT, POLYPHEN, GERP, PhyloP, CADD, ExAC, UniProt, OMIM, IntAct as previously described (Besse et al., 2015; Stiles et al., 2015)
For orthogonal sequence validation and genotyping PARN alleles in additional family members, genomic DNA was isolated from whole blood and the regions corresponding to the PARN p.Y91C and p.(I274*) mutations, the most common TERT promoter variants, and hsa-mir-202, were PCR amplified. The PCR products were analyzed by Sanger sequencing (Eurofins Genomics, LLC). To determine the sequence of the insertion allele, prior to Sanger sequencing, the amplicon encompassing PARN p.(I274*) was cloned using the Zero Blunt™ TOPO™ PCR cloning kit. See Table S2 for oligonucleotide sequences.
Deadenylation assays
PARN deadenylation assays were performed using recombinant proteins as previously described (Henriksson, Nilsson, Wu, Song, & Virtanen, 2010). The kinetic analysis was performed using PARN WT (100 nM) or p.Y91C (30 nM) and A3 trinucleotide substrate. For determining the rate constants under single turnover conditions each reaction contained 10 nM RNA substrate and 30 nM PARN. The reactions were incubated at 30°C for indicated length of time and analyzed as described above. The mathematical model of a multistep reactions with intermediate products is derived in (Kuriyan, Konforti, & Wemmer, 2013). The kobs1 and kobs2 parameters were determined from time course degradation assays of di- or trinucleotide substrates with subsequent curve fitting in Prism 7 (GraphPad) with the intermediate product equation, The given values are the mean ± standard deviation of at least three independent time-course experiments.
Lymphoblastoid cell lines
Lymphoblastoid cell lines (LCLs) were generated by the tissue culture core laboratory within the Department of Molecular and Human Genetics, Baylor College of Medicine. LCLs were cultured in RPMI 1640 medium containing L-glutamine (Invitrogen) and 10% fetal bovine serum in 5% CO2.
Protein extraction and analysis
Cells were lysed in RIPA buffer (50 mM Tris- HCl pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.5% Na deoxycholate, and 0.1% SDS) with 1x protease inhibitor cocktail III (Calbiochem) and centrifuged. The protein concentration of the supernatant was determined using the bicinchoninic acid (BCA) protein assay kit (Pierce). Twenty to 40 μg of protein were loaded per sample. Protein was detected using polyclonal rabbit anti-PARN antibody (Bethyl A303–562A) and β-actin (Sigma-Aldrich A5441).
RNA analyses
To determine if RNA encoding PARN p.(I274*) underwent nonsense-mediated decay, RNA was isolated from whole blood using the RNeasy® kit (QIAGEN) and cDNA synthesized using the qScript Flex cDNA Synthesis Kit (Quanta Biosciences, Beverly, MA). The region surrounding PARN p.Y91C was PCR amplified and analyzed by Sanger sequencing.
PARN gene expression levels were measured by quantitative PCR (qPCR) using a QuantStudio 6 Flex (Applied Biosystem) with PowerUp™ SYBR Green Master Mix (Applied Biosystems, Austin, TX) in triplicate and normalized to GAPDH. For these experiments, total RNA was extracted from LCLs using the RNeasy® kit (QIAGEN) and cDNA synthesized using the qScript Flex cDNA Synthesis Kit (Quanta Biosciences, Beverly, MA) and oligo(dT) primer.
To analyze snoRNAs and scaRNAs, total RNA, including small RNAs, was isolated using the miRNeasy® kit (QIAGEN). Complementary DNAs were prepared with 1–2 μg of RNA using qScript and random or oligo(dT) primer. Quantitative PCR was performed using gene specific primers and PowerUp™ SYBR Green Master Mix (Applied Biosystems™) or Maxima SYBR green master mix (Thermo Scientific) according to the manufacturer’s protocol using the ABI 7300 real time PCR instrument. The relative changes in RNA abundance were calculated by comparative ΔCt method with normalization to GAPDH.
To analyze rRNA by northern blotting, total RNA was extracted using the RNeasy® kit (QIAGEN). Five μg of total RNA per sample were separated on a 1.2% agarose/formaldehyde gel and capillary transferred to a Zeta-Probe cationized nylon membrane (BioRad, 1620165). The membrane was pre-hybridized in ULTRAhybTM buffer (Invitrogen, AM8663) for 45 mins at 42°C and then hybridized with γ32P-ATP end-labeled ITS1–59 probe in ULTRAhyb™ buffer overnight at 65°C. The membrane was exposed to a phosphorimager screen for 4 hours. Signals were quantified using ImageQuant TL 8.2 image analysis software (GE Healthcare). Ratio analysis of multiple precursors was performed as previously described except that the average values of 3 controls were used as control values values for the analysis of LCLs rather than the values of a single control (Wang, Anikin, & Pestov, 2014).
CRISPR-Cas9 guide design and heterozygous PARN cell line generation
The guide RNAs (GGAGGAGCTGAATGATGCTG and CTCACTTACCGAATTAGCAA) were identified using the online software Benchling (www.benchling.com/). The PARN gene sequence (ENSG00000140694) was uploaded and the search for possible guides was performed around exon 12. The guides in plasmid pSpCas9 BB-2A-GFP (Ap311 and Ap312, respectively) and the scrambled sgRNA in pSpCas9 BB-2A-GFP (Ap314) were obtained from GenScript. HCT116 cells were grown in McCoy with 10% FBS and seeded in a 6-well plate at a concentration of 90,000 cells/well. Cells were transfected at 50% confluency with 0.5 μg of Ap311 and Ap312 using Lipofectamine Plus Reagent (Invitrogen) following the manufacturer’s protocol. After 72 hours, the cells were harvested for analysis using the Guide-it Mutation detection Kit (catalog # 631443, Takara Clontech), which uses a resolvase to determine cutting efficiency. Once the efficiency of cleavage was confirmed, the experiment was repeated as described. After 72 hours, GFP positive cells were sorted via flow-cytometry and single cells collected in 92-well plates. Clones from single-cell growth were split into two sister plates. One was cultured while the other was used to identify heterozygous PARN clones by Sanger sequencing. Clones with abnormal chromatograms underwent Zero Blunt™ TOPO™ PCR cloning and Sanger sequencing to determine the genotype. Three clones that underwent transfection but still maintained WT genotype (WT-C1, -C2 and -C3) and one PARN-het clone were used in subsequent experiments.
Transfection with miR mimic RNA
HEK293T were grown in Modified Eagles Media α with 10% FBS and 5% CO2. Cells were seeded in 6-well plates and, when at 60% confluence, transfected with 0.1 μM Pre-miR™ miRNA Precursor hsa-miR-202–5p mimic (Ambion, AM17100), 0.1 μM Pre-miR™ miRNA Precursor random miR mimic (Ambion, AM17111), or transfection reagent alone (DharmaFECT #1, 6 μL in 2 mL). The cells were harvested at 72 hours for western blot analysis.
Statistical analyses
The data are represented as mean ± standard deviation of the values (SD). A p-value <0.05 was considered statistically significant and indicated by an asterisk. For the PARN western blot and qPCR, hTR RH qPCR, and hTR dT qPCR, each biological replicate was normalized to one control individual (128S2 for PARN western and qPCR, RQ4115 for hTR RH and dT qPCRs). One-way Anova with Dunnet’s comparisons were performed on log transformed data for those datasets having a lognormal rather than normal distribution. The log-transformed data were normally distributed. Brown-Forsythe tests showed that the standard deviations were not significantly different between individuals for the PARN western blot or qPCR, or the hTR RH qPCR but were for the hTR dT qPCR. Dunnet’s post-hoc test was performed and p-values adjusted for multiple comparisons. For the H/ACA snoRNA profile, the average of the control individuals (RQ4115, 101F, and 128S2) was used for normalization and a t-test was performed to determine significant differences from the average of controls. For the assays using HCT116 clonal cell lines, the Shapiro-Wilk test was applied to check for normal distribution and the Bartlett test to check for equal variance across samples.
RESULTS
Compound heterozygous PARN variants segregating with HH in family BMF22
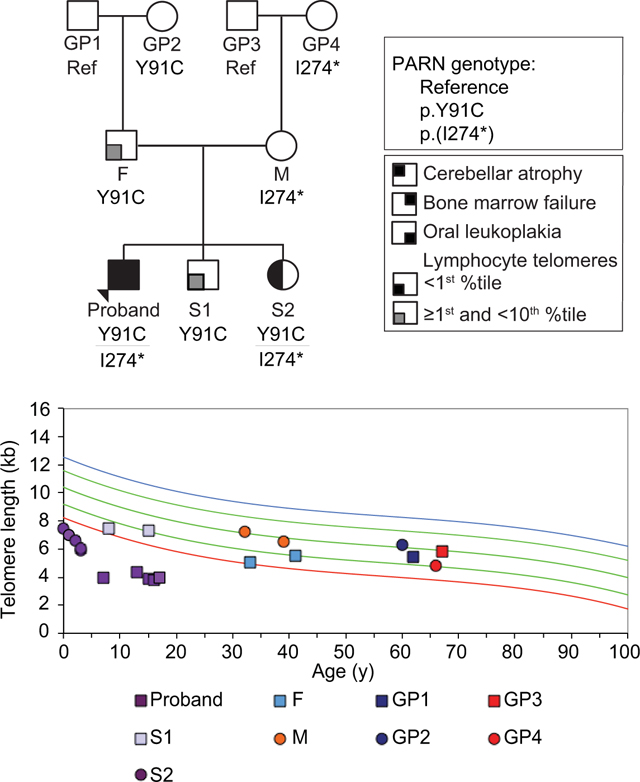
The proband of this study was a male diagnosed with HH, based on clinical features of bone marrow failure, intellectual disability, cerebellar atrophy, and oral leukoplakia, and lymphocyte telomere length <1st percentile (Figures 1A and 1B, Supp. Figure S1 and Table S3). He was 17-years-old at the time of this report. Whole-exome-sequencing (WES) identified two variants in PARN, a variant of uncertain clinical significance (VUS), which mapped to the CAF1 ribonuclease domain (NM_002582.3: c.272A>G; p.Y91C) (Figure 1C, Supp. Figure S2A), and a likely pathogenic 20 bp exonic insertion resulting in an in-frame translation stop codon (NM_002582.3: c.819_820insTAGAAATCATTTCTAGAGTC; p.(I274*)) (Figure 1C, Supp. Figure S2). PCR amplification of the region, blunt-ended cloning, and Sanger sequencing confirmed the sequence of the insertion (Supp. Figure S2B). A portion of the insertion shared microhomology with a nearby 5’ region, suggesting that it was caused by a duplication event.
Figure 1.
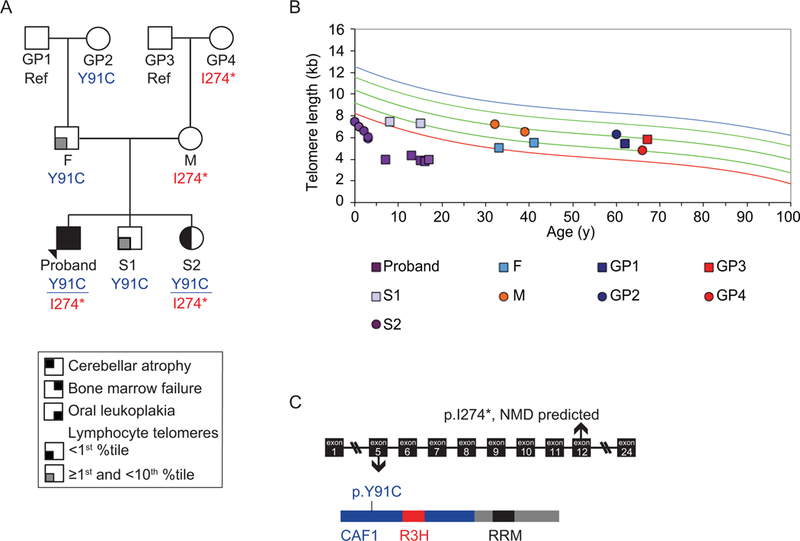
Biallelic variants in PARN segregate with disease. (A) The three-generation pedigree of family BMF22 with the PARN variant segregation (NM_002582.3: c.272A>G; p.Y91C and c.819_820insTAGAAATCATTTCTAGAGTC; p.(I274*)). Arrow indicates the proband. Square indicates male, circle indicates female. Filled quadrant indicates affected with the condition in the key. Ref: reference genotype at the locations of both PARN variants, GP: grandparent, F: father, M: mother, S: sibling. (B) Telomere flow FISH of the lymphocyte subpopulation of peripheral blood. From top to bottom, the colored lines indicate 99th, 90th, 50th, 10th, and 1st percentile telomere lengths. (C) Schematic of the location of the PARN variants in the gene and protein. NMD: nonsense mediated decay. CAF1 (blue) is the ribonuclease domain, and R3H (red) and RRM (black) are RNA binding domains.
Concurrent with these investigations, his younger sister, S2, was also diagnosed with HH based on developmental delays, microcephaly, cerebellar atrophy and telomere length <1st percentile (Figures 1A and 1B, Supp. Figure S1 and Table S3). She was 4-years-old at the time of this report, at which time she had normal peripheral blood counts and indices, except for slightly decreased white blood cell count (4.91 × 103/μl; lower limit of normal range for age 5 × 103/μl). She was also found to carry both PARN variants (Figure 1A, Supp. Figure S2A). Targeted sequencing of the parents and grandparents demonstrated inheritance of the missense p.Y91C variant through the paternal grandmother (GP2), and inheritance of the p.(I274*) insertion through the maternal grandmother (GP4) (Figure 1A, Supp. Figure S2A). The proband’s other sibling, S1, inherited only the missense variant. Only those family members who carried both mutant alleles (the proband and S2) were symptomatic (Figure 1A, Supp. Figure S2A). Siblings of F and M, who were reportedly healthy, are not noted in the pedigree as they were unavailable for analysis with the exception of one sister, BMF22-A, who had limited studies performed (mutation analysis demonstrating the absence of the familial PARN variant and telomere length analysis, which was within normal range; data not shown).
Both PARN p.(I274*) and p.Y91C are damaging
Next, we sought further evidence for the pathogenicity of these variants. We predicted that the RNA encoding the p.(I274*) would undergo nonsense-mediated decay (NMD) because the premature termination codon introduced by the insertion was well upstream of the final exon-exon junction (Figure 1C). To test this, we compared Sanger sequencing of the genomic DNA (gDNA) and complementary DNA (cDNA) at position c.272, within the codon for residue Y91, in the proband, S1, S2, and F, all of whom were heterozygous for the c.272A>G transition. As expected, we found that the reference (A) and variant (G) bases were both clearly demonstrated in the chromatograms of F and S1’s gDNA and cDNA, and the proband and S2’s gDNA (Figure 2A). In contrast, the reference base (A) was barely demonstrated in the chromatograms of the cDNA of the proband and S2, consistent with a specific depletion of the p.(I274*) transcript from the maternally inherited allele by NMD.
Figure 2.
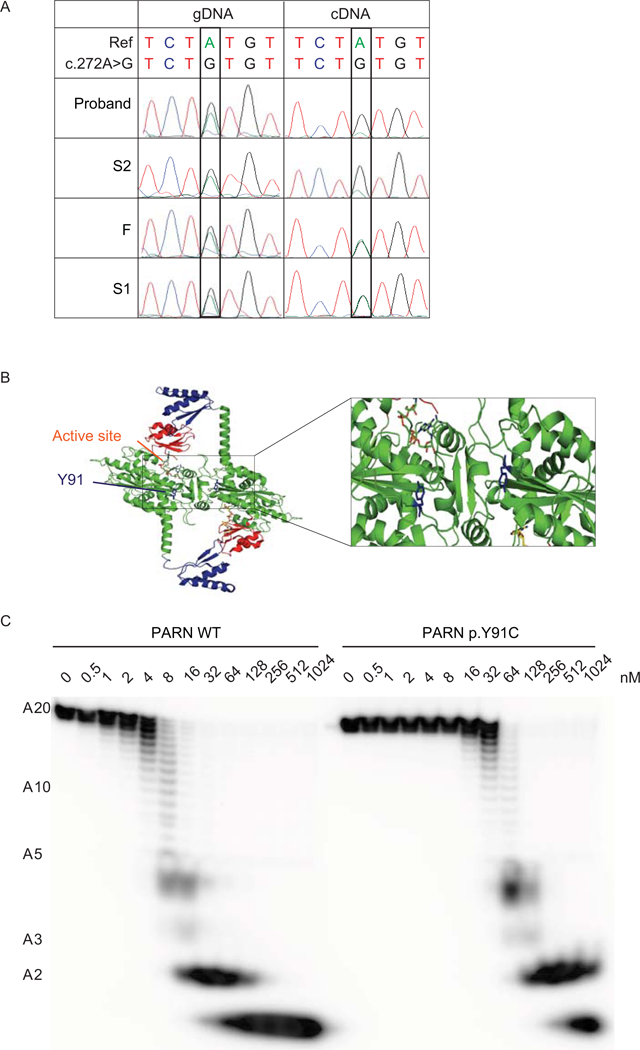
The PARN variant transcript encoding p.(I274*) is subject to NMD and the PARN p.Y91C mutant protein has decreased deadenylation activity. (A) Sanger sequencing of gDNA and cDNA from peripheral blood from the indicated individuals in the region encompassing NM_002582.3: c.272A; p.Y91. (B) Localization of residue Y91 (blue) in the dimeric model of PARN (Virtanen et al., 2013), based on crystal structures PDB: 2A1R (Wu et al., 2005), 2A1S (Wu et al., 2005) and 3D45 (Wu et al., 2009). The expanded view highlights the location of residue Y91 near the active site and the dimer interface in each monomeric subunit of PARN. (C) Deadenylation activity of PARN WT or p.Y91C using the indicated concentrations of recombinant protein expressed and a 5’ end-labeled A20 homopolymeric RNA substrate.
To investigate if the p.Y91C mutation, which mapped near the dimerization interface (Figure 2B), affected the deadenylation activity of PARN, we generated a corresponding mutant PARN polypeptide, PARN p.Y91C, by site-directed mutagenesis, and functionally characterized its deadenylation activity in vitro by incubating it with a 20 nucleotide long homopolymeric adenine substrate (A20). Figure 2C shows that its deadenylation activity was at least 10-fold lower than for the WT PARN polypeptide. We also noted that the deadenylation pattern, i.e. accumulation of A4 and A2 deadenylation intermediates (see (Henriksson et al., 2010)) was not affected when using the mutant PARN p.Y91C polypeptide.
To further analyze the PARN p.Y91C mutant protein, we determined its kinetic rate constants, kobs1 and kobs2, for the complete degradation of an A3 trinucleotide substrate. The kobs1 rate constant describes the release of the 3’-end located adenine residue from the A3 substrate, while the kobs2 rate constant describes the final hydrolysis of the A2 reaction intermediate (see Methods for definitions of kobs1 and kobs2 rate constants). From this analysis we found that the cysteine substitution reduced both kinetic rate constants, kobs1 and kobs2, approximately 30-fold (Table 1, catalytic efficiency ratio). This drop was observed both in reactions performed with Mg2+ or Mn2+ as the divalent metal ion. This showed that the Y91C mutation affected the entire hydrolytic cycle of PARN, which encompasses both one translocation and one hydrolytic event for each round of hydrolysis (Virtanen et al., 2013). Together these studies showed that the Y91C mutated PARN polypeptide was perturbed in its deadenylation activity and suggest that the PARN-mediated deadenylation activity in individuals in this pedigree carrying the mutation was affected.
Table 1.
PARN kinetic rate constants
| kobs1 | kobs2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Divalent metal ion | PARN poly-peptide | Rate kobs1 x 103 (s−1) | Catalytic efficiency x 105 (s−1 M−1) | Catalytic efficiency ratio WT/Y91C | Rate kobs2 x 103 (s−1) | Catalytic efficiency x 105 (s−1 M−1) | Catalytic efficiency ratio WT/Y91C | Rate ratio kobs1/ kobs2 |
| Mg2+ | WT | 22 ± 6 | 7 ± 2 | 30 | 0.2 ± 0.002 | 0.07 ± 0.001 | 31 | 109 |
| Y91C | 2 ± 0.9 | 0.2 ± 0.09 | 0.02 ± 0.005 | 0.002 ± 0.0005 | 111 | |||
| Mn2+ | WT | 14 ± 0.5 | 5 ± 0.2 | 27 | 27 ± 0.1 | 9 ± 0.04 | 30 | 0.5 |
| Y91C | 2 ± 0.9 | 0.2 ± 0.1 | 3 ± 1 | 0.3 ± 0.1 | 0.6 | |||
The same monoallelic PARN variants do not always associate with short telomeres and may be associated with telomere lengthening over time
Having established that both variants were damaging and co-segregated with disease (Figure 1A), we evaluated their effects on telomere length through telomere flow-FISH analysis of peripheral blood leukocytes in BMF22 family members. In Figure 1B, we present the length of the total lymphocyte population, although the trends were similar in granulocytes and subpopulations of lymphocytes (Supp. Figure S3). In the initial set of analyses, among the p.Y91C variant monoallelic carriers (F, S1, and GP2), the lymphocyte telomere lengths in F and S1 were between the 1st and 10th percentiles, whereas the lengths in GP2 were at the 50th percentile, apparently unaffected. Strikingly, M, who carried the p.(I274*) allele subject to NMD, had lymphocyte telomere lengths around the 50th percentile. Thus, the monoallelic variants were not always associated with short telomeres, consistent with prior reports (Dhanraj et al., 2015; Moon et al., 2015; Stuart et al., 2015; Tummala et al., 2015).
Given these results, we next sought to determine leukocyte telomere lengths in the monoallelic PARN variant carriers over time (Figure 1B, Supp. Figure S3, and Table S3). Telomere flow FISH was performed on M, 8 years after her initial testing. Telomere lengths shortened in pan-lymphocytes and each of the lymphocyte subsets, as anticipated, with the percentiles declining to varying extents, although in all cases remaining greater than the 10th percentile and in the memory T and B cells above the 50th percentile.
The results of repeat telomere length analyses of S1 and F, both of whom carried the p.Y91C allele, were different (Figure 1B, Supp. Figure S3A, and Supp. Table S3). F’s second sample, drawn 8 years after the first, surprisingly showed increased telomere length in four of the six subsets (pan-lymphocytes, granulocytes, CD45RA+ (naïve T) cells and CD57+ (NK) cells), with average increases of 63 – 239 bp/year. Despite the increases, F’s telomere lengths remained in the 1st – 9th percentile range except in the naïve T and NK cell populations, which incurred the greatest increases, resulting in lengths in the >10th and <50th percentile range. S1’s granulocyte and NK cell telomere lengths also increased over a 7 year interval, although to a lesser extent than F’s (averages of 66 and 69 bp/year in granulocytes and NK cells, respectively), and his naïve T cell telomere lengths declined only 50 bp total in the 7 year interval compared to a decline of 610 bp in the median (87 bp/year). Nonetheless, S1’s telomere length percentiles remained slightly greater than F’s in all subsets except the NK cells.
We next asked if M’s normal telomere length and F and S1’s increases in telomere lengths were due to a previously reported phenomenon: somatic TERT promoter mutations in individuals with germline monoallelic PARN or TERT variants (Maryoung et al., 2017). These TERT promoter mutations (NC_000005.10(TERT_v002):c.−146C>T and NC_000005.10(TERT_v002):c.−124C>T) are associated with increased TERT expression, telomerase activity, and proliferative capacity (Borah et al., 2015; Chiba et al., 2015). However, there was no TERT promoter variant detected in M, F or S1 (Supp. Figure S3B).
The same biallelic PARN variants may be associated with accelerated telomere shortening or stably, very short telomere lengths over time
The proband and S2 shared biallelic PARN variants and both had severely short telomeres (<1st percentile for age), however, the proband had much shorter telomeres than S2, either objectively or adjusted for age at the time of diagnosis (Figure 1B, Supp. Figure S3A, and Supp. Table S3). This, too, was surprising, so, as with other family members, we performed repeat testing. The proband’s lymphocyte telomeres were stably short (maximum 4.4 and minimum 3.8 kb over an approximately 9 year span), while S2’s lymphocyte telomeres shortened dramatically from 7.5 kb to 5.9 kb in 3 years, a rate greater than expected based on the normal curves. At each of her time points, a complete blood count was obtained, which remained within normal range. Additionally, she reported no significant infections during the time frame. Subsequent testing at 4 years-of-age yielded telomere lengths at 6.1 kb, suggesting a potential stabilization of the rate of attrition.
PARN target RNAs are altered in one individual with biallelic variants, but not the other
In light of the variable telomere length of variant carriers, we proceeded to assess the cellular phenotypes of LCLs derived from BMF22 family members and multiple unique LCL controls. PARN protein level was variable upon biological replicates, even in control LCLs (Figures 3A, Supp. Figure S4). Nonetheless, when we compared all other controls and all family members to control BMF128-S2, PARN was significantly reduced only in the proband, M, and GP4 (p<0.0001, 0.0002, and <0.0001, respectively), as expected since they each carried the insertion (Figure 3B). Surprisingly, despite carrying the insertion, S2’s PARN level was not consistently nor significantly reduced among repeated experiments (p=0.7199) (Figure 3, Supp. Figure S4). The higher PARN protein level in S2 as compared to the other family members carrying the p.(1274*) could not be attributed simply to higher in PARN mRNA level as, while PARN mRNA level, assessed by quantitative reverse transcriptase PCR (qRT-PCR), was significantly lower in M and G4 compared to S2, the level in the proband was comparable to that of S2 (Figure 3C). Sequence analysis of the 5’ and 3’ untranslated regions of the PARN locus for all members of the BMF22 family revealed no variants to account for these differences (data not shown).
Figure 3.
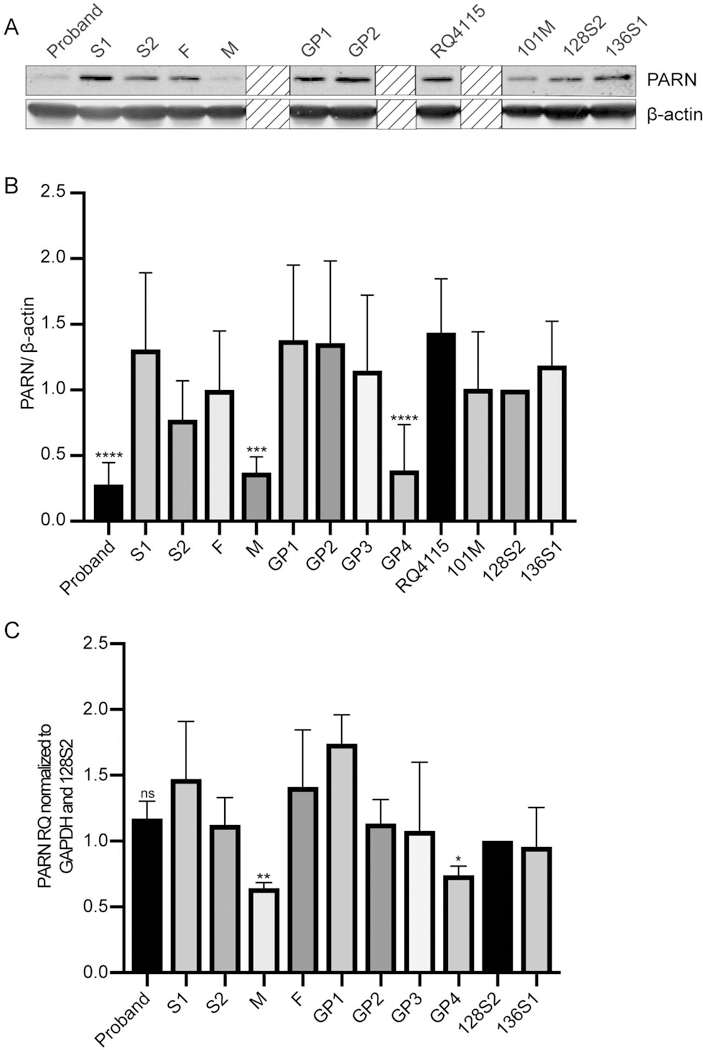
PARN protein levels are variably reduced in LCLs containing the p.(I274*) mutation. (A) Western blot of whole cell extracts of BMF22 family members and unrelated control LCLs (RQ4115, 101M, 128S2, and 136S1). β-actin is a loading control. Boxes with diagonal lines obscure samples from family members or controls for which there were only 1 or 2 samples, and were, therefore, excluded from the analysis. (B) Quantification of western blots in (A) and Figure S4. Bars indicate mean PARN/β-actin ratio +/− standard deviation (SD) compared to 128S2 with N=3–7. ***p<0.001, ****p<0.0001, based on one-way ANOVA of log-transformed data followed by Dunnett’s post-hoc test and adjustment of p-values for multiple comparisons. (C) Analysis of PARN expression in BMF22 family and unrelated controls (128S2 and 136S1) LCLs by qRT-PCR. Complementary DNA was synthesized using oligo(dT) primer and qPCR was performed using primers to the target, PARN, or the control, GAPDH. Bars indicate mean +/− SD compared to 128S2 with N=3. **p=0.0053 and *p=0.0130. ns, not significant. p-values were calculated using one-way ANOVA of followed by Dunnett’s post-hoc test comparing F, M and G4 to S2 and adjustment of p-values for multiple comparisons.
Next we assessed various RNA targets of PARN using qRT-PCR. It was reported previously that DC/HH patients with biallelic PARN variants had increased hTR adenylation and decreased total hTR (Dhanraj et al., 2015; Moon et al., 2015; Tummala et al., 2015). In particular, LCLs derived from a DC/HH patient had reduced hTR level (Tummala et al., 2015). We, therefore, expected to find alterations in the proband and S2. Compared to control RQ4115, only the proband had significantly increased adenylated hTR as measured by qPCR (p=0.0003) (Figure 4A), whereas both the proband and S2 had significantly reduced hTR (p=0.001 and 0.0248, respectively) (Figure 4B).
Figure 4.
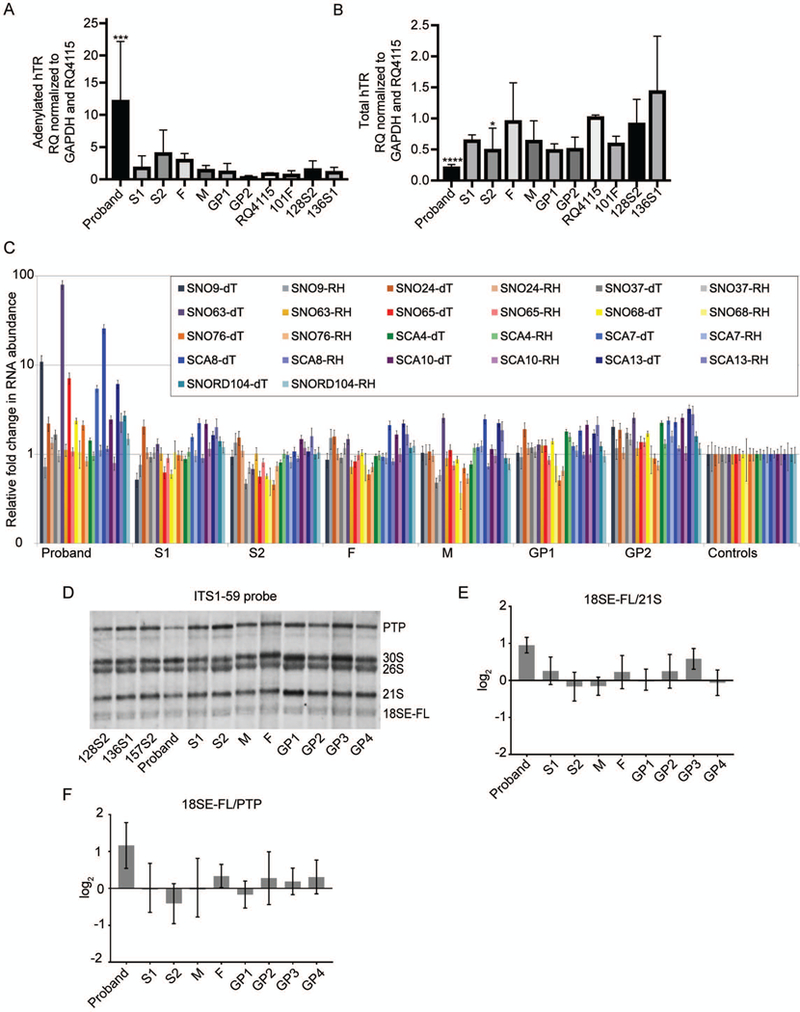
PARN RNA targets are impacted in the proband’s LCLs but not in the other BMF22 family member LCLs. (A) Analysis of adenylated hTR in BMF22 family and unrelated control (RQ4115, 101F, 128S2, and 136S1) LCLs by qRT-PCR. Complementary DNA was synthesized using oligo(dT) primer and qPCR was performed using primers to the target, hTR, or the control, GAPDH. Bars indicate mean +/− SD compared to RQ4115 with N=3–4. ***p=0.001. (B) Analysis of total hTR as in (A) except cDNA was synthesized using random hexamer primers. Bars indicate mean +/− SD compared to RQ4115 with N=3–4. *p<0.05, ****p=0.0001. (C) cDNA was synthesized from oligoadenylated RNA by using oligo(dT) primer or total RNA by using random hexamer primers. Bars indicate mean +/− SD compared to the average of the control individuals (RQ4115, 101F, and 128S2). N=3. (see text for significant p values). (D) Representative northern blot of ribosomal RNA precursor in LCLs using the ITS1–59 probe, which detects 18SE-FL, the untrimmed precursor of 18SE. (E) 18SE-FL/21S RAMP analysis of (D) and two additional biological replicates comparing BMF family member signals to the average signals of the controls 128S2, 136S1, and 157S2. (F) 18SE-FL/PTP RAMP analysis as in (E). In (B), (C), (D), p-values were calculated using one-way ANOVA of log-transformed data followed by Dunnett’s post-hoc test and adjustment of p-values for multiple comparisons. In (E) and (F) one-way ANOVA and Kruskal-Wallis test were used, respectively.
In addition to hTR, PARN deadenylates multiple snoRNAs and scaRNAs (Berndt et al., 2012) and a patient, reported by Dhanraj et al., with biallelic PARN variants and DC/HH was found to have an aberrant snoRNA expression profile (Dhanraj et al., 2015). To determine if this was also the case in LCLs derived from BMF22 family members, we estimated the abundance of the total and adenylated levels of H/ACA type and C/D box type snoRNAs and scaRNAs previously found to be impacted by PARN knockdown (Berndt et al., 2012) or in the patient with biallelic PARN variants reported by Dhanraj et al. (Dhanraj et al., 2015). When compared to an average of controls, the proband had multiple H/ACA snoRNAs demonstrating a marked enrichment of the adenylated form (up to approximately 90-fold) and a slight reduction of abundance (none with less than approximately 0.9-fold) (Figure 4C). In contrast, the snoRNA profile of other family members, including the biallelic carrier S2, was similar to that of controls. The overall snoRNA expression profile of the proband was consistent with the previously reported patient. Of the snoRNAs analyzed, the three with the greatest enrichment of adenylated species were the same three with the highest enrichment in the previously reported patient, and each was significantly increased compared to an average of controls: SNORA9, SNORA63, and SCARNA8 (p=0.003, 0.02093, and 0.0036, respectively).
The above patient studied by Dhanraj, et al., was also suspected to have a defect in ribosome biogenesis because H/ACA snoRNAs are involved in the chemical modification of pre-rRNA, and was found to have a reduction of the mature 80S ribosome (Dhanraj et al., 2015). To further explore the impact of PARN mutation on ribosome biogenesis, we specifically looked at the levels of pre-rRNAs in BMF22 family member LCLs. A single pre-RNA molecule undergoes multiple steps of cleavage, releasing precursors to ultimately generate the mature rRNAs, including 18S rRNA (reviewed in (Henras, Plisson-Chastang, O’Donohue, Chakraborty, & Gleizes, 2015)). These precursors undergo trimming during maturation. The pre-rRNA 18SE-FL is trimmed to 18SE and then to 18S. Analysis of siRNA PARN knockdown cells demonstrates that PARN trims 18SE-FL, facilitating 18S maturation (Ishikawa et al., 2017; Montellese et al., 2017; Tafforeau et al., 2013). Therefore, we performed northern blot analyses using the ITS1–59 probe and Ratio Analysis of Multiple Precursors (RAMP) (Wang et al., 2014) to assess rRNA precursors in LCLs derived from BMF22 family members as compared to control LCLs. The proband LCLs, but not other family member LCLs, had an increase of untrimmed 18SE-FL/PTP and 18SE-FL/21S ratios compared to an average of controls (Figures 4D–4F). These results support the finding that PARN trims 18SE-FL and could contribute to a ribosome defect in patients.
Editing of a single PARN allele is sufficient to shorten telomeres but not to reduce hTR
We were surprised that LCLs derived from monoallelic variant carriers, including M with reduced PARN protein level, did not have altered PARN function as measured by hTR and H/ACA snoRNA adenylation and rRNA maturation. Therefore, we used CRISPR-Cas9 to generate HCT116 cells with a loss-of-function (LOF) mutation in one allele of PARN to model monoallelic PARN variants in an isogenic setting. Initially, we planned to knock-in the insertion allele, and we, therefore, identified guides that efficiently targeted exon 12 of PARN (Supp. Figure S5). Ultimately, we isolated a clone harboring a single c.824_840delinsTTCC mutation following transfection with the synthetic guide RNA (sgRNA)-expressing plasmid Ap312 (see Materials and Methods). Sanger sequencing demonstrated that the other allele was WT at this locus. This heterozygous PARN LOF cell line had a reduction in PARN protein level as expected compared to untransfected cells and WT clones that underwent the same transfection but were unedited (Figures 5A and 5B). Consistent with haploinsufficiency, telomeres shortened in the heterozygous PARN LOF cell line as analyzed by telomere restriction fragment analysis and quantitative FISH (qFISH) (Figures 5C and 5D). Notably, level of adenylated hTR increased in the heterozygous PARN LOF cell line, however, the level of hTR was unchanged (Figures 5E and 5F). Thus, a single LOF mutation can result in altered hTR processing and impaired telomere length maintenance as observed in some, but not all PARN variant carriers. Notably, single LOF mutation did not impact the trimming of 18SE-FL as the untrimmed 18SE-FL/PTP and 18SE-FL/21S ratios were unchanged in the heterozygous PARN LOF cell lines compared to the controls. These results suggest differential effects on hTR vs ribosomes in patients with monoallelic PARN variants.
Figure 5.
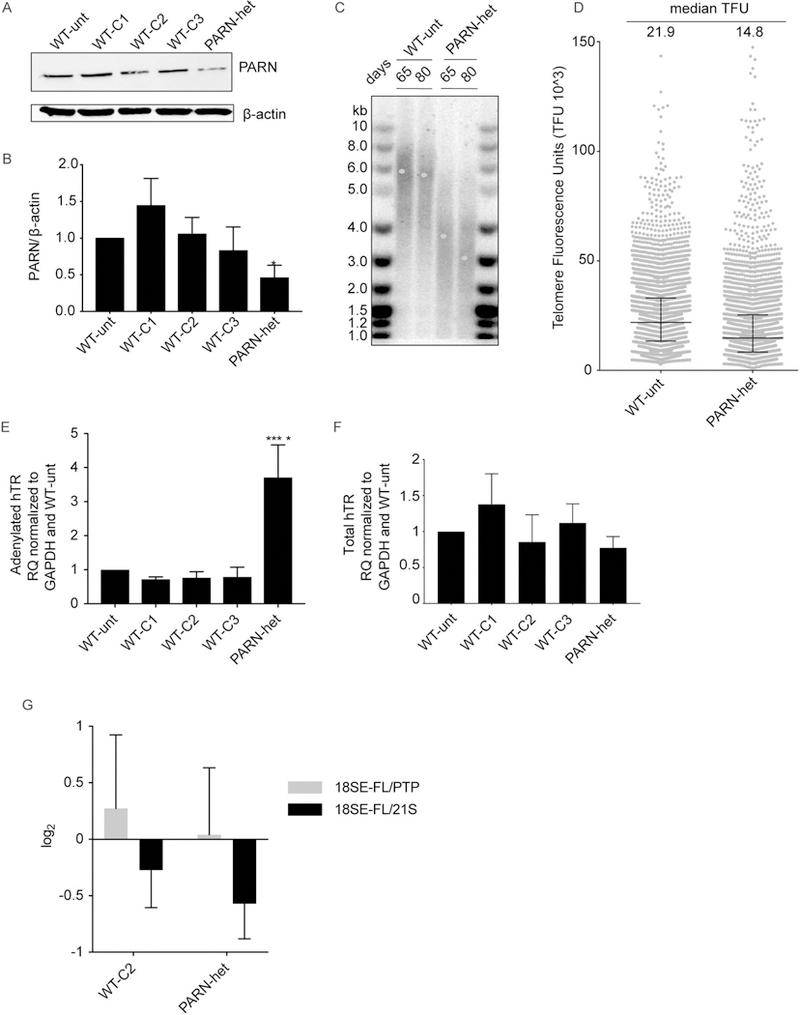
Heterozygous PARN LOF mutation leads to telomere shortening and increased polyadenylated hTR. (A) Western blot of whole cell lysates of clonal cell lines obtained following transfection of HCT116 cells with a construct expressing Cas9 and an sgRNA targeting PARN exon 12. WT-unt, untransfected HCT116 cell; WT-C1, -C2 and -C3, clonal cells lines derived post-transfection that were unedited; and PARN-het, heterozygous for PARN LOF mutation. β-actin served as a loading control. (B) Quantification of (A) and two additional biological replicates. Bars indicate mean PARN/β-actin ratio +/− SD compared to the WT-unt. (C) Telomere restriction fragment length assay of WT-unt and PARN-het cells 65 and 80 days post-isolation of clonal lines, with mean telomere length determined by TeloTool V1.3 and indicated by a blue dot. (D) q-FISH, telomere-length quantification of WT-unt and PARN cells 52 days post-isolation of clonal lines. Median telomere fluorescence units (TFUs) and interquartile ranges are indicated by black solid lines. (E) Analysis of adenylated hTR in WT-unt, untransfected HCT116 cell; WT-C1, -C2 and -C3, clonal cells lines by qRT-PCR as in Figure 4A. GAPDH served as a loading control. Bars indicate mean adenylated hTR/GAPDH ratio +/− SD compared to WT-unt with N=3. (F). Analysis of total hTR by qRT-PCR as in Figure 4B. Bars indicate mean total hTR/GAPDH ratio +/− SD compared to WT-unt with N=3. In (B), (E), and (F), *p <0.05 and ****p<0.0001 based on one-way ANOVA followed by Dunnett’s post-hoc test and adjustment of p-values for multiple comparisons. (G) 18SE-FL/21S and 18SE-FL/PTP RAMP analyses with comparison of WT-C2 and PARN-het to WT-unt. Bars indicate mean +/− SD with N=3.
mir-202–5p as a regulator of PARN
A possible etiology of the incomplete penetrance in PARN heterozygous variant carriers is a relatively common genetic modifier that influences PARN protein level. One such modifier could be a microRNA (miRNA). To investigate this possibility, we utilized TargetScan (http://www.targetscan.org/, release 7.2, default parameters and program options) (Agarwal, Bell, Nam, & Bartel, 2015) to identify miRNAs that might target PARN mRNA. This analysis yielded a single, conserved miRNA, miR-202–5p, with positions 2–8 of the miR-202–5p seed region complementary to nucleotides 1010–1016 of the PARN 3’ untranslated region (UTR) (Context++ score percentile 96) (Figure 6A). To determine if miR-202–5p could down-regulate PARN protein levels, we transfected HEK293T cells with a double-stranded RNA (Ambion Pre-miR ™ miRNA Precursor) designed and chemically modified such that the strand representing miR-202–5p is preferentially utilized in the RNA-induced silencing complex, a Pre-miR™ negative, random sequence control, or transfection reagent alone. In contrast to mock transfected and cells transfected with the negative control Pre- miR™, PARN protein level was significantly lower in cells transfected with mir-202–5p precursor when compared to the untransfected control (p=0.02) (Figures 6B and 6C), suggesting miR-202–5p could influence protein levels in humans.
Figure 6.
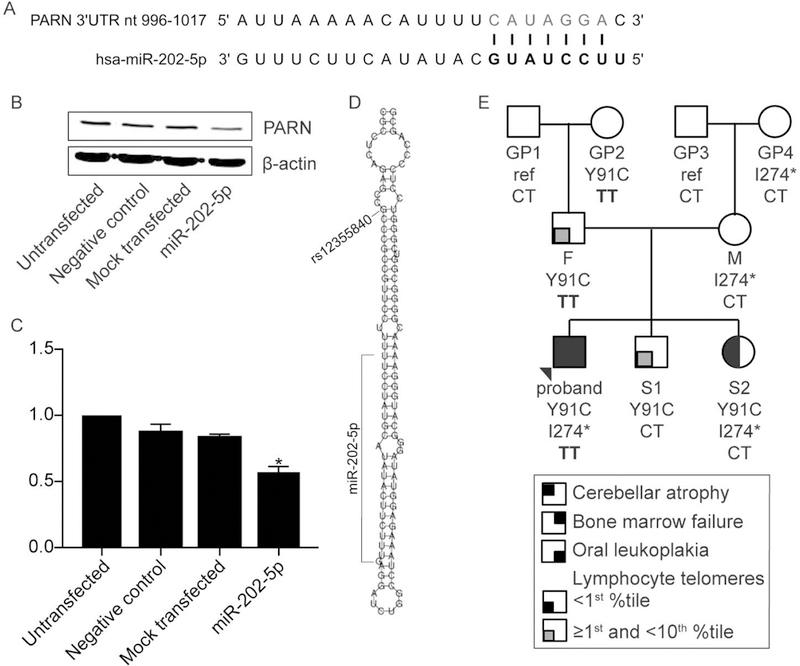
miR-202-5p reduces PARN level. (A) Depiction of the hsa-miR-202–5p binding site in the PARN 3’ UTR. (B) Western blot of whole cell lysates of HCT116 cells that were untransfected, transfected with a Pre-miR™ Precursor random sequence, negative control, mock transfected, or transfected with a Pre-miR™ miRNA Precursor designed to mimic endogenous mature hsa-miR-202–5p (miR-202–5p). β-actin is a loading control. (C) Quantification of the western blot in (A) and two biological replicates. Bars indicate mean +/− SD compared to untransfected cells. **p<0.01 based on Kruskal-Wallis test. (D) Schematic of the location of rs12355840 (NC_000010.11:g.133247608C>T) within hsa-pre-mir-202 and relative to hsa-miR-202–5p. The image was adapted from RNAfold server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). (E) The three-generation pedigree of family BMF22 as in Figure 1A, with MIR202 genotype CT or TT at NC_000010.11:g.133247608.
The single nucleotide polymorphism (SNP) rs12355840 (NC_000010.11:g.133247608C>T) maps to the stem-loop structure of the hsa-pre-mir-202 and is removed during maturation to the mature miR202–5p (Hoffman et al., 2013)(Figure 6D). Both the C (reference) and T (variant) alleles are commonly observed in the Genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org/) (Lek et al., 2016), with frequencies of 0.215 and 0.785, respectively. Interestingly, the presence of C negatively impacts miR-202–5p levels compared to T, likely by destabilizing the stem-loop structure and impairing maturation of the precursor mir to the mature miR-202–5p (Hoffman et al., 2013). Therefore, we considered the possibility that the MIR202 genotype at this position might be a genetic modifier, with the C allele lessening the impact of a given PARN variant on telomere length by lowering miR-202–5p levels and, secondarily, increasing PARN protein levels relative to the T allele. However, because miR-202–5p appears to be predominantly expressed in the male reproductive system (Landgraf et al., 2007), a given MIR202 genotype may primarily impact the telomere length in sperm or during very early development and this may be reflected solely in a father’s offspring. Because the proband and S2 shared the same father, it is unlikely that mir202–5p contributed to the variation in their telomere lengths. Similarly, as GP1 (F’s father) and GP3 (M’s father) both carried the CT alleles, it is unlikely that miR202–5p contributed to the shorter telomeres observed in F than M, even though M had an NMD-inducing mutation.
DISCUSSION
This study represents the first report of a three-generation family, which includes PARN genotyping, telomere length analysis by flow FISH, and biological studies across each generation. The findings highlight several important and challenging aspects of interpreting PARN-related TBDs. First, they reinforce the principle that normal pan-lymphocyte telomere length, including at the mean for a normal population, does not preclude the presence of a pathogenic PARN variant. This was first suggested by the finding that some monoallelic carriers who are parents of biallelic carriers with HH had normal telomere lengths measured by flow-FISH (Burris et al., 2016; Dhanraj et al., 2015; Moon et al., 2015; Tummala et al., 2015). We further demonstrated that M and GP4’s pan-lymphocyte telomere lengths were of normal length (Figure 1B), despite carrying an insertion allele encoding a transcript subject to NMD and having a significant reduction in PARN protein as measured in LCLs. Moreover, we found M’s normal telomere length was not the result of somatic TERT promoter mutations (Supp. Figure S3B). Thus, a single LOF mutation in PARN may not result in haploinsufficiency with respect to telomere length maintenance.
In contrast, F and S1, who carried the p.Y91C allele, had pan-lymphocyte telomere lengths just at or above the 1st percentile upon initial testing and up to the 10th percentile several years later (Figure 1B). One potential explanation for the differential impact of the p.(I274*) and p.Y91C variants is that a reduction in PARN protein level is not as detrimental as the presence of PARN p.Y91C mutant protein, which perhaps elicits a dominant negative effect when dimerized with WT PARN. Arguing against this, however, is the observation that GP2, who also carried the p.Y91C allele, had normal telomere length. In addition, F’s telomere lengths increased in several subsets, most dramatically in NK cells, over time (Supp. Figure S3A). Still, we cannot exclude the possibility that GP2 bore a de novo mutation and there were insufficient generations for telomeres to reach abnormally short lengths, i.e., that there was not yet anticipation. In addition, the presence of multiple splice variants, and insertions and deletions that are predicted to undergo NMD and with evidence of reduced transcript or protein level, coupled with short telomeres in an IPF cohort suggests haploinsufficiency may, indeed, manifest in at least a subset of patients (Stuart et al., 2015).
Since short telomeres are the most common root of the TBDs (Savage, 2018), we propose that the variable impact of a given PARN variant on telomere length in individuals is the basis for incomplete penetrance of IPF and other unclassified interstitial lung diseases observed with PARN families. Thus, it may be useful to use telomere length as the indicator of increased disease risk rather than simply mutation status. Given that the average age of diagnosis of IPF in PARN variant carriers is 64 +/− 8 years (Newton et al., 2016), a rigorous test of this proposal will require long term follow up of PARN families. For example, while neither GP2 (presently age 63 years) nor GP4 (presently age 68 years) have developed symptoms of IPF, consistent with their normal telomere lengths, they may have many additional at-risk years.
Every patient reported to date with PARN biallelic variants and a neurodevelopmental phenotype also had evidence of bone marrow failure (Burris et al., 2016; Dhanraj et al., 2015; Tummala et al., 2015). The youngest of these patients was 3-years-old (Tummala et al., 2015), and the only patient whose follow up over time was reported had the onset of a hematologic phenotype at five-years-old (Burris et al., 2016). Given these reports, age may be a factor contributing to the difference in clinical phenotype between the proband and S2, and this may be reflected in the LCL, with an age-related factor accounting for the reduced PARN level in the LCLs of the proband compared to S2 and, in turn, altering RNAs targeted by PARN only in the proband. The accelerated rate of telomere shortening over the first three years of life also suggests the importance of PARN in telomere length maintenance particularly in early childhood.
Normal hTR levels in LCLs derived from monoallelic PARN variant carriers and in a heterozygous PARN LOF cancer cell line model
Total hTR levels remained intact in LCLs derived from the monoallelic PARN variant carriers with, not only normal, but also short telomere length, which was surprising. It is possible that hTR could be reduced in hematopoietic stem cells, lymphoid progenitors, or peripheral blood lymphocytes and LCLs may not be representative of this or that a biologically relevant but small difference in RNA level is not detectable. However, it is also notable that, in the cancer cell line with a single PARN allele mutated, HCT116 PARN LOF, hTR adenylation was increased but hTR total level was normal. Nonetheless, telomeres shortened. It is possible that monoallelic loss of PARN causes telomere shortening through an RNA other than hTR in HCT116 cells, such as TPP1, as observed in PARN-deficient HT1080 cells (Benyelles et al., 2019), or that increased hTR adenylation is sufficient to shorten telomeres.
PARN deficiency impacts ribosomal rRNA maturation
PARN-deficiency was initially proposed to alter ribosome maturation in patients through its impact on snoRNAs (Dhanraj et al., 2015), which are involved in the chemical modification of pre-rRNA. More recently, it was found that PARN directly trims rRNA precursors and that RNAi depletion causes their accumulation (Montellese et al., 2017). In addition, while this manuscript was in revision, Benyelles et al. (Benyelles et al., 2019) reported accumulation of 18S pre-rRNAs in samples derived from HH individuals, as we do here in our proband (Figures 4D-F), and in a cell line with biallelic PARN knockout. Interestingly, we observed that neither cells with knockout of a single allele (Figure 5G) nor individuals heterozygous for pathogenic PARN variants (Figures 4D-F) have accumulation of 18S pre-rRNA, suggesting a dose-dependent effect. Short telomeres alone appear to be sufficient to lead to an HH phenotype given that, among the genes that are associated with HH, are multiple involved in telomere maintenance but not firmly implicated in ribosome biogenesis or function (e.g., TERT, TINF2, and RTEL1). Nonetheless, these data contribute to the larger question in the field of inherited bone marrow failure syndromes of whether altered ribosomes contribute to DC and HH (Nakhoul et al., 2014).
Regulation of PARN levels
PARN protein was higher in S2 LCL’s than in those of the proband, M, or GP4, all of whom also carried the p.(I274*) variant (Figures 3A-B) and this could not be attributed to elevated PARN mRNA specifically in S2. Indeed, PARN mRNA levels were comparable in S2 and the proband, but reduced in M and GP4 (Figure 3C). The mechanisms underlying these differences remain unclear. Nonetheless, these results underscore the complexity of PARN regulation and the potential impact of PARN variants on telomere length.
We found that exogenous miR-202–5p resulted in a decrease in PARN protein, consistent with miR-202–5p target sequence in the PARN mRNA 3’ UTR (Figures 6A–6C) and suggesting this miRNA might contribute to the regulation of PARN levels in vivo. Because hsa-mir-202 is predominantly expressed in the male reproductive system (Landgraf et al., 2007), it may have its greatest impact on PARN and telomere lengths in male gametes and during the earliest stages of development. While the limited analysis afforded by the small BMF22 family suggests rs12355840 is not a genetic modifier of telomere lengths in the context of PARN mutations, further analysis of larger, multi-generation families may be needed to draw stronger conclusions. Incorporation of paternal age at conception, which has been shown to impact telomere length in offspring (Eisenberg & Kuzawa, 2018), is another variable worth consideration in such larger studies.
A final cautionary note
The PARN insertion encoding p.(I274*) was initially missed in clinical WES analyses of the proband and S2, but was ultimately uncovered due to a high degree of suspicion that biallelic PARN variants were culprit. Clinicians and investigators with unsolved cases of patients for which disease genes are known remain cognizant of the limitations of WES, including the challenge of calling in/dels. In addition, the allele frequency for the PARN c.272A>G; p.Y91C variant in gnomAD is as high as 0.0002115 in Africans and 0.00007992 for the entire cohort. While still very low, the number of individuals with this variant in gnomAD, 22, was notably higher than that for any of the 64 unique variants reported in a 115 member IPF cohort, which included 43 TERT, 6 TERC, 7 RTEL1, and 8 PARN variants (Newton et al., 2016). In that cohort, the number of individuals in gnomAD carrying a given variant ranged from 0 to 7 (median 0), excluding the TERT p.K1050N variant (NM_198253.2:c.3150G>C), which was present in 23 individuals, 20 of which were of Ashkenazi Jewish ancestry. Therefore, functional analyses should be considered for PARN variants of uncertain clinical significance even if present at higher than expected allele frequencies.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the BMF22 family for their participation in this study as well as the healthy individuals who served as controls.
Funding information:
• National Institutes of Health R01HL131744 to AAB; R01NS08372 to PEB; and T32DK060445 and T32GM007330, which supported LMD
• American Society of Hematology Bridge Award to AAB
• Cullen Foundation, which supported LMD
• Linneus Support from the Swedish Research Council to the Uppsala RNA Research Centre to AV
Footnotes
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request. NM_002582.3: c.819_820insTAGAAATCATTTCTAGAGTC; p.(I274*) PARN variant has been submitted to ClinVar (SCV000902457). NM_002582.3: c.272A>G; p.Y91C was previously submitted to ClinVar (VCV000542669) by Invitae Corp.
CONFLICTS OF INTEREST STATEMENT
GA is a part time employee of Repeat Diagnostics, a company providing clinical telomere length measurement services; she also holds a position at the academic institution Terry Fox Laboratory, BC Cancer, Vancouver BC Canada.
REFERENCES
- Agarwal V, Bell GW, Nam JW, & Bartel DP (2015). Predicting effective microRNA target sites in mammalian mRNAs. eLife, 4. doi: 10.7554/eLife.05005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alder JK, Hanumanthu VS, Strong MA, DeZern AE, Stanley SE, Takemoto CM, … Armanios M. (2018). Diagnostic utility of telomere length testing in a hospital-based setting. Proceedings of the National Academy of Sciences of the United States of America, 115(10), E2358–E2365. doi: 10.1073/pnas.1720427115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allsopp RC, & Harley CB (1995). Evidence for a critical telomere length in senescent human fibroblasts. Experimental Cell Research, 219(1), 130–136. doi: 10.1006/excr.1995.1213 [DOI] [PubMed] [Google Scholar]
- Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, … Loyd JE. (2007). Telomerase mutations in families with idiopathic pulmonary fibrosis. New England Journal of Medicine, 356(13), 1317–1326. doi: 10.1056/NEJMoa066157 [DOI] [PubMed] [Google Scholar]
- Ballew BJ, Joseph V, De S, Sarek G, Vannier JB, Stracker T, … Petrini JH. (2013). A recessive founder mutation in regulator of telomere elongation helicase 1, RTEL1, underlies severe immunodeficiency and features of Hoyeraal Hreidarsson syndrome. PLoS Genetics, 9(8), e1003695. doi: 10.1371/journal.pgen.1003695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballew BJ, Yeager M, Jacobs K, Giri N, Boland J, Burdett L, … Savage SA (2013). Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Human Genetics, 132(4), 473–480. doi: 10.1007/s00439-013-1265-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaro PM, Ziegler DS, & Reddel RR (2016). The wide-ranging clinical implications of the short telomere syndromes. Internal Medicine Journal, 46(4), 393–403. doi: 10.1111/imj.12868 [DOI] [PubMed] [Google Scholar]
- Behrens YL, Thomay K, Hagedorn M, Ebersold J, Henrich L, Nustede R, … Gohring G. (2017). Comparison of different methods for telomere length measurement in whole blood and blood cell subsets: Recommendations for telomere length measurement in hematological diseases. Genes, Chromosomes and Cancer, 56(9), 700–708. doi: 10.1002/gcc.22475 [DOI] [PubMed] [Google Scholar]
- Benyelles M, Episkopou H, O’Donohue MF, Kermasson L, Frange P, Poulain F, … Revy P. (2019). Impaired telomere integrity and rRNA biogenesis in PARN-deficient patients and knock-out models. EMBO Molecular Medicine, 11(7), e10201. doi: 10.15252/emmm.201810201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt H, Harnisch C, Rammelt C, Stohr N, Zirkel A, Dohm JC, … Wahle E. (2012). Maturation of mammalian H/ACA box snoRNAs: PAPD5-dependent adenylation and PARN-dependent trimming. RNA, 18(5), 958–972. doi: 10.1261/rna.032292.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertuch AA (2016). The molecular genetics of the telomere biology disorders. RNA Biology, 13(8), 696–706. doi: 10.1080/15476286.2015.1094596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besse A, Wu P, Bruni F, Donti T, Graham BH, Craigen WJ, … Bonnen PE. (2015). The GABA transaminase, ABAT, is essential for mitochondrial nucleoside metabolism. Cell Metabolism, 21(3), 417–427. doi: 10.1016/j.cmet.2015.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borah S, Xi L, Zaug AJ, Powell NM, Dancik GM, Cohen SB, … Cech TR. (2015). Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science, 347(6225), 1006–1010. doi: 10.1126/science.1260200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris AM, Ballew BJ, Kentosh JB, Turner CE, Norton SA, Laboratory NDCGR, … Savage SA. (2016). Hoyeraal-Hreidarsson Syndrome due to PARN Mutations: Fourteen Years of Follow-Up. Pediatric Neurology, 56, 62–68 e61. doi: 10.1016/j.pediatrneurol.2015.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM, & Hockemeyer D (2015). Cancer-associated TERT promoter mutations abrogate telomerase silencing. eLife, 4. doi: 10.7554/eLife.07918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, … Jackson SP. (2003). A DNA damage checkpoint response in telomere-initiated senescence. Nature, 426(6963), 194–198. doi: 10.1038/nature02118 [DOI] [PubMed] [Google Scholar]
- Deng Z, Glousker G, Molczan A, Fox AJ, Lamm N, Dheekollu J, … Tzfati Y. (2013). Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal-Hreidarsson syndrome. Proceedings of the National Academy of Sciences of the United States of America, 110(36), E3408–3416. doi: 10.1073/pnas.1300600110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanraj S, Gunja SM, Deveau AP, Nissbeck M, Boonyawat B, Coombs AJ, … Dror Y. (2015). Bone marrow failure and developmental delay caused by mutations in poly(A)-specific ribonuclease (PARN). Journal of Medical Genetics, 52(11), 738–748. doi: 10.1136/jmedgenet-2015-103292 [DOI] [PubMed] [Google Scholar]
- Dodson L, & Bertuch AA (2018). Dyskeratosis Congenita and the Telomere Biology Disorders In Kupfer GM, Reaman GH, & Smith FO (Eds.), Bone Marrow Failure (pp. 111–136). Cham, Switzerland: Springer International Publishing AG, part of Springer Nature. [Google Scholar]
- Eisenberg DTA, & Kuzawa CW (2018). The paternal age at conception effect on offspring telomere length: mechanistic, comparative and adaptive perspectives. Philosophical Transactions of the Royal Society B, 373(1741). doi: 10.1098/rstb.2016.0442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadalla SM, Khincha PP, Katki HA, Giri N, Wong JY, Spellman S, … Savage SA. (2016). The limitations of qPCR telomere length measurement in diagnosing dyskeratosis congenita. Molecular Genetics & Genomic Medicine, 4(4), 475–479. doi: 10.1002/mgg3.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohring J, Fulcher N, Jacak J, & Riha K (2014). TeloTool: a new tool for telomere length measurement from terminal restriction fragment analysis with improved probe intensity correction. Nucleic Acids Research, 42(3), e21. doi: 10.1093/nar/gkt1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramatges MM, Qi X, Sasa GS, Chen JJ, & Bertuch AA (2013). A homozygous telomerase T-motif variant resulting in markedly reduced repeat addition processivity in siblings with Hoyeraal Hreidarsson syndrome. Blood, 121(18), 3586–3593. doi: 10.1182/blood-2012-08-447755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Rodrigues F, Santana-Lemos BA, Scheucher PS, Alves-Paiva RM, & Calado RT (2014). Direct comparison of flow-FISH and qPCR as diagnostic tests for telomere length measurement in humans. PLoS One, 9(11), e113747. doi: 10.1371/journal.pone.0113747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley CB, Futcher AB, & Greider CW (1990). Telomeres shorten during ageing of human fibroblasts. Nature, 345(6274), 458–460. doi: 10.1038/345458a0 [DOI] [PubMed] [Google Scholar]
- Henras AK, Plisson-Chastang C, O’Donohue MF, Chakraborty A, & Gleizes PE (2015). An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdisciplinary Reviews: RNA, 6(2), 225–242. doi: 10.1002/wrna.1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksson N, Nilsson P, Wu M, Song H, & Virtanen A (2010). Recognition of adenosine residues by the active site of poly(A)-specific ribonuclease. Journal of Biological Chemistry, 285(1), 163–170. doi: 10.1074/jbc.M109.043893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbig U, Jobling WA, Chen BP, Chen DJ, & Sedivy JM (2004). Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Molecular Cell, 14(4), 501–513. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/15149599 [DOI] [PubMed] [Google Scholar]
- Hoffman AE, Liu R, Fu A, Zheng T, Slack F, & Zhu Y (2013). Targetome profiling, pathway analysis and genetic association study implicate miR-202 in lymphomagenesis. Cancer Epidemiology, Biomarkers & Prevention, 22(3), 327–336. doi: 10.1158/1055-9965.EPI-12-1131-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Yoshikawa H, Izumikawa K, Miura Y, Taoka M, Nobe Y, … Takahashi N. (2017). Poly(A)-specific ribonuclease regulates the processing of small-subunit rRNAs in human cells. Nucleic Acids Research, 45(6), 3437–3447. doi: 10.1093/nar/gkw1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannengiesser C, Borie R, Menard C, Reocreux M, Nitschke P, Gazal S, … Crestani B. (2015). Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. European Respiratory Journal. doi: 10.1183/09031936.00040115 [DOI] [PubMed] [Google Scholar]
- Khincha PP, Dagnall CL, Hicks B, Jones K, Aviv A, Kimura M, … Gadalla SM. (2017). Correlation of Leukocyte Telomere Length Measurement Methods in Patients with Dyskeratosis Congenita and in Their Unaffected Relatives. International Journal of Molecular Sciences, 18(8), E1765. doi: 10.3390/ijms18081765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, … Shay JW. (1994). Specific association of human telomerase activity with immortal cells and cancer. Science, 266(5193), 2011–2015. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7605428 [DOI] [PubMed] [Google Scholar]
- Kropski JA, Reiss S, Markin C, Brown KK, Schwartz DA, Schwarz MI, … Cogan JD. (2017). Rare Genetic Variants in PARN Are Associated with Pulmonary Fibrosis in Families. American Journal of Respiratory and Critical Care Medicine, 196(11), 1481–1484. doi: 10.1164/rccm.201703-0635LE [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyan J, Konforti B, & Wemmer D (2013). The Molecules of Life: Physical and Chemical Principles (1 ed): Garland Science, Taylor & Francis Group, LLC [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, … Tuschl T. (2007). A mammalian microRNA expression atlas based on small RNA library sequencing. Cell, 129(7), 1401–1414. doi: 10.1016/j.cell.2007.04.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guen T, Jullien L, Touzot F, Schertzer M, Gaillard L, Perderiset M, … Revy P. (2013). Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Human Molecular Genetics, 22(16), 3239–3249. doi: 10.1093/hmg/ddt178 [DOI] [PubMed] [Google Scholar]
- Lee D, Park D, Park JH, & Shin C (2018). Poly(A)-specific ribonuclease sculpts the 3’ ends of microRNAs. 25(3), 388–405. doi: 10.1261/rna.069633.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, … Exome Aggregation, C. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. doi: 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrone A, Walne A, Tamary H, Masunari Y, Kirwan M, Beswick R, … Dokal I (2007). Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood, 110(13), 4198–4205. doi: 10.1182/blood-2006-12-062851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryoung L, Yue Y, Young A, Newton CA, Barba C, van Oers NS, … Garcia CK. (2017). Somatic mutations in telomerase promoter counterbalance germline loss-of-function mutations. Journal of Clinical Investigation, 127(3), 982–986. doi: 10.1172/JCI91161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montellese C, Montel-Lehry N, Henras AK, Kutay U, Gleizes PE, & O’Donohue MF (2017). Poly(A)-specific ribonuclease is a nuclear ribosome biogenesis factor involved in human 18S rRNA maturation. Nucleic Acids Research, 45(11), 6822–6836. doi: 10.1093/nar/gkx253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon DH, Segal M, Boyraz B, Guinan E, Hofmann I, Cahan P, … Agarwal S. (2015). Poly(A)-specific ribonuclease (PARN) mediates 3’-end maturation of the telomerase RNA component. Nature Genetics, 47(12), 1482–1488. doi: 10.1038/ng.3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD, … Wu JR. (1988). A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proceedings of the National Academy of Sciences of the United States of America, 85(18), 6622–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakhoul H, Ke J, Zhou X, Liao W, Zeng SX, & Lu H (2014). Ribosomopathies: mechanisms of disease. Clinical Medicine Insights: Blood Disorders, 7, 7–16. doi: 10.4137/CMBD.S16952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ND, Dodson LM, Escudero L, Sukumar AT, Williams CL, Mihalek I, … Bertuch AA. (2018). The C-Terminal Extension Unique to the Long Isoform of the Shelterin Component TIN2 Enhances Its Interaction with TRF2 in a Phosphorylation- and Dyskeratosis Congenita Cluster-Dependent Fashion. Molecular and Cellular Biology, 38(12). doi: 10.1128/MCB.00025-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton CA, Batra K, Torrealba J, Kozlitina J, Glazer CS, Aravena C, … Garcia CK. (2016). Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. European Respiratory Journal, 48(6), 1710–1720. doi: 10.1183/13993003.00308-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D, Grenier St-Sauveur V, Bergeron D, Dupuis-Sandoval F, Scott MS, & Bachand F (2015). A Polyadenylation-Dependent 3’ End Maturation Pathway Is Required for the Synthesis of the Human Telomerase RNA. Cell Reports, 13(10), 2244–2257. doi: 10.1016/j.celrep.2015.11.003 [DOI] [PubMed] [Google Scholar]
- Ourliac-Garnier I, & Londono-Vallejo A (2017). Telomere Length Analysis by Quantitative Fluorescent in Situ Hybridization (Q-FISH). Methods in Molecular Biology, 1587, 29–39. doi: 10.1007/978-1-4939-6892-3_3 [DOI] [PubMed] [Google Scholar]
- Palm W, & de Lange T (2008). How shelterin protects mammalian telomeres. Annu. Rev. Genet, 42, 301–334. doi: 10.1146/annurev.genet.41.110306.130350 [DOI] [PubMed] [Google Scholar]
- Petrovski S, Todd JL, Durheim MT, Wang Q, Chien JW, Kelly FL, … Goldstein DB. (2017). An Exome Sequencing Study to Assess the Role of Rare Genetic Variation in Pulmonary Fibrosis. American Journal of Respiratory and Critical Care Medicine, 196(1), 82–93. doi: 10.1164/rccm.201610-2088OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage SA (2018). Beginning at the ends: telomeres and human disease. F1000Research, 7. doi: 10.12688/f1000research.14068.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, & Parker R (2017). PARN Modulates Y RNA Stability and Its 3’-End Formation. Molecular and Cell Biology 37(20), e00264–00217. doi: 10.1128/MCB.00264-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles AR, Ferdinandusse S, Besse A, Appadurai V, Leydiker KB, Cambray-Forker EJ, … Abdenur JE. (2015). Successful diagnosis of HIBCH deficiency from exome sequencing and positive retrospective analysis of newborn screening cards in two siblings presenting with Leigh’s disease. Molecular Genetics and Metabolism, 115(4), 161–167. doi: 10.1016/j.ymgme.2015.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R, … Garcia CK. (2015). Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nature Genetics, 47(5), 512–517. doi: 10.1038/ng.3278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafforeau L, Zorbas C, Langhendries JL, Mullineux ST, Stamatopoulou V, Mullier R, … Lafontaine DL. (2013). The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of Pre-rRNA processing factors. Molecular Cell, 51(4), 539–551. doi: 10.1016/j.molcel.2013.08.011 [DOI] [PubMed] [Google Scholar]
- Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, … Garcia CK. (2007). Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proceedings of the National Academy of Sciences of the United States of America, 104(18), 7552–7557. doi: 10.1073/pnas.0701009104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng CK, Wang HF, Burns AM, Schroeder MR, Gaspari M, & Baumann P (2015). Human Telomerase RNA Processing and Quality Control. Cell Reports, 13(10), 2232–2243. doi: 10.1016/j.celrep.2015.10.075 [DOI] [PubMed] [Google Scholar]
- Tummala H, Walne A, Collopy L, Cardoso S, de la Fuente J, Lawson S, … Dokal I. (2015). Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. Journal of Clinical Investigation, 125(5), 2151–2160. doi: 10.1172/JCI78963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaziri H, Dragowska W, Allsopp RC, Thomas TE, Harley CB, & Lansdorp PM (1994). Evidence for a mitotic clock in human hematopoietic stem cells: loss of telomeric DNA with age. Proceedings of the National Academy of Sciences of the United States of America, 91(21), 9857–9860. doi: 10.1073/pnas.91.21.9857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen A, Henriksson N, Nilsson P, & Nissbeck M (2013). Poly(A)-specific ribonuclease (PARN): an allosterically regulated, processive and mRNA cap-interacting deadenylase. Critical Reviews in Biochemistry and Molecular Biology, 48(2), 192–209. doi: 10.3109/10409238.2013.771132 [DOI] [PubMed] [Google Scholar]
- Walne AJ, Vulliamy T, Kirwan M, Plagnol V, & Dokal I (2013). Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. American Journal of Human Genetics, 92(3), 448–453. doi: 10.1016/j.ajhg.2013.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Anikin L, & Pestov DG (2014). Two orthogonal cleavages separate subunit RNAs in mouse ribosome biogenesis. Nucleic Acids Research, 42(17), 11180–11191. doi: 10.1093/nar/gku787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright WE, Piatyszek MA, Rainey WE, Byrd W, & Shay JW (1996). Telomerase activity in human germline and embryonic tissues and cells. Developmental Genetics, 18(2), 173–179. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=8934879 [DOI] [PubMed] [Google Scholar]
- Wu M, Nilsson P, Henriksson N, Niedzwiecka A, Lim MK, Cheng Z, … Song H (2009). Structural basis of m(7)GpppG binding to poly(A)-specific ribonuclease. Structure, 17(2), 276–286. doi: 10.1016/j.str.2008.11.012 [DOI] [PubMed] [Google Scholar]
- Wu M, Reuter M, Lilie H, Liu Y, Wahle E, & Song H (2005). Structural insight into poly(A) binding and catalytic mechanism of human PARN. EMBO Journal, 24(23), 4082–4093. doi: 10.1038/sj.emboj.7600869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoda M, Cifuentes D, Izumi N, Sakaguchi Y, Suzuki T, Giraldez AJ, & Tomari Y (2013). Poly(A)-specific ribonuclease mediates 3’-end trimming of Argonaute2-cleaved precursor microRNAs. Cell Reports, 5(3), 715–726. doi: 10.1016/j.celrep.2013.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.