

Abstract
A fundamental challenge in chemical biology and medicine is to understand and expand the fraction of the human proteome that can be targeted by small molecules. We recently described a strategy that integrates fragment-based ligand discovery with chemical proteomics to furnish global portraits of reversible small molecule-protein interactions in human cells. Excavating clear structure-activity relationships from these “ligandability” maps, however, was confounded by the distinct physicochemical properties and corresponding overall protein-binding potential of individual fragments. Here, we describe a compelling solution to this problem by introducing a next-generation set of fully functionalized fragments (FFFs) differing only in absolute stereochemistry. Using these enantiomeric probe pairs, or “enantioprobes”, we identify numerous stereoselective protein-fragment interactions in cells and show that these interactions occur at functional sites on proteins from diverse classes. Our findings thus indicate that incorporating chirality into FFF libraries provides a robust and streamlined method to discover ligandable proteins in cells.
Graphical Abstract
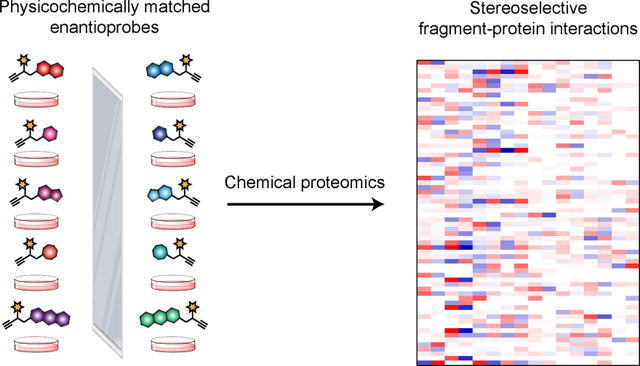
Chemical probes are versatile tools to interrogate the functions of proteins in biological systems and complement genetic approaches1 by producing reversible and graded gains or losses of protein activity, as well as, in certain instances, neo-functional outcomes2–5. Small molecules also represent a principal category of clinically approved drugs, and quality chemical probes are needed to pharmacologically validate novel targets on the path to developing therapeutic agents.
Despite their basic and translational value, chemical probes are lacking for the vast majority of human proteins6. Methods for the discovery of new chemical probes often rely on high-throughput screening (HTS) of large libraries (~106) of relatively high molecular weight and structurally diverse compounds against individual proteins (target-based) or cellular systems (phenotype-based)7,8. Hits from such libraries can often be challenging to optimize due to their structural complexity and suboptimal ligand efficiencies9. Further, many proteins are problematic to express, purify, and format for in vitro HTS, especially if they are parts of large complexes and/or remain poorly characterized in terms of biochemical function. These challenges underscore the need for new methods that can more broadly assess the ‘ligandability’ (i.e., ability to bind small molecules) of the human proteome in native biological systems.
Fragment-based ligand discovery (FBLD) has emerged as a versatile approach for the discovery of atom-efficient, small-molecule binders for a wide range of proteins10–12. However, due to the generally low affinity of fragment hits and the biophysical methods typically used for their discovery (e.g., NMR, surface plasmon resonance, isothermal calorimetry), FBLD has been mostly limited to the study of purified proteins in vitro11. We recently introduced a strategy that integrates FBLD with chemical proteomics to globally assess small molecule-protein interactions in human cells13. Using a specialized library of fully functionalized fragment (FFF) probes, which possess variable fragment binding elements coupled to photoreactive and bioorthogonal reporter groups, we mapped >2000 reversible fragment-protein interactions in human cells and showed that these interactions can be advanced into more potent and selective compounds capable of modulating the activity of proteins in cells13.
The fragment binding elements in our initial studies were selected based on their representation in drug-like molecules14 and were accordingly diverse in structure and physicochemical properties. As a consequence, we found that individual FFF probes showed substantial differences in their overall proteomic interaction profiles, which made for complicated structure-activity relationships (SARs) requiring careful manual review to identify fragment-protein interactions that reflected authentic recognition events (versus simply correlating with the overall proteomic interaction profiles of the FFF probes). We describe herein a general strategy to address this confounding bottleneck in the form of a next-generation set of FFF probes consisting of physicochemically matched fragment pairs differing only in absolute stereochemistry. The stereoselective engagement of protein targets is a feature of numerous chemical probes and drugs15–19 and we reasoned that this outcome, measured on a proteome-wide scale, would provide instant evidence of specific interactions between small-molecule fragments and proteins in cells. Using a set of eight pairs of enantiomeric FFF probes – or “enantioprobes” – we expeditiously identify >170 stereochemistry-dependent small molecule-protein interactions in human cells. The enantioprobe targets span diverse structural and functional classes and include proteins that lack chemical probes. We validate enantioprobe interactions for several recombinantly expressed proteins and show that the interactions occur at functionally relevant sites on these proteins. Finally, we describe a quantitative, multiplexed workflow capable of performing up to five enantioprobe pair comparisons in a single experiment, thereby greatly increasing the throughput and dimensionality of fragment-based ligand discovery in cells.
Results
Design and initial proteomic profiling of enantioprobes
Our original set of FFF probes were designed to contain: 1) a “variable” recognition element consisting of structurally diverse small-molecule fragments intended to promote interactions with distinct proteins in human cells; and 2) a structurally minimized “constant” region bearing a photoactivatable diazirine group and alkyne handle, which together enabled UV light-induced covalent modification and detection, enrichment, and identification of fragment-interacting protein targets (Fig. 1a)13. Here, we reasoned that the introduction of stereochemistry into FFF probe design could furnish pairs of compounds that display equivalent physicochemical properties and gross overall protein binding in cells, but differ in their stereoselective interactions with authentic small molecule-binding pockets in proteins. The preferential enrichment of proteins by one member of an enantioprobe pair would then constitute instant evidence of ligandability for these proteins.
Figure 1. Enantioprobes for mapping stereoselective protein-small molecule fragment interactions in human cells.

a, Structures of enantioprobes, which consist of a “variable” element of stereopure fragment pairs (enclosed box) and a “constant” region containing a diazirine photoreactive group and a clickable alkyne handle. b, Gel-based profiling of enantioprobe-protein interactions in human cells. HEK293T cells were treated with enantioprobes (20 μM) for 30 min, photocrosslinked, lysed, and proteomes conjugated to an azide-rhodamine tag using CuAAC chemistry and analyzed by SDS-PAGE and in-gel fluorescent scanning. Red asterisks mark representative stereoselective enantioprobe-protein interactions. Gel image reflects representative results from two independently performed experiments.
We synthesized a library of eight enantioprobe pairs, where members of each pair differ only in absolute stereochemistry of the fragment recognition element (Fig. 1a). We then qualitatively assessed the overall proteomic interaction profiles for enantioprobes using established SDS-PAGE methods13. In brief, we treated HEK293T cells with each enantioprobe (20 μM, 30 min) followed by exposure to UV light (365 nm, 10 min), harvesting, lysis, coupling of probe-modified proteins to an azide-rhodamine reporter tag using copper-catalyzed azide-alkyne cycloaddition chemistry (CuAAC)20, and visualization of these proteins by SDS-PAGE and in-gel fluorescence scanning. As expected, we observed substantial differences in protein interactions across the enantioprobe pairs, with one probe pair ((R) and (S)-6) exhibiting much greater overall protein labeling compared to others (Fig. 1b). Encouragingly, however, the (R) and (S) members within each enantioprobe pair showed similar overall proteomic labeling with the exception of select proteins that exhibited stereochemistry-dependent (“stereoselective”) interactions (Fig. 1b and Supplementary Fig. 1, red asterisks). The enantioprobe pairs also showed clear increases in protein labeling across a test concentration range of 5–100 μM (Supplementary Fig. 1), indicating good cell permeability, and virtually all of these protein labeling events were dependent on UV light exposure (Supplementary Fig. 1). We next turned our attention to mapping enantioprobe-protein interactions in human cells by quantitative mass spectrometry (MS)-based proteomics.
Global maps of enantioprobe-protein interactions in human cells
We evaluated two complementary cell types for enantioprobe interactions by quantitative MS-based proteomics: 1) primary human peripheral blood mononuclear cells (PBMCs); and 2) HEK239T cells. The selection of these cell types afforded an opportunity to directly compare enantioprobe profiles to those generated with the original set of FFF probes (generated in HEK293T cells)13 and extend our understanding of fragment ligandability to primary human immune cells. Both cell types were treated with equal concentrations of (R)- or (S)-compounds from each enantioprobe pair (200 μM, 30 min) and then exposed to UV light to induce photocrosslinking of enantioprobe-bound proteins, lysed, and enantioprobe-labeled proteins conjugated to an azide-biotin tag by CuAAC chemistry, enriched by streptavidin, and analyzed by MS-based proteomics, where stereoselective interactions were quantified by isotopic labeling using either reductive dimethylation (ReDiMe) with heavy or light formaldehyde (PBMCs)21,22 or SILAC (stable isotope labeling by amino acids in cell culture; HEK293T cells)23 (Fig. 2a). For PBMCs, these experiments were performed in replicate in both isotopic directions (heavy vs light and light vs heavy) to furnish four independent experiments for each enantioprobe pair, and, for HEK293T cells, a subset of the enantioprobe pairs was examined. We operationally defined a protein as engaging in a “stereoselective” interaction if it showed preferential enrichment by an average value of > 2.5-fold by one member of an enantioprobe pair in either PBMCs or HEK293T cells. For the SILAC studies in HEK293T cells, we also performed control experiments where the heavy- and light-labeled cells were treated with the same enantioprobe to ensure that, under these conditions, enantioprobe-enriched proteins showed a ratio of ~1.0. (Supplementary Fig. 2, Supplementary Dataset 1, and Supplementary Dataset 2).
Figure 2. MS-based profiling of enantioprobe-protein interactions in human cells.

a, Schematic workflow for identifying stereoselective enantioprobe-protein interactions in human cells. b, Heatmap showing relative protein enrichment ratios for pairwise comparisons of (R) and (S) enantioprobes (200 μM each) in both isotopic directions in human peripheral blood mononuclear cells (PBMCs). White signals in the heatmap either correspond to proteins with ratio values of ~ 1 or proteins that were not enriched and quantified with the indicated enantioprobe pair. (R)* and (S)*- represent (R,R) and (S,S)- for enantioprobe 7. c, Representative scatter plot showing protein enrichment ratios for (R)-1 versus (S)-1 in PBMCs. Proteins enriched > 2.5-fold by one enantiomer over the other are considered stereoselective targets. Red and blue protein targets show stereoselective interactions with (R)-1 and (S)-1, respectively. Data reflect an average of at least two independently performed experiments for each isotopic direction that provided similar results (see Supplementary Dataset 2). d, Similar stereoselective interactions are observed in different cell types. Plot depicts Log2 values of protein enrichment ratios for (R)-1/(S)-1 in HEK293T cells (x-axis) versus PBMCs (y-axis). The graph contains 812 total quantified proteins. r values are Pearson correlation coefficients. Data reflect an average of two independently performed experiments that provided similar results (see Supplementary Dataset 2). e, Number of stereoselective protein interactions found for each enantioprobe pair in PBMCs. f, Number of proteins showing stereoselective interactions with the indicated number of enantioprobe pairs in PBMCs. g, Quantity of aggregate spectral counts for PYGB (left graph) and ABRAXAS2 (right graph) enriched by each enantioprobe in PBMCs.
In total, 176 proteins showed stereoselective interactions with one or more enantioprobe pairs, which included 119 proteins identified in PBMCs (Fig. 2b, Supplementary Dataset 1, and Supplementary Dataset 2) and 108 proteins identified in HEK293T cells (Supplementary Fig. 2, Supplementary Dataset 1, and Supplementary Dataset 2). We observed similar numbers of stereoselective protein interactions for each member of an enantioprobe pair (Fig. 2c and Supplementary Fig. 2), indicating equivalent potential for the R or S-enantiomer to preferentially enrich proteins. Proteins identified in both PBMCs and HEK293T cells generally showed consistent profiles across the cell types, that is, stereoselective interactions identified in PBMCs were also observed in HEK293Ts and vice versa (Fig. 2d and Supplementary Fig. 2). The enantioprobe pairs displayed considerable differences in their total number of stereoselective interactions with the human proteome (Fig. 2e), and, notably, these profiles were unrelated to the extent of overall protein labeling displayed by the probes (Fig. 1b). This result suggests that stereoselective interactions are based on factors beyond the general protein binding potential of a given fragment structure.
The majority of proteins showing stereoselective interactions (>80%) did so with only one of the enantioprobe pairs (Fig. 2f and Supplementary Fig. 2). Embedded within this specificity were multiple profiles, including proteins that were enriched by several enantioprobe pairs, but stereoselectively by one pair, as well as proteins that showed strong enrichment predominantly with a single enantioprobe across the entire probe set (Fig. 2g). Proteins showing stereoselective interactions with enantioprobes spanned diverse functional and structural classes (Fig. 3a, Supplementary Table 1, and Supplementary Dataset 1). Perhaps unsurprisingly, many of the enantioprobe targets were enzymes, including kinases, methyltransferases, and various metabolic enzymes (Fig. 3a, Supplementary Table 1, and Supplementary Dataset 1), likely reflecting the high potential for these proteins to specifically bind small molecules. We also, however, observed stereoselective interactions for various scaffolding/adaptor proteins and transcriptional regulators – classes that have been historically considered challenging to target with small molecules (Fig. 3a, Supplementary Table 1, and Supplementary Dataset 1). To the extent that the magnitude of stereoselective enrichment is predictive of a robust small-molecule interaction, we further noted that high stereoselective enrichment (ratio values > 4.0 in pairwise comparisons of R vs S enantioprobes) was observed for several scaffolding/adaptor proteins and transcriptional regulators (Supplementary Table 1 and Supplementary Dataset 1). Enantioprobe targets that were observed in PBMCs, but not HEK293T cells, tended to correspond to immune-enriched proteins (e.g., IRAK324, PARP10 25) (Supplementary Dataset 1). We finally observed limited overlap of enantioprobe targets with proteins that demonstrated ligandability in previous chemical proteomic studies using cysteine-26,27 or lysine-reactive28 electrophilic fragments (Fig. 3b), indicating that non-covalent and covalent fragments generally interact with distinct sets of proteins in human cells. Moreover, while many of the enantioprobe targets were also enriched by members of the original FFF probe set13, these previous profiles often did not provide useful SAR information, either reflecting substantial enrichment by all of the FFF probes or mirroring the respective global protein interaction footprints of these probes (Supplementary Fig. 2).
Figure 3. Characterization of stereoselective protein targets of enantioprobes.

a, Functional classes of stereoselective protein targets of enantioprobes in PBMCs and HEK293T cells. b, Fraction of stereoselective protein targets of enantioprobes showing evidence of ligandability with cysteine and/or lysine-reactive fragments, as determined previously26–28. The left graph includes all stereoselective targets; the right graph shows only those stereoselective targets with quantified cysteines and/or lysines in previous studies26–28. c-f, Top: Confirmation of stereoselective enantioprobe-protein interactions with recombinantly expressed proteins. RPS6KA3 (c), PACSIN2 (d), SMYD3 (e), and UNC119B (f) were recombinantly expressed with FLAG epitope tags by transient transfection in HEK293T cells, and transfected cells were then treated with the indicated concentrations of enantioprobes, photocrosslinked, lysed, and proteomes conjugated to an azide-rhodamine tag by CuAAC chemistry and analyze by SDS-PAGE and in-gel fluorescence scanning. Gel images reflect representative results from two independently experiments. Bottom left: Extracted MS1 chromatograms of representative tryptic peptides for endogenous forms of the protein targets in HEK293T cells or PBMCs treated with indicated enantioprobes (200 μM). Bottom right: quantification of protein labeling by the indicated enantioprobes derived from gel-based profiles show in Top section. Data reflect two independently performed experiments. Confirmation of additional stereoselective interactions shown in Supplementary Fig 3.
Enantioprobes engage functionally relevant sites on proteins
We next sought to confirm stereoselective interactions for representative proteins targeted by diverse enantioprobes and originating from different functional classes, including enzymes (kinase, RPS6KA3; methyltransferase, SMYD3; and a metabolic enzyme; DCTPP1), a lipid-binding protein (UNC119B), a transporter (TSPO), a membrane-binding/adaptor protein (PACSIN2), a transcriptional regulator (HDGF), and an uncharacterized protein (TTC38). Each protein was recombinantly expressed with a FLAG epitope tag in HEK293T cells by transient transfection, and 48 h later, cells were treated with the indicated enantioprobe pair (5–80 μM, unless otherwise indicated) followed by photocrosslinking with UV light, CuAAC coupling to an azide-rhodamine tag, and visualization of protein labeling by SDS-PAGE and in-gel fluorescence scanning. All experiments also included mock-transfected cells as a control. Each recombinantly expressed protein displayed stereoselective interactions with the enantioprobes that mirrored the preferential labeling of the endogenous forms of these proteins in PBMCs and or HEK293T cells (Fig. 3c–f and Supplementary Fig. 3). Most of these stereoselective interactions could be detected with 5–10 μM of the preferred enantioprobe and were preserved across the entire enantioprobe concentration range (Fig. 3c–f and Supplementary Fig. 3).
Some of the protein targets have known ligands, which afforded an opportunity to test whether the enantioprobes and ligands share a common binding site on these proteins. Four representative proteins were selected for analysis – 1) the lysine methyltransferase SMYD3, a target of (R)-1 and (R)-5 that binds both cofactors (SAM, SAH) and the synthetic inhibitor EPZ03168629,30; 2) the lipid-binding protein UNC119B, a target of (R)-1 and (R)-5 that binds the natural product squarunkin A31; 3) the sterol transporter TSPO, a target of (R,R)-7 that binds the synthetic ligand PK 1119532,33, which is used to image brain injury and inflammation34,35; and 4) the uncharacterized protein TTC38, a target of (S)-4 that has been found to bind the histone deacetylase (HDAC) inhibitor panobinostat36. In all four cases, we found that enantioprobe interactions with both the endogenous and recombinantly expressed protein targets were competitively blocked by increasing concentrations of ligand, as measured by MS-based (endogenous protein) and gel-based (recombinant protein) methods (Fig. 4a, b and Supplementary Fig. 4). We further assessed the relative magnitude of target enrichment and competition measured with pure enantioprobes vs racemic mixtures of these probes. As expected, racemic mixtures of enantioprobes show substantially reduced enrichment of stereoselective protein targets compared to the preferred enantioprobes (Supplementary Fig. 4). More interestingly, however, these experiments also uncovered qualitative differences in how individual protein targets interact with enantioprobes. We found, for instance, that the competitive inhibitor EPZ031686 blocked the labeling of SMYD3 by the preferred enantioprobe (R)-1, but not the non-preferred enantioprobe (S)-1 (Supplementary Fig. 4). In contrast, squarunkin A blocked the interaction of UNC119B with both the preferred and non-preferred enantioprobes ((R)-1 and (S)-1, respectively; Supplementary Fig. 4). These data indicate that, for some protein targets (e.g., UNC119B), the magnitude of observed stereoselectivity reflects a difference in specific interactions with both enantioprobes, while, for other proteins (e.g., SMYD3), the degree of stereoselectivity may be even greater than that experimentally measured, being instead suppressed by low-level nonspecific interactions with the non-preferred enantioprobe.
Figure 4. Stereoselective interactions occur at functional and druggable sites on protein targets of enantioprobes.

a-b, Left: Structure of competitor ligands (a) EPZ031686; (b) Squarunkin A. Middle: Waterfall plots of competitive blockade of enantioprobe (200 μM) interactions with endogenous protein targets for corresponding ligands (20 μM) in HEK293T cells. Data reflect average values from two independently performed experiments that provided similar results (see Supplementary Dataset 2). Right: Gel-based profiles of competitive blockade of enantioprobe interactions with recombinantly expressed protein targets for corresponding ligands in transfected HEK293T cells. Gel images reflect representative results from two independently performed experiments. c, Structure of SMYD3 in complex with EPZ031686 (shown as stick model; PDB 5CCM) highlighting (R)-1-modified tryptic peptide (aa 255–265, light red; predicted probe-modified residues D255-Y257, dark red). d, Predicted binding modes of enantioprobe (R)-1 (top, green sticks) or (S)-1 (bottom, cyan sticks) to SMYD3, as determined by docking simulations, superimposed on the co-crystallized EPZ030456 inhibitor-SMYD3 complex (brown sticks). Predicted hydrogen bonds between (R)-1 or (S)-1 and T184 of SMYD3 are depicted as red dashed lines. e, Structure of UNC119A in complex with a myristoylated peptide from NPHP3 (yellow; PDB 5L7K) highlighting (R)-1-modified tryptic peptide (aa 227–235, light red, predicted probe-modified residues S227-Y230, dark red).
For SMYD3, only EPZ031686, but not SAM or SAH, blocked (R)-1 interactions (Fig. 4a). Structural studies have shown that EPZ031686 binds in the lysine substrate binding pocket of SMYD3 and noncompetitively with SAM29. These data suggested that (R)-1 may also bind to the lysine substrate pocket, which we confirmed by mapping the site of (R)-1 photolabeling on SMYD3 by quantitative MS. Using previously described protocols13,37,38, we identified a single tryptic peptide in SMYD3 that was photolabeled by (R)-1 – R.DQYCFECDCFR.C (amino acids (aa) 255–265) – with the predicted site(s) of photoreactivity being residues D255-Y257. These residues are located within 3.6 angstroms of an EPZ031686 analog in the SMYD3 co-crystal structure (Fig. 4c), and Y257 specifically has been found to interact with the methylated lysine in substrate co-crystal structures39. Quantitative MS-based proteomics further demonstrated that photolabeling of the aa 255–265 peptide by (R)-1 was blocked by co-incubation with EPZ031686 and was not observed with the inactive enantioprobe (S)-1 (Fig. 4c). To better understand the molecular basis for the stereoselective interaction of (R)-1 with SMYD3, we performed conventional docking simulations, which revealed that, when binding freely to SMYD3, (R)-1 can adapt two major energetically equivalent poses that would engage the SMYD3 pocket in different ways – one placing the diazirine 4.5 Å from the amide of Q256, and the other positioning the diazirine 3.1 Å from the Oη of Y257 (Supplementary Fig. 4). The latter binding mode, in addition to placing the diazirine closer the mapped region of (R)-1 labeling of SMYD3, also matched more closely the molecular envelope of other co-crystallized SMYD3 inhibitors (e.g., EPZ030456, PDB 5CCM; an oxindole screening hit, PDB 5CCL), with the (R)-1 amide overlapping with the inhibitor amide and hydrogen-bonding with T184, and the (R)-1 aromatic ring overlapping with the azabicyclic ring of EPZ031686 to engage a hydrophobic region (Fig. 4d, top).
The docking results also helped to explain the stereoselectivity of the (R)-1-SMYD3 interaction, as, while (S)-1 was able to reproduce a similar binding mode, the inverted chiral center reduced the quality of the docking match by placing the hydrophobic phenyl ring outside the molecular envelope of the inhibitor EPZ030456 and toward hydrophilic side chains T184 and E192 (Fig. 4d, bottom). Finally, to further support these conventional docking results, we also performed covalent docking studies, where we simulated the conformational rearrangements occurring when the (R)-1 diazirine reacts with the Y257 side chain of SMYD3. These covalent docking experiments predicted that minimal molecular rearrangements would be required to accommodate a reaction between (R)-1 and Y257 Oη of SMYD3 (Supplementary Fig. 4). These molecular modeling findings, taken together, suggest that the stereoselective interaction between (R)-1 and SMYD3 reflects a preferred binding mode for this chemical probe over the enantiomer (S)-1.
We also mapped the primary sites of enantioprobe labeling for UNC119B (Fig. 4e) and TSPO (Supplementary Fig. 4) and confirmed the stereoselectivity of these labeling events and their blockade by treatment with competitive ligands (squarunkin A and PK 11195, respectively). For UNC119B, (R)-1 modified the tryptic peptide containing residues 227–236 (R.SDSFYFVDNK.L) with predicted sites of labeling spanning S227-Y231 (Fig. 4e). These residues represent a highly conserved stretch of amino acids in UNC119 proteins that are within 2.5 angstroms of a fatty acylated peptide in a co-crystal structure with the related protein UNC119A40, and S229 is predicted to hydrogen bond with squarunkin A in a docking model of the natural product bound to UNC119A31 (Fig. 4e). For TSPO, (R,R)-7 modified the N-terminal peptide (aa 2–24, M.APPWVPAMGFTLAPSLGCFVGSR.F) with the principle site of labeling being C19 (Supplementary Fig. 4). In the solution structure of mouse TSPO, the corresponding residue (G19) is 2.8 angstroms away from the ligand PK 1119532.
Taken together, our follow-up studies on representative targets indicate that enantioprobes engage functionally relevant and druggable sites on diverse classes of proteins. We also noted that our chemical proteomic studies with enantioprobes identified additional, unanticipated targets for some of the tested small-molecule competitors. For instance, EPZ031686 blocked (R)-1 interactions with the solute carrier SLC35F2 and the peptidase PRCP (Fig. 4a), while PK 11195 decreased (R,R)-7 interactions with the lipid-binding protein ABHD5 (Supplementary Fig. 4). While ABHD5 also showed independent evidence of stereoselective interactions with enantioprobes (Supplementary Dataset 1), SLC35F2 and PRCP did not (Supplementary Dataset 1), suggesting that these latter proteins may specifically bind enantioprobes, but without stereochemical preference. Motivated to explore this general concept further, as well as to increase the throughput and information content of our chemical proteomic experiments, we set out to create a multiplexed platform for the streamlined analysis of enantioprobe-protein interactions in human cells.
Multiplexed analysis of enantioprobe-protein interactions in cells
While we were generally satisfied with the sensitivity and robustness of our chemical proteomic experiments using SILAC or ReDiMe as quantitative MS-based measurement protocols of enantioprobe-protein interactions, we also recognized that the pairwise nature of these comparisons had drawbacks. Prominently, the limited throughput prevented a deeper exploration of SAR both within and across enantioprobe pairs. For instance, a protein that interacts specifically, but without stereo-preference with both enantioprobes in a pairwise comparison is difficult to distinguish from a non-specific interaction, as both outcomes furnish an enrichment ratio of ~1.0. And, relatedly, the stochastic nature of protein identification events in untargeted MS-based proteomic experiments hindered confident assignment of proteins that selectively interacted with one or a subset of enantioprobes across different experiments. Finally, the throughput of pairwise comparisons also becomes restrictive when attempting to compare the protein interaction profiles of several enantioprobes under various conditions (e.g., different cell types, probe concentrations, etc.).
We considered that many of the aforementioned challenges could be addressed by analyzing enantioprobes with a multiplexed approach for quantitative MS-based proteomics that uses isobaric tandem mass tags (TMT)41–43. In this workflow, up to 10 separate populations of cells are each treated with an enantioprobe (200 μM, 30 min), photocrosslinked, lysed, conjugated to biotin azide via CuAAC, enriched and trypsinized as described above. Tryptic peptides stemming from each treatment group are then labeled with a TMT tag of equivalent parent mass, but differentiable by MS3-derived fragmentation products, combined, and analyzed in a single MS experiment (Fig. 5a).43 Applying 10-plex TMT, we compared the protein interaction profiles of four enantioprobe pairs (Supplementary Dataset 3), alongside a previously described methyl control probe13 (in duplicate), in human PBMCs and HEK293T cells (Fig. 5b and Supplementary Fig. 5). We required that at least three unique peptides were quantified for each protein to interpret stereoselective interactions with enantioprobes, and stereoselective interactions were defined as those displaying > 2.5-fold differential enrichment between (R) and (S) members of at least one enantioprobe pair, along with > 5-fold enrichment over the methyl control probe.
Figure 5. Multiplexed MS-based quantification for expedited discovery of stereoselective protein-enantioprobe interactions.

a, Schematic of TMT-based workflow for mapping enantioprobe-protein interactions in a multi (10)-plex format. b, Heatmap depicting TMT quantification of stereoselective protein targets in PBMCs. Relative enrichment ratios are calculated as a percent of maximum signal per protein. c, Similar profiles are found for stereoselective protein targets of enantioprobes in pairwise (ReDiMe) versus multiplexed (TMT) experiments (top panels). Multiplexed experiments also enable the identification of proteins that interact with enantioprobes in a chemotype-selective manner (bottom panels). White signals in the heatmap either correspond to proteins with ratio values of ~ 1 or proteins that were not enriched and quantified with the indicated enantioprobe pair. d, Representative scatter plots showing the correlation between pairwise (x-axis) and multiplexed (y-axis) experiments performed with enantioprobes (R/S)-1 and (R/S)-2. Left graph contains 1095 total quantified proteins; right graph contains 1005 total quantified proteins. r values are Pearson correlation coefficients. Data reflect an average of two independently performed experiments that provided similar results (see Supplementary Dataset 2 and Supplementary Dataset 3). e, Concentration-dependent profiles for representative stereoselective enantioprobe-protein interactions as determined by multiplexed experiments of PBMCs treated with 0, 5, 20, 50, 100 and 200 μM of the indicated enantioprobe pair. Data reflect two independently performed experiments.
We observed a robust overall correlation between the enantioprobe profiles quantified by multiplexed (TMT-based) versus pairwise (ReDiMe/SILAC) comparative proteomic experiments (Fig. 5c, d and Supplementary Fig. 5), and the vast majority (> 85%) of enantioprobe targets identified in pairwise comparisons showed consistent stereoselective interactions in multiplexed experiments (Supplementary Datasets 1–3). Another 115 stereoselective interactions were mapped by multiplexing, and these newly discovered events mostly corresponded to proteins that were not quantified in pairwise experiments performed with the relevant enantioprobe pair(s). In addition to recapitulating and extending the stereoselective enantioprobe-protein interactions discovered in pairwise experiments, the multiplexed method also illuminated proteins that showed enrichment by one or more enantioprobe pairs, but without stereopreference. Examples included CYP27A1 and TLR8, which interacted preferentially with the (R)/(S)-2 and (R)/(S)-3 probe pairs, respectively, over the other enantioprobes (Fig. 5c, lower panels). We interpret these enrichment profiles to also reflect specific probe-protein interactions, where the SAR across the enantioprobe set is driven by chemotype rather than stereotype.
We reasoned that the greater sample capacity afforded by multiplexing could also provide an efficient means of assessing the relative potency of enantioprobe-protein interactions by comparing protein enrichment profiles across several probe concentrations. We performed a proof-of-principle experiment with a representative enantioprobe pair – (S)-3 and (R)-3 – tested at five different concentrations (5, 20, 50, 100 and 200 μM) in human PBMCs (Supplementary Datasets 1 and 3). The concentration-dependent profiles revealed that previously mapped enantioprobe targets maintained stereoselective interactions across the entire probe concentration range (e.g., see IRAK3 and PARP10 in Fig. 5e; also see Supplementary Fig. 6). Some interactions further showed evidence of saturated enrichment at lower concentrations of the preferred enantioprobe (e.g., TTC38, Fig. 5e), possibly reflecting higher affinity binding events. Consistent with this hypothesis, we found that the non-crosslinkable analogue of (S)-4, but not (R)-4, competitively blocked enantioprobe binding to TTC38 (Supplementary Fig. 6), reflecting the stereoselective enrichment profile for this protein with the (S)-4 and (R)-4 enantioprobe pair (Supplementary Fig. 3 and Supplementary Dataset 1). Saturated enrichment was also observed for certain proteins that did not show stereopreference between (S)-3 and (R)-3 (e.g., SLC25A20, Supplementary Fig. 6). Finally, a third type of profile was observed, albeit rarely, where a protein displayed saturated enrichment with both (S)-3 and (R)-3, but the absolute signal plateaued at different values between the enantioprobes (e.g., PTGR2, Supplementary Fig. 6). This outcome might reflect cases where equivalent binding is observed for both enantioprobes, but one of the probes generates a greater amount of photoadduct with the protein target (see Discussion below).
Taken together, these data indicate that the mapping of fragment-protein interactions in cells can be efficiently performed proteome-wide using multiplexing MS-based methods to expedite the discovery of ligandable proteins with a rich body of integrated information on SAR and potency.
Discussion
Efforts to expand the proportion of the human proteome that can be targeted by chemical probes would benefit from methods capable of evaluating small molecule-protein interactions on a global scale in native biological systems. We previously described a chemical proteomic strategy to perform fragment-based ligand discovery experiments in human cells13. The initial set of fully functionalized (clickable, photoreactive) fragments (FFFs) uncovered many new small molecule-protein interactions, some of which were advanced to selective and cell-active chemical probes. Nonetheless, we also found that individual FFFs showed substantially different overall protein interaction profiles in human cells, which complicated the assignment of small molecule-protein interactions displaying authentic SARs (vs nonspecific binding to the FFFs). The enantioprobes described herein offer a general solution to this challenge by specifying ligandable proteins as those showing differential interactions with physicochemically matched compounds differing only in absolute stereochemistry. We identified many such stereoselective interactions across diverse functional and structural protein classes and verified several using recombinantly expressed proteins. Importantly, in each case where a protein target had an established ligand, we found that this ligand blocked enantioprobe binding. These results indicate that stereoselective interactions of enantioprobes often occur at functional sites on proteins. If this principle generalizes across the broader set of enantioprobe targets identified herein, it highlights the potential of fragment-based screening in cells to serve as a foundation for the pursuit of chemical probes that perturb the function of a wide range of proteins.
There are some important considerations when considering the broader implementation of enantioprobes for mapping protein ligandability in biological systems. First, we emphasize that a substantial fraction of enantioprobe targets showed stereoselective interactions with only a single (R)/(S) probe pair. We interpret this result to indicate that, with our modest set of eight enantioprobe pairs, we are vastly under-sampling the proportion of human proteins that have the capacity to show stereoselective interactions with small-molecule fragments. Future attention should thus be given to expanding the size and structural diversity of the enantioprobe library, as well as to applying these probes in more diverse cell types to survey a broader fraction of the human proteome. Such experiments may also uncover context-dependent enantioprobe-protein interactions, if, for instance, a protein’s participation in a dynamic complex or its reversible post-translational modification state affects enantioprobe interactions. There may be further technical reasons why some stereoselective enantioprobe-protein interactions are overlooked in our chemical proteomic experiments. For instance, interactions that are too low in binding affinity may not provide for sufficient enrichment of proteins for detection by MS-based proteomics, while other stereoselective interactions may be masked by multiple binding sites of a given enantioprobe on the same protein. We also admit that, in most cases, we do not know with certainty whether stereoselective interactions between an enantioprobe and a protein reflect preferential binding versus photoreactivity (i.e., the extent of carbene adduct with a protein target following photoexcitation of the diazirine). In some cases, it is conceivable that both members of an enantioprobe pair bind equivalently to a protein target, but one probe produces a greater yield of photoadduct with the protein. While this SAR outcome would ultimately need to be clarified to guide efforts toward more advanced chemical probes that display higher affinity and selectivity for individual protein targets, we posit that stereoselective binding and stereoselective photoreactivity are equivalently useful parameters for identifying novel druggable sites in the proteome, as both would likely require specific interactions with a protein to discriminate between an enantioprobe pair. Finally, our data highlight the value of incorporating TMT-based multiplexing readouts into enantioprobe profiling experiments, which greatly expedited the discovery of stereoselective interactions without substantial losses in sensitivity or accuracy. Moreover, these multiplexing experiments provide additional SAR information by identifying proteins that interact in a chemoselective, rather than stereoselective, manner with the enantioprobe set.
Projecting forward, we envision several exciting pursuits with enantioprobes that should address fundamental questions about the ligandability of the human proteome. For instance, will the stereoselective interactions displayed by fragment enantioprobes be retained as these ligands are elaborated into more advanced chemical probes, or, alternatively, will the preferential interaction with a single stereocenter dissipate in importance as additional recognition elements are built into the probes? Toward this end, we note that the enantioprobes offer a convenient target engagement assay for assessing competitive binding of elaborated analogues in cells, and that several of the enantiopure fragment recognition groups deployed herein are poised for direct modification using synthetic methodologies such as C-H bond activation chemistry44–46. Second, would more structurally complex enantioprobes identify ligandable proteins that, for instance, do not display sufficient binding affinity to simple fragment probes? Finally, what fraction of stereoselective interactions observed proteome-wide occur at functional sites on proteins? Here, we admit that a complete answer is not likely to be soon forthcoming, as we are dependent on both mapping the sites of enantioprobe binding, a still technically challenging task, and the availability of protein structures to predict functional pockets. Consider TTC38, for instance, a poorly characterized protein that has been previously identified as an off-target of the HDAC inhibitor panobinostat36 and found herein to display stereoselective interaction with the (S)-4 probe that was blocked by panobinostat. We would presume that the site of binding of panobinostat and probe (S)-4 is relevant to TTC38 function, but absent a structure or, for that matter, even a biochemical activity for the protein, this conclusion is premature. Of course, ligands that are found to bind silent sites on proteins can still be converted into “functional” chemical probes that promote protein degradation using PROTAC-like technologies47–49.
In summary, our findings demonstrate that enantioprobes offer a highly efficient way to discover small molecule-protein interactions in human cells. Differentiating proteins based on stereoselective interactions with otherwise physicochemically equivalent fragment probes offers instant evidence of authentic ligandability. These stereoselective interactions can then form the basis for pursuit of more advanced chemical probes targeting a diverse range of proteins for basic and translational research purposes.
Methods
A detailed Methods section is provided in the Supplementary Information.
Data availability
All data associated with this study are available in the published article and its supplementary information. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE50 partner repository with the dataset identifier PXD015104. All other raw data are available upon request.
Supplementary Material
Acknowledgements
We thank P.S. Baran for helpful discussions, PX. Shen for assistance in compounds characterization, and R. Park for technical advices on proteomics data analysis. The authors acknowledge the Scripps Normal Blood Donor Services, NMR, MS and Automated Synthesis core facilities. This work was supported by the NIH (CA231991, CA211526, and DK114785).
Footnotes
Competing Interests
The authors declare competing financial interests. B.F.C. is a founder and advisor to Vividion Therapeutics, a biotechnology company interested in using chemical proteomic methods to develop small-molecule drugs to treat human disease. C.G.P. serves as a consultant for Vividion Therapeutics.
References
- 1.Knight ZA & Shokat KM Chemical genetics: where genetics and pharmacology meet. Cell 128, 425–430, doi: 10.1016/j.cell.2007.01.021 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Belshaw PJ, Ho SN, Crabtree GR & Schreiber SL Controlling protein association and subcellular localization with a synthetic ligand that induces heterodimerization of proteins. P Natl Acad Sci USA 93, 4604–4607, doi:DOI 10.1073/pnas.93.10.4604 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lai AC & Crews CM Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov 16, 101–114, doi: 10.1038/nrd.2016.211 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu G et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309, doi: 10.1126/science.1244917 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schreiber S A chemical biology view of bioactive small molecules and a binder-based approach to connect biology to precision medicines. bioRxiv (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oprea TI et al. Unexplored therapeutic opportunities in the human genome. Nature Reviews Drug Discovery 17, 317–332, doi: 10.1038/nrd.2018.14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schenone M, Dancik V, Wagner BK & Clemons PA Target identification and mechanism of action in chemical biology and drug discovery. Nat Chem Biol 9, 232–240, doi: 10.1038/Nchembio.1199 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swinney DC & Anthony J How were new medicines discovered? Nature Reviews Drug Discovery 10, 507–519, doi: 10.1038/nrd3480 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Hajduk PJ & Greer J A decade of fragment-based drug design: strategic advances and lessons learned. Nat Rev Drug Discov 6, 211–219, doi: 10.1038/nrd2220 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Bembenek SD, Tounge BA & Reynolds CH Ligand efficiency and fragment-based drug discovery. Drug Discov Today 14, 278–283, doi: 10.1016/j.drudis.2008.11.007 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Scott DE, Coyne AG, Hudson SA & Abell C Fragment-based approaches in drug discovery and chemical biology. Biochemistry 51, 4990–5003, doi: 10.1021/bi3005126 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Erlanson DA, Fesik SW, Hubbard RE, Jahnke W & Jhoti H Twenty years on: the impact of fragments on drug discovery. Nature Reviews Drug Discovery 15, 605–619, doi: 10.1038/nrd.2016.109 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Parker CG et al. Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 168, 527–541 e529, doi: 10.1016/j.cell.2016.12.029 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Welsch ME, Snyder SA & Stockwell BR Privileged scaffolds for library design and drug discovery. Curr Opin Chem Biol 14, 347–361, doi: 10.1016/j.cbpa.2010.02.018 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agranat I, Caner H & Caldwell J Putting chirality to work: the strategy of chiral switches. Nat Rev Drug Discov 1, 753–768, doi: 10.1038/nrd915 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Nguyen LA, He H & Pham-Huy C Chiral drugs: an overview. Int J Biomed Sci 2, 85–100 (2006). [PMC free article] [PubMed] [Google Scholar]
- 17.Zanos P et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533, 481–486, doi: 10.1038/nature17998 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sui JJ, Zhang JH, Ching CB & Chen WN Expanding proteomics into the analysis of chiral drugs. Mol Biosyst 5, 603–608, doi: 10.1039/b903858b (2009). [DOI] [PubMed] [Google Scholar]
- 19.Sanna MG et al. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol 2, 434–441, doi: 10.1038/nchembio804 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Rostovtsev VV, Green LG, Fokin VV & Sharpless KB A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed Engl 41, 2596–2599, doi: (2002). [DOI] [PubMed] [Google Scholar]
- 21.Boersema PJ, Raijmakers R, Lemeer S, Mohammed S & Heck AJ Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc 4, 484–494, doi: 10.1038/nprot.2009.21 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Hsu JL, Huang SY, Chow NH & Chen SH Stable-isotope dimethyl labeling for quantitative proteomics. Anal Chem 75, 6843–6852, doi: 10.1021/ac0348625 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Ong SE et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1, 376–386, doi: 10.1074/mcp.M200025-MCP200 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Hubbard LL & Moore BB IRAK-M regulation and function in host defense and immune homeostasis. Infect Dis Rep 2, doi: 10.4081/idr.2010.e9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verheugd P et al. Regulation of NF-κB signalling by the mono-ADP-ribosyltransferase ARTD10. Nature Communications 4, 1683, doi: 10.1038/ncomms2672 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Backus KM et al. Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574, doi: 10.1038/nature18002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bar-Peled L et al. Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 171, 696–709, doi: 10.1016/j.cell.2017.08.051 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hacker SM et al. Global profiling of lysine reactivity and ligandability in the human proteome. Nat Chem 9, 1181–1190, doi: 10.1038/Nchem.2826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitchell LH et al. Novel Oxindole Sulfonamides and Sulfamides: EPZ031686, the First Orally Bioavailable Small Molecule SMYD3 Inhibitor. ACS Med Chem Lett 7, 134–138, doi: 10.1021/acsmedchemlett.5b00272 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Aller GS et al. Structure-Based Design of a Novel SMYD3 Inhibitor that Bridges the SAM-and MEKK2-Binding Pockets. Structure 24, 774–781, doi: 10.1016/j.str.2016.03.010 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Mejuch T et al. Small-Molecule Inhibition of the UNC119-Cargo Interaction. Angew Chem Int Ed Engl 56, 6181–6186, doi: 10.1002/anie.201701905 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Jaremko L, Jaremko M, Giller K, Becker S & Zweckstetter M Structure of the mitochondrial translocator protein in complex with a diagnostic ligand. Science 343, 1363–1366, doi: 10.1126/science.1248725 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le Fur G et al. Peripheral benzodiazepine binding sites: effect of PK 11195, 1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide. I. In vitro studies. Life Sci 32, 1839–1847 (1983). [DOI] [PubMed] [Google Scholar]
- 34.Owen DRJ & Matthews PM Imaging Brain Microglial Activation Using Positron Emission Tomography and Translocator Protein-Specific Radioligands. Int Rev Neurobiol 101, 19–39, doi: 10.1016/B978-0-12-387718-5.00002-X (2011). [DOI] [PubMed] [Google Scholar]
- 35.Rupprecht R et al. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nature Reviews Drug Discovery 9, 971–988, doi: 10.1038/nrd3295 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Becher I et al. Thermal profiling reveals phenylalanine hydroxylase as an off-target of panobinostat. Nat Chem Biol 12, 908–+, doi: 10.1038/Nchembio.2185 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Niphakis MJ et al. A Global Map of Lipid-Binding Proteins and Their Ligandability in Cells. Cell 161, 1668–1680, doi: 10.1016/j.cell.2015.05.045 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weerapana E, Speers AE & Cravatt BF Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP) - a general method for mapping sites of probe modification in proteomes. Nature Protocols 2, 1414–1425, doi: 10.1038/nprot.2007.194 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Fu WQ et al. Structural Basis for Substrate Preference of SMYD3, a SET Domain-containing Protein Lysine Methyltransferase. J Biol Chem 291, 9173–9180, doi: 10.1074/jbc.M115.709832 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaiswal M et al. Novel Biochemical and Structural Insights into the Interaction of Myristoylated Cargo with Unc119 Protein and Their Release by Arl2/3. J Biol Chem 291, 20766–20778, doi: 10.1074/jbc.M116.741827 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thompson A et al. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 75, 1895–1904, doi: 10.1021/ac0262560 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Dayon L et al. Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal Chem 80, 2921–2931, doi: 10.1021/ac702422x (2008). [DOI] [PubMed] [Google Scholar]
- 43.McAlister GC et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 86, 7150–7158, doi: 10.1021/ac502040v (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ming S et al. Modular, Stereocontrolled Cβ–H/Cα–C Activation of Alkyl Carboxylic Acids. (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tran AT & Yu JQ Practical Alkoxythiocarbonyl Auxiliaries for Iridium(I)-Catalyzed C-H Alkylation of Azacycles. Angew Chem Int Ed Engl 56, 10530–10534, doi: 10.1002/anie.201704755 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jain P, Verma P, Xia GQ & Yu JQ Enantioselective amine alpha-functionalization via palladium-catalysed C-H arylation of thioamides. Nat Chem 9, 140–144, doi: 10.1038/Nchem.2619 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deshaies RJ Protein degradation: Prime time for PROTACs (vol 11, pg 634, 2015). Nat Chem Biol 11, 887–887, doi: 10.1038/nchembio1115-887b (2015). [DOI] [PubMed] [Google Scholar]
- 48.Neklesa TK, Winkler JD & Crews CM Targeted protein degradation by PROTACs. Pharmacol Therapeut 174, 138–144, doi: 10.1016/j.pharmthera.2017.02.027 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Winter GE et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381, doi: 10.1126/science.aab1433 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perez-Riverol Y et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Research 47, D442–D450, doi: 10.1093/nar/gky1106 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are available in the published article and its supplementary information. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE50 partner repository with the dataset identifier PXD015104. All other raw data are available upon request.